
Differential Effects of Histone Deacetylases on the Expression of NKG2D Ligands and NK Cell-Mediated Anticancer Immunity in Lung Cancer Cells

Abstract
:1. Introduction
2. Results
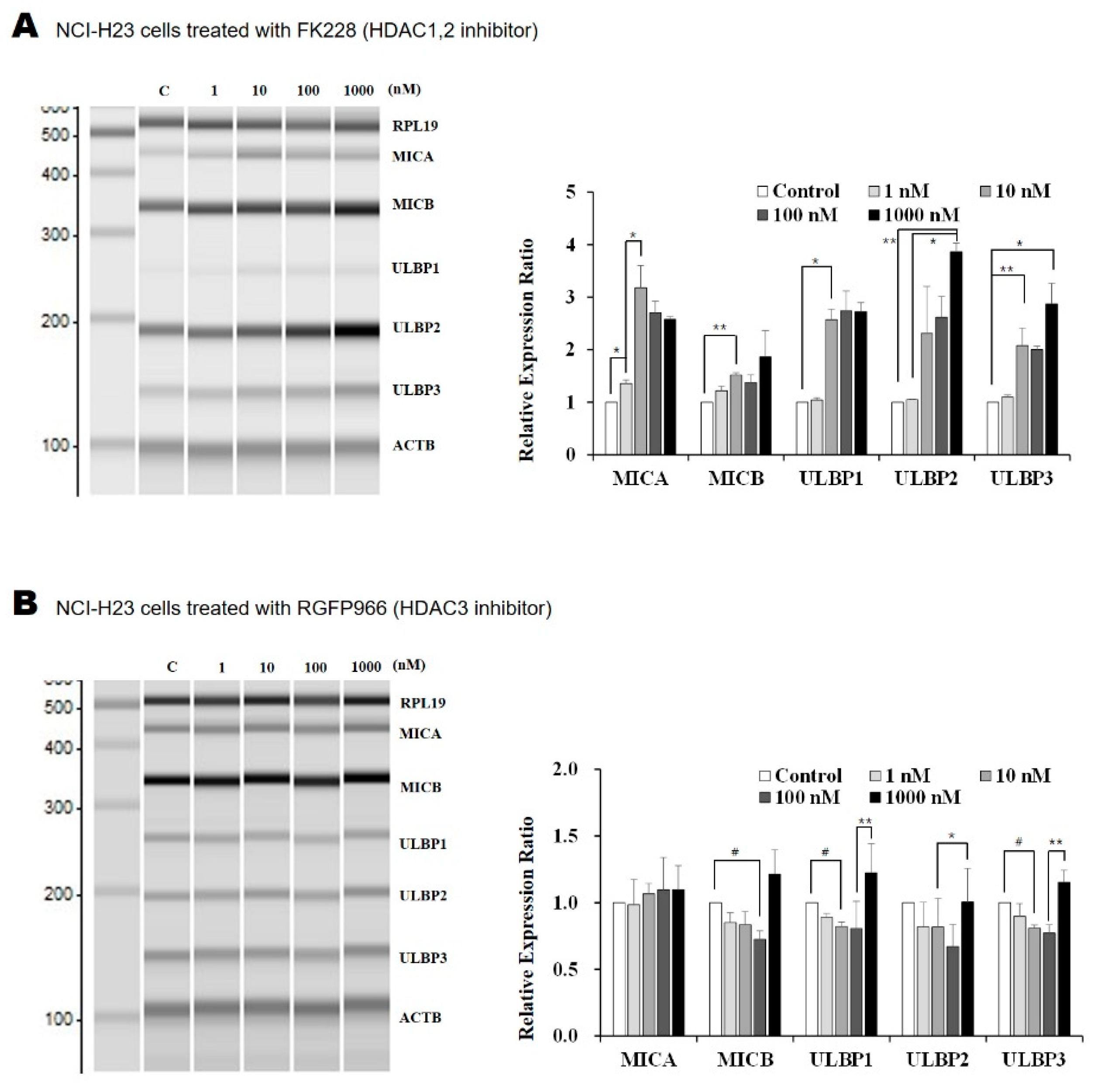
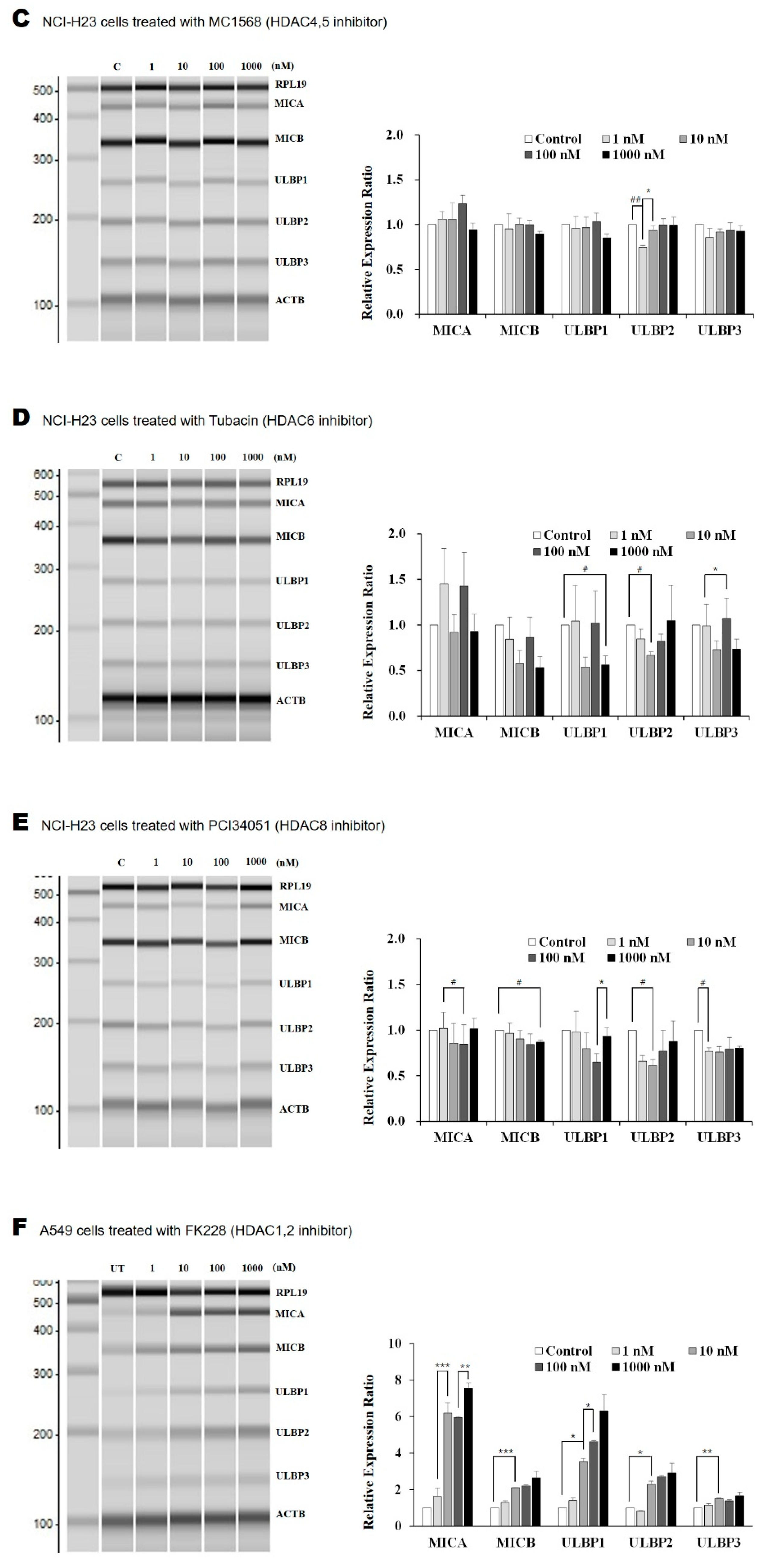
2.1. The HDAC1/2 Inhibitor, FK228, Increased Transcription of NKG2D Ligands in Lung Cancer Cells
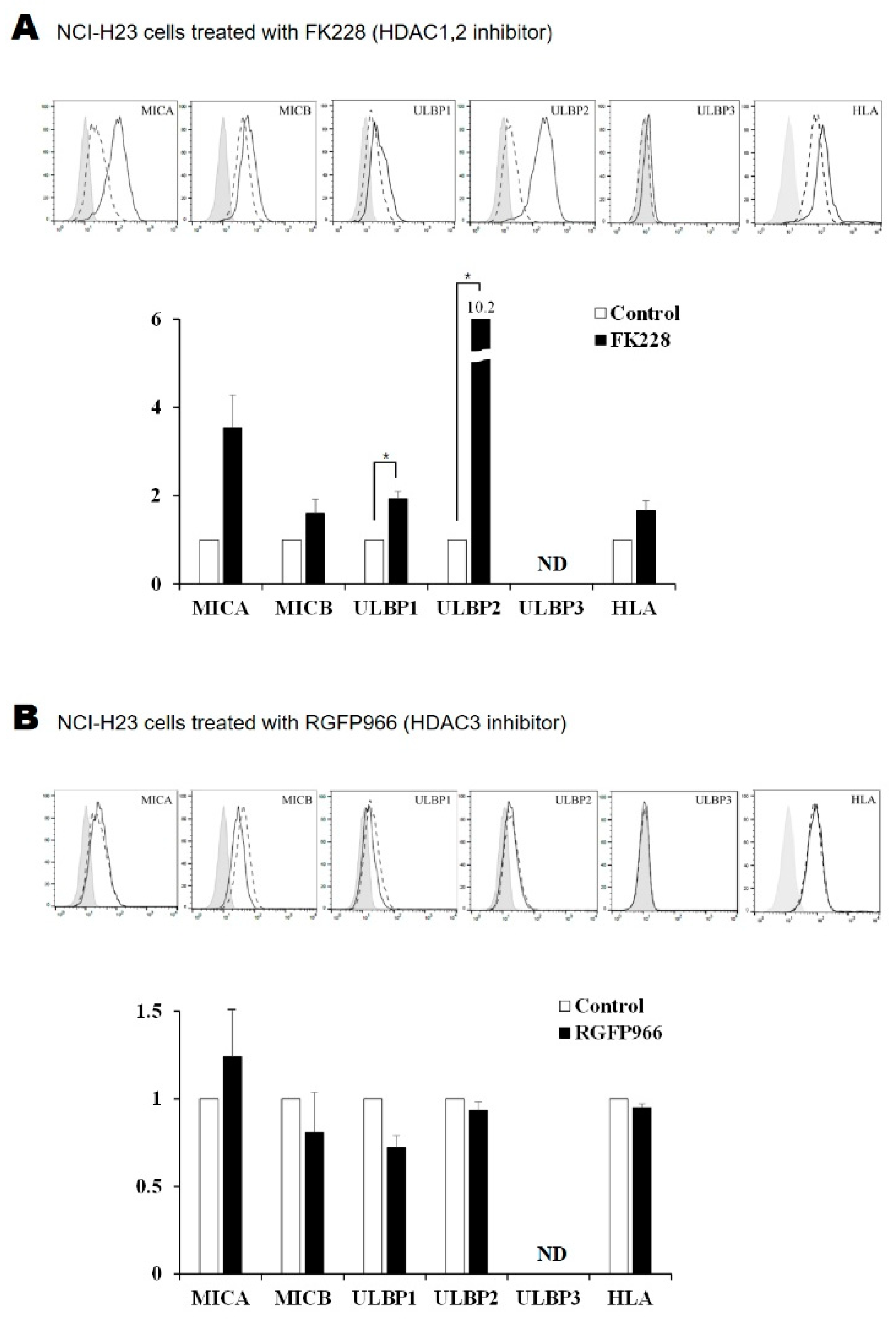
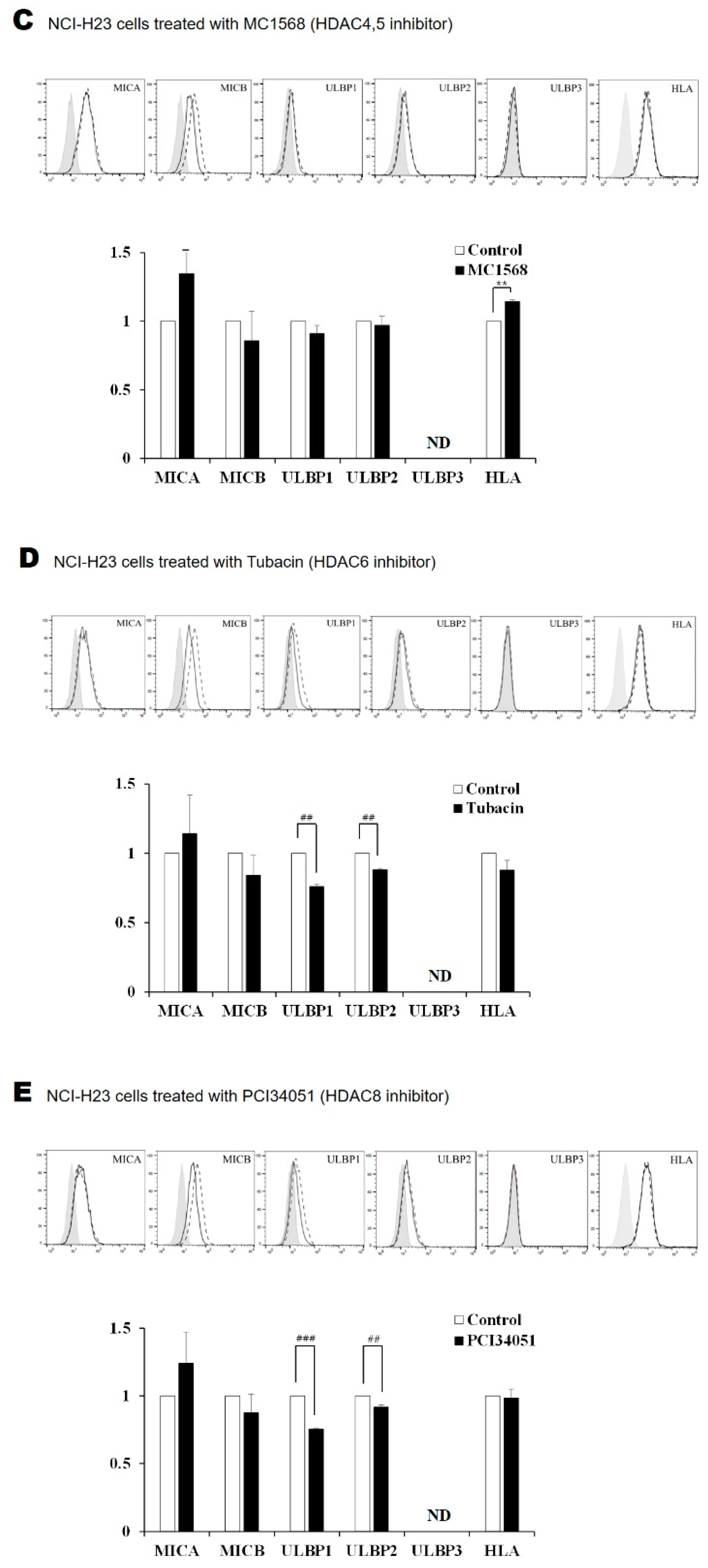
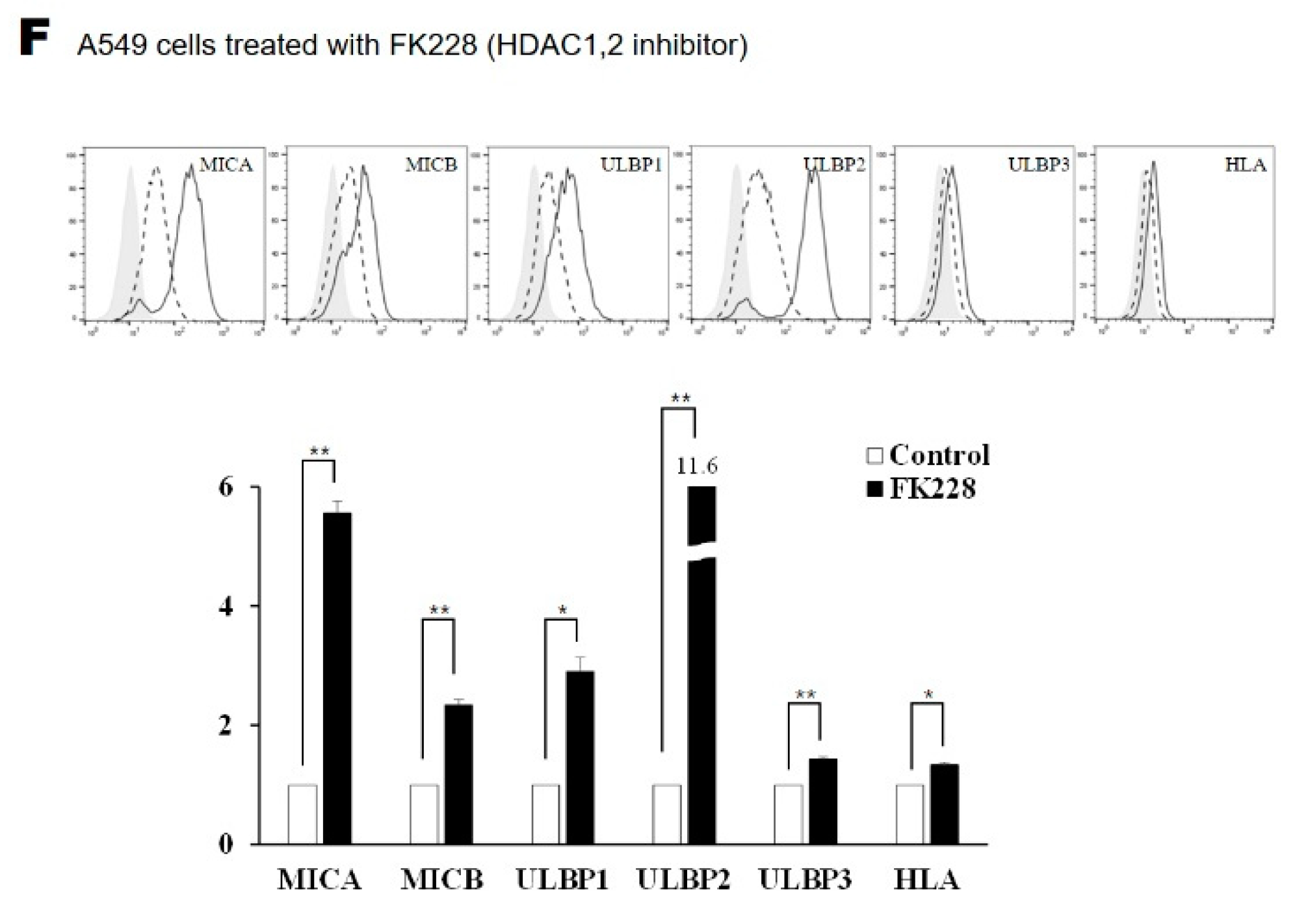
2.2. Surface Expression of NKG2D Ligands in Lung Cancer Cells Is Increased by FK228 Treatment
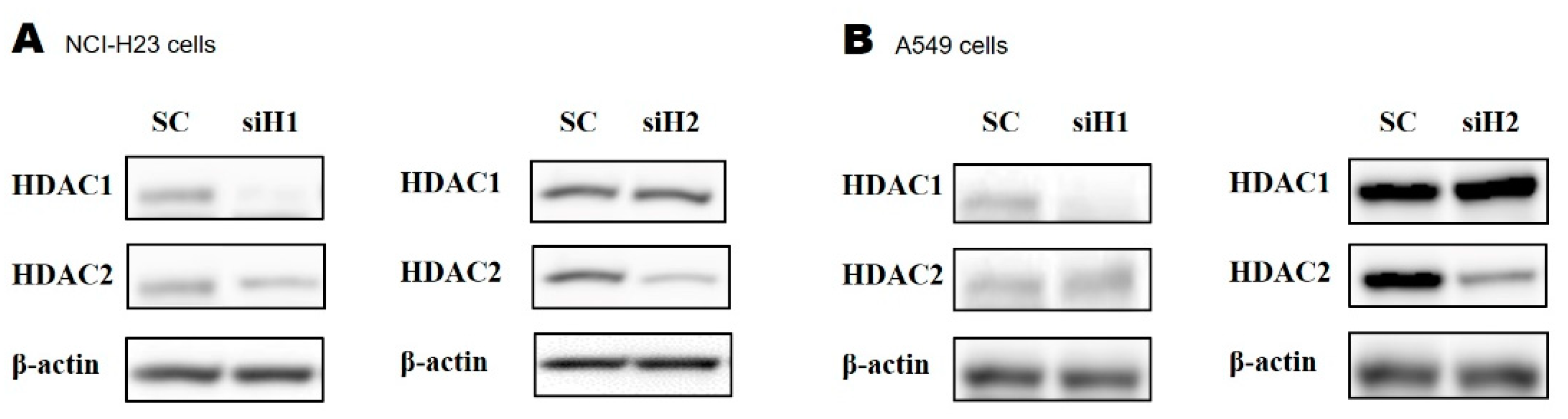
2.3. Silencing HDAC1 or HDAC2 Using Small Interfering RNA (siRNA)
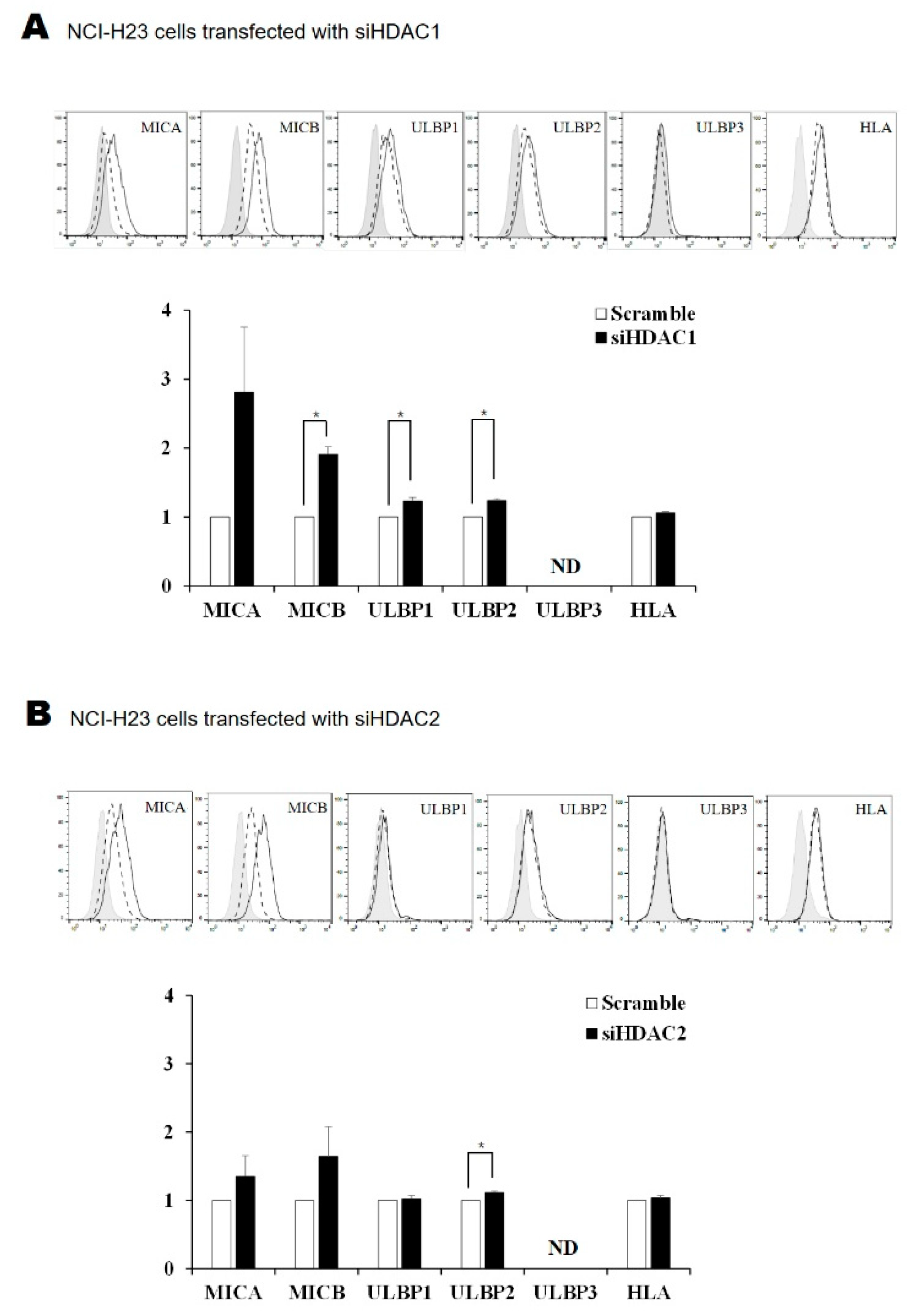
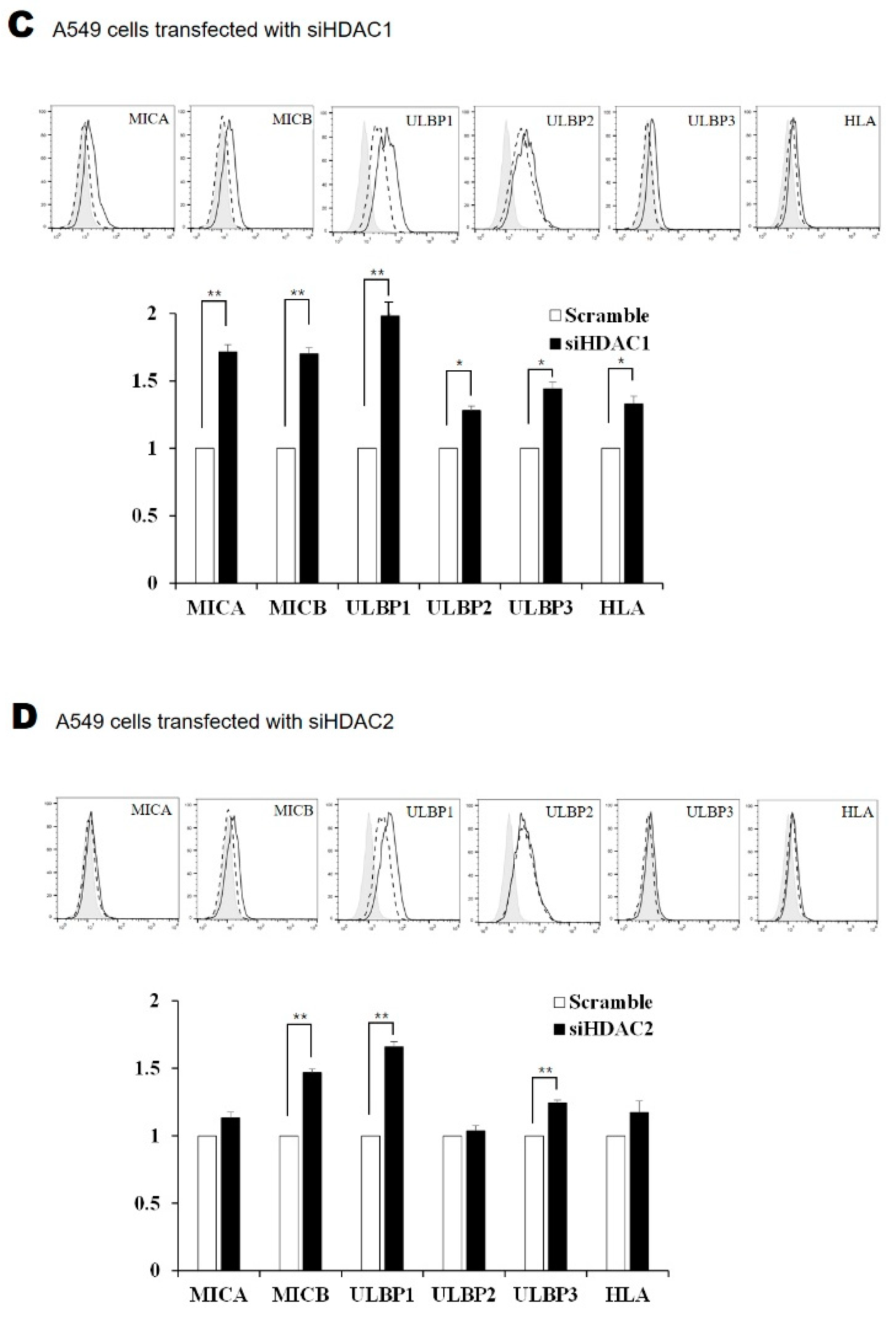
2.4. Silencing HDAC1 and HDAC2 Increased the Surface Expression of NKG2D Ligands in Lung Cancer Cells
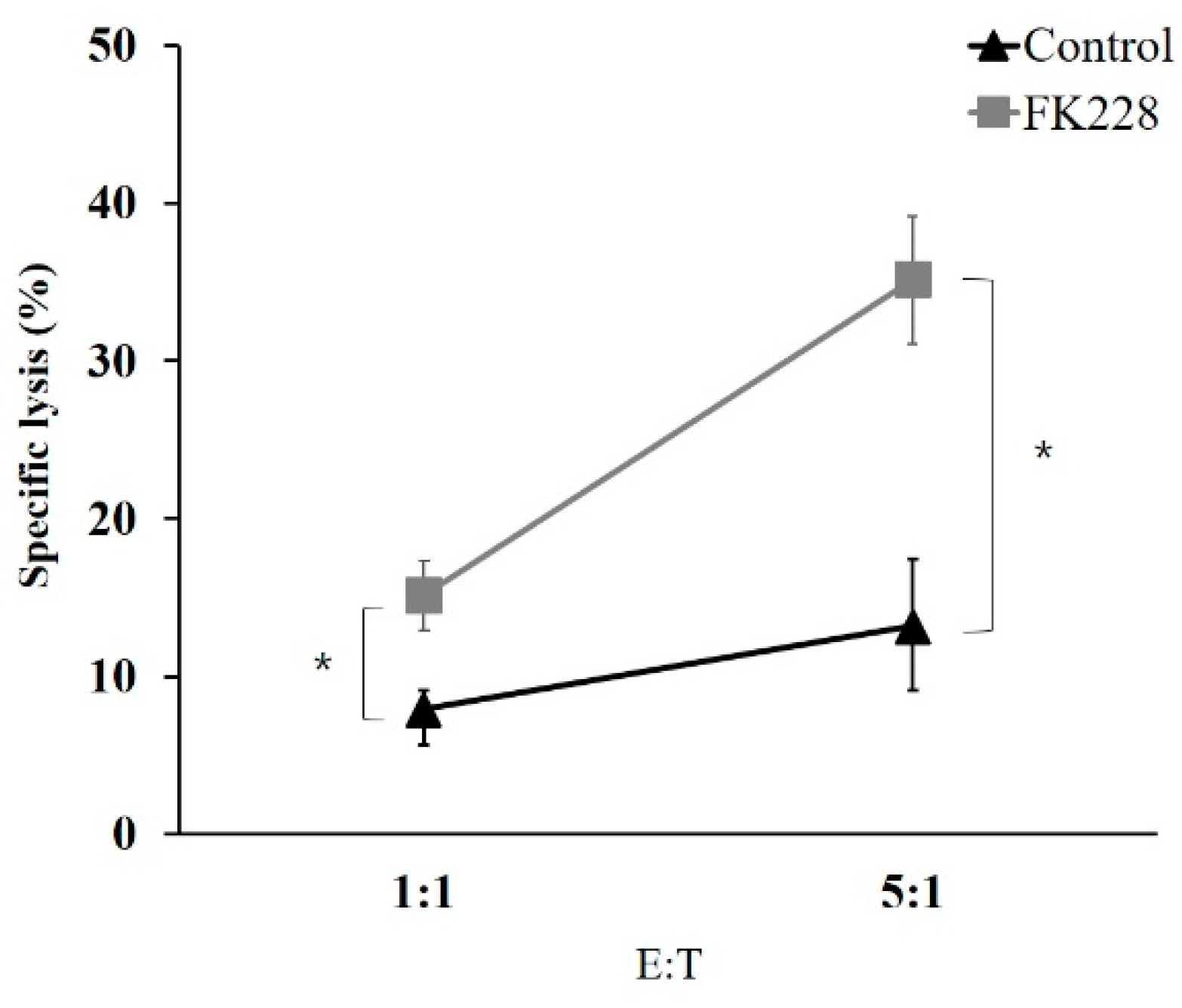
2.5. The HDAC1/2 Inhibitor, FK228, Increases the Susceptibility of NCI-H23 Cells to NK Cells
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Reagents
4.2. Total RNA Extraction and Multiplex Reverse Transcription PCR (RT-PCR)
4.3. Flow Cytometry
4.4. Flow Cytometry Analysis of the NK Cell-Mediated Cytotoxicity Assay
