 Jinxin Liu1†
Jinxin Liu1† Mengmeng Shi1,2†
Mengmeng Shi1,2† Zhaolei Zhang1,2
Zhaolei Zhang1,2 Hongbo Xie2
Hongbo Xie2 Weijun Kong3
Weijun Kong3 Qiuling Wang1
Qiuling Wang1 Xinlei Zhao1
Xinlei Zhao1 Chunying Zhao2Yulin Lin1
Chunying Zhao2Yulin Lin1 Xiaoxia Zhang4*
Xiaoxia Zhang4* Linchun Shi1*
Linchun Shi1*- 1Key Laboratory of Chinese Medicine Resources Conservation, State Administration of Traditional Chinese Medicine of the People’s Republic of China, Engineering Research Center of Chinese Medicine Resource of Ministry of Education, Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing, China
- 2Hebei Key Laboratory of Study and Exploitation of Chinese Medicine, Chengde Medical University, Chengde, China
- 3School of Traditional Chinese Medicine, Capital Medical University, Beijing, China
- 4State Key Laboratory of Systematic and Evolutionary Botany, Institute of Botany, Chinese Academy of Sciences, Beijing, China
Atractylodes species are widely distributed across East Asia and are cultivated as medicinal herbs in China, Japan, and Korea. Their unclear morphological characteristics and low levels of genetic divergence obscure the taxonomic relationships among these species. In this study, 24 plant samples were collected representing five species of Atractylodes located in China; of these, 23 belonged to members of the A. lancea complex. High-throughput sequencing was used to obtain the concatenated nrDNA sequences (18S-ITS1-5.8S-ITS2-28S) and plastid genomes. The concatenated nrDNA sequence lengths for all the Atractylodes species were 5,849 bp, and the GC content was 55%. The lengths of the whole plastid genome sequences ranged from 152,138 bp (A. chinensis) to 153,268 bp (A. lancea), while their insertion/deletion sites were mainly distributed in the intergenic regions. Furthermore, 33, 34, 36, 31, and 32 tandem repeat sequences, as well as 30, 30, 29, 30, and 30 SSR loci, were detected in A. chinensis, A. koreana, A. lancea, A. japonica, and A. macrocephala, respectively. In addition to these findings, a considerable number of heteroplasmic variations were detected in the plastid genomes, implying a complicated phylogenetic history for Atractylodes. The results of the phylogenetic analysis involving concatenated nrDNA sequences showed that A. lancea and A. japonica formed two separate clades, with A. chinensis and A. koreana constituting their sister clade, while A. lancea, A. koreana, A. chinensis, and A. japonica were found based on plastid datasets to represent a mixed clade on the phylogenetic tree. Phylogenetic network analysis suggested that A. lancea may have hybridized with the common ancestor of A. chinensis and A. japonica, while ABBA–BABA tests of SNPs in the plastid genomes showed that A. chinensis was more closely related to A. japonica than to A. lancea. This study reveals the extensive discordance and complexity of the relationships across the members of the A. lancea complex (A. lancea, A. chinensis, A. koreana, and A. japonica) according to cytonuclear genomic data; this may be caused by interspecific hybridization or gene introgression.
Introduction
Atractylodes, mainly distributed across East Asia, is a perennial herb from the Asteraceae family. The dried rhizomes of Atractylodes plants have been used for more than 2,000 years as traditional herbal medicines (“cangzhu” and “baizhu” in China, and so-jutsu and byaku-jutsu in Japan) to treat gastroduodenal diseases and colds (Yin et al., 2015; Deng et al., 2016). Due to the limitations of wild resources, the plants of Atractylodes have been cultivated in China since the 1980s, and production of their dried rhizome has now reached 5000 tons per year. However, the cultivation of particular species of Atractylodes presents the challenge of heterogeneous germplasm, which is mainly caused by high variability and continuous variation in morphological features among individual plants (Deng et al., 2016). In 1959, A. chinensis DC. was considered an independent species in Northeast Medicinal Flora (Liu, 1959) and was divided into several variants, such as A. chinensis var. koreana (Nakai) Chu, A. chinensis DC. var. simplicifolia (Loes.) Chu, and A. chinensis DC. var. liaotungensis Kitag. In 1981, Shi (Zhu, 1981) considered pinnatipartite leaves to be a volatile mutation, and further claimed, following an in-depth analysis of documented specimens and the literature, that the labels A. chinensis and A. lancea were actually used to refer to a single species. In 1987, A. chinensis was treated as a synonym of A. lancea according to the law of priority nomenclature in Flora Reipublicae Popularis Sinicae (Lin and Shi, 1987). In 2011, according to Flora of China, A. japonica was also treated as a synonym of A. lancea; at present, A. lancea is an overcrowded group whose leaves are described as undivided or divided almost to base into 3-5(-9) pinnately arranged segments (Shi, 2011). The nomenclatural history of Atractylodes is illustrated in Supplementary Figure 1. Although several molecular markers have been used to investigate the taxonomic and phylogenetic characteristics of Atractylodes (Peng et al., 2012; Kim et al., 2016), there is still taxonomic controversy surrounding this genus, arising primarily from the interspecific and intraspecific taxonomic treatment of the A. lancea complex, consisting of A. japonica, A. koreana, A. lancea, and A. chinensis.
