
Stereoselective Synthesis of 1-Substituted Homotropanones, including Natural Alkaloid (−)-Adaline

1
Departamento de Química Orgánica, Facultad de Ciencias, Universidad de Alicante, Apdo. 99, 03080 Alicante, Spain
2
Instituto de Síntesis Orgánica (ISO), Universidad de Alicante, Apdo. 99, 03080 Alicante, Spain
3
Centro de Innovación en Química Avanzada (ORFEO-CINQA), Universidad de Alicante, Apdo. 99, 03080 Alicante, Spain
*
Author to whom correspondence should be addressed.
Molecules 2023, 28(5), 2414; https://doi.org/10.3390/molecules28052414
Submission received: 16 February 2023
/
Revised: 1 March 2023
/
Accepted: 3 March 2023
/
Published: 6 March 2023
(This article belongs to the Special Issue A Journey of Organic Chemistry in Spain)
Abstract
:The stereocontrolled synthesis of 1-substituted homotropanones, using chiral N-tert-butanesulfinyl imines as reaction intermediates, is described. The reaction of organolithium and Grignard reagents with hydroxy Weinreb amides, chemoselective N-tert-butanesulfinyl aldimine formation from keto aldehydes, decarboxylative Mannich reaction with β-keto acids of these aldimines, and organocatalyzed L-proline intramolecular Mannich cyclization are key steps of this methodology. The utility of the method was demonstrated with a synthesis of the natural product (−)-adaline, and its enantiomer, (+)-adaline.
1. Introduction
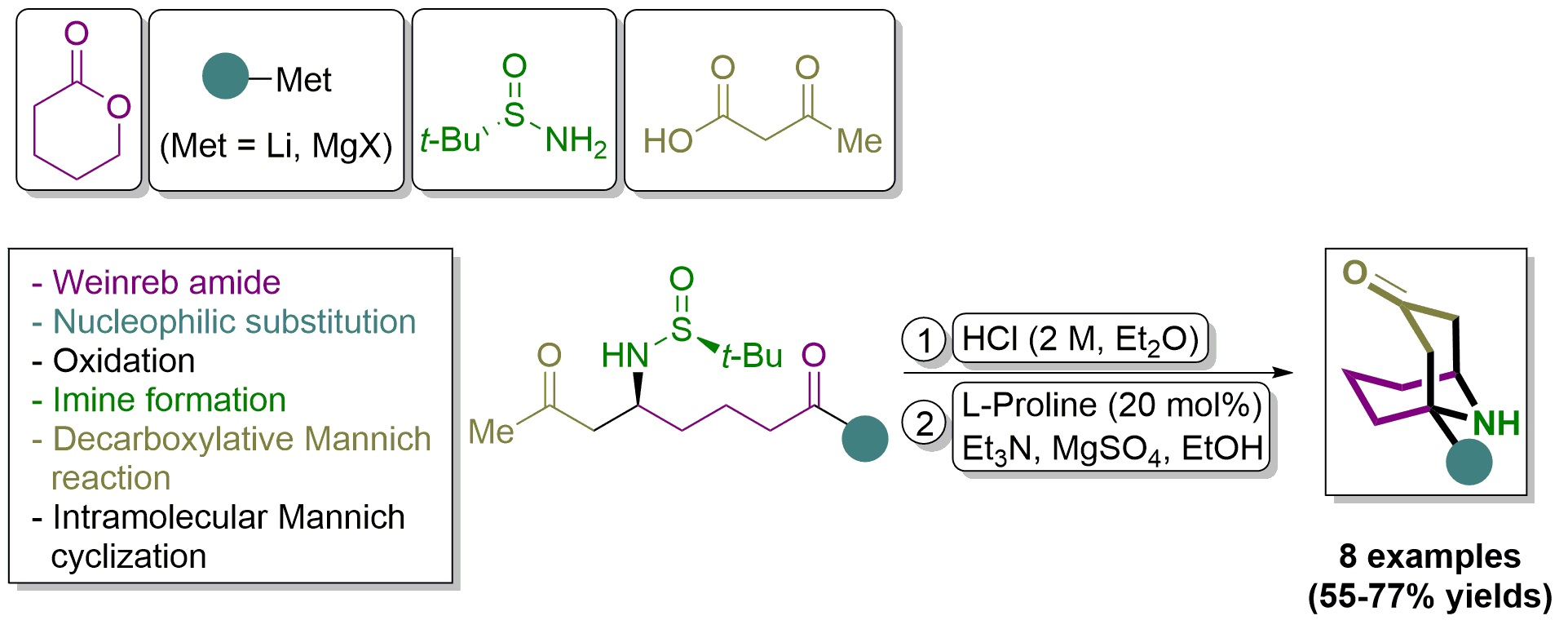
Bicyclic alkaloids are significantly represented in nature and display a wide range of biological activities. Among them, compounds with a bridgehead nitrogen atom 1-azabicyclo[4.3.0]nonanes and 1-azabicyclo[4.4.0]decanes, the so-called indolizidines and quinolizidines [1], respectively, are more abundant than other azabicyclic systems with the nitrogen atom bonded to both bridgehead carbon atoms, such as 8-azabicyclo[3.2.1]octanes and 9-azabicyclo[3.3.1]nonanes, which are the basic skeletons of tropane [2] and homotropane alkaloids [3]. Representative tropane alkaloids include hyoscyamine, scopolamine, cocaine and calystegine A3, among others (Figure 1). Despite these alkaloids having a similar structure, they differ in biological activity, and have been used in the treatment of different diseases. For instance, hyoscyamine is an antimuscarinic, scopolamine is anticholinergic, calystegine A3 is employed to combat type 2 diabetes, and cocaine, an addictive stimulant drug, binds to the dopamine transporter, blocking the removal of dopamine from the synapse, producing an amplified signal to the receiving neurons. Homotropane alkaloids are less abundant than their homologous tropane derivatives, (+)-euphococcinine and (−)-adaline being the most significant (Figure 1). These compounds were found in the defensive fluid deployed by ladybirds to repel predators when disturbed. Precisely, (+)-euphococcinine was first isolated from the Australian coastal plant Euphorbia atoto [4] and was also found in the defense secretion of ladybirds Cryptolaemus montrouzieri [5] and Epilachna varivestis [6].
On the other hand, (−)-adaline was isolated from ladybird Adalia bipunctata [7] and Cryptolaemus moutrouzieri secretions [5]. It was recently found that (−)-adaline can target nicotinic acetylcholine receptors (nAChRs), acting as receptor antagonists, and also as an open channel blocker of nAChRs [8].
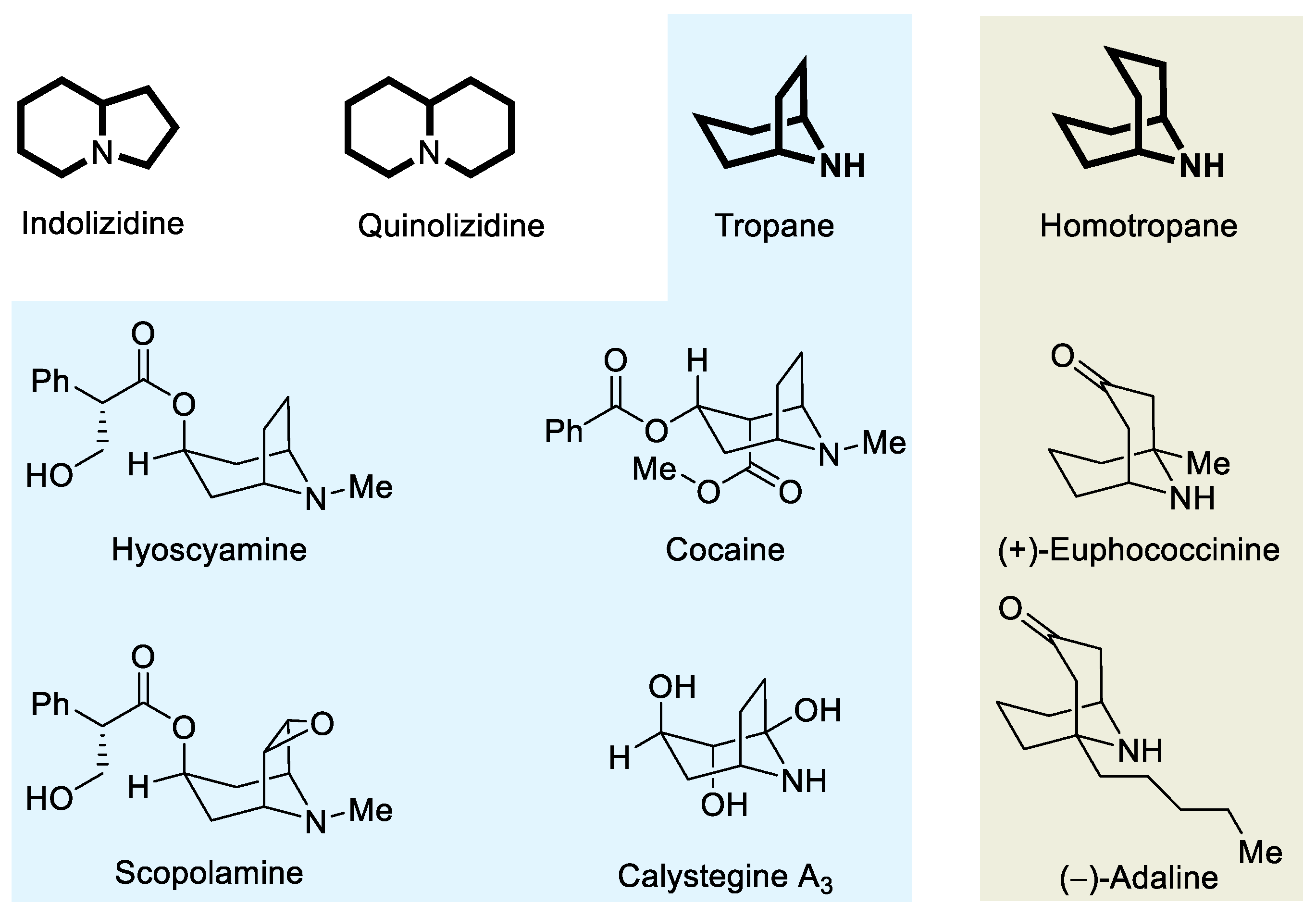
A wide variety of synthetic strategies have been employed in the synthesis of adaline and euphococcinine [3,9]. Recent approaches to these bicyclic systems are summarized in Scheme 1. It should be mentioned that the Yu synthesis, starting from 3,4-dihydro-2-ethoxy-2H-pyran in a 6-step sequence, involves an intramolecular allylation of a cyclic imine as a key step [10]. The stereoselective synthesis of these alkaloids was also accomplished by Spino et al., taking advantage of p-menthane-3-carboxaldehyde as a chiral auxiliary. In the last step of this synthesis, the treatment with copper chloride of an isocyanate intermediate promotes an intramolecular conjugate addition of the nitrogen atom to the cyclic enone system [11]. A double cyclization through a four-step cascade reaction comprising N-desulfinylation, ketal hydrolysis, intramolecular imine formation and Mannich cyclization took place in the synthesis of (+)-adaline and (+)-euphococcinine reported by Davis and Edupuganti, upon treatment of a N-sulfinylamino ketone ketal with ammonium acetate and acetic acid [12]. More recently, Prasad and Khandare reported a four-step synthesis of these bicyclic homotropinone alkaloids, involving as key steps a diastereoselective addition of a Wittig phosphorene to a chiral N-tert-butanesulfinyl ketimine, a ring-closing metathesis, and a final intramolecular Michael reaction [13] (Scheme 1).
The diastereoselective additions of different types of nucleophiles to chiral imines are recurrently used to access compounds with a nitrogen atom bonded to a stereogenic center. Of special relevance in this respect are N-tert-sulfinyl imines [14,15], in which the sulfinyl group plays a fundamental role in controlling the stereochemical pathway of these additions, which are highly stereospecific since the configuration of the sulfur atom determines the configuration of the newly formed stereocenter. Our group has studied in deep both the indium-mediated allylation [16] and the decarboxylative Mannich coupling of β-keto acids with N-tert-sulfinyl imines [17], and the application of the resulting homoallylamine derivatives [18,19,20,21] and β-amino ketones [22,23], respectively, to the synthesis of natural products.