4.5. Silencing HDAC1 or HDAC2 Using siRNA Transfection
4.6. Western Blot Analysis
4.7. Statistical Analysis
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Dela Cruz, C.S.; Tanoue, L.T.; Matthay, R.A. Lung cancer: Epidemiology, etiology, and prevention. Clin. Chest. Med. 2011, 32, 605–644. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soo, R.A.; Chen, Z.; Yan Teng, R.S.; Tan, H.L.; Iacopetta, B.; Tai, B.C.; Soong, R. Prognostic significance of immune cells in non-small cell lung cancer: Meta-analysis. Oncotarget 2018, 9, 24801–24820. [Google Scholar] [CrossRef] [Green Version]
- Jamieson, A.M.; Diefenbach, A.; McMahon, C.W.; Xiong, N.; Carlyle, J.R.; Raulet, D.H. The role of the NKG2D immunoreceptor in immune cell activation and natural killing. Immunity 2002, 17, 19–29. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, S.; Lopez-Soto, A.; Suarez-Alvarez, B.; Lopez-Vazquez, A.; Lopez-Larrea, C. NKG2D ligands: Key targets of the immune response. Trends Immunol. 2008, 29, 397–403. [Google Scholar] [CrossRef]
- Park, S.W.; Bae, J.H.; Kim, S.D.; Son, Y.O.; Kim, J.Y.; Park, H.J.; Lee, C.H.; Park, D.Y.; Kim, J.Y.; Lee, M.K.; et al. Comparison of level of NKG2D ligands between normal and tumor tissue using multiplex RT-PCR. Cancer Investig. 2007, 25, 299–307. [Google Scholar] [CrossRef]