The rapid development of next-generation sequencing (NGS) technology (McPherson et al., 2013), coupled with powerful bioinformatic tools (Liu et al., 2012; Shi et al., 2019), has made it possible to study the genomic evolution and interspecific relationships of organisms according to whole plastid genomic data. Substantial nucleotide substitution, indel events, and structural rearrangements have been found in plastid genomes, indicating that the whole plastid genome contains a significant amount of phylogenetic information (Liu et al., 2018). Additionally, plant plastids display intraspecific heteroplasmic variation, a term which refers to the presence of nonidentical plastid molecules in a cell or organism (Scarcelli et al., 2016). Previous studies have used NGS methods to detect heteroplasmy in the plastid genome of Astragalus membranaceus, which could explain why the de novo genome assembly program has failed to assemble the genome in heterogeneous regions (Lei et al., 2016). Moreover, intra-individual polymorphism can provide new evidence that can be used in evolutionary and classification analysis (Sun et al., 2019). In addition, extensive phylogenetic discordance among nuclear and organellar phylogenies has been found in the genus Sphagnum; this has been caused by incomplete lineage sorting (ILS) following the rapid radiation of the genus, rather than by post-speciation introgression (Meleshko et al., 2021).
In 2020, Wang et al. used the plastomes and nuclear sequences of Atractylodes lancea, A. chinensis, and A. macrocephala to reconstruct the phylogenetic relationships of these three species (Wang et al., 2020). Phylogeny analysis using the plastid data indicated that A. lancea and A. chinensis are more closely related to one another than to A. macrocephala. Interestingly, this study further observed intra-individual polymorphism of SLD5, such that SLD5 has two haplotypes in A. macrocephala, of which one can be found in A. lancea and, separately, the other can be found in A. chinensis. This intra-individual polymorphism was taken to imply that A. macrocephala may be a hybrid of A. lancea and A. chinensis, or the result of introgressive hybridization (Wang et al., 2020). Subsequently, analysis of six plastid genomes of Atractylodes (A. chinensis, A. koreana, A. lancea, A. macrocephala, A. japonica, and A. carlinoides) has revealed that the phylogenetic relationship within Atractylodes is complex (Wang et al., 2021). The results indicated that A. japonica and A. lancea are clustered into a subclade, while A. chinensis and A. koreana are clustered into another subclade. The abovementioned studies have confirmed that plastid genome analysis is a valuable tool for the phylogenetic study of Atractylodes, and additional specimens should be collected to obtain further evidence on the complex evolutionary history of A. lancea (Wang et al., 2021).
In this study, we collected 24 plant samples, 23 of which represented species of A. lancea complex. High-throughput sequencing was used to obtain the concatenated nrDNA sequences (18S-ITS1-5.8S-ITS2-28S) and plastid genomes. Our analyses showed that A. chinensis was more closely related to A. japonica than to A. lancea. Furthermore, extensive discordance and complex relationships across the genus of Atractylodes were revealed through analysis of the cytonuclear genomic data.
Materials and methods
Sample collection
For this study, 24 samples representing specimens of Atractylodes lancea, A. chinensis, A. macrocephala, A. japonica, and A. koreana were collected, of which six samples were collected from wild regions and 18 samples from cultivated regions (Supplementary Figure 2). All the samples were morphologically authenticated by Chunying Zhao (Chengde Medical University), Xinlei Zhao (Institute of Medicinal Plant Development, CAMS), Yulin Lin (Institute of Medicinal Plant Development, CAMS), and Qiuling Wang (Institute of Medicinal Plant Development, CAMS). Detailed information is provided in Supplementary Table 1 and Supplementary Figure 3. In addition, data relating to 20 samples representing six species of genus Atractylodes and ten species of outgroup taxa were downloaded from GenBank (Supplementary Table 2).
DNA extraction, library preparation, and high-throughput sequencing
Total genomic DNA extraction was performed on the leaf tissues using a modified CTAB method. The quantity and quality of the DNA were determined using Qubit 4.0 (Thermo Fisher Scientific Inc., USA). The sequencing library (~350 bp) was constructed using purified DNA and a TruSeq DNA PCR-Free High Throughput Library Prep Kit (Illumina USA). An Illumina NovaSeq platform was employed to conduct high-throughput sequencing. The raw data were deposited in the Sequence Read Archive (SRA) under BioProject accession number PRJNA682118. The final plastid genomes and concatenated nrDNA sequences of the A. lancea, A. chinensis, A. macrocephala, A. japonica, and A. koreana specimens were assembled, annotated, and submitted to GenBank (Supplementary Table 1).
