
In Vitro and In Silico Evaluation of Cholinesterase Inhibition by Alkaloids Obtained from Branches of Abuta panurensis Eichler

 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
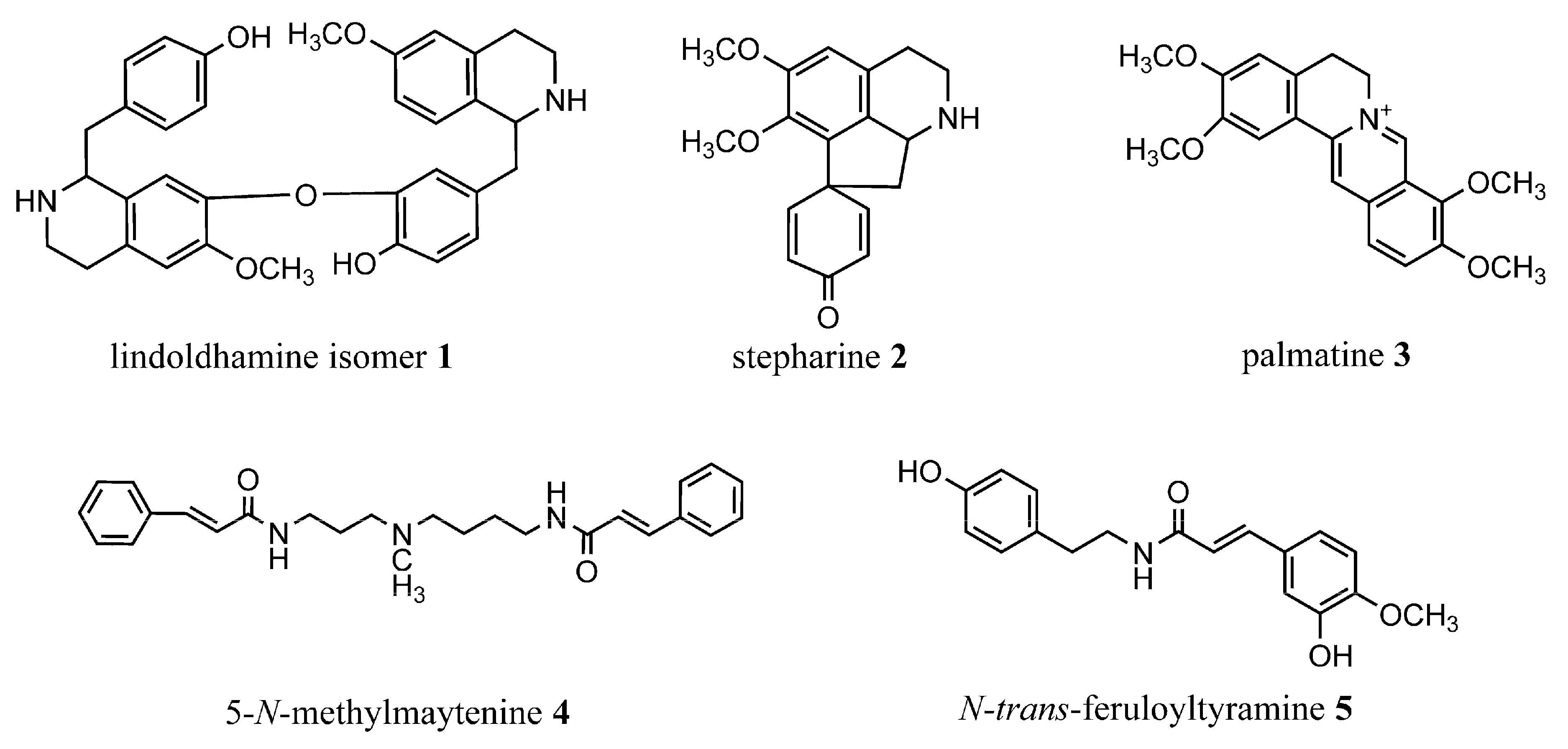
2.1. Spectral Data
2.2. Acetylcholinesterase Inhibition Assay
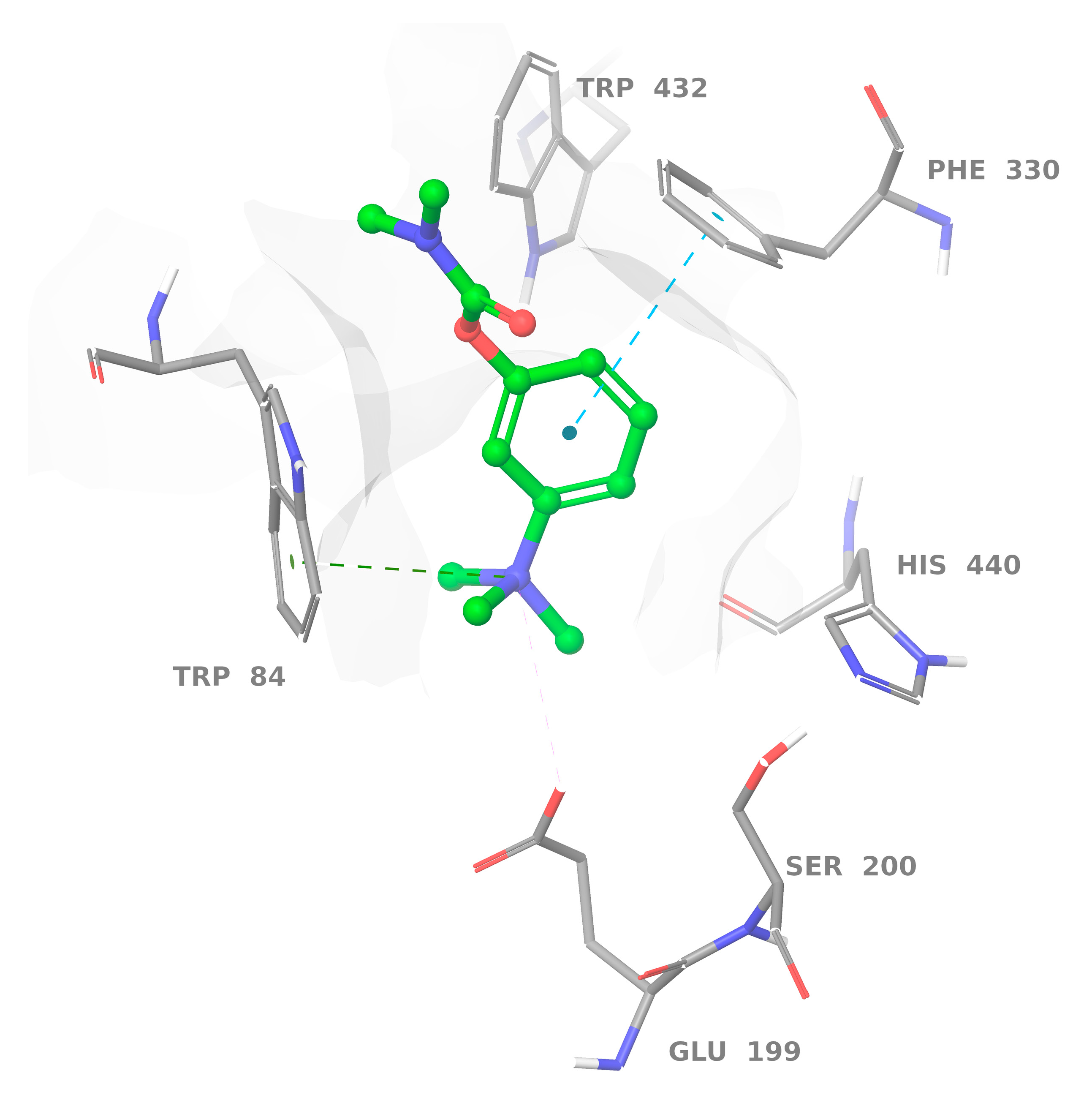
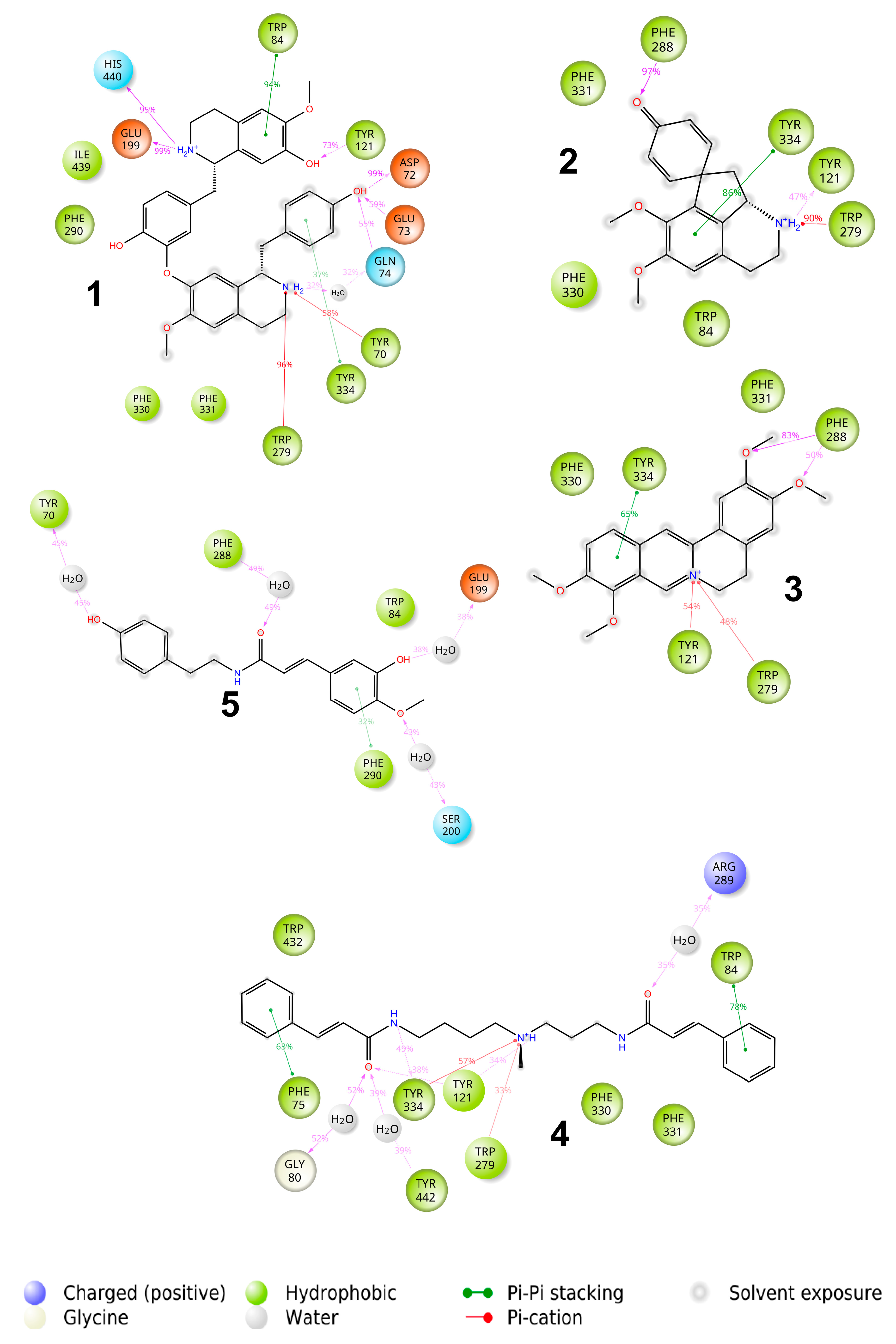
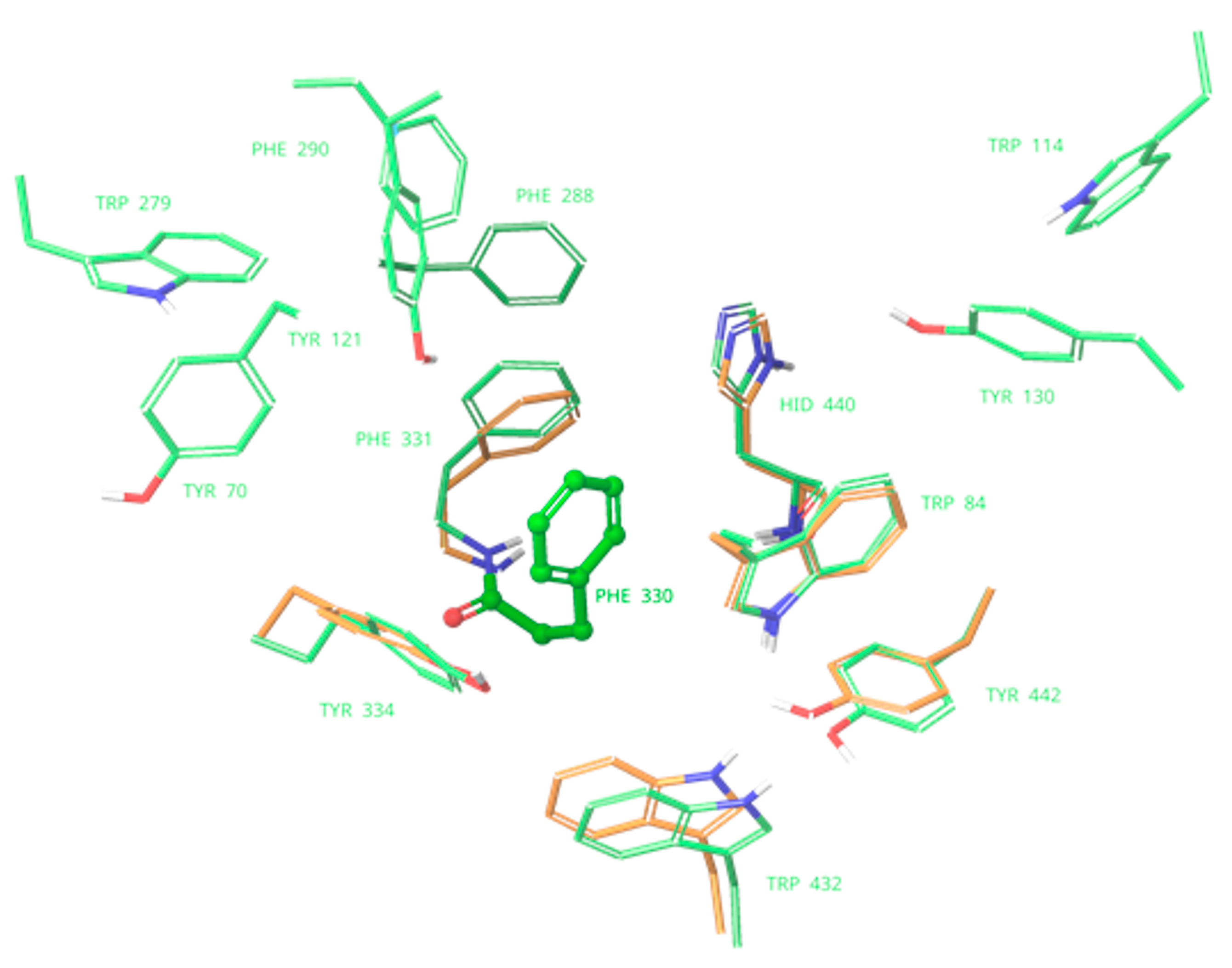
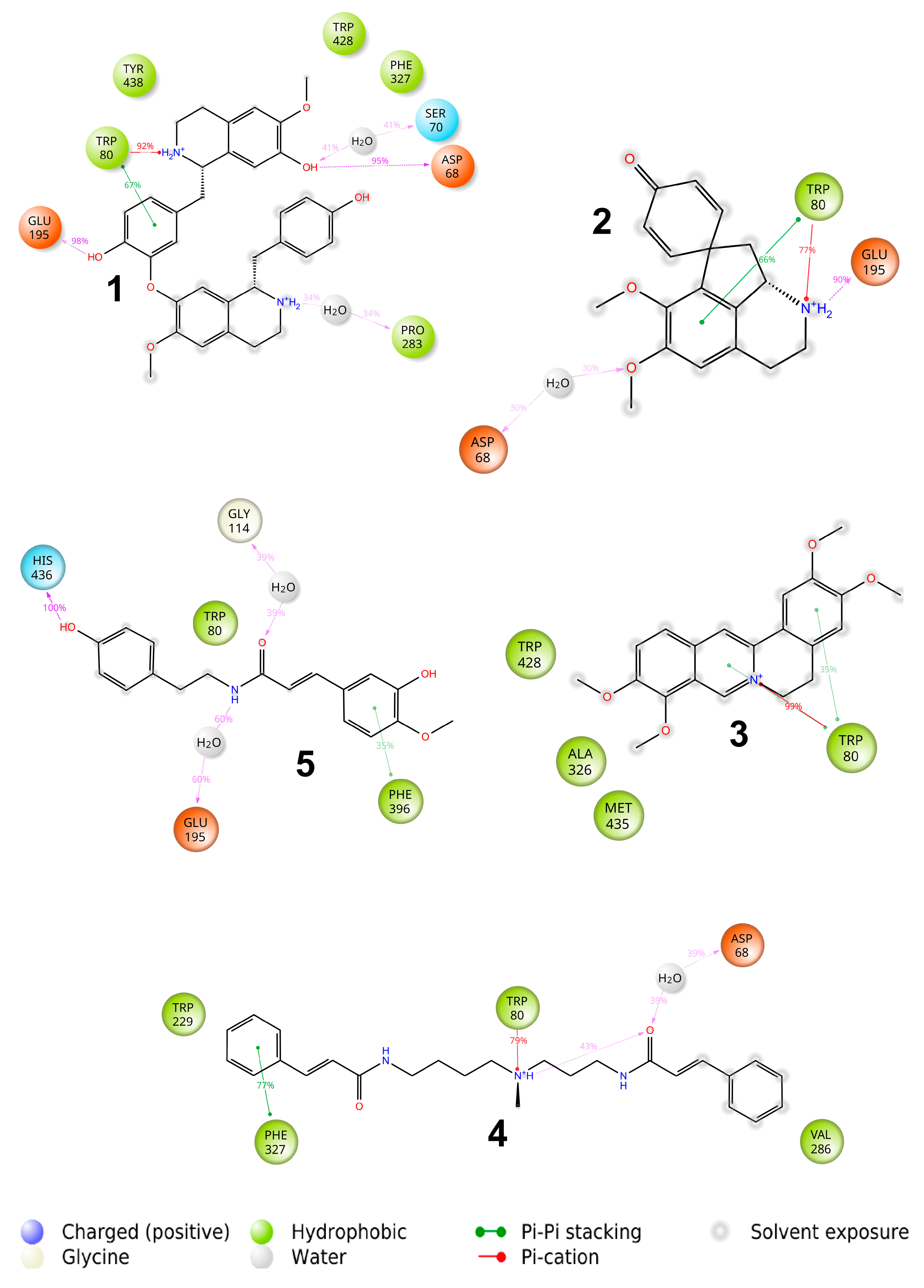
2.3. Binding with Acetylcholinesterase of Tetronarce californica (TcAChE)
2.4. Binding with Human Butyrylcholinesterase (BChE)
3. Materials and Methods
3.1. Chemicals
3.2. Plant Material
3.3. Extraction
3.4. HPLC-APCI-MS Analysis
3.5. Semi-Preparative HPLC Analysis
3.6. High-Resolution Mass Spectrometry
3.7. 1D and 2D NMR Spectroscopy
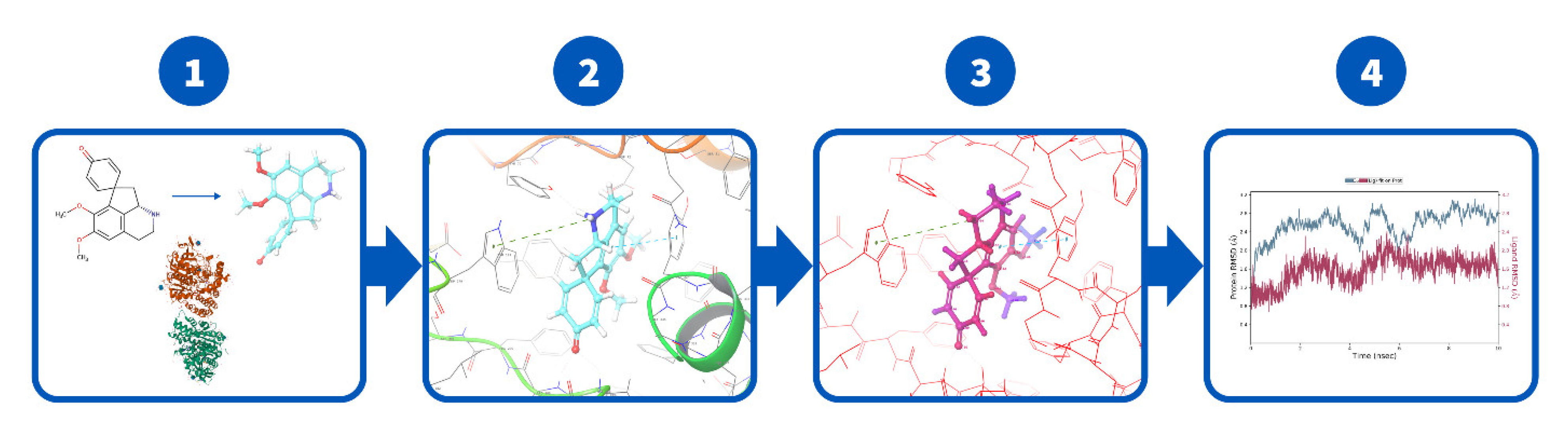
3.8. Docking and MD Procedures
3.9. Acetylcholinesterase Inhibition Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Barbosa-Filho, J.; da-Cunha, E.V.L.; Gray, A.I. Alkaloids of the Menispermaceae. Alkaloids Chem. Biol. 2000, 54, 1–190. [Google Scholar] [CrossRef]
- Manu, A.; Tanvi, S.; Anu, D.; Neeraj, B.; Ahmad, S.A. An Inside Review of Cissampelos Pareira LINN: A Potential Medicinal Plant of India. Int. Res. J. Pharm. 2012, 3, 38–41. [Google Scholar]
