
Harnessing Greenhouse Gases Absorption by Doped Fullerenes with Externally Oriented Electric Field

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
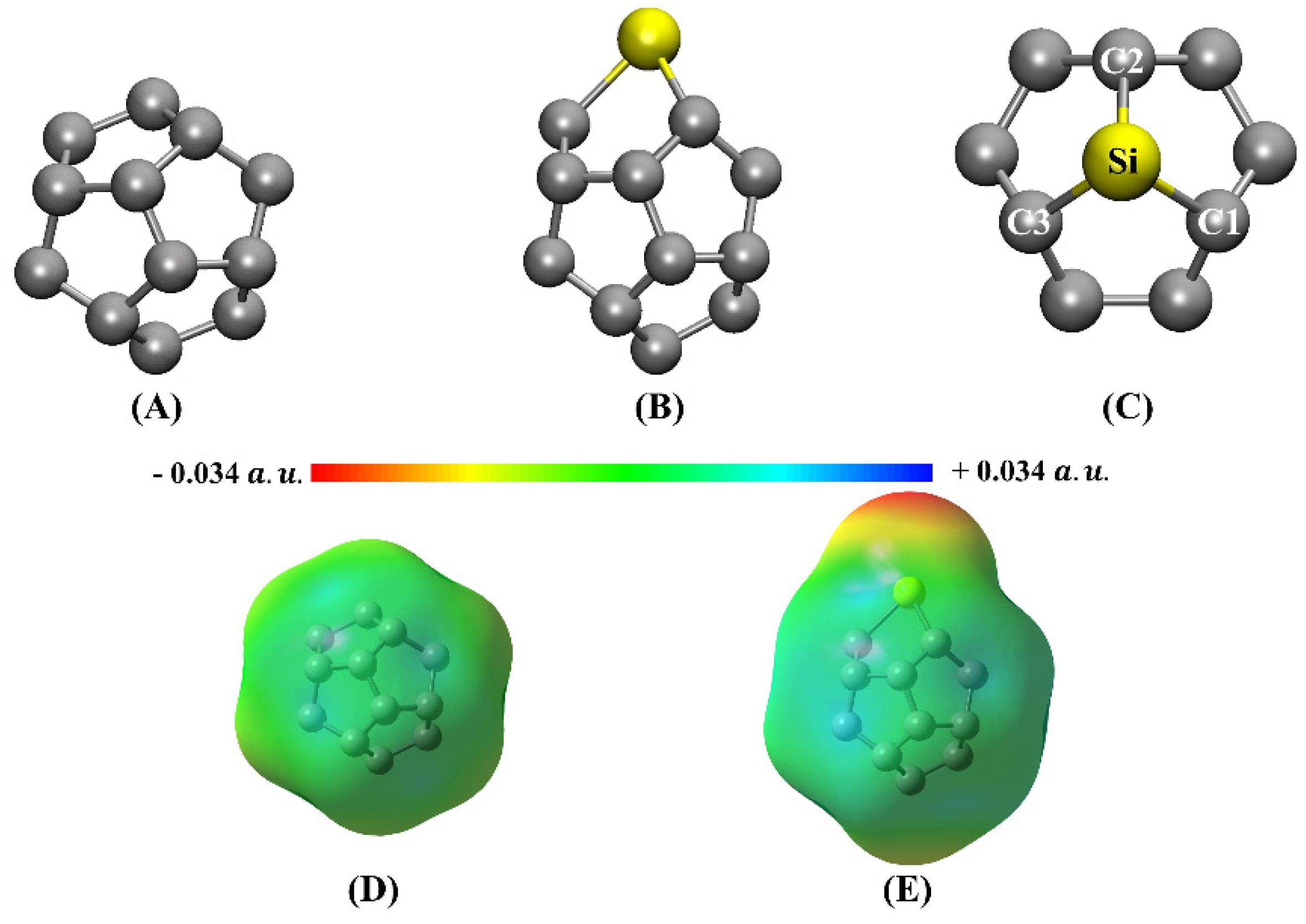
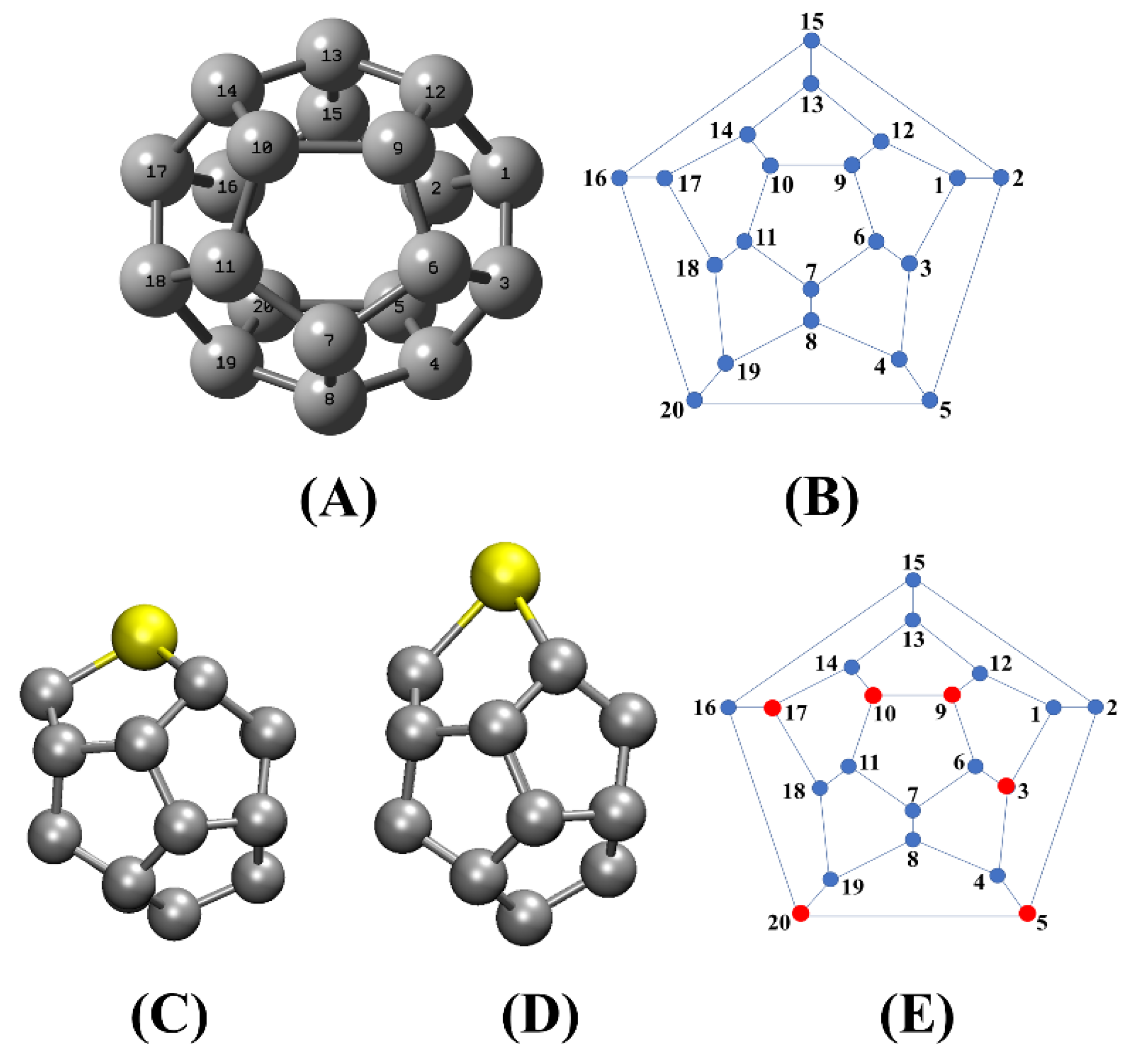
3.1. Doping Effects on the Electronic and Structural Properties of Fullerenes
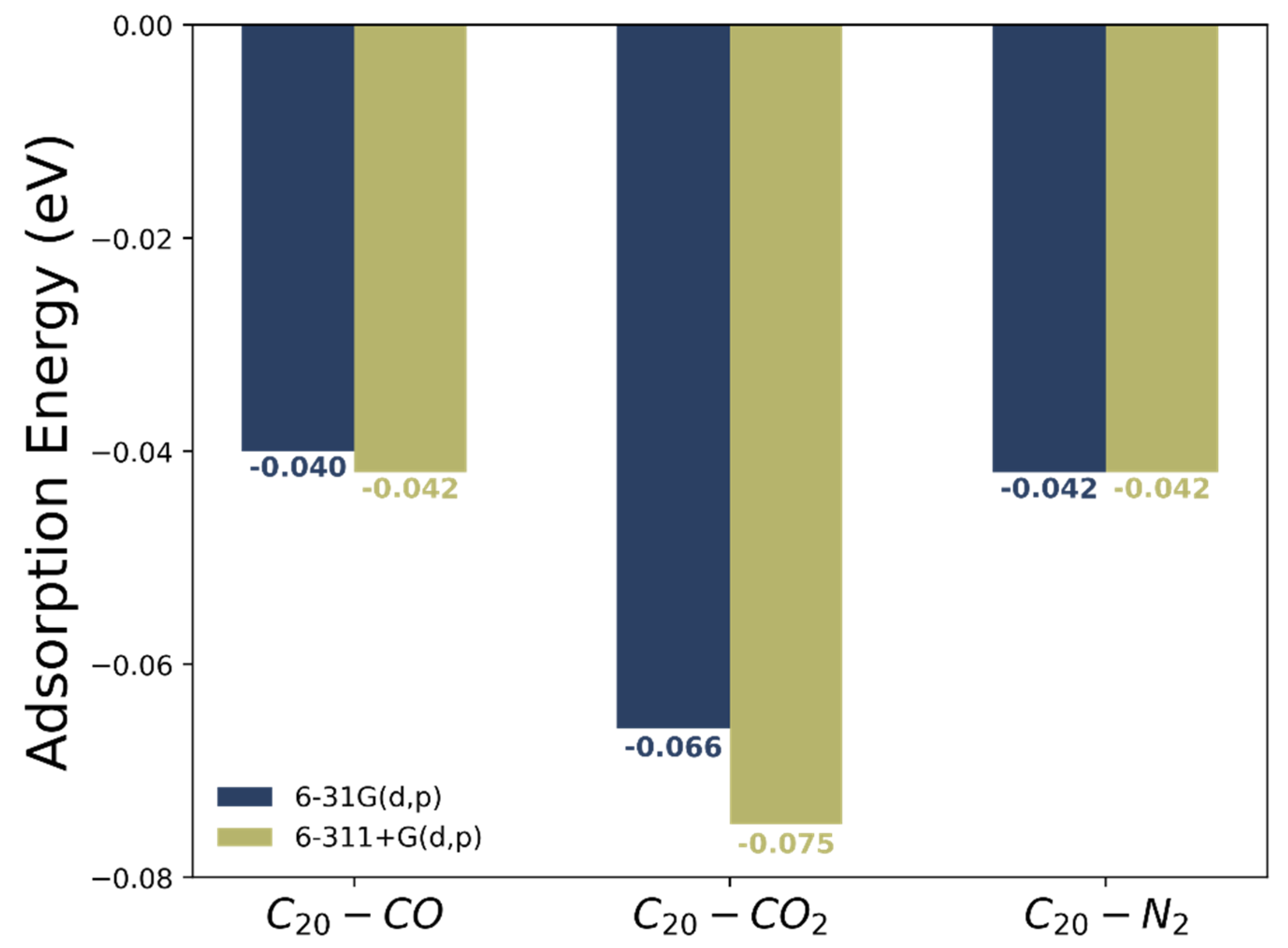
3.2. Adsorption Energies between C20 Fullerene and CO, CO2 and N2 Molecules
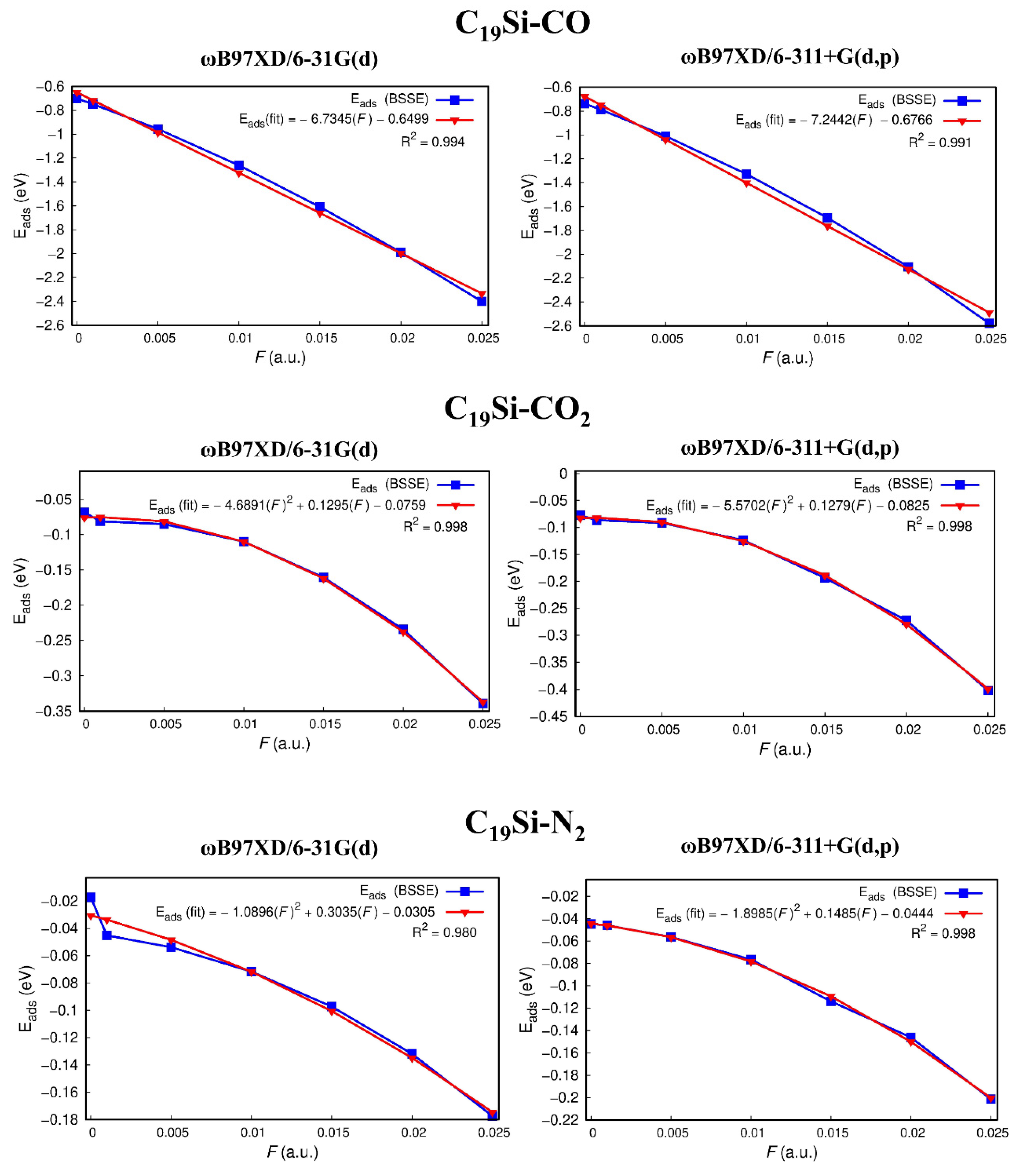
3.3. Electric Field Effect on the Adsorption Energies
3.4. Intermolecular Interaction Characterization under EOEF Influence
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, C.M.; Chen, T.C.; Yang, Y.C.; Hsiao, M.C.; Meyyappan, M.; Lai, C.S. Ultraviolet illumination effect on monolayer graphene-based resistive sensor for acetone detection. Vacuum 2017, 140, 89–95. [Google Scholar] [CrossRef]
- Nomani, M.W.K.; Shishir, R.; Qazi, M.; Diwan, D.; Shields, V.B.; Spencer, M.G.; Tompa, G.S.; Sbrockey, N.M.; Koley, G. Highly sensitive and selective detection of NO2 using epitaxial graphene on 6H-SiC. Sens. Actuators B Chem. 2010, 150, 301–307. [Google Scholar] [CrossRef]
- Romero, H.E.; Joshi, P.; Gupta, A.K.; Gutierrez, H.R.; Cole, M.W.; Tadigadapa, S.A.; Eklund, P.C. Adsorption of ammonia on graphene. Nanotechnology 2009, 20, 245501. [Google Scholar] [CrossRef] [PubMed]
- Justino, C.I.L.; Gomes, A.R.; Freitas, A.C.; Duarte, A.C.; Rocha-Santos, T.A.P. Graphene based sensors and biosensors. TrAC Trends Anal. Chem. 2017, 91, 53–66. [Google Scholar] [CrossRef]
- Feng, X.; Zhang, Y.; Zhou, J.; Li, Y.; Chen, S.; Zhang, L.; Ma, Y.; Wang, L.; Yan, X. Three-dimensional nitrogen-doped graphene as an ultrasensitive electrochemical sensor for the detection of dopamine. Nanoscale 2015, 7, 2427–2432. [Google Scholar] [CrossRef]
- Li, X.; Zhao, H.; Shi, L.; Zhu, X.; Lan, M.; Zhang, Q.; Hugh Fan, Z. Electrochemical sensing of nicotine using screen-printed carbon electrodes modified with nitrogen-doped graphene sheets. J. Electroanal. Chem. 2017, 784, 77–84. [Google Scholar] [CrossRef]
- Kroto, H.W.; Walton, D.R.M. Stable derivatives of small fullerenes. Chem. Phys. Lett. 1993, 214, 353–356. [Google Scholar] [CrossRef]
- Osawa, E.; Kroto, H.W.; Fowler, P.W.; Wasserman, E. The evolution of the football structure for the C60 molecule: A retrospective. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 1993, 343, 1–8. [Google Scholar] [CrossRef]
- Kroto, H.W. The stability of the fulerenes Cn, with n = 24, 28, 32, 36, 50, 60 and 70. Nature 1987, 329, 529–531. [Google Scholar] [CrossRef]