Assembly, annotation, and characterization of the concatenated nrDNA and plastid genome sequences
The sequencing adapter and low-quality reads were filtered using Trimmomatic v0.38 (Bolger et al., 2014). Whole plastid genomes were assembled via the organelle assembler NOVOPlasty v4.2.1 (Jin et al., 2020) and GetOrganelle (Jin et al., 2020). The plastid genome sequence of A. lancea (accession number: NC_037483) was selected as a reference in the NOVOPlasty configuration file. CpGAVAS2 (www.herbalgenomics.org/cpgavas2) with default parameters was used to annotate the protein-coding, rRNA, and tRNA genes of the plastid genome and to facilitate visualization (Shi et al., 2019), with the initial annotations being edited manually using the Apollo genome editor. A circular map was generated using OrganellarGenomeDRAW (OGDRAW) (Greiner et al., 2019). The concatenated nrDNA sequences (18S, ITS1, 5.8S, ITS2, and 28S nrDNA) were assembled using Getorganelle and compared with the nuclear ribosomal RNA database to obtain the annotation results. The codon usage and relative synonymous codon usage (RSCU) of the plastid genomes were calculated using CodonW (http://codonw.sourceforge.net/).
Analysis of repeat structures and intraspecific variation in the plastid genomes
The REPuter (Kurtz et al., 2001) program was used to identify four types of sequence repeats, including forward (F), reverse (R), complementary (C), and palindromic (P). The minimum repeat size for oligonucleotide repeats was set at 30 bp, with a Hamming distance of 3 (i.e., a sequence identity of 90%). Tandem repeats were analyzed using the TRF (Benson, 1999) software with default parameters. Simple sequence repeats (SSRs) were detected using the MIcroSAtellite identification tool (MISA, available online: http://pgrc.ipk-gatersleben.de/misa/) (Beier et al., 2017) with minimum repeat thresholds of 10, 6, 5, 5, 5, and 5 for mono-, di-, tri-, tetra-, penta-, and hexa-nucleotide SSRs, respectively. Intraspecific variations were detected by first mapping the reads to reference sequences using Bowtie2 (Langmead and Salzberg, 2012), and subsequently conducting analysis using our local python program and visualizing the sequences using an integrative genomics viewer.
Comparative analysis of the plastid genomes
The mVISTA program (http://genome.lbl.gov/vista/mvista/submit.shtml) was used in Shuffle-LAGAN mode for the comparative analysis of divergence regions with default parameters, and A. chinensis was used as a reference. Intra- and inter-distances were analyzed in accordance with procedures reported in our previous study (Chen et al., 2010). Mauve (Darling et al., 2004), a system used to construct multiple genome alignments in the presence of large-scale evolutionary events, was used to identify locally collinear blocks (LCBs) of Atractylodes species. The contraction and expansion of the IR boundaries between the four main parts of the genome (LSC/IRb/SSC/IRa) were visualized using IRscope (https://irscope.shinyapps.io/irapp/).
Phylogenetic and gene introgression analysis
A total of 44 Asteraceae whole plastid genomes and concatenated nrDNA sequences were used for phylogenetic analysis. Lactuca raddeana and Ainsliaea latifolia were used as the outgroup species (Supplementary Table 2). Each region was first aligned using MUSCLE v3.8 (Edgar, 2004) and then concatenated to form six matrices, namely 1) a dataset of concatenated nrDNA sequences (aligned length 5,855 bp), 2) a dataset of 73 conserved protein-coding sequences (aligned length 61,413 bp), 3) a dataset of 95 common genes including rRNA and tRNA genes (aligned length 83,547 bp), 4) a dataset of 88 intergenic spacer regions (IGS, aligned length 37,809 bp), 5) a dataset of 73 protein sequences (aligned length 20,393 bp), and 6) the dataset of the whole plastid genomes (aligned length 121,356 bp). The maximum likelihood (ML) phylogenetic trees of the six matrices were constructed using RAxML v8.2.12 (Stamatakis, 2014) with 1000 bootstrap replicates. The GTRGAMMA substitution model was applied to the protein-coding genes, genes, IGS, and whole plastid genomes, while PROGAMMAAUTO was applied to protein sequences. The parameters for this analysis included “raxmlHPC-PTHREADS-SSE3 -f a -N 1000 -m GTRGAMMA -x 551314260 -p 551314260 -T 20”. Tree visualization was performed using MEGA X (Kumar et al., 2018). Finally, the topologies recovered after analysis of the plastid and nrDNA data were compared using the dendextend package (Galili, 2015).
The analysis of phylogenetic networks was carried out using PhyloNet v.3.8.21 (Wen et al., 2018) with the command ‘InferNetwork_MPL’ using gene tree topologies estimated by IQ-TREE2 (Minh et al., 2020); subsequently, the phylogenetic networks in the form of Rich Newick strings were visualized in Dendroscope3 (Huson and Scornavacca, 2012). The D-statistic (ABBA–BABA) method in Dsuite (Malinsky et al., 2021) was used to test for introgression events using SNP data on Atractylodes plastid genomes.