- Ribeiro, J.E.L.D.S.; Hopkins, M.J.G.; Vicentini, A.; Sothers, C.A.; Costa, M.A.S.; Brito, J.M.; Souza, M.A.D.; Martins, L.H.P.; Lohmann, L.G.; Assunção, P.A.C.L.; et al. Menispermaceae. In Flora da Reserva Ducke: Guia de Identificacão das Plantas Vasculares de Uma Floresta de Terra-Firme na Amazônia Central; INPA, Ed.; INPA-DFID: Manaus, Brasil, 1999; p. 793. [Google Scholar]
- Menachery, M.D. The Alkaloids of South American Menispermaceae. In Alkaloids: Chemical and Biological Perspectives; S.W. Pelletier: New York, NY, USA, 1996; pp. 269–302. [Google Scholar]
- Rocha, A.I.D.; Luz, A.I.R.; Silva, M.F.D. A Presença de Alcaloides Em Espécies Botânicas Da Amazônia-Menispermaceae. Acta Amaz. 1984, 14, 244–254. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.H.; Li, H.M.; He, J.; Deng, X.; Peng, L.Y.; Gao, L.H.; Zhao, Q.S.; Li, R.T.; Wu, X. De New Alkaloids Sinomacutines A-E, and Cephalonine-2-O-β-d-Glucopyranoside from Rhizomes of Sinomenium Acutum. Tetrahedron 2014, 70, 8893–8899. [Google Scholar] [CrossRef]