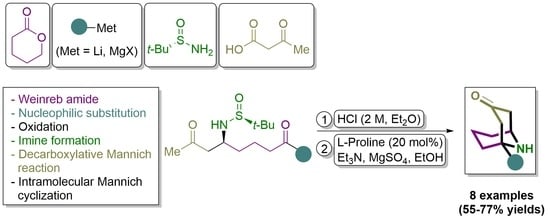
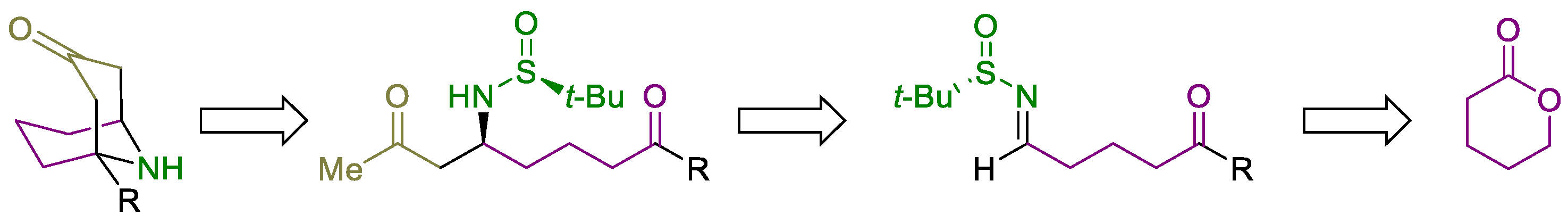
Continuing our interest in the use of N-tert-butanesulfinyl imines as electrophiles [24,25,26,27,28], we decided to explore new synthetic pathways to access 1-substituted homotropanones in an enantioenriched form involving these chiral imines. Sequential decarboxylative Mannich reaction of a chiral N-tert-butanesulfinyl keto aldimine and a β-keto acid, followed by an organocatalyzed intramolecular Mannich reaction, upon desulfinylation, are key steps in the synthetic strategy we have envisioned for the synthesis of these bicyclic homotropanones, which is closely related to the strategy reported by Davis [12] (Scheme 2).
2. Results and Discussion
2.1. Synthesis of N-tert-Butanesulfinyl Keto Aldimines 5
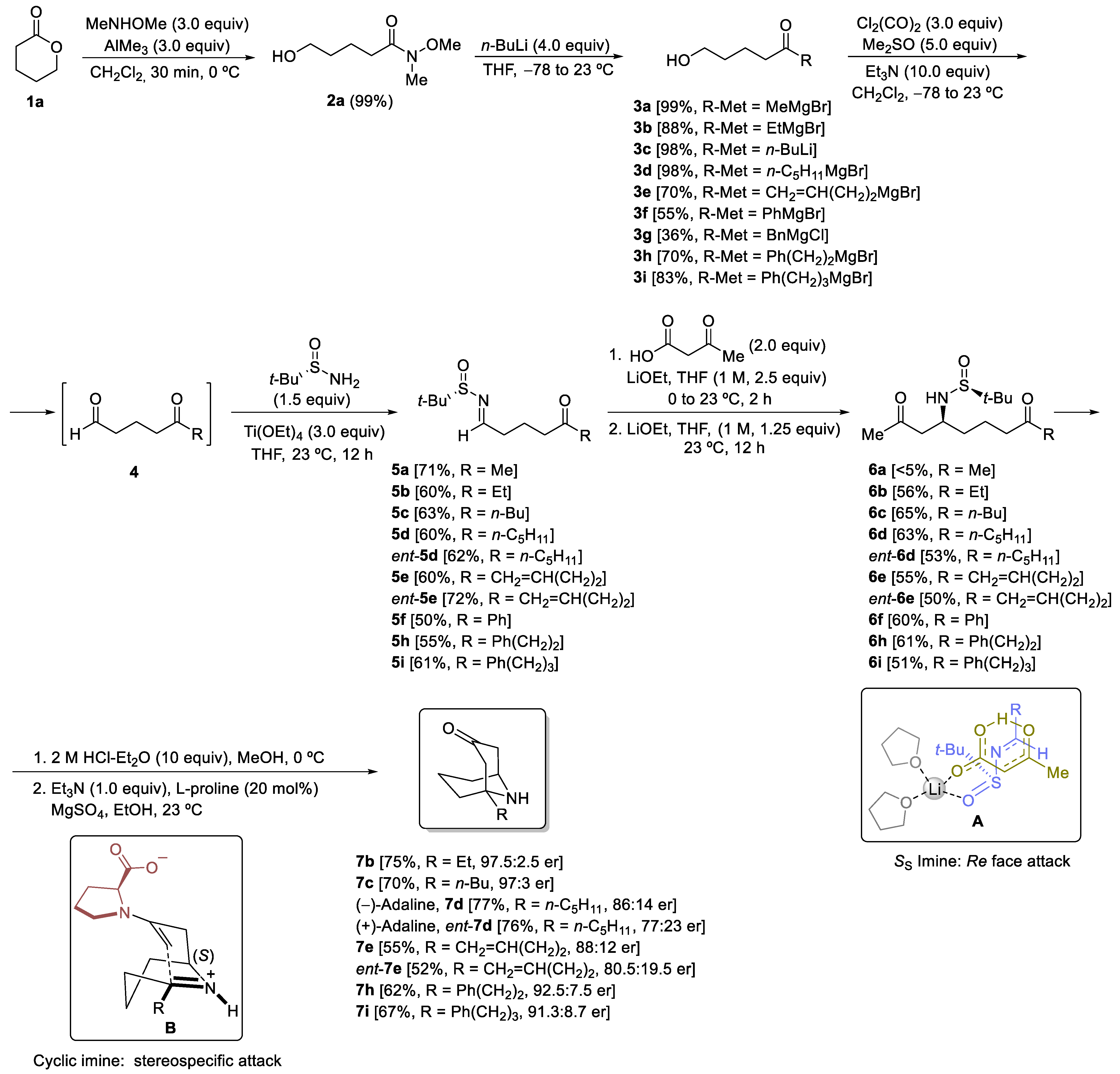
The synthesis of the target 1-substituted homotropanones started with the nucleophilic opening of δ-valerolactone (1a) upon treatment with N,O-dimethylhydroxylamine in the presence of trimethylaluminum in dichloromethane [29]. The corresponding Weinreb δ-hydroxyamide 2a was formed in an almost quantitative yield. Further reaction with an excess of the corresponding organolithium or Grignard reagent led to the formation of δ-hydroxyketones 3 in excellent yields, in general. Structural diversity is introduced at this step of the here reported methodology. Only ketones 3f and 3g were obtained in moderate and low yields, respectively (55 and 36%). Phenylmagnesium bromide and benzylmagnesium chloride were the organometallic reagents involved in the synthesis of 3f and 3g, respectively (Scheme 3).
Chiral N-tert-butanesulfinyl imino ketones 5 were key synthetic intermediates in the strategy depicted in Scheme 2. They were prepared in a two-step process from hydroxyketones 3. First, Swern oxidation of alcohols 3 produced keto aldehydes 4. These reaction products were not isolated nor characterized. After the work-up, and removal of the solvents, the crude reaction mixtures were directly treated with (S)-tert-butanesulfinamide in the presence of titanium tetraethoxide, in THF, at room temperature. At this point, it merits mentioning that despite having two carbonyl groups (aldehyde and ketone) working under these reaction conditions, aldimines 5 were exclusively formed. It is known that more demanding reaction conditions are necessary for the synthesis of ketimines, which can be prepared with the same reagents and solvents, but working at higher temperatures, at least reflux of THF (Scheme 3) [30]. Chiral sulfinyl imino ketones ent-5d and ent-5e were also prepared to work with (R)-tert-butanesulfinamide in the aldimine formation step. The expected reaction products were isolated after two synthetic operations in fairly good yields, ranging from 50 to 71% (Scheme 3).
2.2. Synthesis of N-tert-Butanesulfinyl Amino Diketones 6
The next step in the proposed synthetic pathway to the target 9-azabicyclo[3.3.1]nonan-3-ones comprises a base-promoted decarboxylative-Mannich coupling of 3-oxobutanoic acid and N-tert-butanesulfinyl keto aldimines 5. This methodology was developed in our research group [17], and we found that the resulting amino diketone derivatives were formed in a highly diastereoselective manner (>95:5 dr). We observed that yields were significantly improved when after 2 h of reaction, 1.5 equivalents of the base were added to the reaction mixture. Concerning the stereochemical pathway, the nucleophilic addition of the enolate took place to the Re face of the imine with SS configuration. This result was rationalized considering an eight-membered cyclic transition state A, which was also supported in DFT calculations (Scheme 3). In these transformations, yields ranged from 51 to 65%. Unfortunately, and after many attempts with varying reaction conditions, methyl ketone derivative 5a led to the expected amino diketone 6a in an extremely low yield. Only traces of this compound were detected from the 1H-NMR spectrum of the reaction crude (Scheme 3).
2.3. Synthesis of 1-Substituted 9-Azabicyclo[3.3.1]nonan-3-Ones 7
An intramolecular Mannich cyclization was the last step of the synthesis of compounds 7 from amino diketone derivatives 6. We found that the treatment of compounds 6 first with a solution of hydrogen chloride in diethyl ether, in methanol as solvent, led to the free amine hydrochloride, which in a second step participated in a L-proline organocatalyzed intramolecular Mannich reaction, involving a six-membered cyclic imine initially formed, and the enolizable methyl group of the methyl ketone of the L-proline enamine. The intramolecular Mannich cyclization took place in a stereospecific manner, involving an iminium enamine carboxylate as a reaction intermediate, through, probably, a working model of type B depicted on Scheme 3. The last step of this synthesis is rather similar to the one reported by Davis and Edupuganti in their synthesis of (+)-adaline and (+)-euphococcinine [12]. They found ammonium acetate and acetic acid at 75 °C the optimal conditions to transform a N-sulfinylamino ketone ketal into the corresponding 9-azabicyclo[3.3.1]nonan-3-one. When we applied these conditions to amino diketone derivative 6d, natural product (−)-adaline 7d was isolated in 63%, a lower yield than the one we found under the L-proline organocatalyzed cyclization (77%, Scheme 3). The overall yield in this double cyclization transformation ranged from 55 to 77%. Importantly, we could prepare both enantiomers of these homotropanones by choosing the appropriate tert-butanesulfinamide enantiomer in the aldimine step formation, as it was exemplified in the synthesis of alkaloid (−)-adaline (7d) and its enantiomer (+)-adaline (ent-7d).