- Huergo-Zapico, L.; Acebes-Huerta, A.; Lopez-Soto, A.; Villa-Alvarez, M.; Gonzalez-Rodriguez, A.P.; Gonzalez, S. Molecular Bases for the Regulation of NKG2D Ligands in Cancer. Front. Immunol. 2014, 5, 106. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.S.; Heo, W.; Son, C.H.; Kang, C.D.; Park, Y.S.; Bae, J. Upregulation of Myc promotes the evasion of NK cellmediated immunity through suppression of NKG2D ligands in K562 cells. Mol. Med. Rep. 2019, 20, 3301–3307. [Google Scholar]
- Nausch, N.; Cerwenka, A. NKG2D ligands in tumor immunity. Oncogene 2008, 27, 5944–5958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stern-Ginossar, N.; Mandelboim, O. An integrated view of the regulation of NKG2D ligands. Immunology 2009, 128, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.N.; Tomasi, T.B. Histone deacetylase regulation of immune gene expression in tumor cells. Immunol. Res. 2008, 40, 164–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, J.; Dubin, S.; Dananberg, A.; Quabius, E.S.; Fritsch, J.; Dowds, C.M.; Saxena, A.; Chitadze, G.; Lettau, M.; Kabelitz, D. Histone Deacetylase Inhibitor Modulates NKG2D Receptor Expression and Memory Phenotype of Human Gamma/Delta T Cells Upon Interaction With Tumor Cells. Front. Immunol. 2019, 10, 569. [Google Scholar] [CrossRef] [Green Version]