- Semwal, D.K.; Semwal, R.B. Efficacy and Safety of Stephania Glabra: An Alkaloid-Rich Traditional Medicinal Plant. Nat. Prod. Res. 2015, 29, 396–410. [Google Scholar] [CrossRef] [PubMed]
- Rosario, S.L.; da SiIva, A.J.R.; Parente, J.P. Alkamides from Cissampelos Glaberrima. Planta Med. 1996, 62, 376–377. [Google Scholar] [CrossRef]
- Cava, M.P.; Saa, J.M.; Lakshmikantham, M.V.; Mitchell, M.J. Panurensine and Norpanurensine, New Bisbenzylisoquinoline Alkaloids from Abuta Panurensis. J. Org. Chem. 1975, 40, 2647–2649. [Google Scholar] [CrossRef]
- Da Silva Mesquita, R.; Kyrylchuk, A.; Costa de Oliveira, R.; Costa Sá, I.S.; Coutinho Borges Camargo, G.; Soares Pontes, G.; Moura Araújo da Silva, F.; Saraiva Nunomura, R.D.C.; Grafov, A. Alkaloids of Abuta Panurensis Eichler: In Silico and in Vitro Study of Acetylcholinesterase Inhibition, Cytotoxic and Immunomodulatory Activities. PLoS ONE 2020, 15, e0239364. [Google Scholar] [CrossRef]
- Cometa, M.F.; Fortuna, S.; Palazzino, G.; Volpe, M.T.; Rengifo Salgado, E.; Nicoletti, M.; Tomassini, L. New Cholinesterase Inhibiting Bisbenzylisoquinoline Alkaloids from Abuta Grandifolia. Fitoterapia 2012, 83, 476–480. [Google Scholar] [CrossRef]