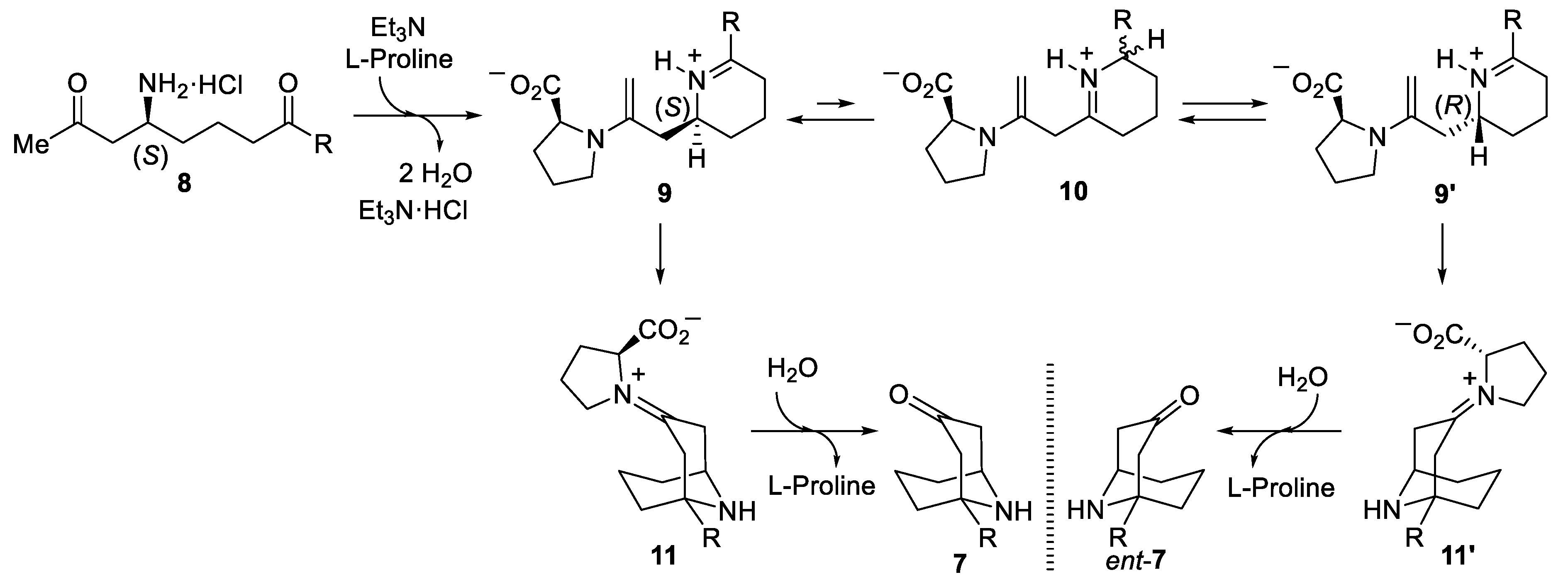
The optical purity of compounds 7 was determined by GC using a column packet with a chiral stationary phase. Relatively high enantiomeric ratios were observed for compounds 7b, 7c, 7h and 7i. However, poorer enantioselectivities were found for pentyl and but-3-enyl substituted homotropanones 7d and 7e (Scheme 3). Importantly, the starting amino diketone derivatives 6 were almost enantiopure (>95:5 dr), and it seems that the stereochemical integrity of these compounds was eroded in the double cyclization process. For that reason, and in order to rationalize the stereochemical outcome, we proposed the mechanism depicted in Scheme 4. First, removing the tert-butanesulfinyl group was carried out under acidic conditions to produce the ammonium chloride 8, which upon treatment with triethylamine and L-proline led to the iminium-enamine intermediate 9. This compound is formed by a double condensation involving on one side the amine functionality and the carbonyl group, leading to the six-membered cyclic imine, and on the other side, the remaining carbonyl and the L-proline, forming the corresponding enamine. At this stage, isomerization could take place in some extension in the high polar reaction medium to give iminium 10, losing the stereochemical integrity of the stereogenic center as a consequence. Intermediate 10 could be in equilibrium with iminium 9 and 9′, which can participate in a second cyclization by nucleophilic attack of the enamine to the electrophilic carbon of the iminium through a transition state of type B (Scheme 3), leading to the formation of homotropanones 7 and ent-7, respectively (Scheme 4).
2.4. Synthesis of 1-Substituted 8-Azabicyclo[3.2.1]octane 16
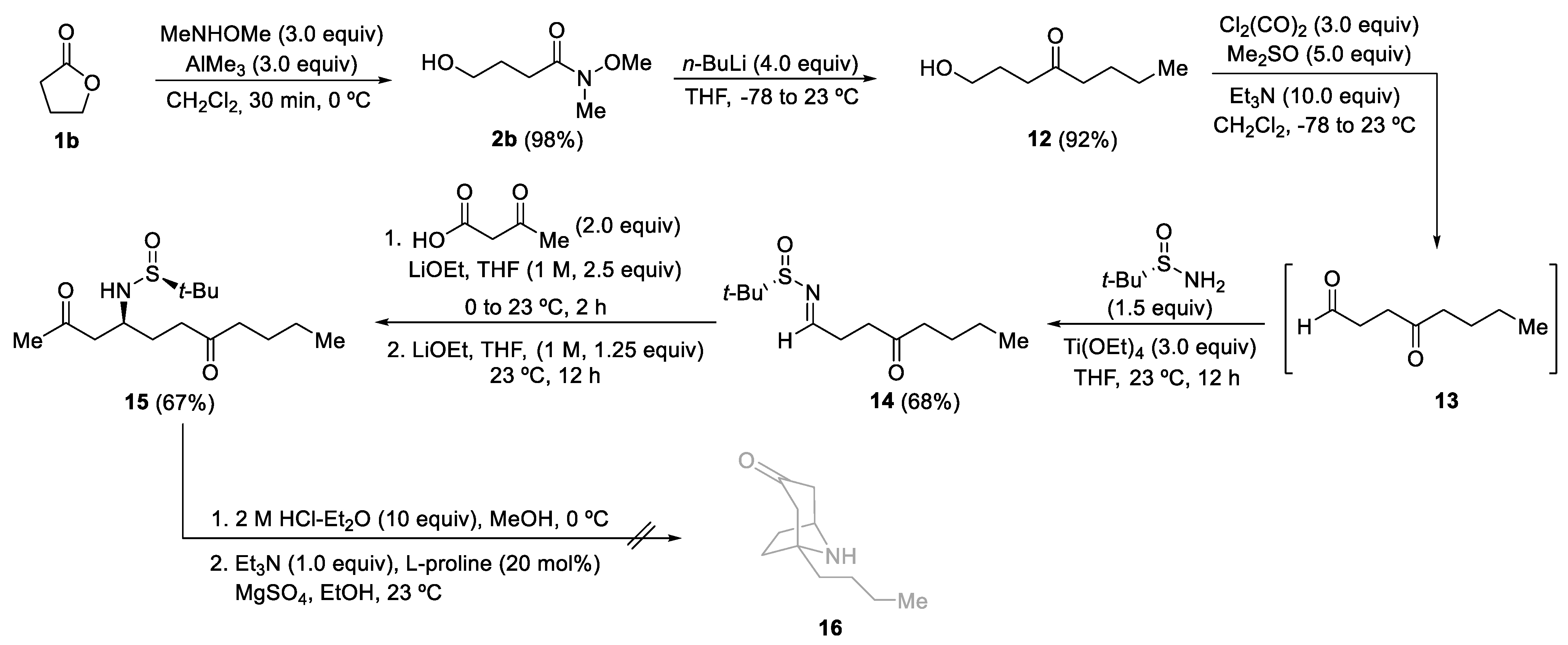
Taking advantage of the methodology developed for the synthesis of homotropanones 7, we envisioned a synthesis under the same reaction conditions of more abundant natural tropanones, starting in this case from γ-butyrolactone 1b. The formation of Weinreb amide 2b from 1b and hydroxy ketone 12 from 2b, using n-buthyllithium, took place in high yields. Swern oxidation of alcohol 12, followed by the formation of the imine 14, proceeded in 68% overall yield. The next step was the base-promoted decarboxylative-Mannich coupling of 3-oxobutanoic acid and N-tert-butanesulfinyl keto aldimine 14. The expected aminodiketone derivative 15 was isolated in 67% yield. However, the L-proline organocatalyzed intramolecular Mannich cyclization, under the reaction conditions that worked well for compounds 6, did not provide tropanone derivative 16 (Scheme 5). Complex reaction mixtures were always obtained even varying (solvent, temperature, stoichiometry) these optimal reaction conditions.
3. Materials and Methods
3.1. General Information
Reagents and solvents were of reagent grade and purchased from commercial suppliers [Sigma-Aldridh (Saint Louis, MO, USA), Fisher Scientific (Kandel, Germany)] and used as received. (S)- and (R)-tert-butanesulfinamide were a gift from Medalchemy S.L. (Alicante, Spain) (>99% ee by chiral HPLC on a Chiracel AS column, 90:10 n-hexane/i-PrOH, 1.2 mL/min, λ = 222 nm).
Optical rotations were measured using a Jasco P-1030 polarimeter (Jasco, Tokyo, Japan) with a thermally jacketed 5 cm cell at approximately 23 °C, and concentrations (c) are given in g/100 mL. Low-resolution mass spectra (LRMS) were obtained in the electron impact mode (EI) with an Agilent MS5973N spectrometer with a SIS (Scientific Instrument Services) direct insertion probe (73DIP-1) at 70 eV and with an Agilent GC/MS5973N spectrometer in the electron impact mode (EI) at 70 eV. In both cases, fragment ions are given in m/z with relative intensities (%) in parentheses. High-resolution mass spectra (HRMS) were also carried out in the electron impact mode (EI) at 70 eV on an Agilent 7200 spectrometer equipped with a time of flight (TOF) analyzer, and the samples were introduced through a direct insertion probe or through an Agilent GC7890B (Agilent, Santa Clara, CA, USA). NMR spectra were recorded at 300 or 400 MHz for 1H NMR and at 75 or 100 MHz for 13C NMR with a Bruker AV300 Oxford or a Bruker AV400 spectrometers (Bruker, Karlsruhe, Germany), respectively, using CDCl3 as solvent, and TMS as internal standard (0.00 ppm). The data are reported as: (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet or unresolved, br s = broad signal, coupling constant(s) in Hz, integration). 13C NMR spectra were recorded with 1H-decoupling at 100 MHz and referenced to CDCl3 at 77.16 ppm. The DEPT-135 experiments were performed to assign CH, CH2 and CH3.
TLCs were performed on prefabricated Merck (Sigma-Aldrich, Saint Louis, MO, USA) aluminum plates with silica gel 60 coated with fluorescent indicator F254 and were visualized with phosphomolybdic acid (PMA) stain. The Rf values were calculated under these conditions. Flash chromatography was carried out on handpacked columns of silica gel 60 (230–400 mesh). GC-MS analysis were carried out in an Agilent 6890N spectrometer with FID detector, helium gas transportation (2 mL/min), injection pressure: 12 psi, temperature in detection with an injection blocks: 270 °C, column type HP-1 (12 m long, 0.22 mm internal diameter, 0.25 μm thickness methylsilicone rubber and OV-101 stationary phase). Temperature programs: (A) initial temperature (60 °C) for 3 min, heating 15 °C/min until final temperature (270 °C), final temperature (270 °C) for 10 min or (B) initial temperature (80 °C) for 5 min, heating 15 °C/min until final temperature (270 °C), final temperature (270 °C) for 10 min.
3.2. Preparation and Characterization of Compounds
3.2.1. Synthesis of Weinreb’s Amides 2
General Procedure. A solution of N,O-dimethylhydroxylamine hydrochloride (5.30 g, 54.0 mmol) in dry dichloromethane (30.0 mL) was stirred at −78 °C for 15 min. Then, a 2.0 M solution of AlMe3 in toluene (27.0 mL, 54.0 mmol) was slowly added over 1 h. The reaction mixture was stirred for 12 h and allowed to warm up until it reached 23 °C. After that, the reaction flask was cooled down at 0 °C, and the corresponding lactone 1 (18.0 mmol) was added. The resulting reaction mixture was stirred for 30 min at the same temperature, and for 2 h at 23 °C. After that, it was hydrolyzed with an aqueous solution (10.0 mL) of Rochelle’s salt (7.0 g). The resulting suspension was filtered through celite and repeatedly washed with dichloromethane (3 × 10 mL). Then, the solvent was removed under vacuum (15 Torr, <30 °C), giving rise to the expected compounds 2, which was used in the next reaction step without the need for further purification.
5-Hydroxy-N-methoxy-N-methylpentanamide (2a) [31]. Following the general procedure, compound 2a was obtained from δ-valerolactone (1a) as a colorless oil (2.870 g, 17.82 mmol, 99%): C7H15NO3; Rf 0.33 (hexane/EtOAc 1:4); 1H NMR (400 MHz, CDCl3) δ 3.68 (s, 3H), 3.63 (t, J = 6.3 Hz, 2H), 3.17 (s, 3H), 2.46 (t, J = 7.2 Hz, 2H), 1.78–1.66 (m, 2H), 1.66–1.52 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 174.7 (C), 62.0 (CH2), 61.2 (CH3), 32.3 (CH2), 32.2 (CH3), 31.3 (CH2), 20.4 (CH2); LRMS (EI) m/z 162 (M+ + 1, 3%), 144 (3), 101 (91), 83 (51), 61 (99), 57 (27), 55 (100).
4-Hydroxy-N-methoxy-N-methylbutanamide (2b) [32]. Following the general procedure, compound 2b was obtained from γ-butyrolactone (1b) as a colorless oil (2.593 g, 17.64 mmol, 98%): C6H13NO3; Rf 0.36 (hexane/EtOAc 1:4); 1H NMR (400 MHz, CDCl3) δ 3.68 (s, 3H), 3.67 (t, J = 6.3 Hz, 3H), 3.17 (s, 3H), 2.57 (t, J = 7.0 Hz, 2H), 1.94–1.82 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 174.7 (C), 62.1 (CH2), 61.2 (CH3), 32.12 (CH3), 28.9 (CH2), 27.3 (CH2); LRMS (EI) m/z 148 (M+ + 1, 1%), 87 (41), 69 (38), 61 (100).