Results
The structure of the Atractylodes plastid genome and concatenated nrDNA sequences
The plastid genome length of Atractylodes ranged from 152,138 bp (A. chinensis) to 153,268 bp (A. lancea). The greatest length variations among individuals of A. chinensis, A. koreana, and A. lancea were 956 bp, 983 bp, and 1000 bp, respectively. Each plastid genome displayed a typical quadripartite structure, consisting of a large single-copy (LSC) region (83,206~84,293 bp), a small single-copy (SSC) region (18,604~18,698 bp), and a pair of inverted repeat (IR) regions (IRa and IRb) (25,137~25,185 bp). The GC content varied from 37.69% to 37.77% and was higher in the IR regions (about 43%) than in the LSC and SSC regions (about 35% and 31%) in all species (Figure 1, Supplementary Table 3). Moreover, several long-term indels were verified by genome mapping, including the 989 bp deletion of the plastid ndhC_trnV-UAC IGS in A. lancea (HPAB0031), A. chinensis (HPAB0001, HPAB0003, and HPAB0006), and A. koreana (HPAB0010). Length differences in coding genes such as rpoB and ycf2 were mainly due to the occurrence of repeat units in the sequence. A 6 bp insertion unit (TTAACC) of rpoB was found in A. lancea (HPAB0027), while a 9 bp deletion unit of ycf2 was present in A. chinensis (HPAB0001).

Figure 1 Plastid genome map of Atractylodes species. Gene locations outside the outer rim are transcribed in a counterclockwise direction, while genes inside are transcribed in a clockwise direction. The colored bars indicate known functional family differences. The dashed gray area in the inner circle shows the proportional GC content of the corresponding genes. LSC, large single-copy region; SSC, small single-copy region; IR, inverted repeat region.
A total of 113 unique genes were annotated in the plastid genomes, consisting of 80 protein-coding genes, 29 tRNA genes, and four rRNA genes (rrn23S, rrn16S, rrn5S, and rrn4.5S). Of these, 82 genes (61 protein-coding genes and 21 tRNA genes) were located in the LSC region. The SSC region contained 12 protein-coding genes and one tRNA gene (trnL-UAG). Furthermore, 17 genes contained introns: 14 (nine protein-coding and five tRNA genes) contained one intron, and three (rps12, ycf3, and clpP) contained two introns (Supplementary Table 4). Small exons were also annotated in the petB, petD, and rpl16 genes, with lengths of 6 bp, 8 bp, and 9 bp, respectively. Finally, rps12 was identified as a trans-splicing gene.
The length of the concatenated nrDNA sequence was 5,849 bp; this sequence consisted of five parts. The lengths of 18S, ITS1, 5.8S, ITS2, and 28S were 1,809 bp, 259 bp, 158 bp, 229 bp, and 3,394 bp, respectively. The GC content varied between 55.43% and 55.62% and was higher in the ITS regions (about 63%, including ITS1 and ITS2) than in the 18S and 28S regions (about 49% and 57%) in all samples (Supplementary Table 5).
Codon usage in the Atractylodes plastid genomes
The amino acid frequency, codon usage, and relative synonymous codon usage (RSCU) of 80 protein-coding regions in all Atractylodes species were analyzed using Codon W. RSCU values ranged from 63.98 to 64.05; the number of codons ranged from 22,846 (HPAB0022) to 22,873 (HPAB0023) in 26 species; and the number of amino acids ranged from 21,706 (HPAB0023) to 22,398 (HPAB0016). Of these codons, leucine (2,275 ~ 2,338 codons) was the most abundant amino acid, with a frequency of 9.95 ~ 10.22%, while the proportion of cysteine (471 ~ 486 codons) was 2.06 ~ 2.13%; AGA (encoding arginase) and CGC (encoding arginase) were the most and least used codons, respectively (Supplementary Table 6). Almost all the amino acids had more than one synonymous codon; the exceptions were methionine and tryptophan. Furthermore, 31 codons displayed RSCU values exceeding 1. Most of the biased codons were used with A or T bases as the third codons. ATG and TGG, encoding methionine and tryptophan, exhibited no bias (RSCU = 1.00) (Supplementary Table 6).
Three types of starting codons were detected in 80 protein-coding genes. Of these, 77 genes used ATG as start codons, while two (ndhD, psbL) used ACG and one (rps19) used GTG. TAA, TAG, and TGA were present as stop codons in these genes. The most used stop codon was TAA at 55.56%, followed by TAG (25.92%) and TGA (18.52%). ndhF was the only gene that used both TAA and TGA as stop codons; of this gene, eight samples (HPAB0003, HPAB0006, HPAB0009, HPAB0010, HPAB0011, HPAB0016, HPAB0018, and HPAB0031) showed a preference for TAA and 18 utilized TGA.
The SSR units and repeat structures of Atractylodes plastid genomes
SSRs were detected in 24 Atractylodes plastid genome sequences (Supplementary Table 7). Specifically, 30, 30, 29, 30, and 30 SSR loci were detected in the Atractylodes chinensis, A. koreana, A. lancea, A. japonica, and A. macrocephala plastid genomes, respectively. A large proportion of the SSRs were distributed in the LSC region (85.23%), with 12 in the SSC region and 10 in the IR regions. Polyadenine (poly-A) (34.70%, 10~16) and polythymine (poly-T) (60.41%, 10~21) represented the dominant repeats.