- Murebwayire, S.; Ingkaninan, K.; Changwijit, K.; Frédérich, M.; Duez, P. Triclisia Sacleuxii (Pierre) Diels (Menispermaceae), a Potential Source of Acetylcholinesterase Inhibitors. J. Pharm. Pharmacol. 2008, 61, 103–107. [Google Scholar] [CrossRef]
- Houghton, P.J.; Ren, Y.; Howes, M.J. Acetylcholinesterase Inhibitors from Plants and Fungi. Nat. Prod. Rep. 2006, 23, 181–199. [Google Scholar] [CrossRef] [PubMed]
- Nabavi, S.M.E.U.; Rastrelli, L.; Sobarzo-Sánchez, E. Aporphines and Alzheimer’s Disease: Towards a Medical Approach Facing the Future. Curr. Med. Chem. 2019, 26, 3253–3259. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.W.; Cai, L.; Fang, Y.S.; Xiao, H.; Li, Z.J.; Ding, Z.T. Proaporphine and Aporphine Alkaloids with Acetylcholinesterase Inhibitory Activity from Stephania Epigaea. Fitoterapia 2015, 104, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, Y.B.; Arzamascev, E.V.; Mironova, M.I.; Eliseev, A.T.; Bogonatova, L.N. Remedies for treatment of traumatic and postoperative damage of peripheral nervous system. Patent USSR 1713151, 1986. Available online: https://patents.su/0-1713151-sredstvo-dlya-lecheniya-travmaticheskikh-i-posleoperacionnykh-povrezhdenijj-perifericheskojj-nervnojj-sistemy.html (accessed on 12 May 2022).