- Balasubramanian, S.; Ramos, J.; Luo, W.; Sirisawad, M.; Verner, E.; Buggy, J.J. A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas. Leukemia 2008, 22, 1026–1034. [Google Scholar] [CrossRef] [PubMed]
- Furumai, R.; Matsuyama, A.; Kobashi, N.; Lee, K.H.; Nishiyama, M.; Nakajima, H.; Tanaka, A.; Komatsu, Y.; Nishino, N.; Yoshida, M.; et al. FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases. Cancer Res. 2002, 62, 4916–4921. [Google Scholar] [PubMed]
- Malvaez, M.; McQuown, S.C.; Rogge, G.A.; Astarabadi, M.; Jacques, V.; Carreiro, S.; Rusche, J.R.; Wood, M.A. HDAC3-selective inhibitor enhances extinction of cocaine-seeking behavior in a persistent manner. Proc. Natl. Acad. Sci. USA 2013, 110, 2647–2652. [Google Scholar] [CrossRef] [Green Version]
- Griffin, E.A.; Melas, P.A.; Zhou, R.; Li, Y.; Mercado, P.; Kempadoo, K.A.; Stephenson, S.; Colnaghi, L.; Taylor, K.; Hu, M.C.; et al. Prior alcohol use enhances vulnerability to compulsive cocaine self-administration by promoting degradation of HDAC4 and HDAC5. Sci. Adv. 2017, 3, e1701682. [Google Scholar] [CrossRef] [Green Version]