- Mojica, M.; Alonso, J.A.; Méndez, F. Synthesis of fullerenes. J. Phys. Org. Chem. 2013, 26, 526–539. [Google Scholar] [CrossRef]
- Silva, R.A.L.; de Brito, S.F.; Machado, D.F.S.; Carvalho-Silva, V.H.; de Oliveira, H.C.B.; Ribeiro, L. The influence of the configuration of the (C70)2 dimer on its rovibrational spectroscopic properties: A theoretical survey. J. Mol. Model. 2018, 24, 235. [Google Scholar] [CrossRef] [PubMed]
- Mahdavifar, Z.; Nomresaz, Z.; Shakerzadeh, E. Hetero-fullerenes C59M (M = B, Al, Ga, Ge, N, P, As) for sulfur dioxide gas sensing: Computational approach. Chem. Phys. 2020, 530, 110606. [Google Scholar] [CrossRef]
- Heidari, M.; Janjanpour, N.; Vakili, M.; Daneshmehr, S.; Jalalierad, K.; Alipour, F. Study of the Ionization Potential, Electron Affinity and HOMO-LUMO Gaps in the Smal Fullerene Nanostructures. Chem. Rev. Lett. 2018, 1, 45–48. [Google Scholar]
- Motlagh, N.M.; Rouhani, M.; Mirjafary, Z. Aminated C20 fullerene as a promising nanosensor for detection of A-234 nerve agent. Comput. Theor. Chem. 2020, 1186, 112907. [Google Scholar] [CrossRef]
- Metin, T.; Parlak, C.; Alver, Ö.; Tepe, M. Hydrogen storage and electronic properties of C20, C15M5 and H2@C15M5 (M = Al, Si, Ga, Ge) nanoclusters. J. Mol. Struct. 2021, 1247, 131272. [Google Scholar] [CrossRef]
- Khan, A.A.; Ahmad, I.; Ahmad, R. Influence of electric field on CO2 removal by P-doped C60-fullerene: A DFT study. Chem. Phys. Lett. 2020, 742, 137155. [Google Scholar] [CrossRef]
- Stankevich, I.V.; Nikerov, M.V.; Bochvar, D.A. The Structural Chemistry of Crystalline Carbon: Geometry, Stability, and Electronic Spectrum. Russ. Chem. Rev. 1984, 53, 640–655. [Google Scholar] [CrossRef]
- Prinzbach, H.; Weller, A.; Landenberger, P.; Wahl, F.; Wörth, J.; Scott, L.T.; Gelmont, M.; Olevano, D.; Issendorff, B.V. Gas-phase production and photoelectron spectroscopy of the smallest fullerene, C20. Nature 2000, 407, 60–63. [Google Scholar] [CrossRef]
- Guo, B.C.; Kerns, K.P.; Castleman, A.W. Ti8C12+-metallo-carbohedrenes: A new class of molecular clusters? Science 1992, 255, 1411–1413. [Google Scholar] [CrossRef]
- Slanina, Z.; Adamowicz, L. MNDO study of charged complexes of dodecahedron-shaped C20 with Li. J. Mol. Struct. THEOCHEM 1993, 281, 33–37. [Google Scholar] [CrossRef]
- Magoulas, K.; Tassios, D. Thermophysical properties of n-Alkanes from C1 to C20 and their prediction for higher ones. Fluid Phase Equilib. 1990, 56, 119–140. [Google Scholar] [CrossRef]