- Swaffar, D.S.; Holley, C.J.; Fitch, R.W.; Elkin, K.R.; Zhang, C.; Sturgill, J.P.; Menachery, M.D. Phytochemical Investigation and In Vitro Cytotoxic Evaluation of Alkaloids from Abuta Rufescens. Planta Med. 2012, 78, 230–232. [Google Scholar] [CrossRef] [Green Version]
- Rojas, Y.; Soto, R.; Amaya, E.; Retuerto, F.; Fuertes, C.M. Efecto Antitumoral de Los Alcaloides Hidrosolubles de Abuta Grandifolía (MART) Satidwith, En Línea Celular HEP-2. Cienc. Investig. 2004, 7, 22–26. [Google Scholar] [CrossRef]
- Stevigny, C.; Bailly, C.; Quetin-Leclercq, J. Cytotoxic and Antitumor Potentialities of Aporphinoid Alkaloids. Curr. Med. Chem. Agents 2005, 5, 173–182. [Google Scholar] [CrossRef] [Green Version]
- Chang, F.; Hwang, T.; Yang, Y.; Li, C. Anti-Inflammatory and Cytotoxic Diterpenes from Formosan Polyalthia Longifolia Var. Pendula. Planta Med. 2006, 72, 1344–1347. [Google Scholar] [CrossRef]
- Vieira, G.C.; Gadelha, F.A.A.F.; Pereira, R.F.; Ferreira, L.K.D.P.; Barbosa-Filho, J.M.; Bozza, P.T.; Piuvezam, M.R. Warifteine, an Alkaloid of Cissampelos Sympodialis, Modulates Allergic Profile in a Chronic Allergic Rhinitis Model. Brazilian J. Pharmacogn. 2018, 28, 50–56. [Google Scholar] [CrossRef]
- Duté, P.; Weber, J.F.; Fournet, A.; Cavé, A.; Bruneton, J. Bis-Benzylisoquinoline Alkaloids from Abuta Pahni. Phytochemistry 1987, 26, 2136–2137. [Google Scholar] [CrossRef]
- Lavault, M.; Bruneton, J.; Cavé, A.; Chan, K.C.; Deverre, J.R.; Sevenet, T.; Guinaudeau, H. Alcaloides Bisbenzylisoquinoleiques de Albertisia Cf. A. Papuana. Can. J. Chem. 1987, 65, 343–347. [Google Scholar] [CrossRef]
- Malca Garcia, G.R.; Hennig, L.; Shelukhina, I.V.; Kudryavtsev, D.S.; Bussmann, R.W.; Tsetlin, V.I.; Giannis, A. Curare Alkaloids: Constituents of a Matis Dart Poison. J. Nat. Prod. 2015, 78, 2537–2544. [Google Scholar] [CrossRef] [PubMed]
- Hagel, J.M.; Facchini, P.J. Benzylisoquinoline Alkaloid Metabolism: A Century of Discovery and a Brave New World. Plant Cell Physiol. 2013, 54, 647–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steele, J.C.P.; Simmonds, M.S.J.; Veitch, N.C.; Warhurst, D.C. Evaluation of the Anti-Plasmodial Activity of Bisbenzylisoquinoline Alkaloids from Abuta Grandifolia. Planta Med. 1999, 65, 413–416. [Google Scholar] [CrossRef] [PubMed]
- Alamzeb, M.; Omer, M.; Ur-Rashid, M.; Raza, M.; Ali, S.; Khan, B.; Ullah, A. NMR, Novel Pharmacological and in Silico Docking Studies of Oxyacanthine and Tetrandrine: Bisbenzylisoquinoline Alkaloids Isolated from Berberis Glaucocarpa Roots. J. Anal. Methods Chem. 2018, 2018, 7692913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shangguan, Y.; He, J.; Kang, Y.; Wang, Y.; Yang, P.; Guo, J.; Huang, J. Structural Characterisation of Alkaloids in Leaves and Roots of Stephania Kwangsiensis by LC-QTOF-MS. Phytochem. Anal. 2018, 29, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Thuy, T.T.; Porzel, A.; Franke, K.; Wessjohann, L.; Sung, T. Van Isoquinolone and Protoberberine Alkaloids from Stephania Rotunda. Pharmazie 2005, 60, 701–704. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Song, N.; Lu, T.; Pan, Y.; Song, J.; Chen, G.; Sun, L.; Li, N. Rapid Characterization of TCM Qianjinteng by UPLC-QTOF-MS and Its Application in the Evaluation of Three Species of Stephania. J. Pharm. Biomed. Anal. 2018, 156, 284–296. [Google Scholar] [CrossRef]
- Bajpai, V.; Singh, A.; Chandra, P.; Negi, M.P.S.; Kumar, N.; Kumar, B. Analysis of Phytochemical Variations in Dioecious Tinospora Cordifolia Stems Using HPLC/QTOF MS/MS and UPLC/QqQLIT-MS/MS. Phytochem. Anal. 2016, 27, 92–99. [Google Scholar] [CrossRef]
- Bala, M.; Verma, P.K.; Awasthi, S.; Kumar, N.; Lal, B.; Singh, B. Chemical Prospection of Important Ayurvedic Plant Tinospora Cordifolia by UPLC-DAD-ESI-QTOF-MS/MS and NMR. Nat. Prod. Commun. 2015, 10, 43–48. [Google Scholar] [CrossRef] [Green Version]
- Boonen, J.; Sharma, V.; Dixit, V.K.; Burvenich, C.; De Spiegeleer, B. LC-MS N-Alkylamide Profiling of an Ethanolic Anacyclus Pyrethrum Root Extract. Planta Med. 2012, 78, 1787–1795. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.M.; El-Fishawy, A.M.; Slatikin, D.J.; Schiff, P.L., Jr. Quaternary Alkaloids of Tinospora Capillipes. Planta Med. 1984, 50, 88–90. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.; Yadav, A.; Dabur, R. Interactions of a Medicinal Climber Tinospora Cordifolia with Supportive Interspecific Plants Trigger the Modulation in Its Secondary Metabolic Profiles. Sci. Rep. 2019, 9, 14327. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Shi, Q.; Shi, P.; Zhang, W.; Cheng, Y. Characterization of Isoquinoline Alkaloids, Diterpenoids and Steroids in the Chinese Herb Jin-Guo-Lan (Tinospora Sagittata and Tinospora Capillipes) by High-Performance Liquid Chromatography/Electrospray Ionization with Multistage Mass Spectrometry. Rapid Commun. Mass Spectrom. 2006, 20, 2328–2342. [Google Scholar] [CrossRef]
- Deevanhxay, P.; Suzuki, M.; Maeshibu, N.; Li, H.; Tanaka, K.; Hirose, S. Simultaneous Characterization of Quaternary Alkaloids, 8-Oxoprotoberberine Alkaloids, and a Steroid Compound in Coscinium Fenestratum by Liquid Chromatography Hybrid Ion Trap Time-of-Flight Mass Spectrometry. J. Pharm. Biomed. Anal. 2009, 50, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Hocquemiller, R.; Cave, A.; Fournet., A. La Saulatine, Alcaloide Isoquinoléique Original Isole de Abuta Bullata. J. Nat. Prod. 1984, 47, 539–540. [Google Scholar] [CrossRef]