- Haggarty, S.J.; Koeller, K.M.; Wong, J.C.; Grozinger, C.M.; Schreiber, S.L. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc. Natl. Acad. Sci. USA 2003, 100, 4389–4394. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold. Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Ropero, S.; Esteller, M. The role of histone deacetylases (HDACs) in human cancer. Mol. Oncol. 2007, 1, 19–25. [Google Scholar] [CrossRef]
- Chen, H.P.; Zhao, Y.T.; Zhao, T.C. Histone deacetylases and mechanisms of regulation of gene expression. Crit. Rev. Oncog. 2015, 20, 35–47. [Google Scholar] [CrossRef]
- Kulka, L.A.M.; Fangmann, P.V.; Panfilova, D.; Olzscha, H. Impact of HDAC Inhibitors on Protein Quality Control Systems: Consequences for Precision Medicine in Malignant Disease. Front. Cell Dev. Biol. 2020, 8, 425. [Google Scholar] [CrossRef]
- Zhao, G.X.; Pan, H.; Ouyang, D.Y.; He, X.H. The critical molecular interconnections in regulating apoptosis and autophagy. Ann. Med. 2015, 47, 305–315. [Google Scholar] [CrossRef]
- Yang, X.J.; Seto, E. HATs and HDACs: From structure, function and regulation to novel strategies for therapy and prevention. Oncogene 2007, 26, 5310–5318. [Google Scholar] [CrossRef] [PubMed]