3.2.2. Synthesis of Hydroxy Ketones 3 and 12
General Procedure. A solution of the corresponding Weireb’s amide 2 (6.0 mmol) in dry THF (50.0 mL) was stirred at −78 °C for 15 min. Then, a solution of the corresponding organolithium or Grignard reagent (24.0 mmol) was slowly added. The reaction mixture was stirred and allowed to warm up until it reached 23 °C for 15 h. After that, it was hydrolyzed with water (10 mL), extracted with ethyl acetate (3 × 20 mL), dried over magnesium sulfate, and the solvent was evaporated under a vacuum (15 Torr). The residue was pure enough to be used in the following step for compounds 3a–d and 3i. The residue for compounds 3e–h and 12 was purified by column chromatography (silica gel, hexane/EtOAc) to yield pure products 3 and 12.
6-Hydroxyhexan-2-one (3a). Following the general procedure, compound 3a was obtained from amide 2a as a colorless oil (689.1 mg, 5.94 mmol, 99%): C6H12O2; Rf 0.35 (hexane/EtOAc 1:1); 1H NMR (400 MHz, CDCl3) δ 3.62 (t, J = 6.3 Hz, 2H), 2.49 (t, J = 7.1 Hz, 2H), 2.15 (s, 3H), 1.74–1.61 (m, 3H), 1.61–1.49 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 209.4 (C), 62.1, 43.2 (CH2), 32.0 (CH2), 29.8 (CH3), 19.8 (CH2); LRMS (EI) m/z 116 (M+, 1%), 98 (99), 83 (54), 58 (47), 56 (41), 55 (100); HRMS (EI-TOF) Calcd for C6H12O2 [M+]: 116.0884, found: 116.0889.
7-Hydroxyheptan-3-one (3b). Following the general procedure, compound 3b was obtained from amide 2a as a colorless oil (686.4 mg, 5.28 mmol, 88%): C7H14O2; Rf 0.40 (hexane/EtOAc 1:1); 1H NMR (400 MHz, CDCl3) δ 3.61 (t, J = 6.3 Hz, 2H), 2.48–2.40 (m, 4H), 1.70–1.50 (m, 5H), 1.04 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 212.1 (C), 62.1 (CH2), 41.9 (CH2), 35.9 (CH2), 32.1 (CH2), 19.9 (CH2), 7.8 (CH3); LRMS (EI) m/z 112 (M+-H2O, 27%), 101 (22), 83 (40), 57 (100), 55 (70); HRMS (EI-TOF) Calcd for C7H14O2 [M+]: 130.0994, found: 130.0990.
1-Hydroxynonan-5-one (3c). Following the general procedure, compound 3c was obtained from amide 2a as a colorless oil (929.1 mg, 5.88 mmol, 98%): C9H18O2; Rf 0.42 (hexane/EtOAc 1:1); 1H NMR (400 MHz, CDCl3) δ 3.62 (t, J = 6.2 Hz, 2H), 2.51–2.36 (m, 4H), 1.72–1.62 (m, 3H), 1.62–1.47 (m, 4H), 1.36–1.21 (m, 2H), 0.90 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 211.8 (C), 62.2 (CH2), 42.6 (CH2), 42.3 (CH2), 32.1 (CH2), 26.0 (CH2), 22.35 (CH2), 19.7 (CH2), 13.87 (CH3); LRMS (EI) m/z 140 (M+-H2O, 18%), 116 (32), 111 (16), 101 (29), 98 (70), 85 (100), 83 (63), 57 (91), 55 (85); HRMS (EI-TOF) Calcd for C9H18O2 [M+]: 158.1307, found: 158.1295.
1-Hydroxydecan-5-one (3d). Following the general procedure, compound 3d was obtained from amide 2a as a colorless oil (1.011 g, 5.88 mmol, 98%): C10H20O2; Rf 0.48 (hexane/EtOAc 1:1); 1H NMR (400 MHz, CDCl3) δ 3.63 (t, J = 6.1 Hz, 2H), 2.61–2.22 (m, 4H), 1.83 (s, 1H), 1.71–1.51 (m, 6H), 1.34–1.21 (m, 4H), 0.89 (t, J = 6.9 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 211.7 (C), 62.3 (CH2), 42.8 (CH2), 42.3 (CH2), 32.15 (CH2), 31.4 (CH2), 23.6 (CH2), 22.5 (CH2), 19.7 (CH2), 13.9 (CH3); LRMS (EI) m/z 154 (M+-H2O, 15%), 111 (24), 99 (100), 98 (90), 83 (89), 71 (85), 55 (94); HRMS (EI-TOF) Calcd for C10H20O2 [M+]: 172.1463, found: 172.1446.
9-Hydroxynon-1-en-5-one (3e). Following the general procedure, compound 3e was obtained from amide 2a as a colorless oil (655.2 mg, 4.20 mmol, 70%): C9H16O2; Rf 0.41 (hexane/EtOAc 1:1); 1H NMR (400 MHz, CDCl3) δ 5.82–5.77 (m, 1H), 5.12–4.81 (m, 2H), 3.61 (t, J = 6.2 Hz, 2H), 2.67–2.39 (m, 4H), 2.39–2.23 (m, 2H), 2.04 (s, 1H), 1.77–1.58 (m, 2H), 1.62–1.45 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 210.6 (C), 137.2 (CH), 115.3 (CH2), 62.3 (CH2), 42.5 (CH2), 41.9 (CH2), 32.2 (CH2), 27.9 (CH2), 19.8 (CH2); LRMS (EI) m/z 138 (M+-H2O, 46%), 123 (18), 101 (23), 96 (27), 83 (52), 67 (18), 55 (100); HRMS (EI-TOF) Calcd for C9H14O [M+-H2O]: 138.1045, found: 138.1041.
5-Hydroxy-1-phenylpentan-1-one (3f). Following the general procedure, compound 3f was obtained from amide 2a as a colorless oil (587.4 mg, 3.30 mmol, 55%): C11H14O2; Rf 0.44 (hexane/EtOAc 1:1); 1H NMR (400 MHz, CDCl3) δ 8.06–7.91 (m, 2H), 7.67–7.50 (m, 1H), 7.52–7.41 (m, 2H), 3.79–3.63 (m, 2H), 3.05 (t, J = 7.1 Hz, 2H), 1.92–1.79 (m, 2H), 1.74 (s, 1H), 1.71–1.61 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 200.5 (C), 137.2 (C), 133.1 (CH), 128.7 (CH), 128.2 (CH), 62.55 (CH2), 38.25 (CH2), 32.4 (CH2), 20.40 (CH2); LRMS (EI) m/z 178 (M+, 1%), 119 (20), 101 (45), 91 (40), 77 (100), 65 (25), 55 (65); HRMS (EI-TOF) Calcd for C11H14O2 [M+]: 178.0994, found: 178.0992.
7-Hydroxy-1-phenylheptan-3-one (3h). Following the general procedure, compound 3h was obtained from amide 2a as a colorless oil (848.4 mg, 4.20 mmol, 70%): C13H18O2; Rf 0.45 (hexane/EtOAc 1:1); 1H NMR (400 MHz, CDCl3) δ 7.39–7.13 (m, 5H), 3.61 (t, J = 6.3 Hz, 2H), 2.92 (t, J = 7.6 Hz, 2H), 2.45 (t, J = 7.1 Hz, 2H), 1.71–1.61 (m, 3H), 1.61–1.49 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 210.3 (C), 141.1 (C), 128.5 (CH), 128.3 (CH), 126.1 (CH), 62.3 (CH2), 44.25 (CH2), 42.55 (CH2), 32.1 (CH2), 29.8 (CH2), 19.7 (CH2); LRMS (EI) m/z 206 (M+, 3%), 188 (54), 133 (37), 105 (75), 104 (28), 91 (100), 83 (19), 55 (22); HRMS (EI-TOF) Calcd for C13H18O2 [M+]: 206.1307, found: 206.1307.
8-Hydroxy-1-phenyloctan-4-one (3i). Following the general procedure, compound 3i was obtained from amide 2a as a colorless oil (1.095 g, 4.98 mmol, 83%): C14H20O2; Rf 0.50 (hexane/EtOAc 1:1); 1H NMR (400 MHz, CDCl3) δ 7.41–7.08 (m, 5H), 3.62 (t, J = 6.2 Hz, 2H), 2.69–2.56 (m, 2H), 2.49–2.36 (m, 3H), 2.02–1.84 (m, 4H), 1.79–1.45 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 211.2 (C), 141.6 (C), 128.5 (CH), 128.5 (CH), 128.4 (CH), 126.0 (CH), 62.2 (CH2), 42.35 (CH2), 41.9 (CH2), 35.1 (CH2), 32.1 (CH2), 25.2 (CH2), 19.7 (CH2); LRMS (EI) m/z 202 (M+-H2O, 22%), 116 (20), 104 (44), 98 (100), 91 (38), 83 (12), 55 (13); HRMS (EI-TOF) Calcd for C14H20O2 [M+]: 220.1463, found: 220.1463.
1-Hydroxyoctan-4-one (12). Following the general procedure, compound 12 was obtained from amide 2b as a colorless oil (795.0 mg, 5.52 mmol, 92%): C8H16O2; Rf 0.45 (hexane/EtOAc 1:1); 1H NMR (400 MHz, CDCl3) δ 3.64 (t, J = 6.1 Hz, 2H), 2.56 (t, J = 6.9 Hz, 2H), 2.43 (t, J = 7.5 Hz, 2H), 1.89–1.80 (m, 3H), 1.66–1.46 (m, 2H), 1.35–1.27 (m, 2H), 0.94–0.85 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 212.2 (C), 62.15 (CH2), 42.7 (CH2), 39.5 (CH2), 26.6 (CH2), 26.0 (CH2), 22.4 (CH2), 13.89 (CH3); LRMS (EI) m/z 144 (M+, 2%), 126 (13), 102 (45), 97 (29), 87 (99), 85 (85), 69 (46), 58 (93), 57 (100); HRMS (EI-TOF) Calcd for C8H16O2 [M+]: 144.1150, found: 144.1153.
3.2.3. Synthesis of N-tert-Butanesulfinyl Keto Aldimines 5 and 14
General Procedure. A solution of oxalyl chloride (1.143 g, 0.772 mL, 9.0 mmol) in dry dichloromethane (20.0 mL) was stirred at −78 °C for 15 min. Then, DMSO (1.171 g, 1.065 mL, 15.0 mmol) was added dropwise. The reaction mixture was stirred for 5 min at the same temperature, and after that, a solution of the corresponding hydroxy ketone 3 or 12 (3.0 mmol) in dichloromethane (10.0 mL) was added. The resulting mixture was stirred for 15 min, and after that, Et3N (3.218 g, 4.432 mL, 31.8 mmol) was slowly added over 10 min. The reaction mixture was allowed to warm up and stirred for 2 h. Then, dichloromethane (20.0 mL) and a NH4Cl saturated aqueous solution (20.0 mL) were sequentially added. The aqueous layer was extracted with dichloromethane (3 × 25 mL), and the combined organic phases were washed with brine, dried over magnesium sulfate, and the solvent was evaporated under vacuum (15 Torr). The resulting keto aldehydes 4 were not isolated nor characterized and were used in the next step, the formation of the sulfinyl imine. A solution of the corresponding crude reaction mixture 4, (S)- or (R)-tert-butanesulfinamide 1 (0.435 g, 3.6 mmol) and titanium tetraoxide (1.605 g, 1.507 mL, 7.2 mmol) in dry THF (5.0 mL) was stirred for 12 h at 23 °C. Then the resulting mixture was hydrolyzed with brine (2.0 mL), filtered through a celite pad, and washed with ethyl acetate (3 × 10 mL). The solvent was evaporated under vacuum (15 Torr), and the residue was purified by column chromatography (silica gel, hexane/EtOAc) to yield pure products 5 and 14.