In addition to the SSRs, 33, 34, 36, 31, and 32 tandem repeat sequences were detected in the A. chinensis, A. koreana, A. lancea, A. japonica, and A. macrocephala plastid genomes, respectively. The tandem repeat lengths were 9~45 bp, and most were located in the IGS regions of the genomes. In addition, 1, 3, 5, 5, 5, 47, and 25 tandem repeats were found in the atpI, ndhF, rpoC2, rps18, petD, ycf1, and ycf2 coding regions, respectively (Figure 2, Supplementary Table 8). Finally, an average of 42 repeat structures were revealed for each species, including F, P, R, and C repeats. P was the most common repeat type, accounting for 48.8~53.2% of all repeats, followed by F (41.9~51.2%), C (6.9%), and R (2.3%) (Figure 2, Supplementary Table 9).

Figure 2 The types and distribution of repeat sequences in 24 Atractylodes plastid genomes. (A) The number of each of the four repeat types (F, forward; P, palindrome; R, reverse; C, complement). (B) The distribution of repeat sequences across three regions: LSC, SSC, and IR.
Variation in Atractylodes plastid genome sequences and concatenated nrDNA sequences
The 24 plastid genome sequences were compared to identify differences using the online software platform mVISTA, with the A. chinensis plastome as the reference genome (Figure 3, Supplementary Figure 4). Highly conservative regions were evident across most of the plastid genomes, and only three relatively high-variability regions were identified in the whole genome (Figure 3). Subsequently, Mauve was used to identify local collinear blocks (LCBs) of Atractylodes plastid genomes (Figure 4, Supplementary Figure 5); the A. chinensis genome is shown at the top as the reference genome. These species showed a consistent sequence order in all the genes. The collinear blocks of all the plastid genomes, including the LSC, SSC, and IR regions, revealed relatively high levels of conservation, with no gene rearrangement.
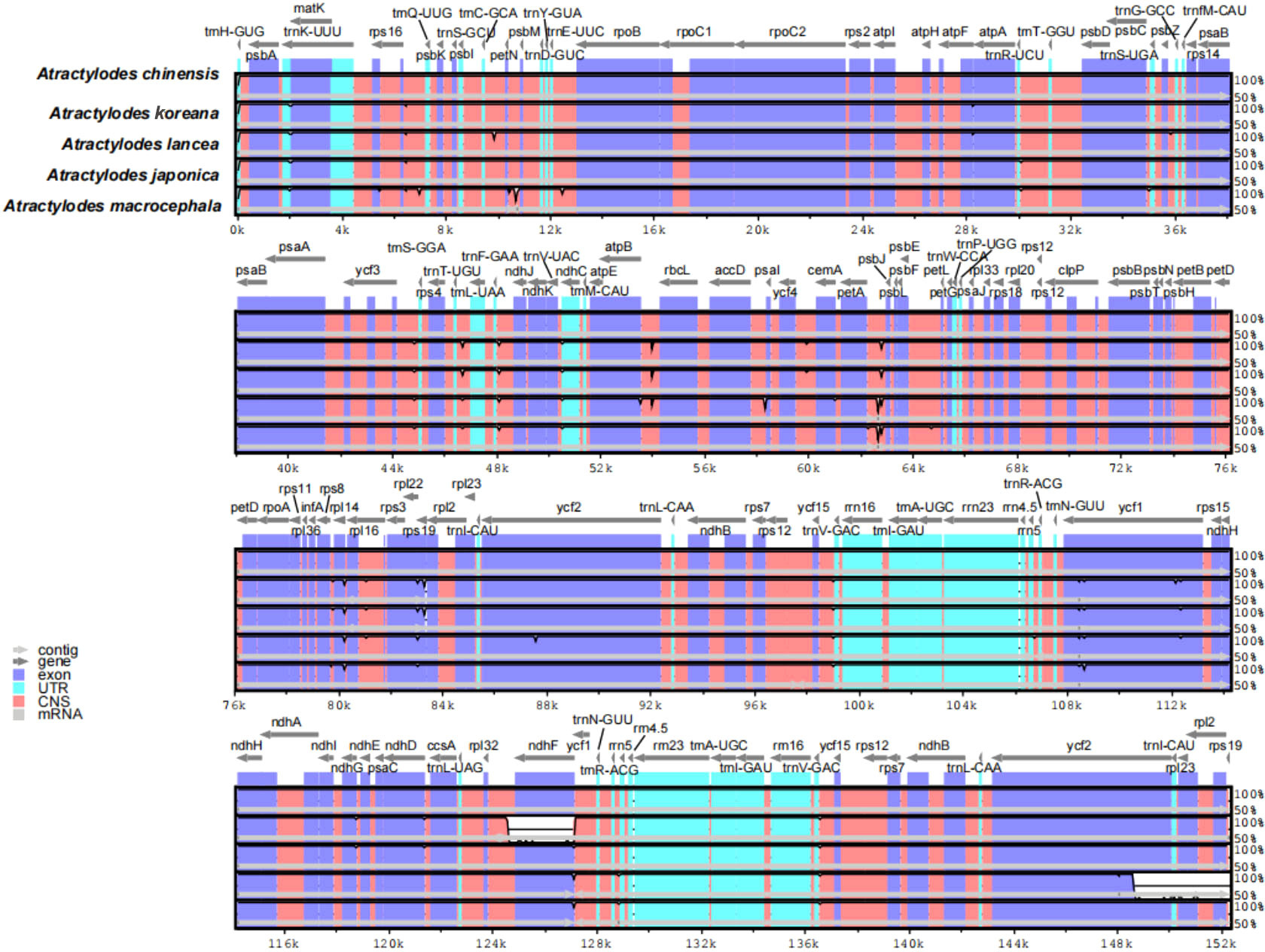
Figure 3 A comparison of the five plastid genomes, with A. chinensis as a reference, using the mVISTA alignment program. The grey arrows above the alignments show the orientation of the genes. The violet blocks indicate exons, the cyan blocks denote introns, and the salmon blocks signify conserved non-coding sequences (CNS). The y-axis indicates the identity percentage, ranging from 50% to 100%, while the x-axis represents sequence length.