- Shigematsu, N.; Ueda, H.; Takase, S.; Tanaka, H.; Yamamoto, K.; Tada, T. FR901228, a novel antitumor bicyclic depsipeptide produced by Chromobacterium violaceum No. 968. II. Structure determination. J. Antibiot. 1994, 47, 311–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueda, H.; Manda, T.; Matsumoto, S.; Mukumoto, S.; Nishigaki, F.; Kawamura, I.; Shimomura, K. FR901228, a novel antitumor bicyclic depsipeptide produced by Chromobacterium violaceum No. 968. III. Antitumor activities on experimental tumors in mice. J. Antibiot. 1994, 47, 315–323. [Google Scholar] [CrossRef] [Green Version]
- Ueda, H.; Nakajima, H.; Hori, Y.; Fujita, T.; Nishimura, M.; Goto, T.; Okuhara, M. FR901228, a novel antitumor bicyclic depsipeptide produced by Chromobacterium violaceum No. 968. I. Taxonomy, fermentation, isolation, physico-chemical and biological properties, and antitumor activity. J. Antibiot. 1994, 47, 301–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wells, C.E.; Bhaskara, S.; Stengel, K.R.; Zhao, Y.; Sirbu, B.; Chagot, B.; Cortez, D.; Khabele, D.; Chazin, W.J.; Cooper, A.; et al. Inhibition of histone deacetylase 3 causes replication stress in cutaneous T cell lymphoma. PLoS ONE 2013, 8, 68915. [Google Scholar] [CrossRef] [PubMed]