(S)-N-(5-Oxohex-1-ylidene)-tert-butanesulfinamide (5a). Following the general procedure, compound 5a was obtained from hydroxy ketone 3a as a yellow oil (462.4 mg, 2.13 mmol, 71%): C10H19NO2S; Rf 0.33 (hexane/EtOAc 3:1); [α]20D +183.1 (c 1.03, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 8.07 (t, J = 4.3 Hz, 1H), 2.61–2.50 (m, 4H), 2.15 (s, 3H), 2.00–1.86 (m, 2H), 1.20 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 207.9 (C), 168.8 (CH), 56.7 (C), 42.6 (CH2), 35.3 (CH2), 30.1 (CH2), 22.5 (CH3), 19.4 (CH2); LRMS (EI) m/z 160 (M+-C4H9, 4%), 112 (25), 103 (39), 70 (18), 57 (100), 55 (13), 43 (80), 41 (15); HRMS (EI-TOF) Calcd for C6H10NO2S [M+-C4H9] 160.0432, found 160.0422.
(S)-N-(5-Oxohep-1-ylidene)-tert-butanesulfinamide (5b). Following the general procedure, compound 5b was obtained from hydroxy ketone 3b as a yellow oil (416.0 mg, 1.80 mmol, 60%): C11H21NO2S; Rf 0.35 (hexane/EtOAc 3:1); [α]20D +198.2 (c 0.84, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 8.07 (t, J = 4.3 Hz, 1H), 2.71–2.47 (m, 4H), 2.48–2.35 (m, 2H), 2.05–1.85 (m, 2H), 1.19 (s, 9H), 1.06 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 210.8 (C), 168.9 (CH), 56.7 (C), 41.3 (CH2), 36.1 (CH2), 35.4 (CH2), 22.4 (CH3), 19.45 (CH2), 7.1 (CH3); LRMS (EI) m/z 175 (M+-C4H8, 5%), 127 (25), 111 (23), 57 (100), 56 (10), 55 (11), 43 (13), 41 (22); HRMS (EI-TOF) Calcd for C7H13NO2S [M+-C4H8] 175.0667, found 175.0667.
(S)-N-(5-Oxonon-1-ylidene)-tert-butanesulfinamide (5c). Following the general procedure, compound 5c was obtained from hydroxy ketone 3c as a yellow oil (489.7 mg, 1.89 mmol, 63%): C13H25NO2S; Rf 0.40 (hexane/EtOAc 3:1); [α]20D +131.6 (c 1.50, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 8.07 (t, J = 4.3 Hz, 1H), 2.59–2.47 (m, 4H), 2.40 (t, J = 7.4 Hz, 2H), 2.00–1.85 (m, 2H), 1.60–1.49 (m, 2H), 1.37–1.25 (m, 2H), 1.20 (s, 9H), 0.91 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 210.5 (C), 168.9 (CH), 56.7 (C), 42.8 (CH2), 41.7 (CH2), 35.4 (CH2), 26.1 (CH2), 22.5 (CH3), 19.5 (CH2), 14.0 (CH3); LRMS (EI) m/z 203 (M+-C4H8, 13%), 155 (75), 139 (12), 113 (17), 103 (14), 98 (14), 85 (13), 70 (14), 57 (100), 56 (24), 55 (15), 43 (14), 41 (32); HRMS (EI-TOF) Calcd for C9H17NO2S [M+-C4H8] 203.0980, found 203.0987.
(S)-N-(5-Oxodec-ylidene)-tert-butanesulfinamide (5d). Following the general procedure, compound 5d was obtained from hydroxy ketone 3d as a yellow oil (491.6 mg, 1.80 mmol, 60%): C14H27NO2S; Rf 0.42 (hexane/EtOAc 3:1); [α]20D +153.6 (c 2.83, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 8.07 (t, J = 4.3 Hz, 1H), 2.62–2.45 (m, 4H), 2.39 (t, J = 7.4 Hz, 2H), 2.02–1.82 (m, 2H), 1.58 (s, 2H), 1.41–1.23 (m, 4H), 1.20 (s, 9H), 0.89 (t, J = 7.0 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 210.45 (C), 168.9 (CH), 56.7 (C), 43.0 (CH2), 41.7 (CH2), 35.4 (CH2), 31.5 (CH2), 23.7 (CH2), 22.55 (CH3), 19.4 (CH2), 14.0 (CH3); LRMS (EI) m/z 217 (M+-C4H8, 12%), 169 (75), 153 (15), 113 (12), 103 (16), 99 (15), 98 (18), 70 (19), 57 (100), 56 (33), 55 (20), 43 (34), 41 (32); HRMS (EI-TOF) Calcd for C10H19NO2S [M+-C4H8] 217.1136, found 217.1134.
(R)-N-(5-Oxodec-ylidene)-tert-butanesulfinamide (ent-5d). Following the general procedure, compound ent-5d was obtained from hydroxy ketone 3d as a yellow oil (508.0 mg, 1.86 mmol, 62%). Physical and spectroscopic data were found to be the same as for 5d. [α]20D −183.1 (c 0.98, CH2Cl2).
(S)-N-(5-Oxonon-8-en-1-ylidene)-tert-butanesulfinamide (5e). Following the general procedure, compound 5e was obtained from hydroxy ketone 3e as a yellow oil (462.8 mg, 1.80 mmol, 60%): C13H23NO2S; Rf 0.41 (hexane/EtOAc 3:1); [α]20D +181.3 (c 0.95, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 8.07 (t, J = 4.3 Hz, 1H), 5.87–5.67 (m, 1H), 5.07–4.89 (m, 2H), 2.60–2.44 (m, 6H), 2.40–2.26 (m, 2H), 2.02–1.86 (m, 2H), 1.19 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 209.2 (C), 168.7 (CH), 136.95 (CH), 115.3 (CH2), 56.6 (C), 41.9 (CH2), 41.7 (CH2), 35.2 (CH2), 27.7 (CH2), 22.3 (CH3), 19.2 (CH2); LRMS (EI) m/z 201 (M+-C4H8, 21%), 153 (95), 137 (13), 111 (10), 103 (20), 99 (16), 98 (13), 96 (14), 57 (100), 56 (26), 55 (40), 41 (24); HRMS (EI-TOF) Calcd for C9H15NO2S [M+-C4H8] 201.0823, found 201.0823.
(R)-N-(5-Oxonon-8-en-1-ylidene)-tert-butanesulfinamide (ent-5e). Following the general procedure, compound ent-5e was obtained from hydroxy ketone 3e as a yellow oil (555.3 mg, 2.16 mmol, 72%). Physical and spectroscopic data were found to be the same as for 5e. [α]20D −223.2 (c 1.02, CH2Cl2).
(S)-N-(5-Oxo-5-phenylpent-1-ylidene)-tert-butanesulfinamide (5f). Following the general procedure, compound 5f was obtained from hydroxy ketone 3f as a yellow oil (419.1 mg, 1.50 mmol, 50%): C15H21NO2S; Rf 0.33 (hexane/EtOAc 3:1); [α]20D +164.7 (c 2.01, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 8.13 (t, J = 4.2 Hz, 1H), 8.03–7.90 (m, 2H), 7.64–7.54 (m, 1H), 7.54–7.42 (m, 2H), 3.18–3.01 (m, 2H), 2.70–2.60 (m, 2H), 2.20–2.02 (m, 2H), 1.20 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 199.4 (C), 168.9 (CH), 136.9 (C), 133.3 (CH), 128.8 (CH), 128.1 (CH), 56.7 (C), 37.6 (CH2), 35.5 (CH2), 22.5 (CH3), 19.9 (CH2); LRMS (EI) m/z 223 (M+-C4H8, 14%), 176 (12), 175 (100), 133 (12), 105 (68), 103 (23), 77 (52), 73 (10), 70 (21), 57 (82), 56 (29), 51 (11), 43 (14), 41 (24); HRMS (EI-TOF) Calcd for C11H13NO2S [M+-C4H8] 223.0667, found 223.0665.
(S)-N-(5-Oxo-7-phenylhep-1-ylidene)-tert-butanesulfinamide (5h). Following the general procedure, compound 5h was obtained from hydroxy ketone 3h as a yellow oil (507.2 mg, 1.65 mmol, 55%): C17H25NO2S; Rf 0.35 (hexane/EtOAc 3:1); [α]20D +195.5 (c 1.91, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 8.06 (t, J = 4.3 Hz, 1H), 7.34–7.25 (m, 2H), 7.25–7.15 (m, 3H), 2.92 (t, J = 7.6 Hz, 2H), 2.80–2.70 (m, 2H), 2.57–2.43 (m, 4H), 2.01–1.86 (m, 2H), 1.20 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 209.25 (C), 168.8 (CH), 141.0 (C), 128.6 (CH), 128.45 (CH), 128.4 (CH), 126.3 (CH2), 56.72 (C), 44.5 (CH2), 42.0 (CH2), 35.3 (CH2), 29.9 (CH2), 22.5 (CH3), 19.3 (CH2); LRMS (EI) m/z 251 (M+-C4H8, 15%), 204 (14), 203 (94), 187 (13), 105 (36), 103 (21), 99 (25), 98 (15), 91 (91), 77 (10), 70 (12), 57 (100), 56 (32), 55 (15), 43 (11), 41 (23); HRMS (EI-TOF) Calcd for C13H15NO [M+-C4H10OS] 201.1154, found 201.1152.
(S)-N-(5-Oxo-8-phenyloct-1-ylidene)-tert-butanesulfinamide (5i). Following the general procedure, compound 5i was obtained from hydroxy ketone 3i as a yellow oil (588.1 mg, 1.83 mmol, 61%): C18H27NO2S; Rf 0.39 (hexane/EtOAc 3:1); [α]20D +256.8 (c 1.93, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 8.07 (t, J = 4.3 Hz, 1H), 7.34–7.25 (m, 2H), 7.25–7.14 (m, 3H), 2.63 (t, J = 7.5 Hz, 2H), 2.58–2.49 (m, 2H), 2.50–2.44 (m, 2H), 2.46–2.38 (m, 2H), 1.99–1.83 (m, 4H), 1.20 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 209.9 (C), 168.7 (CH), 141.50 (C), 128.5 (CH), 128.4 (CH), 126.0 (CH), 56.61 (C), 42.0 (CH2), 41.6 (CH2), 35.25 (CH2), 35.1 (CH2), 25.2 (CH2), 22.35 (CH3), 19.26 (CH2); LRMS (EI) m/z 265 (M+-C4H8, 19%), 218 (13), 217 (85), 201 (17), 147 (11), 126 (10), 113 (93), 105 (15), 104 (58), 103 (22), 98 (14), 91 (65), 70 (12), 57 (100), 56 (25), 55 (13), 41 (22); HRMS (EI-TOF) Calcd for C14H19NO2S [M+-C4H8] 265.1136, found 265.1128.
(S)-N-(4-Oxooct-1-ylidene)-tert-butanesulfinamide (14). Following the general procedure, compound 14 was obtained from hydroxy ketone 12 as a yellow oil (500.4 mg, 2.04 mmol, 68%): C12H23NO2S; Rf 0.33 (hexane/EtOAc 3:1); [α]20D +124.3 (c 1.10, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 8.11 (t, J = 2.6 Hz, 1H), 2.96–2.63 (m, 4H), 2.52–2.38 (m, 2H), 1.61–1.47 (m, 2H), 1.37–1.25 (m, 2H), 1.15 (s, 9H), 0.91 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 208.8 (C), 167.8 (CH), 56.7 (C), 42.7 (CH2), 37.0 (CH2), 29.9 (CH2), 25.9 (CH2), 22.3 (CH3), 13.8 (CH2); LRMS (EI) m/z 189 (M+-C4H8, 11%), 141 (70), 127 (15), 99 (21), 98 (14), 83 (16) 57 (100), 56 (31), 55 (14), 41 (32); HRMS (EI-TOF) Calcd for C8H14NO2S [M+-C4H9] 188.0762, found 188.0753.
3.2.4. Synthesis of N-tert-Butanesulfinyl Amino Diketones 6 and 15
General Procedure. To a solution of 3-oxobutanoic acid (40.9 mg, 0.044 mL, 0.4 mmol) in THF (2.0 mL) at 0 °C was added a 1.0 M solution of LiOEt in THF (0.50 mL, 0.5 mmol). The reaction mixture was allowed to reach 10 °C, and the corresponding N-tert-butanesulfinyl keto aldimine 5 or 14 (0.2 mmol) was added, and stirring was continued for 2 h. If the starting imine 5 or 14 was not consumed after 2 h (TLC), a 1.0 M solution of LiOEt in THF (0.25 mL, 0.25 mmol) was added, and the resulting mixture was stirred for 12 h at 23 °C. After that, the reaction was hydrolyzed with water (10 mL), extracted with ethyl acetate (3 × 10 mL), dried over magnesium sulfate, and the solvent was evaporated (15 Torr). The residue was purified by column chromatography (silica gel, hexane/EtOAc) to yield pure products 6 and 15.