- Deng, H.T.; Guo, B.C.; Kerns, K.P.; Castleman, A.W. Formation and stability of metallocarbohedrenes: TixMyC12(x + y = 8, M = Nb, Ta, Y, and Si). Int. J. Mass Spectrom. Ion Process. 1994, 138, 275–281. [Google Scholar] [CrossRef]
- Hassanpour, A.; Nezhad, P.D.K.; Hosseinian, A.; Ebadi, A.; Ahmadi, S.; Ebrahimiasl, S. Characterization of IR spectroscopy, APT charge, ESP maps, and AIM analysis of C20 and its C20-nAln heterofullerene analogous (n = 1–5) using DFT. J. Phys. Org. Chem. 2021, 34, e4198. [Google Scholar] [CrossRef]
- Huda, M.N.; Ray, A.K. Evolution of SiC nanocluster from carbon fullerene: A density functional theoretic study. Chem. Phys. Lett. 2008, 457, 124–129. [Google Scholar] [CrossRef]
- Salem, M.A.; Katin, K.P.; Kaya, S.; Kochaev, A.I.; Maslov, M.M. Interaction of dopants and functional groups adsorbed on the carbon fullerenes: Computational study. Phys. E Low-Dimens. Syst. Nanostruct. 2020, 124, 114319. [Google Scholar] [CrossRef]
- Rad, A.S.; Aghaei, S.M.; Aali, E.; Peyravi, M. Study on the electronic structure of Cr- and Ni-doped fullerenes upon adsorption of adenine: A comprehensive DFT calculation. Diam. Relat. Mater. 2017, 77, 116–121. [Google Scholar] [CrossRef]
- Bai, H.; Wu, Y.; Qiao, W.; Ji, Y. Theoretical investigations on the geometrical structures, energies, and electronic properties of the heterofullerenes made of the smallest fullerene. Fuller. Nanotub. Carbon Nanostruct. 2015, 23, 399–405. [Google Scholar] [CrossRef]
- Koohi, M.; Soleimani Amiri, S.; Shariati, M. Silicon impacts on structure, stability and aromaticity of C20-nSin heterofullerenes (n = 1–10): A density functional perspective. J. Mol. Struct. 2017, 1127, 522–531. [Google Scholar] [CrossRef]
- Ajeel, F.N.; Mohammed, M.H.; Khudhair, A.M. Tuning the electronic properties of the fullerene C20 cage via silicon impurities. Russ. J. Phys. Chem. B 2017, 11, 850–858. [Google Scholar] [CrossRef]
- Alver, Ö.; Parlak, C.; Umar, Y.; Ramasami, P. DFT/QTAIM analysis of favipiravir adsorption on pristine and silicon doped C20 fullerenes. Main Group Met. Chem. 2019, 42, 143–149. [Google Scholar] [CrossRef]
- Rouhani, M. Theoretical investigation about the adsorption of the Sarin nerve agent on C20 fullerene and its boron-doped derivative. Dispos. Am. 2018, 15, 39–46. [Google Scholar] [CrossRef]
- Ahmadi, R.; Jalali Sarvestani, M.R. Adsorption of proline amino acid on the surface of fullerene (C20) and boron nitride cage (B12N12): A comprehensive DFT study. Q. J. Iran. Chem. Commun. 2019, 7, 344–351. [Google Scholar] [CrossRef]