- Scognamiglio, A.; Nebbioso, A.; Manzo, F.; Valente, S.; Mai, A.; Altucci, L. HDAC-class II specific inhibition involves HDAC proteasome-dependent degradation mediated by RANBP2. Biochim. Biophys. Acta 2008, 1783, 2030–2038. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, A.; Melesina, J.; Kolbinger, F.R.; Oehme, I.; Senger, J.; Witt, O.; Sippl, W.; Jung, M. Targeting histone deacetylase 8 as a therapeutic approach to cancer and neurodegenerative diseases. Future Med. Chem. 2016, 8, 1609–1634. [Google Scholar] [CrossRef]
- Rettig, I.; Koeneke, E.; Trippel, F.; Mueller, W.C.; Burhenne, J.; Kopp-Schneider, A.; Fabian, J.; Schober, A.; Fernekorn, U.; von Deimling, A.; et al. Selective inhibition of HDAC8 decreases neuroblastoma growth in vitro and in vivo and enhances retinoic acid-mediated differentiation. Cell Death Dis. 2015, 6, e1657. [Google Scholar] [CrossRef]
- Son, C.H.; Keum, J.H.; Yang, K.; Nam, J.; Kim, M.J.; Kim, S.H.; Kang, C.D.; Oh, S.O.; Kim, C.D.; Park, Y.S.; et al. Synergistic enhancement of NK cell-mediated cytotoxicity by combination of histone deacetylase inhibitor and ionizing radiation. Radiat. Oncol. 2014, 9, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| The Classification of HDACs and Their Selective Inhibitors | ||||
|---|---|---|---|---|