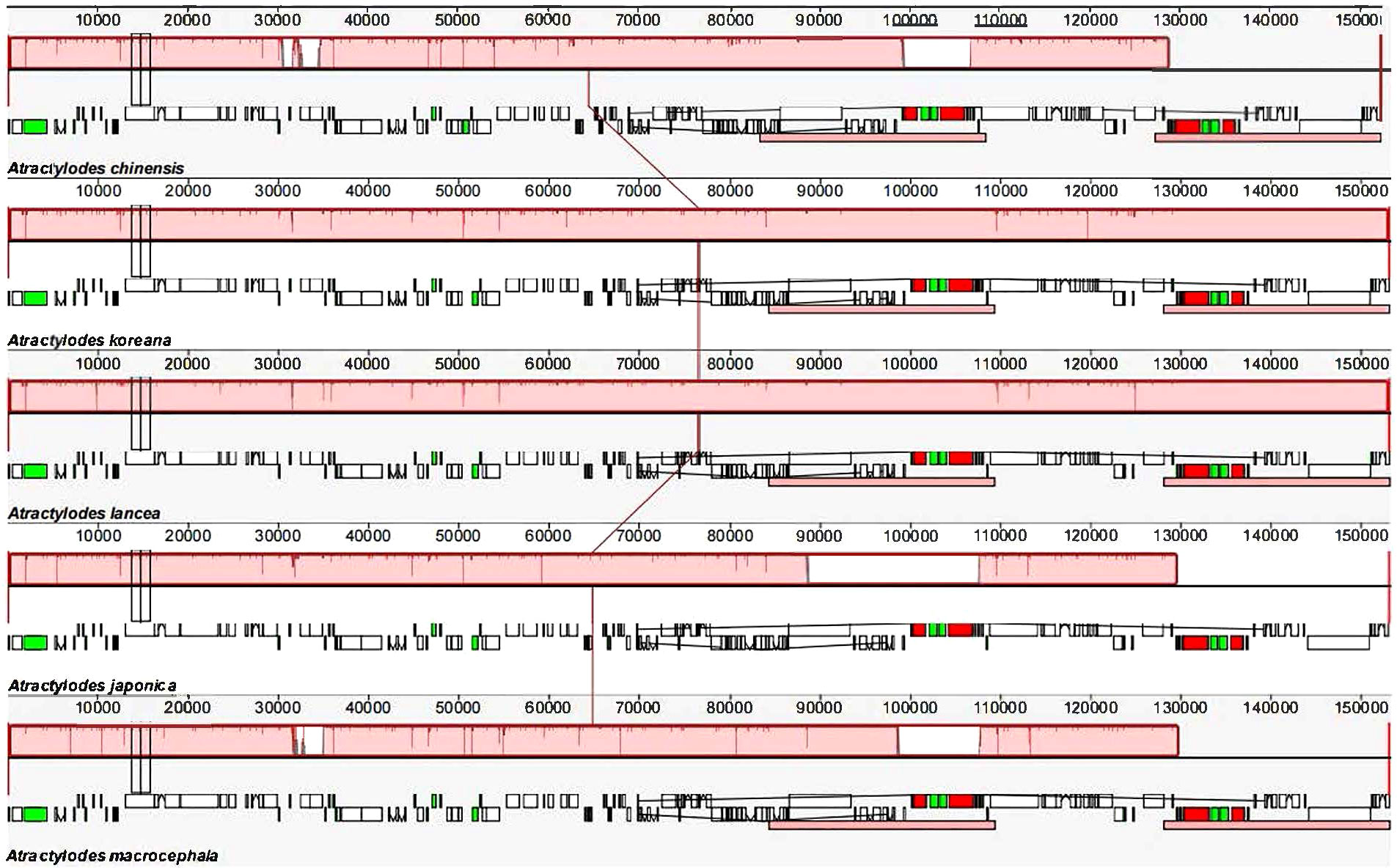
Figure 4 A comparison of the whole plastid genomes of the Atractylodes species using the Mauve algorithm. The red LCBs indicate syntenic regions, while the histograms within each block represent the degree of sequence similarity. rRNA, protein-coding, and tRNA gene annotations are denoted by red, white, and green boxes, respectively.