- Vessally, E.; Siadati, S.A.; Hosseinian, A.; Edjlali, L. Selective sensing of ozone and the chemically active gaseous species of the troposphere by using the C20 fullerene and graphene segment. Talanta 2017, 162, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Carratu, M.; Chen, Q.; Cotti, G.; Hazucha, M.; Jantunen, M.; Lahmann, E.; Lauriola, P.; Mathieu-Nolf, M.; Pankow, D.; Penney, D. Environmental Health Criteria 213 Carbon Monoxide; World Health Organisation: Geneva, Switzerland, 1999; pp. 1–101. [Google Scholar]
- Léonard, N.G.; Dhaoui, R.; Chantarojsiri, T.; Yang, J.Y. Electric Fields in Catalysis: From Enzymes to Molecular Catalysts. ACS Catal. 2021, 11, 10923–10932. [Google Scholar] [CrossRef]
- Yu, S.; Vermeeren, P.; Hamlin, T.A.; Bickelhaupt, F.M. How Oriented External Electric Fields Modulate Reactivity. Chem.— Eur. J. 2021, 27, 5683–5693. [Google Scholar] [CrossRef]
- Sun, K.-B.; Zhang, S.-H.; Ren, F.-D.; Hao, Y.-P.; Ba, S.H. Theoretical prediction of the trigger linkage, cage strain, and explosive sensitivity of CL-20 in the external electric fields. J. Mol. Model. 2021, 27, 85. [Google Scholar] [CrossRef]
- Li, Y.; Chen, J.; Zhao, C. Influence of external electric field on the electronic structure and optical properties of pyrite. RSC Adv. 2017, 7, 56676–56681. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.A.; Ahmad, R.; Ahmad, I.; Su, X. Selective adsorption of CO2 from gas mixture by P-decorated C24N24 fullerene assisted by an electric field: A DFT approach. J. Mol. Graph. Model. 2021, 103, 107806. [Google Scholar] [CrossRef]
- Esrafili, M.D. Electric field assisted activation of CO2 over P-doped graphene: A DFT study. J. Mol. Graph. Model. 2019, 90, 192–198. [Google Scholar] [CrossRef]
- Song, N.; Wang, Y.; Gao, H.; Jiang, W.; Zhang, J.; Xu, B.; Sun, Q.; Jia, Y. Electric field improved hydrogen storage of Ca-decorated monolayer MoS2. Phys. Lett. Sect. A Gen. At. Solid State Phys. 2015, 379, 815–819. [Google Scholar] [CrossRef]
- Liu, H.; Lee, J.Y. Electric field effects on the adsorption of CO on a graphene nanodot and the healing mechanism of a vacancy in a graphene nanodot. J. Phys. Chem. C 2012, 116, 3034–3041. [Google Scholar] [CrossRef]
- Sun, Q.; Zhou, J.; Wang, Q.; Jena, P. Electric field enhanced hydrogen storage on polarizable materials substrates. ACS Natl. Meet. B. Abstr. 2010, 107, 2801–2806. [Google Scholar]
- Baggio, A.R.; Machado, D.F.S.; Carvalho-Silva, V.H.; Paterno, L.G.; de Oliveira, H.C.B.; Kummel, A.C.; Trogler, W.C.; Sanyal, B.; Eriksson, O.; Puglia, C.; et al. Rovibrational spectroscopic constants of the interaction between ammonia and metallo-phthalocyanines: A theoretical protocol for ammonia sensor design. Phys. Chem. Chem. Phys. 2017, 19, 10843–10853. [Google Scholar] [CrossRef]