- Yu, L.L.; Li, R.T.; Ai, Y.B.; Liu, W.; Deng, Z.S.; Zou, Z.M. Protoberberine Isoquinoline Alkaloids from Arcangelisia Gusanlung. Molecules 2014, 19, 13332–13341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, H.A.; Min, B.-S.; Yokozawa, T.; Lee, J.-H.; Kim, Y.S.; Choi, J.S. Anti-Alzheimer and Antioxidant Activities of Coptidis Rhizoma Alkaloids. Biol. Pharm. Bull. 2009, 32, 1433–1438. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Zhou, S.; Zhang, M.; Feng, J.; Wang, S.; Wang, D.; Geng, Y.; Wang, X. An In Vitro AChE Inhibition Assay Combined with UF-HPLC-ESI-Q-TOF/MS Approach for Screening and Characterizing of AChE Inhibitors from Roots of Coptis Chinensis Franch. J. Pharm. Biomed. Anal. 2016, 120, 235–240. [Google Scholar] [CrossRef]
- Rabelo, D.D.M.; Pinheiro, M.L.B.; Barison, A.; Salomé, K.S.; Costa, E.V.; Silva, F.M.; Chaves, Y.O.; Bastos, I.D.S. Isoquinoline Alkaloids and Investigation of the Antibacterial and Antiplasmodial Activities of Guatteria Citriodora (Annonaceae). Quim. Nova 2014, 37, 1453–1458. [Google Scholar] [CrossRef]
- Qing, Z.-X.; Huang, J.-L.; Yang, X.-Y.; Liu, J.-H.; Cao, H.-L.; Xiang, F.; Cheng, P.; Zeng, J.-G. Anticancer and Reversing Multidrug Resistance Activities of Natural Isoquinoline Alkaloids and Their Structure-Activity Relationship. Curr. Med. Chem. 2018, 25, 5088–5114. [Google Scholar] [CrossRef]
- Cavin, A.; Hostettmann, K.; Dyatmyko, W.; Potterat, O. Antioxidant and Lipophilic Constituents of Tinospora Crispa. Planta Med. 1998, 64, 393–396. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, N.; Yonemitsu, M.; Kimura, T. Studies on the Constituents of the Stems of Tinospora Tuberculata Beumée. I. N-Trans- and N-Cis-Feruloyl Tiramine, and a New Phenolic Glucoside, Tinotuberide. Chem. Pharm. Bull. 1983, 31, 156–161. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.C.; Kingston, D.G.I. Two Phenolic Amides from Cocculus Diversifolius. Kor. J. Pharmacogn. 1995, 26, 273–275. [Google Scholar]
- Lopatriello, A.; Previtera, R.; Pace, S.; Werner, M.; Rubino, L.; Werz, O.; Taglialatela-scafati, O.; Forino, M. Phytochemistry NMR-Based Identi Fi Cation of the Major Bioactive Molecules from an Italian Cultivar of Lycium Barbarum. Phytochemistry 2017, 144, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Amaro, C.A.B.; González-Cortazar, M.; Herrera-Ruiz, M.; Román-Ramos, R.; Aguilar-Santamaría, L.; Tortoriello, J.; Jiménez-Ferrer, E. Hypoglycemic and Hypotensive Activity of a Root Extract of Smilax Aristolochiifolia, Standardized on N-Trans-Feruloyl-Tyramine. Molecules 2014, 19, 11366–11384. [Google Scholar] [CrossRef] [Green Version]
- Murray, A.P.; Faraoni, M.B.; Castro, M.J.; Alza, N.P.; Cavallaro, V. Natural AChE Inhibitors from Plants and Their Contribution to Alzheimer’s Disease Therapy. Curr. Neuropharmacol. 2013, 11, 388–413. [Google Scholar] [CrossRef] [Green Version]
- Giacobini, E. Cholinesterase Inhibitors: New Roles and Therapeutic Alternatives. Pharmacol. Res. 2004, 50, 433–440. [Google Scholar] [CrossRef]
- Greig, N.H.; Lahiri, D.K.; Sambamurti, K. Butyrylcholinesterase: An Important New Target in Alzheimer’s Disease Therapy. Int. Psychogeriatr. 2002, 14, 77–91. [Google Scholar] [CrossRef]
- Ballard, C.G. Advances in the Treatment of Alzheimer’s Disease: Benefits of Dual Cholinesterase Inhibition. Eur. Neurol. 2002, 47, 64–70. [Google Scholar] [CrossRef]
- Shim, H.J.; Lee, J.Y.; Kim, B.; Hong, J. General Fragmentations of Alkaloids in Electrospray Ionization Tandem Mass Spectrometry. Mass Spectrom. Lett. 2013, 4, 79–82. [Google Scholar] [CrossRef] [Green Version]
- Soares, E.R.; Da Silva, F.M.A.; De Almeida, R.A.; De Lima, B.R.; Da Silva Filho, F.A.; Barison, A.; Koolen, H.H.F.; Pinheiro, M.L.B.; De Souza, A.D.L. Direct Infusion ESI-IT-MSn Alkaloid Profile and Isolation of Tetrahydroharman and Other Alkaloids from Bocageopsis Pleiosperma Maas (Annonaceae). Phytochem. Anal. 2015, 26, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Stévigny, C.; Jiwan, J.L.H.; Rozenberg, R.; De Hoffmann, E.; Quetin-Leclercq, J. Key Fragmentation Patterns of Aporphine Alkaloids by Electrospray Ionization with Multistage Mass Spectrometry. Rapid Commun. Mass Spectrom. 2004, 18, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Clericuzio, M.; Tabasso, S.; Garbarino, J.A.; Piovano, M.; Cardile, V.; Russo, A.; Vidari, G. Non-Phenolic Dicinnamamides from Pholiota Spumosa: Isolation, Synthesis and Antitumour Activity. European J. Org. Chem. 2007, 2007, 5551–5559. [Google Scholar] [CrossRef]
- Schlittler, V.E.; Spitaler, U.; Weber, N. Uber Die Synthesen von Maytenin, N-Methylspermidin Und N-Methylmaytenin. Helv. Chim. Acta 1973, 56, 1097–1099. [Google Scholar] [CrossRef] [PubMed]