The nrDNA sequences exhibited 53 variation sites and 25 parsimony-informative sites, accounting for 0.91% and 0.43% of the total nrDNA sequences, respectively. The majority of the variation sites were located in the ITS1 and ITS2 regions. The average inter-specific distance, average theta prime, and smallest inter-specific distance were used to characterize the inter-specific divergence, taking values of 0.0027, 0.0027, and 0.0014, respectively. The intra-specific variation was determined according to the average intra-specific difference, theta, and average coalescent depth, which yielded values of 0.0003, 0.0008, and 0.0012, respectively. DNA barcoding gaps were clearly present for species A. carlinoides, A. macrocephala, A. japonica, and A. lancea, whereas the relationship between A. chinensis and A. koreana could not be resolved.
Contraction and expansion of the IR region in Atractylodes plastid genome sequences
The circular structure of the plastid genome was highly conserved and generated four boundaries in the IR, LSC, and SSC regions. As the genome evolved, the contraction and expansion of the IR boundary produced different plastid genome sizes and altered certain gene locations. The rps19 gene crossed the LSC and IRb regions in all the species. Although the ycf1 gene was distributed across the SSC and IRb regions, most of the genes were located in the SSC region, with minor differences in length. Four indels were identified via multiple sequence alignment in the ycf1 gene. Specifically, a TTTGAA insertion was detected in positions 4,435-4,440 in A. chinensis and A. macrocephala; an AAGACGAA deletion occurred at positions 800-808 in A. chinensis; an AAATAC deletion was evident at positions 4,290-4,295 in A. lancea; and finally, an AAGACGAAG insertion was detected at positions 791-799 in A. macrocephala. The ndhF gene and the ycf1 pseudogene were detected at the junction of SSC and IRa. The ndhF gene was mainly located in the SSC region but spanned the junction 15 bp into the IRB region. The ycf1 pseudogene was entirely located in the IRa region. Additionally, the rps19 pseudogene and trnH gene were located at the junction of the IRa and LSC regions, while rps19 spanned the junction 1 bp into the LSC region (Figure 5, Supplementary Figure 6).
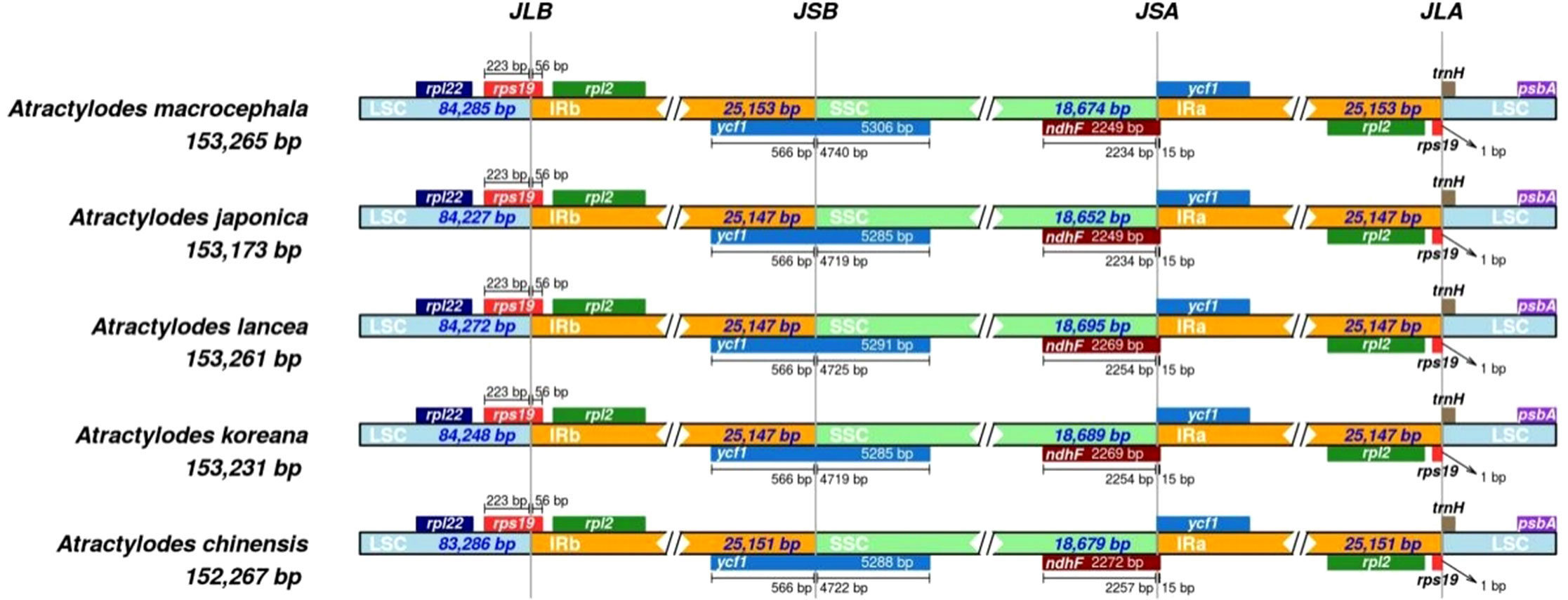
Figure 5 A comparison of the LSC and IRb border region and the SSC and IRa border region for the five Atractylodes species. JLB: junction of LSC and IRb; JSA: junction of SSC and IRa. JSB: junction of IRb and SSC; JLA: junction of IRa and LSC.