- Ader, M.; Thomazo, C.; Sansjofre, P.; Busigny, V.; Papineau, D.; Laffont, R.; Cartigny, P.; Halverson, G.P. Interpretation of the nitrogen isotopic composition of Precambrian sedimentary rocks: Assumptions and perspectives. Chem. Geol. 2016, 429, 93–110. [Google Scholar] [CrossRef] [Green Version]
- Nibe, M.; Svanborg, M.; Lemos-silva, R.; Da, D.A.; Filho, S.; Jørgensen, B.; Albrektsen, O.; Kjelstrup-hansen, J. Work function difference of naphthyl end-capped oligothiophene in different crystal alignments studied by Kelvin probe force microscopy. Org. Electron. 2021, 89, 106060. [Google Scholar] [CrossRef]
- Nemati-Kande, E.; Abbasi, M.; Doust Mohammadi, M. DFT, QTAIM and NBO Investigation of the Interaction of Rare Gases with Pristine and Decorated Boron Nitride Nanotube. ChemistrySelect 2018, 3, 9833–9840. [Google Scholar] [CrossRef]
- Agheli, Z.; Pordel, M.; Beyramabadi, S.A. Synthesis, characterization, optical properties, computational characterizations, QTAIM analysis and cyclic voltammetry of new organic dyes for dye-sensitized solar cells. J. Mol. Struct. 2020, 1202, 127228. [Google Scholar] [CrossRef]
- Acharya, C.K.; Turner, C.H. Effect of an electric field on the adsorption of metal clusters on boron-doped carbon surfaces. J. Phys. Chem. C 2007, 111, 14804–14812. [Google Scholar] [CrossRef]
- Boys, S.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Oliveira, B.G.; Araújo, R.C.M.U.; Ramos, M.N. A topologia molecular QTAIM e a descrição mecânico-quântica de ligações de hidrogênio e ligações de di-hidrogênio. Quim. Nova 2010, 33, 1155–1162. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Essén, H. The characterization of atomic interactions. J. Chem. Phys. 1984, 80, 1943. [Google Scholar] [CrossRef]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.; Beratan, D.N.; Yang, W. NCIPLOT: A Program for Plotting Noncovalent Interaction Regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing noncovalent interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Javan, M.B.; Ganji, M.D. Theoretical investigation on the encapsulation of atomic hydrogen into heterofullerene nanocages. Curr. Appl. Phys. 2013, 13, 1525–1531. [Google Scholar] [CrossRef]
- Shariatinia, Z.; Shahidi, S. A DFT study on the physical adsorption of cyclophosphamide derivatives on the surface of fullerene C60 nanocage. J. Mol. Graph. Model. 2014, 52, 71–81. [Google Scholar] [CrossRef]