- Galarce-Bustos, O.; Pavón-Pérez, J.; Henríquez-Aedo, K.; Aranda, M. An Improved Method for a Fast Screening of α-Glucosidase Inhibitors in Cherimoya Fruit (Annona Cherimola Mill.) Applying Effect-Directed Analysis via High-Performance Thin-Layer Chromatography-Bioassay-Mass Spectrometry. J. Chromatogr. A 2019, 1608, 460415. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Q.-S.; Xu, L.-L.; Zhang, J.-Y.; Wang, Z.-J.; Jiang, Y.-Y.; Liu, B.; Jiao, Q.-S.; Xu, L.-L.; Zhang, J.-Y.; Wang, Z.-J.; et al. Rapid Characterization and Identification of Non-Diterpenoid Constituents in Tinospora Sinensis by HPLC-LTQ-Orbitrap MSn. Molecules 2018, 23, 274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oukoloff, K.; Coquelle, N.; Bartolini, M.; Naldi, M.; Le Guevel, R.; Bach, S.; Josselin, B.; Ruchaud, S.; Catto, M.; Pisani, L.; et al. Design, Biological Evaluation and X-Ray Crystallography of Nanomolar Multifunctional Ligands Targeting Simultaneously Acetylcholinesterase and Glycogen Synthase Kinase-3. Eur. J. Med. Chem. 2019, 168, 58–77. [Google Scholar] [CrossRef] [Green Version]
- Catto, M.; Pisani, L.; de la Mora, E.; Belviso, B.D.; Mangiatordi, G.F.; Pinto, A.; Palma, A.D.; Denora, N.; Caliandro, R.; Colletier, J.-P.; et al. Chiral Separation, X-Ray Structure, and Biological Evaluation of a Potent and Reversible Dual Binding Site AChE Inhibitor. ACS Med. Chem. Lett. 2020, 11, 869–876. [Google Scholar] [CrossRef]
- Galdeano, C.; Coquelle, N.; Cieslikiewicz-Bouet, M.; Bartolini, M.; Pérez, B.; Clos, M.; Silman, I.; Jean, L.; Colletier, J.-P.; Renard, P.-Y.; et al. Increasing Polarity in Tacrine and Huprine Derivatives: Potent Anticholinesterase Agents for the Treatment of Myasthenia Gravis. Molecules 2018, 23, 634. [Google Scholar] [CrossRef] [Green Version]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Rosenberry, T.; Brazzolotto, X.; Macdonald, I.; Wandhammer, M.; Trovaslet-Leroy, M.; Darvesh, S.; Nachon, F. Comparison of the Binding of Reversible Inhibitors to Human Butyrylcholinesterase and Acetylcholinesterase: A Crystallographic, Kinetic and Calorimetric Study. Molecules 2017, 22, 2098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Chen, L.; Sun, J. Oxoisoaporphine Alkaloids: Prospective Anti-Alzheimer’s Disease, Anticancer, and Antidepressant Agents. ChemMedChem 2018, 13, 1262–1274. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.; Sorribas, A.; Howes, M.-J.R. Natural Products as a Source of Alzheimer’s Drug Leads. Nat. Prod. Rep. 2011, 28, 48–77. [Google Scholar] [CrossRef] [PubMed]
- Miller-Fleming, L.; Olin-Sandoval, V.; Campbell, K.; Ralser, M. Remaining Mysteries of Molecular Biology: The Role of Polyamines in the Cell. J. Mol. Biol. 2015, 427, 3389–3406. [Google Scholar] [CrossRef] [PubMed]
- Gilad, G.M.; Gilad, V.H. Early Polyamine Treatment Enhances Survival of Sympathetic Neurons after Postnatal Axonal Injury or Immunosympathectomy. Dev. Brain Res. 1988, 38, 175–181. [Google Scholar] [CrossRef]
- Morrison, L.D.; Kish, S.J. Brain Polyamine Levels Are Altered in Alzheimer’s Disease. Neurosci. Lett. 1995, 197, 5–8. [Google Scholar] [CrossRef]
- Yadav, M.; Parle, M.; Jindal, D.K.; Sharma, N. Potential Effect of Spermidine on GABA, Dopamine, Acetylcholinesterase, Oxidative Stress and Proinflammatory Cytokines to Diminish Ketamine-Induced Psychotic Symptoms in Rats. Biomed. Pharmacother. 2018, 98, 207–213. [Google Scholar] [CrossRef]
- Greig, N.H.; Utsuki, T.; Yu, Q.; Zhu, X.; Holloway, H.W.; Perry, T.; Lee, B.; Ingram, D.K.; Lahiri, D.K. A New Therapeutic Target in Alzheimer’s Disease Treatment: Attention to Butyrylcholinesterase. Curr. Med. Res. Opin. 2001, 17, 159–165. [Google Scholar] [CrossRef]
- Cheng, Y.; Li, H.L.; Zhou, Z.W.; Long, H.Z.; Luo, H.Y.; Wen, D.D.; Cheng, L.; Gao, L.C. Isoliensinine: A Natural Compound with “Drug-Like” Potential. Front. Pharmacol. 2021, 12, 852. [Google Scholar] [CrossRef]
- Lin, Z.; Wang, H.; Fu, Q.; An, H.; Liang, Y.; Zhang, B.; Hashi, Y.; Chen, S. Simultaneous Separation, Identification and Activity Evaluation of Three Butyrylcholinesterase Inhibitors from Plumula Nelumbinis Using on-Line HPLC-UV Coupled with ESI-IT-TOF-MS and BChE Biochemical Detection. Talanta 2013, 110, 180–189. [Google Scholar] [CrossRef]
- Singh, N.; Sharma, B. Toxicological Effects of Berberine and Sanguinarine. Front. Mol. Biosci. 2018, 5, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, H.F.; Shen, L. Berberine: A Potential Multipotent Natural Product to Combat Alzheimer’s Disease. Molecules 2011, 16, 6732–6740. [Google Scholar] [CrossRef] [PubMed]
- Fontes Barbosa, M.; Benatti Justino, A.; Machado Martins, M.; Roberta Anacleto Belaz, K.; Barbosa Ferreira, F.; Junio de Oliveira, R.; Danuello, A.; Salmen Espindola, F.; Pivatto, M. Cholinesterase Inhibitors Assessment of Aporphine Alkaloids from Annona Crassiflora and Molecular Docking Studies. Bioorg. Chem. 2022, 120, 105593. [Google Scholar] [CrossRef] [PubMed]
- Sichaem, J.; Tip-pyang, S.; Lugsanangarm, K. Bioactive Aporphine Alkaloids from the Roots of Artabotrys Spinosus: Cholinesterase Inhibitory Activity and Molecular Docking Studies. Nat. Prod. Commun. 2018, 13, 1279–1282. [Google Scholar] [CrossRef] [Green Version]
- Šali, A.; Blundell, T.L. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Schrödinger Release 2020-4: Small-Molecule Drug Discovery Suite, Schrödinger, LLC, New York, NY. 2020. Available online: https://www.schrodinger.com/products/maestro (accessed on 12 May 2022).