| Zinc-dependent | Class I | HDAC1 | FK228 | Resveratrol |
| HDAC2 | ||||
| HDAC3 | RGFP966 | |||
| HDAC8 | RGFP966 | |||
| Class IIa | HDAC4 | MC1568 | ||
| HDAC5 | ||||
| HDAC7 | ||||
| HDAC9 | ||||
| Class IIb | HDAC6 | Tubacin | ||
| HDAC10 | ||||
| Class IV | HDAC11 | |||
| DAD-dependent | Class III | Sirtuin | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cho, H.; Son, W.-C.; Lee, Y.-S.; Youn, E.J.; Kang, C.-D.; Park, Y.-S.; Bae, J. Differential Effects of Histone Deacetylases on the Expression of NKG2D Ligands and NK Cell-Mediated Anticancer Immunity in Lung Cancer Cells. Molecules 2021, 26, 3952. https://doi.org/10.3390/molecules26133952
Cho H, Son W-C, Lee Y-S, Youn EJ, Kang C-D, Park Y-S, Bae J. Differential Effects of Histone Deacetylases on the Expression of NKG2D Ligands and NK Cell-Mediated Anticancer Immunity in Lung Cancer Cells. Molecules. 2021; 26(13):3952. https://doi.org/10.3390/molecules26133952
Chicago/Turabian StyleCho, Haeryung, Woo-Chang Son, Young-Shin Lee, Eun Jung Youn, Chi-Dug Kang, You-Soo Park, and Jaeho Bae. 2021. "Differential Effects of Histone Deacetylases on the Expression of NKG2D Ligands and NK Cell-Mediated Anticancer Immunity in Lung Cancer Cells" Molecules 26, no. 13: 3952. https://doi.org/10.3390/molecules26133952