- Silva, R.A.L.; da Silva Filho, D.A.; Moberg, M.E.; Pappenfus, T.M.; Janzen, D.E. Halogen Interactions in Halogenated Oxindoles: Crystallographic and Computational Investigations of Intermolecular Interactions. Molecules 2021, 26, 5487. [Google Scholar] [CrossRef]
- Bai, H.; Ji, W.; Liu, X.; Wang, L.; Yuan, N.; Ji, Y. Doping the buckminsterfullerene by substitution: Density functional theory studies of C59X (X = B, N, Al, Si, P, Ga, Ge, and As). J. Chem. 2012, 2013, 571709. [Google Scholar] [CrossRef] [Green Version]
- Simeon, T.M.; Yanov, I.; Leszczynski, J. Ab initio quantum chemical studies of fullerene molecules with substitutes C59X [X = Si, Ge, Sn], C59X−[X = B, Al, Ga, In], and C59X+ [X = N, P, As, Sb]. Int. J. Quantum Chem. 2005, 105, 429–436. [Google Scholar] [CrossRef]
- Paul, D.; Deb, J.; Bhattacharya, B.; Sarkar, U. Density functional theory study of pristine and transition metal doped fullerene. AIP Conf. Proc. 2017, 1832, 1–4. [Google Scholar] [CrossRef]
- Dheivamalar, S.; Sugi, L. Density functional theory (DFT) investigations on doped fullerene with heteroatom substitution. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2015, 151, 687–695. [Google Scholar] [CrossRef]
- Aihara, J.I. Reduced HOMO-LUMO Gap as an Index of Kinetic Stability for Polycyclic Aromatic Hydrocarbons. J. Phys. Chem. A 1999, 103, 7487–7495. [Google Scholar] [CrossRef]
- Etminan, N.; Yoosefian, M.; Raissi, H.; Hakimi, M. Solvent effects on the stability and the electronic properties of histidine/Pd-doped single-walled carbon nanotube biosensor. J. Mol. Liq. 2016, 214, 313–318. [Google Scholar] [CrossRef]
- Behjatmanesh-Ardakani, R.; Pourroustaei-Ardakani, F.; Taghdiri, M.; Kotena, Z.M. DFT-B3LYP study of interactions between host biphenyl-1-aza-18-crown-6 ether derivatives and guest Cd2+: NBO, NEDA, and QTAIM analyses. J. Mol. Model. 2016, 22, 149. [Google Scholar] [CrossRef] [PubMed]
- Saadat, K.; Tavakol, H. An exceptional functionalization of doped fullerene observed via theoretical studies on the interactions of sulfur-doped fullerenes with halogens and halides. RSC Adv. 2015, 5, 55227–55237. [Google Scholar] [CrossRef]
- Tariq, S.; Khalid, M.; Raza, A.R.; Rubab, S.L.; de Alcântara Morais, S.F.; Khan, M.U.; Tahir, M.N.; Braga, A.A.C. Experimental and computational investigations of new indole derivatives: A combined spectroscopic, SC-XRD, DFT/TD-DFT and QTAIM analysis. J. Mol. Struct. 2020, 1207, 127803. [Google Scholar] [CrossRef]
- Grabowski, S.J. What Is the Covalency of Hydrogen Bonding? Chem. Rev. 2011, 111, 2597–2625. [Google Scholar] [CrossRef]