(SS,S)-4-(tert-Butanesulfinylamino)decane-2,8-dione (6b). Following the general procedure, compound 6b was obtained from keto aldimine 5b as a yellow oil (32.4 mg, 0.112 mmol, 56%): C14H27NO3S; Rf 0.31 (hexane/EtOAc 1:3); [α]20D +48.0 (c 0.38, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 3.98 (d, J = 9.0 Hz, 1H), 3.63–3.35 (m, 1H), 2.95–2.71 (m, 2H), 2.44–2.34 (m, 4H), 2.14 (s, 3H), 1.77–1.39 (m, 4H), 1.19 (s, 9H), 1.04 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 211.2 (C), 208.0 (C), 55.9 (C), 53.4 (CH), 48.9 (CH2), 41.6 (CH2), 35.95 (CH2), 35.0 (CH2), 31.0 (CH3), 22.6 (CH3), 20.25 (CH2), 7.8 (CH3); LRMS (EI) m/z 233 (M+-C4H8, 2%), 215 (19), 175 (11), 167 (19), 127 (100), 111 (11), 109 (10), 57 (74), 56 (14), 43 (35), 41 (16); HRMS (EI-TOF) Calcd for C14H27NO3S [M+] 289.1712, found 289.1699.
(SS,S)-4-(tert-Butanesulfinylamino)dodecane-2,8-dione (6c). Following the general procedure, compound 6c was obtained from keto aldimine 5c as a yellow oil (41.2 mg, 0.130 mmol, 65%): C16H31NO2S; Rf 0.36 (hexane/EtOAc 1:3); [α]20D +47.3 (c 1.39, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 4.02 (d, J = 9.0 Hz, 1H), 3.52–3.48 (m, 1H), 3.05–2.66 (m, 2H), 2.47–2.33 (m, 4H), 2.15 (s, 3H), 1.64–1.44 (m, 5H), 1.38–1.22 (m, 3H), 1.21 (s, 9H), 0.90 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 211.1 (C), 208.3 (C), 56.1 (C), 53.6 (CH), 49.0 (CH2), 42.8 (CH2), 42.1 (CH2), 35.1 (CH2), 31.1 (CH3), 26.1 (CH2), 22.8 (CH3), 22.5 (CH2), 20.3 (CH2), 14.0 (CH3); LRMS (EI) m/z 261 (M+-C4H8, 2%), 243 (39), 221 (10), 203 (13), 195 (16), 156 (20), 155 (100), 153 (10), 147 (12), 139 (17), 113 (16), 97 (13), 85 (35), 73 (15), 70 (10), 57 (60), 56 (14), 43 (29), 41 (19); HRMS (EI-TOF) Calcd for C12H23NO3S [M+-C4H8] 261.1399, found 261.1384.
(SS,S)-4-(tert-Butanesulfinylamino)tridecane-2,8-dione (6d). Following the general procedure, compound 6d was obtained from keto aldimine 5d as a yellow oil (41.8 mg, 0.126 mmol, 63%): C17H33NO3S; Rf 0.39 (hexane/EtOAc 1:3); [α]20D +47.1 (c 1.01, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 4.01 (d, J = 9.0 Hz, 1H), 3.53–3.49 (m, 1H), 2.97–2.73 (m, 2H), 2.46–2.32 (m, 4H), 2.15 (s, 3H), 1.67–1.41 (m, 5H), 1.44–1.21 (m, 5H), 1.21 (s, 9H), 0.89 (t, J = 7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 210.95 (C), 208.1 (C), 55.9 (C), 53.4 (CH), 48.9 (CH2), 42.9 (CH2), 42.0 (CH2), 35.0 (CH2), 31.4 (CH2), 31.0 (CH3), 23.5 (CH2), 22.65 (CH3), 22.4 (CH2), 20.2 (CH2), 13.89 (CH3); LRMS (EI) m/z 275 (M+-C4H8, 1%), 257 (22), 170 (12), 169 (100), 153 (17), 99 (26), 71 (13), 57 (33), 56 (14), 43 (34), 41 (12); HRMS (EI-TOF) Calcd for C17H33NO3S [M+] 311.2181, found 331.2182.
(RS,R)-4-(tert-Butanesulfinylamino)tridecane-2,8-dione (ent-6d). Following the general procedure, compound ent-6d was obtained from keto aldimine ent-5d as a yellow oil (35.1 mg, 0.106 mmol, 53%). Physical and spectroscopic data were found to be the same as for 6d. [α]20D −49.3 (c 0.89, CH2Cl2).
(SS,S)-4-(tert-Butanesulfinylamino)dodec-11-ene-2,8-dione (6e). Following the general procedure, compound 6e was obtained from keto aldimine 5e as a yellow oil (34.7 mg, 0.110 mmol, 55%): C16H29NO3S; Rf 0.38 (hexane/EtOAc 1:3); [α]20D +52.9 (c 0.52, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 5.79 (dd, J = 17.0, 10.3 Hz, 1H), 5.09–4.93 (m, 2H), 4.09 (d, J = 9.1 Hz, 1H), 3.56–3.44 (m, 1H), 2.85 (qd, J = 17.9, 5.2 Hz, 2H), 2.49 (t, J = 7.4 Hz, 2H), 2.42 (t, J = 6.8 Hz, 2H), 2.37–2.26 (m, 2H), 2.15 (s, 3H), 1.81–1.41 (m, 4H), 1.21 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 209.9 (C), 208.2 (C), 137.1 (CH), 115.4 (CH2), 56.1 (C), 53.6 (CH), 49.0 (CH2), 42.2 (CH2), 41.95 (CH2), 35.1 (CH2), 31.1 (CH3), 27.85 (CH2), 22.8 (CH3), 20.3 (CH2); LRMS (EI) m/z 259 (M+-C4H8, 11%), 241 (42), 201 (11), 193 (19), 177 (54), 154 (11), 153 (86), 141 (29), 137 (15), 135 (17), 111 (14), 99 (11), 97 (17), 95 (23), 83 (100), 69 (11), 57 (86), 56 (26), 55 (84), 43 (48), 41 (30); HRMS (EI-TOF) Calcd for C12H21NO3S [M+-C4H8] 259.1242, found 259.1235.
(RS,R)-4-(tert-Butanesulfinylamino)dodec-11-ene-2,8-dione (ent-6e). Following the general procedure, compound ent-6e was obtained from keto aldimine ent-5e as a yellow oil (31.5 mg, 0.100 mmol, 50%). Physical and spectroscopic data were found to be the same as for 6e. [α]20D −54.6 (c 0.89, CH2Cl2).
(SS,S)-5-(tert-Butanesulfinylamino)-1-phenyloctane-1,7-dione (6f). Following the general procedure, compound 6f was obtained from keto aldimine 5f as a yellow oil (40.5 mg, 0.120 mmol, 60%): C18H27NO3S; Rf 0.31 (hexane/EtOAc 3:1); [α]20D +46.2 (c 1.14, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 7.95–7.82 (m, 2H), 7.57–7.31 (m, 3H), 4.07–3.98 (m, 1H), 3.59–3.44 (m, 1H), 2.93 (t, J = 6.9 Hz, 2H), 2.84–2.80 (m, 2H), 2.09 (s, 3H), 1.94–1.44 (m, 4H), 1.14 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 208.0 (C), 199.8 (C), 136.8 (C), 133.0 (CH), 128.6 (CH), 127.9 (CH), 55.9 (C), 53.4 (CH), 48.9 (CH2), 37.85 (CH2), 35.05 (CH2), 30.9 (CH3), 22.6 (CH3), 20.6 (CH2); LRMS (EI) m/z 281 (M+-C4H8, 5%), 199 (20), 175 (11), 105 (100), 77 (17), 57 (20), 43 (11); HRMS (EI-TOF) Calcd for C14H19NO3S [M+-C4H8] 281.1086, found 281.1076.
(SS,S)-4-(tert-Butanesulfinylamino)-10-phenyldecane-2,8-dione (6h). Following the general procedure, compound 6h was obtained from keto aldimine 5h as a yellow oil (44.6 mg, 0.122 mmol, 61%): C20H31NO3S; Rf 0.34 (hexane/EtOAc 1:3); [α]20D +39.7 (c 0.58, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 7.36–7.06 (m, 5H), 4.03 (d, J = 9.1 Hz, 1H), 3.48 (d, J = 4.8 Hz, 1H), 2.93–2.87 (m, 2H), 2.91–2.74 (m, 2H), 2.75–2.66 (m, 2H), 2.40 (t, J = 6.8 Hz, 2H), 2.15 (s, 3H), 1.79–1.59 (m, 2H), 1.62–1.48 (m, 2H), 1.20 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 209.7 (C), 208.1 (C), 141.0 (C), 128.5 (CH), 128.3 (CH), 126.1 (CH), 56.0 (C), 53.45 (CH), 48.9 (CH2), 44.3 (CH2), 42.3 (CH2), 34.9 (CH2), 31.0 (CH3), 29.7 (CH2), 22.7 (CH3), 20.15 (CH2); LRMS (EI) m/z 292 (M+-C4H9O, 6%), 291 (30), 243 (19), 204 (16), 203 (100), 187 (14), 133 (19), 105 (44), 103 (10), 99 (15), 97 (13), 91 (74), 57 (51), 56 (19), 43 (35), 41 (16); HRMS (EI-TOF) Calcd for C16H22NO3S [M+-C4H8] 308.1320, found 308.1324.
(SS,S)-4-(tert-Butanesulfinylamino)-11-phenylundecane-2,8-dione (6i). Following the general procedure, compound 6i was obtained from ketoaldimine 5i as a yellow oil (38.7 mg, 0.102 mmol, 51%): C21H33NO3S; Rf 0.35 (hexane/EtOAc 1:3); [α]20D +46.8 (c 1.49, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 7.37–7.06 (m, 5H), 3.99 (d, J = 9.1 Hz, 1H), 3.55–3.40 (m, 1H), 2.97–2.74 (m, 2H), 2.61 (t, J = 7.6 Hz, 2H), 2.46–2.33 (m, 4H), 2.14 (s, 3H), 1.98–1.82 (m, 2H), 1.75–1.38 (m, 4H), 1.20 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 210.4 (C), 208.1 (C), 141.5 (C), 128.5 (CH), 128.4 (CH), 126.0 (CH), 55.9 (C), 53.4 (CH), 48.9 (CH2), 42.1 (CH2), 42.0 (CH2), 35.1 (CH2), 35.0 (CH2), 31.0 (CH3), 25.2 (CH2), 22.7 (CH3), 20.2 (CH2); LRMS (EI) m/z 323 (M+-C4H8, 1%), 305 (51), 218 (18), 217 (100), 201 (26), 153 (27), 147 (60), 113 (78), 105 (20), 104 (67), 91 (31), 91 (89), 70 (15), 57 (92), 56 (27), 43 (66), 41 (33); HRMS (EI-TOF) Calcd for C17H24NO3S [M+-C4H8] 322.1520, found 322.1524.
(SS,S)-4-(tert-Butanesulfinylamino)undecane-2,7-dione (15). Following the general procedure, compound 15 was obtained from keto aldimine 14 as a yellow oil (40.6 mg, 0.134 mmol, 67%): C15H29NO2S; Rf 0.29 (hexane/EtOAc 1:3); [α]20D +39.3 (c 0.80, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 4.05 (d, J = 9.6 Hz, 1H), 3.61–3.38 (m, 1H), 2.88 (qd, J = 17.9, 5.0 Hz, 2H), 2.63–2.49 (m, 2H), 2.44–2.34 (m, 2H), 2.16 (s, 3H), 1.89–1.77 (m, 2H), 1.64–1.47 (m, 2H), 1.33–1.26 (m, 2H), 1.22 (s, 9H), 0.90 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 210.7 (C), 208.2 (C), 56.1 (C), 53.6 (CH), 49.4 (CH2), 42.8 (CH2), 39.45 (CH2), 31.1 (CH3), 29.4 (CH2), 26.1 (CH2), 22.8 (CH3), 22.4 (CH2), 13.9 (CH3); LRMS (EI) m/z 247 (M+-C4H8, 8%), 204 (12), 198 (25), 155 (26), 140 (20), 113 (28), 111 (17), 105 (100), 57 (80), 56 (18), 43 (19); HRMS (EI-TOF) Calcd for C11H19NO3S [M+-C4H10] 245.1086, found 245.1086.