- Schrödinger Release 2020-4: Protein Preparation Wizard; Epik, Schrödinger, LLC, New York, NY. 2020. Available online: https://www.schrodinger.com/science-articles/protein-preparation-wizard (accessed on 12 May 2022).
- Schrödinger Release 2020-4: Impact, Schrödinger, LLC, New York, NY. 2020. Available online: https://www.schrodinger.com/products/maestro (accessed on 12 May 2022).
- Schrödinger Release 2020-4: Prime, Schrödinger, LLC, New York, NY. 2020. Available online: https://www.schrodinger.com/products/prime (accessed on 12 May 2022).
- Schrödinger Release 2020-4: LigPrep, Schrödinger, LLC, New York, NY. 2020. Available online: https://www.schrodinger.com/products/ligprep (accessed on 12 May 2022).
- Roos, K.; Wu, C.; Damm, W.; Reboul, M.; Stevenson, J.M.; Lu, C.; Dahlgren, M.K.; Mondal, S.; Chen, W.; Wang, L.; et al. OPLS3e: Extending Force Field Coverage for Drug-Like Small Molecules. J. Chem. Theory Comput. 2019, 15, 1863–1874. [Google Scholar] [CrossRef]
- Schrödinger Release 2020-4: Induced Fit Docking Protocol; Glide, Schrödinger, LLC, New York, NY. 2020. Available online: https://www.schrodinger.com/products/glide (accessed on 12 May 2022).
- Sherman, W.; Beard, H.S.; Farid, R. Use of an Induced Fit Receptor Structure in Virtual Screening. Chem. Biol. Drug Des. 2006, 67, 83–84. [Google Scholar] [CrossRef]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel Procedure for Modeling Ligand/Receptor Induced Fit Effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef]
- Schrödinger Release 2020-4: Maestro-Desmond Interoperability Tools, Schrödinger, LLC, New York, NY. 2020. Available online: https://www.schrodinger.com/products/desmond (accessed on 12 May 2022).
- Schrödinger Release 2020-4: Desmond Molecular Dynamics System, Schrödinger, LLC, New York, NY. 2020. Available online: https://www.schrodinger.com/products/desmond (accessed on 12 May 2022).
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. A New and Rapid Colorimetric Determination of Acetylcholinesterase Activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Senol, F.S.; Orhan, I.E.; Ustun, O. In Vitro Cholinesterase Inhibitory and Antioxidant Effect of Selected Coniferous Tree Species. Asian Pac. J. Trop. Med. 2015, 8, 269–275. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | IC50 (µM) | ΔG(AChE) | ΔG(BChE) |
|---|---|---|---|
| lindoldhamine isomer 1 | 39.38 ± 0.08 | −114.60 | −110.15 |
| stepharine 2 | 61.24 ± 0.03 | −68.87 | −66.01 |
| palmatine 3 | 35.25 ± 0.04 | −74.87 | −82.64 |
| 5-N-methylmaytenine 4 | 19.55 ± 0.09 | −82.01 | −68.49 |
| N-trans-feruloyltyramine 5 | not active | −68.43 | −67.20 |
| neostigmine (positive control) | 3.72 ± 0.03 | −61.71 | −32.18 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
da Silva Mesquita, R.; Kyrylchuk, A.; Cherednichenko, A.; Costa Sá, I.S.; Macedo Bastos, L.; Moura Araújo da Silva, F.; Saraiva Nunomura, R.d.C.; Grafov, A. In Vitro and In Silico Evaluation of Cholinesterase Inhibition by Alkaloids Obtained from Branches of Abuta panurensis Eichler. Molecules 2022, 27, 3138. https://doi.org/10.3390/molecules27103138
da Silva Mesquita R, Kyrylchuk A, Cherednichenko A, Costa Sá IS, Macedo Bastos L, Moura Araújo da Silva F, Saraiva Nunomura RdC, Grafov A. In Vitro and In Silico Evaluation of Cholinesterase Inhibition by Alkaloids Obtained from Branches of Abuta panurensis Eichler. Molecules. 2022; 27(10):3138. https://doi.org/10.3390/molecules27103138
Chicago/Turabian Styleda Silva Mesquita, Rochelly, Andrii Kyrylchuk, Anton Cherednichenko, Ingrity Suelen Costa Sá, Lílian Macedo Bastos, Felipe Moura Araújo da Silva, Rita de Cássia Saraiva Nunomura, and Andriy Grafov. 2022. "In Vitro and In Silico Evaluation of Cholinesterase Inhibition by Alkaloids Obtained from Branches of Abuta panurensis Eichler" Molecules 27, no. 10: 3138. https://doi.org/10.3390/molecules27103138