- Lipkowski, P.; Grabowski, S.J.; Robinson, T.L.; Leszczynski, J. Properties of the C−H···H Dihydrogen Bond: An ab Initio and Topological Analysis. J. Phys. Chem. A 2004, 108, 10865–10872. [Google Scholar] [CrossRef]
- Bianchi, R.; Gervasio, G.; Marabello, D. Experimental electron density analysis of Mn2(CO)10: Metal-metal and metal-ligand bond characterization. Inorg. Chem. 2000, 39, 2360–2366. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.S.V.; Raghavendra, V.; Subramanian, V. Bader’s Theory of Atoms in Molecules (AIM) and its Applications to Chemical Bonding. J. Chem. Sci. 2016, 128, 1527–1536. [Google Scholar] [CrossRef]
- Matta, C.F. Hydrogen-hydrogen bonding: The non-electrostatic limit of closed-shell interaction between two hydro. In Hydrogen Bonding—New Insights; Grabowski, S., Ed.; Springer: Łódz, Poland, 2006; pp. 337–375. ISBN 1402048521. [Google Scholar]
- Berryman, V.E.J.; Whalley, Z.J.; Shephard, J.J.; Ochiai, T.; Price, A.N.; Arnold, P.L.; Parsons, S.; Kaltsoyannis, N. Computational analysis of M-O covalency in M(OC6H5)4 (M = Ti, Zr, Hf, Ce, Th, U). Dalt. Trans. 2019, 48, 2939–2947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziółkowski, M.; Grabowski, S.J.; Leszczynski, J. Cooperativity in hydrogen-bonded interactions: Ab initio and “atoms in molecules” analyses. J. Phys. Chem. A 2006, 110, 6514–6521. [Google Scholar] [CrossRef] [PubMed]
- Berryman, V.E.J.; Shephard, J.J.; Ochiai, T.; Price, A.N.; Arnold, P.L.; Parsons, S.; Kaltsoyannis, N. Quantum chemical topology and natural bond orbital analysis of M-O covalency in M(OC6H5)4 (M = Ti, Zr, Hf, Ce, Th, Pa, U, Np). Phys. Chem. Chem. Phys. 2020, 22, 16804–16812. [Google Scholar] [CrossRef] [PubMed]
- Astakhov, A.V.; Chernyshev, V.M. Molecular structure of 3-amino[1,2,4]triazolo-[4,3-a]pyrimidin-5-one in various tautomeric forms: Investigation by DFT and Qtaim methods. Chem. Heterocycl. Compd. 2014, 50, 319–326. [Google Scholar] [CrossRef]
- Baryshnikov, G.V.; Minaev, B.F.; Minaeva, V.A.; Baryshnikova, A.T. Structure and intramolecular stabilization of geometric isomers of Bi- and trithiazolidine-4-ones and their methyl derivatives: A DFT and QTAIM study. J. Struct. Chem. 2012, 53, 428–435. [Google Scholar] [CrossRef]
- Baryshnikov, G.V.; Minaev, B.F.; Korop, A.A.; Minaeva, V.A.; Gusev, A.N. Structure of zinc complexes with 3-(pyridin-2-yl)-5-(arylideneiminophenyl)-1H-1,2,4-triazoles in different tautomeric forms: DFT and QTAIM study. Russ. J. Inorg. Chem. 2013, 58, 928–934. [Google Scholar] [CrossRef]