3.2.5. Synthesis of 1-Substituted 9-Azabicyclo[3.3.1]nonan-3-ones 7
General Procedure. To a solution of the corresponding amino diketone derivative 6 (0.2 mmol) in MeOH (2.0 mL), a 2.0 M solution of HCl was added into Et2O (1.0 mL, 2.0 mmol) at 0 °C. After stirring for 30 min at this temperature, the disappearance of the starting reagent and the subsequent formation of the ammonium chloride was monitored by TLC. After that, the solvents were removed under vacuum (15 Torr), and EtOH, 2.0 mL), triethylamine (20.3 g, 0.028 mL, 0.2 mmol), L-proline (4.6 mg, 0.04 mmol) and anhydrous magnesium sulfate (24.0 mg, 0.2 mmol) were successively added to the flask containing the dry ammonium salt, and the resulting reaction mixture was stirred for 12 h at 23 °C. After that, the reaction was hydrolyzed with a sodium bicarbonate saturated aqueous solution (10 mL), extracted with dichloromethane (3 × 10 mL), dried over magnesium sulfate, and the solvents were evaporated (15 Torr). The residue was purified by column chromatography (silica gel, hexane/EtOAc) to yield pure products 7.
(1R,5S)-1-Ethyl-9-azabicyclo[3.3.1]nonan-3-one (7b). Following the general procedure, compound 7b was obtained from amino diketone derivative 6b as a yellow oil (25.1 mg, 0.150 mmol, 75%): C10H17NO; 2.5:97.5 er [GC (CP-Chirasil-Dex CB column, Tinlet = 275 °C, Tdetector = 250 °C, Tcolumn = 70 °C and 70–200 °C (4 °C/min), p = 101 kPa): tminor = 26.75 min, tmajor = 26.89 min]; Rf 0.55 (CH2Cl2/EtOH 10:1); [α]20D −5.6 (c 1.23, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 3.78 (s, 1H), 2.70 (dd, J = 16.3, 6.8 Hz, 1H), 2.43 (d, J = 16.9 Hz, 2H), 2.30 (d, J = 16.5 Hz, 1H), 1.94–1.77 (m, 1H), 1.77–1.35 (m, 6H), 1.22–1.08 (m, 2H), 0.94 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 210.2 (C), 55.6 (C), 50.6 (CH2), 49.9 (CH), 46.2 (CH2), 36.7 (CH2), 35.7 (CH2), 31.2 (CH2), 17.70 (CH2), 7.15 (CH3); LRMS (EI) m/z 167 (M+, 43%), 125 (31), 124 (100), 110 (53), 96 (33), 82 (20); HRMS (EI-TOF) Calcd for C10H17NO [M+] 167.1310, found 167.1311.
(1R,5S)-1-Butyl-9-azabicyclo[3.3.1]nonan-3-one (7c). Following the general procedure, compound 7c was obtained from amino diketone derivative 6c as a yellow oil (27.4 mg, 0.140 mmol, 70%): C12H21NO; 93:7 er [GC (CP-Chirasil-Dex CB column, Tinlet = 275 °C, Tdetector = 250 °C, Tcolumn = 70 °C and 70–200 °C (4 °C/min), p = 101 kPa): tmajor = 35.84 min, tminor = 36.64 min]; Rf 0.56 (CH2Cl2/EtOH 10:1); [α]20D −8.0 (c 0.51, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 3.87–3.69 (m, 2H), 2.72 (dd, J = 16.4, 6.8 Hz, 1H), 2.50–2.30 (m, 3H), 1.73–1.47 (m, 6H), 1.38–1.21 (m, 6H), 0.99–0.87 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 209.8 (C), 55.4 (C), 50.7 (CH2), 49.5 (CH), 45.7 (CH2), 43.6 (CH2), 35.8 (CH2), 30.75 (CH2), 24.7 (CH2), 23.1 (CH2), 17.45 (CH2), 14.0 (CH3); LRMS (EI) m/z 195 (M+, 21%), 166 (22), 153 (100), 152 (77), 138 (33), 110 (58), 96 (96); HRMS (EI-TOF) Calcd for C12H21NO [M+] 195.1623, found 195.1633.
(1R,5S)-1-Pentyl-9-azabicyclo[3.3.1]nonan-3-one, (−)-Adaline (7d). Following the general procedure, compound 7d was obtained from amino diketone derivative 6d as a yellow oil (32.2 mg, 0.154 mmol, 77%): C13H23NO; 86:14 er [GC (CP-Chirasil-Dex CB column, Tinlet = 275 °C, Tdetector = 250 °C, Tcolumn = 70 °C and 70–200 °C (4 °C/min), p = 101 kPa): tmajor = 38.21 min, tminor = 38.90 min]; Rf 0.57 (CH2Cl2/EtOH 10:1); [α]20D −8.6 (c 1.49, CH2Cl2) [lit. [33] −11.4 (c 0.74, CHCl3)]; 1H NMR (400 MHz, CDCl3) δ 3.74 (s, 1H), 2.65 (dd, J = 16.2, 6.7 Hz, 1H), 2.49–2.34 (m, 2H), 2.29 (d, J = 16.1 Hz, 1H), 1.82–1.13 (m, 15H), 0.95–0.84 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 210.3 (C), 55.3 (C), 51.0 (CH2), 49.6 (CH), 46.0 (CH2), 44.1 (CH2), 36.1 (CH2), 32.4 (CH2), 31.0 (CH2), 22.6 (CH2), 22.3 (CH2), 17.65 (CH2), 14.1 (CH3); LRMS (EI) m/z 209 (M+, 13%), 180 (17), 166 (99), 153 (87), 138 (21), 124 (16), 110 (53), 96 (100); HRMS (EI-TOF) Calcd for C13H23NO [M+] 209.1780, found 209.1777.
(1S,5R)-1-Pentyl-9-azabicyclo[3.3.1]nonan-3-one, (+)-Adaline (ent-7d). Following the general procedure, compound ent-7b was obtained from amino diketone derivative ent-6d as a yellow oil (31.8 mg, 0.152 mmol, 76%). Physical and spectroscopic data were found to be the same as for 7d. 23:77 er [GC (CP-Chirasil-Dex CB column, Tinlet = 275 °C, Tdetector = 250 °C, Tcolumn = 70 °C and 70–200 °C (4 °C/min), p = 101 kPa): tminor = 38.50 min, tmajor = 38.87 min]; [α]20D +8.0 (c 2.70, CH2Cl2).
(1S,5S)-1-(But-3-en-1-yl)-9-azabicyclo[3.3.1]nonan-3-one (7e). Following the general procedure, compound 7e was obtained from amino diketone derivative 6e as a yellow oil (21.3 mg, 0.110 mmol, 55%): C12H19NO; 88:12 er [GC (CP-Chirasil-Dex CB column, Tinlet = 275 °C, Tdetector = 250 °C, Tcolumn = 70 °C and 70–200 °C (4 °C/min), p = 101 kPa): tmajor = 36.31 min, tminor = 37.02 min]; Rf 0.55 (CH2Cl2/EtOH 10:1); [α]20D −8.5 (c 1.25, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 5.90–5.70 (m, 1H), 5.15–4.89 (m, 2H), 4.71–4.67 (m, 2H), 3.80 (t, J = 5.3 Hz, 1H), 2.84–2.60 (m, 1H), 2.55–2.31 (m, 3H), 2.20–2.06 (m, 2H), 1.78–1.46 (m, 7H); 13C NMR (100 MHz, CDCl3) δ 209.4 (C), 138.0 (CH), 115.3 (CH2), 55.5 (C), 50.7 (CH2), 49.4 (CH), 45.6 (CH2), 42.75 (CH2), 35.8 (CH2), 30.7 (CH2), 27.1 (CH2), 17.5 (CH2); LRMS (EI) m/z 193 (M+, 11%), 150 (100), 136 (93), 122 (35), 110 (44), 96 (51), 82 (38), 67 (22), 55 (39); HRMS (EI-TOF) Calcd for C12H19NO [M+] 193.1467, found 193.1471.
(1R,5R)-1-(But-3-en-1-yl)-9-azabicyclo[3.3.1]nonan-3-one (ent-7e). Following the general procedure, compound ent-7e was obtained from amino diketone derivative ent-6e as a yellow oil (20.1 mg, 0.104 mmol, 52%). Physical and spectroscopic data were found to be the same as for 7e. 19.5:80.5 er [GC (CP-Chirasil-Dex CB column, Tinlet = 275 °C, Tdetector = 250 °C, Tcolumn = 70 °C and 70–200 °C (4 °C/min), p = 101 kPa): tminor = 36.61 min, tmajor = 37.06 min]; [α]20D + 7.9 (c 1.57, CH2Cl2).
(1S,5S)-1-(2-Phenylethyl)-9-azabicyclo[3.3.1]nonan-3-one (7h). Following the general procedure, compound 7h was obtained from amino diketone derivative 6h as a yellow oil (30.2 mg, 0.124 mmol, 62%): C16H21NO; 92.5:7.5 er [GC (CP-Chirasil-Dex CB column, Tinlet = 275 °C, Tdetector = 250 °C, Tcolumn = 70 °C and 70–200 °C (4 °C/min), p = 101 kPa): tmajor = 53.44 min, tminor = 53.70 min]; Rf 0.58 (CH2Cl2/EtOH 10:1); [α]20D −5.5 (c 1.70, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 7.43–7.03 (m, 5H), 3.73 (s, 1H), 2.78–2.09 (m, 7H), 1.86–1.43 (m, 8H); 13C NMR (100 MHz, CDCl3) δ 210.5 (C), 141.9 (C), 128.7 (CH), 128.4 (CH), 126.15 (CH), 55.2 (C), 51.4 (CH2), 49.75 (CH), 46.4 (CH2), 46.3 (CH2), 36.4 (CH2), 31.35 (CH2), 29.3 (CH2), 17.81 (CH2); LRMS (EI) m/z 243 (M+, 73%), 200 (47), 186 (100), 184 (42), 91 (61); HRMS (EI-TOF) Calcd for C16H21NO [M+] 243.1623, found 243.1621.
(1R,5S)-1-(3-Phenylpropyl)-9-azabicyclo[3.3.1]nonan-3-one (7i). Following the general procedure, compound 7i was obtained from amino diketone derivative 6i as a yellow oil (34.5 mg, 0.134 mmol, 67%): C17H23NO; 91.3:8.7 er [GC (CP-Chirasil-Dex CB column, Tinlet = 275 °C, Tdetector = 250 °C, Tcolumn = 100 °C and 100–200 °C (4 °C/min), p = 101 kPa): tmajor = 66.97 min, tminor = 67.30 min]; Rf 0.61 (CH2Cl2/EtOH 10:1); [α]20D −7.8 (c 0.69, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 7.42–7.02 (m, 5H), 3.82–3.59 (m, 1H), 2.68–2.12 (m, 7H), 1.76–1.35 (m, 10H); 13C NMR (100 MHz, CDCl3) δ 211.1 (C), 142.0 (C), 128.45 (CH), 125.97 (CH), 54.75 (C), 51.5 (CH2), 49.7 (CH), 46.6 (CH2), 44.1 (CH2), 36.5 (CH2), 36.3 (CH2), 31.5 (CH2), 24.7 (CH2), 17.8 (CH2); LRMS (EI) m/z 257 (M+, 5%), 214 (24), 154 (10), 153 (100), 110 (22), 96 (89), 91 (20); HRMS (EI-TOF) Calcd for C17H23NO [M+] 257.1780, found 257.1780.
Copies of 1H-NMR, 13C-NMR, DEPT spectra of compounds 5, 6, 7, 14 and 15, as well as chiral GC chromatograms of compounds 7 are available is Supplementary Materials.
4. Conclusions
Homotropanones with substituents at C-1 position can be accessed in a stereoselective fashion from δ-valerolactone. Formation of the Weinreb amide resulting from the nucleophilic opening of the lactone, followed by successive alcohol oxidation, N-tert-butanesulfinyl keto aldimine formation, decarboxylative Mannich reaction involving 3-oxobutanoic acid, and an intramolecular L-proline organocatalyzed Mannich cyclization, are the sequential steps in these transformations, which proceeded in fair to good yields. The stereochemical outcome is determined by the configuration of the sulfur atom of the sulfinyl group of the chiral sulfinyl imines, as it has been exemplified in the synthesis of both enantiomers of the adaline alkaloid. Unfortunately, tropanones could not be synthesized following the same sequence of reactions when starting from γ-butyrolactone, since the final L-proline organocatalyzed cyclization failed to achieve the desired compound.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules28052414/s1, Copies of 1H-NMR, 13C-NMR, DEPT spectra of compounds 5, 6, 7, 14 and 15. Chiral GC chromatograms of compounds 7.
Author Contributions
S.H.-I. and A.S. performed chemical synthesis experiments, analyzed results, and wrote the manuscript. Author Contributions: M.Y. and F.F. designed chemical synthesis, analyzed results, and wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Spanish Ministerio de Economía y Competitividad (CTQ2017-85093-P), Ministerio de Ciencia, Innovación y Universidades (RED2018-102387-T, PID2019-107268GB-100), and the University of Alicante (VIGROB-068).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in this article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Michael, J.P. Indolizidine and quinolizidine alkaloids. Nat. Prod. Rep. 2007, 24, 191–222. [Google Scholar] [CrossRef]
- Kohnen-Johannsen, K.L.; Kayser, O. Tropane alkaloids: Chemistry, pharmacology, biosynthesis and production. Molecules 2019, 24, 796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lima, D.J.P.; Santana, A.E.G.; Birkett, M.A.; Porto, R.S. Recent progress in the synthesis of homotropane alkaloids adaline, euphococcinine and N-methyleuphococcinine. Beilstein J. Org. Chem. 2021, 17, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Breer, H.; Sattelle, D.B. Molecular-properties and functions of insect acetylcholine-receptors. J. Insect Physiol. 1987, 33, 771–790. [Google Scholar] [CrossRef]
- Kalamida, D.; Poulas, K.; Avramopoulou, V.; Fostieri, E.; Lagoumintzis, G.; Lazaridis, K.; Sideri, A.; Zouridakis, M.; Tzartos, S.J. Muscle and neuronal nicotinic acetylcholine receptors—Structure, function and pathogenicity. FEBS J. 2007, 274, 3799–3845. [Google Scholar] [CrossRef]
- Changeux, J.-P.; Edelstein, S.J. Allosteric receptors after 30 years. Neuron 1998, 21, 959–980. [Google Scholar] [CrossRef] [Green Version]
- Wooltorton, J.R.A.; Pidoplichko, V.I.; Broide, R.S.; Dani, J.A. Differential desensitization and distribution of nicotinic acetylcholine receptor subtypes in midbrain dopamine areas. J. Neurosci. 2003, 23, 3176–3185. [Google Scholar] [CrossRef]
- Richards, D.P.; Patel, R.N.; Duce, I.R.; Khambay, B.P.S.; Birkett, M.A.; Pickett, J.A.; Mellor, I.R. (−)-Adaline from the Adalia Genus of ladybirds is a potent antagonist of insect and specific mammalian nicotinic acetylcholine receptors. Molecules 2022, 27, 7074. [Google Scholar] [CrossRef]
- Glisan King, A.; Meinwald, J. Review of the defensive chemistry of coccilellids. Chem. Rev. 1996, 96, 1105–1122. [Google Scholar] [CrossRef]
- Lee, B.; Kwon, J.K.; Yu, C.-M. Asymmetric sequential allylic transfer strategy for the synthesis of (−)-adaline and (−)-euphococcinine. Synlett 2009, 2009, 1498–1500. [Google Scholar]
- Arbour, M.; Roy, S.; Godbout, C.; Spino, C. Stereoselective synthesis of (+)-euphococcinine and (−)-adaline. J. Org. Chem. 2009, 74, 3806–3814. [Google Scholar] [CrossRef] [PubMed]
- Davis, F.A.; Edupuganti, R. N-Sulfinyl β-amino ketone ketals. (−)-Euphococcinine and (−)-adaline. Org. Lett. 2010, 12, 848–851. [Google Scholar] [CrossRef]
- Khandare, S.P.; Prasad, K.R. Four-step total synthesis of (+)-euphococcinine and (±)-adaline. J. Org. Chem. 2021, 86, 12285–12291. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, F.; Botuha, C.; Chemla, F.; Pérez-Luna, A. tert-Butanesulfinimines: Structure, synthesis and synthetic applications. Chem. Soc. Rev. 2009, 38, 1162–1186. [Google Scholar] [CrossRef]
- Robak, M.A.T.; Herbage, M.A.; Ellman, J.A. Synthesis and applications of tert-butanesulfinamide. Chem. Rev. 2010, 110, 3600–3740. [Google Scholar] [CrossRef] [PubMed]
- Foubelo, F.; Yus, M. Diastereoselective indium-promoted allylation of chiral N-sulfinyl imines. Eur. J. Org. Chem. 2014, 2014, 485–491. [Google Scholar] [CrossRef]
- Lahosa, A.; Soler, T.; Arrieta, A.; Cossío, F.P.; Foubelo, F.; Yus, M. Stereoselective coupling of N-tert-butanesulfinyl aldimines and β-keto acids: Access to β-amino ketones. J. Org. Chem. 2017, 82, 7481–7491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Gómez, J.C.; Medjahdi, M.; Foubelo, F.; Yus, M. Stereoselective α-aminoallylation of aldehydes with chiral tert-butanesulfinamides and allyl bromides. J. Org. Chem. 2010, 75, 6308–6311. [Google Scholar] [CrossRef] [PubMed]
- Medjahdi, M.; González-Gómez, J.C.; Foubelo, F.; Yus, M. Concise route to (+)-aphanorphine. Eur. J. Org. Chem. 2011, 2011, 2230–2234. [Google Scholar] [CrossRef]
- Bosque, I.; González-Gómez, J.C.; Foubelo, F.; Yus, M. Straightforward access to enantioenriched 2-allylpiperidine: Application to the synthesis of alkaloids. J. Org. Chem. 2012, 77, 780–784. [Google Scholar] [CrossRef]
- Bosque, I.; González-Gómez, J.C.; Guijarro, A.; Foubelo, F.; Yus, M. Concise total synthesis and stereochemical analysis of tetraponerines T3 and T4. J. Org. Chem. 2012, 77, 10340–10346. [Google Scholar] [CrossRef] [PubMed]
- Lahosa, A.; Yus, M.; Foubelo, F. Enantiodivergent approach to the synthesis of cis-2,6-disubstituted piperidin-4-ones. J. Org. Chem. 2019, 84, 7331–7341. [Google Scholar] [CrossRef] [PubMed]
- Sirvent, A.; Hernández-Ibáñez, S.; Yus, M.; Foubelo, F. Pyrrolidine and indolizidine alkaloids from chiral N-tert-butanesulfinyl imines derived from 4-halobutanal. Synthesis 2021, 53, 1749–1759. [Google Scholar]
- Mendes, J.A.; Merino, P.; Soler, T.; Salustiano, E.J.; Costa, P.R.R.; Yus, M.; Foubelo, F.; Buarque, C.D. Enantioselective synthesis, DFT calculations and preliminary antineoplastic activity of dibenzo 1-azaspiro[4.5]decanes on drug resistant leukemias. J. Org. Chem. 2019, 84, 2219–2233. [Google Scholar] [CrossRef] [Green Version]
- Maciá, E.; Foubelo, F.; Yus, M. Indium mediated allylation of N-tert-butanesulfinyl imines with 1,3-dibromopropene: Stereoselective synthesis of aziridines. Heterocycles 2019, 99, 248–266. [Google Scholar]
- Hernández-Ibáñez, S.; Barros, O.S.R.; Lahosa, A.; García-Muñoz, M.J.; Benlahrech, M.; Behloul, C.; Foubelo, F.; Yus, M. Stereoselective synthesis of 5-(1-aminoalkyl)-2-pyrrolidones and 1,7-diazaspiro[4.5]decane-2,8-diones from chiral N-tert-butanesulfinyl imines and ethyl 4-nitrobutanoate. Tetrahedron 2020, 76, 130842. [Google Scholar] [CrossRef]
- Sirvent, A.; García-Muñoz, M.J.; Yus, M.; Foubelo, F. Stereoselective synthesis of tetrahydroisoquinolines from chiral 4-azaocta-1,7-diynes and 4-azaocta-1,7-enynes. Eur. J. Org. Chem. 2020, 2020, 113–126. [Google Scholar] [CrossRef]
- Sirvent, A.; Foubelo, F.; Yus, M. Stereoselective synthesis of δ- and ε-amino ketone derivatives from N-tert-butanesulfinyl aldimines and functionalized organolithium compounds. Molecules 2021, 26, 6503. [Google Scholar] [CrossRef]
- Davies, S.G.; Goodwin, C.J.; Hepworth, D.; Roberts, P.M.; Thomson, J.E. On the origins of diastereoselectivity in the alkylation of enolates derived from N-1-(1′-naphthyl)ethyl-O-tert-butylhydroxamates: Chiral Weinreb amide equivalents. J. Org. Chem. 2010, 75, 1214–1227. [Google Scholar] [CrossRef]
- Cogan, D.A.; Ellman, J.A. Asymmetric synthesis of α,α-dibranched amines by the trimethylaluminum-mediated 1,2-addition of organolithiums to tert-butanesulfinyl ketimines. J. Am. Chem. Soc. 1999, 121, 268–269. [Google Scholar] [CrossRef]
- Fenster, E.; Fehl, C.; Aubé, J. Use of a tandem Prins/Friedel–Crafts reaction in the construction of the indeno-tetrahydropyridine core of the haouamine alkaloids: Formal synthesis of (−)-haouamine A. Org. Lett. 2011, 13, 2614–2617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerr, W.J.; Morrison, A.J.; Paterson, L.C. Synthesis of α-methylene propellanone via the strategic employment of metal-mediated cyclisation chemistry. Tetrahedron 2015, 71, 5356–5361. [Google Scholar] [CrossRef] [Green Version]
- Itoh, T.; Yamazaki, N.; Kibayashi, C. Asymmetric synthesis of (−)-adaline. Org. Lett. 2002, 4, 2469–2472. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Representative tropane and homotropane alkaloids.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Scheme 2.
Retrosynthetic analysis for 1-substituted homotropanones.

Scheme 3.
Synthesis of 1-substituted 9-azabicyclo[3.3.1]nonan-3-ones 7 from δ-valerolactone 1a.

Scheme 4.
Mechanism to rationalize the stereochemical outcome of the intramolecular double cyclization of compounds 6 to give homotropanones 7.
Scheme 4.
Mechanism to rationalize the stereochemical outcome of the intramolecular double cyclization of compounds 6 to give homotropanones 7.

Scheme 5.
An attempt to synthesize 1-substituted 8-azabicyclo[3.2.1]octan-3-one 16 from γ-butyrolactone 1b.
Scheme 5.
An attempt to synthesize 1-substituted 8-azabicyclo[3.2.1]octan-3-one 16 from γ-butyrolactone 1b.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hernández-Ibáñez, S.; Sirvent, A.; Yus, M.; Foubelo, F. Stereoselective Synthesis of 1-Substituted Homotropanones, including Natural Alkaloid (−)-Adaline. Molecules 2023, 28, 2414. https://doi.org/10.3390/molecules28052414
AMA Style
Hernández-Ibáñez S, Sirvent A, Yus M, Foubelo F. Stereoselective Synthesis of 1-Substituted Homotropanones, including Natural Alkaloid (−)-Adaline. Molecules. 2023; 28(5):2414. https://doi.org/10.3390/molecules28052414
Chicago/Turabian StyleHernández-Ibáñez, Sandra, Ana Sirvent, Miguel Yus, and Francisco Foubelo. 2023. "Stereoselective Synthesis of 1-Substituted Homotropanones, including Natural Alkaloid (−)-Adaline" Molecules 28, no. 5: 2414. https://doi.org/10.3390/molecules28052414