- Ayarde-Henríquez, L.; Guerra, C.; Duque-Noreña, M.; Rincón, E.; Pérez, P.; Chamorro, E. Are There Only Fold Catastrophes in the Diels-Alder Reaction between Ethylene and 1,3-Butadiene? J. Phys. Chem. A 2021, 125, 5152–5165. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Clarendon Press: Oxford, UK, 1990; ISBN 9780198558651. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MK | ||||||
|---|---|---|---|---|---|---|
| C19Si (1) | 1.033 | −1012.718 | −7.237 | −2.439 | 4.798 | ~0 |
| C19Si (2) | 1.044 | −1012.718 | −7.237 | −2.439 | 4.798 | ~0 |
| C19Si (3) | −0.174 | −1012.777 | −7.765 | −2.047 | 5.718 | 16.784 |
| C19Si (4) | 1.033 | −1012.718 | −7.237 | −2.439 | 4.798 | ~0 |
| C19Si (5) | −0.173 | −1012.777 | −7.763 | −2.047 | 5.716 | 16.590 |
| C19Si (6) | 1.044 | −1012.718 | −7.237 | −2.438 | 4.799 | ~0 |
| C19Si (7) | 1.034 | −1012.718 | −7.237 | −2.436 | 4.801 | ~0 |
| C19Si (8) | 1.034 | −1012.718 | −7.237 | −2.436 | 4.801 | ~0 |
| C19Si (9) | −0.185 | −1012.777 | −7.763 | −2.048 | 5.716 | 16.555 |
| C19Si (10) | −0.185 | −1012.777 | −7.763 | −2.047 | 5.716 | 16.590 |
| C19Si (11) | 1.033 | −1012.718 | −7.237 | −2.439 | 4.798 | ~0 |
| C19Si (12) | 1.033 | −1012.718 | −7.237 | −2.447 | 4.790 | ~0 |
| C19Si (13) | 1.045 | −1012.718 | −7.237 | −2.436 | 4.801 | ~0 |
| C19Si (14) | 1.044 | −1012.718 | −7.237 | −2.439 | 4.798 | ~0 |
| C19Si (15) | 1.045 | −1012.718 | −7.237 | −2.436 | 4.801 | ~0 |
| C19Si (16) | 1.033 | −1012.718 | −7.237 | −2.447 | 4.790 | ~0 |
| C19Si (17) | −0.178 | −1012.777 | −7.765 | −2.047 | 5.718 | 16.784 |
| C19Si (18) | 1.044 | −1012.718 | −7.237 | −2.439 | 4.798 | ~0 |
| C19Si (19) | 1.044 | −1012.718 | −7.237 | −2.439 | 4.798 | ~0 |
| C19Si (20) | −0.162 | −1012.777 | −7.765 | −2.044 | 5.722 | 16.696 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lemos Silva, R.A.; Scalabrini Machado, D.F.; Nunes Rodrigues, N.M.; de Oliveira, H.C.B.; Ribeiro, L.; da Silva Filho, D.A. Harnessing Greenhouse Gases Absorption by Doped Fullerenes with Externally Oriented Electric Field. Molecules 2022, 27, 2968. https://doi.org/10.3390/molecules27092968
Lemos Silva RA, Scalabrini Machado DF, Nunes Rodrigues NM, de Oliveira HCB, Ribeiro L, da Silva Filho DA. Harnessing Greenhouse Gases Absorption by Doped Fullerenes with Externally Oriented Electric Field. Molecules. 2022; 27(9):2968. https://doi.org/10.3390/molecules27092968
Chicago/Turabian StyleLemos Silva, Rodrigo A., Daniel F. Scalabrini Machado, Núbia Maria Nunes Rodrigues, Heibbe C. B. de Oliveira, Luciano Ribeiro, and Demétrio A. da Silva Filho. 2022. "Harnessing Greenhouse Gases Absorption by Doped Fullerenes with Externally Oriented Electric Field" Molecules 27, no. 9: 2968. https://doi.org/10.3390/molecules27092968