Heteroplasmic variations in the whole plastid genome
A total of 64 heteroplasmic variations were detected in the whole plastid genome of Atractylodes (Supplementary Table 10). This heteroplasmy consisted primarily of two types, namely heteroplasmy in insertion/deletions (indels) and heteroplasmy in single nucleotide polymorphism sites (SNPs). For example, an ndhF gene sequence deletion of 20 bp in length was identified in eight samples (HPAB0003, HPAB0006, HPAB0009, HPAB0010, HPAB0011, HPAB0016, HPAB0018, and HPAB0031) via multiple sequence alignment. Further genome mapping simultaneously detected two types of reads (insertion and deletion) in the intra-plastid genome, such as in sample HPAB0016. A further example is that the base R representing adenine (A) and guanine (G) was found in the assembly result for HPAB0001. Subsequently, a total of 235 reads related to this region were extracted from the shotgun sequencing data, indicating the simultaneous detection of G (185) and A (50) at the corresponding site (Supplementary Table 10).
Phylogenetic tree, phylogenetic network, and gene introgression analyses
The phylogenetic relationships of Atractylodes were analyzed using six data sets of concatenated nrDNA sequences, 73 conserved plastid protein-coding gene sequences, 95 plastid common gene sequences, 88 plastid IGS regions, 73 plastid protein sequences, and the whole plastid genome sequences (Supplementary Table 11). The resulting phylogenetic trees showed that Atractylodes was a monophyletic clade related to Tugarinovia mongolica. The concatenated nrDNA sequences and plastid phylogenetic analyses indicated that A. carlinoides separated from the rest of the Atractylodes species with a high bootstrap value. A. macrocephala alone formed a relatively independent clade, with the A. lancea complex as a sister group of this.
The phylogenetic tree generated for the concatenated nrDNA sequence dataset showed that the A. lancea complex was divided into three subclades: A. lancea, A. japonica, and A. chinensis–A. koreana. The A. lancea clade included ten samples (HPAB0011, HPAB0012, HPAB0018, HPAB0021, HPAB0022, HPAB0023, HPAB0026, HPAB0028, HPAB0031, and MG874804). The A. japonica clade included three samples (HPAB0015, MW301112, and MT834523), which were outside the A. chinensis–A. koreana clade with a bootstrap value of 91. The most controversial aspect of the tree was the A. chinensis–A. koreana clade, which included 17 A. chinensis and A. koreana samples (Figure 6, Supplementary Figure 7).

Figure 6 A comparison of the topologies recovered via plastid data (left) and via concatenated nrDNA sequences (right). A. lancea are shown in red, A. japonica in blue, and the A. chinensis–A. koreana clade in black.
The phylogenetic trees for the five different plastid datasets presented similar topologies (Supplementary Figures 8-Supplementary Figure 12). The whole plastid genome dataset yielded better-supported trees than the other four datasets. In this tree, the A. lancea complex was divided into one small and one large clade. The small clade was a sister of A. macrocephala and contained two A. chinensis samples (HPAB0003 and HPAB0006), one A. lancea sample (HPAB0031), and two A. koreana samples (HPAB0010 and HPAB0016) with a bootstrap value of 100. The remaining 25 samples, consisting of nine A. lancea, three A. japonica, four A. koreana, and nine A. chinensis, formed a large clade with a bootstrap value of 55. The phylogeny of the four species in this large clade was ambiguous and could not be clearly resolved (Figure 6). Phylogenetic analyses for the nrDNA and whole plastid genome indicated plastid and nuclear discordance.
PhyloNet was used to further assess putative hybridization events in the phylogeny. The analysis indicated that certain loci in the genome of Atractylodes shared a most recent common ancestor with loci in that of Arctium lappa, and others shared a most recent common ancestor with loci in that of Tugarinovia mongolica; this was the case in all networks allowing 1-4 reticulations (Figure 7). When 4 reticulations were allowed, the resulting network showed that A. lancea may have hybridized with the common ancestor of A. chinensis and A. japonica. The results of ABBA–BABA tests showed that A. chinensis was more closely related to A. japonica than to A. lancea (Supplementary Table 12).
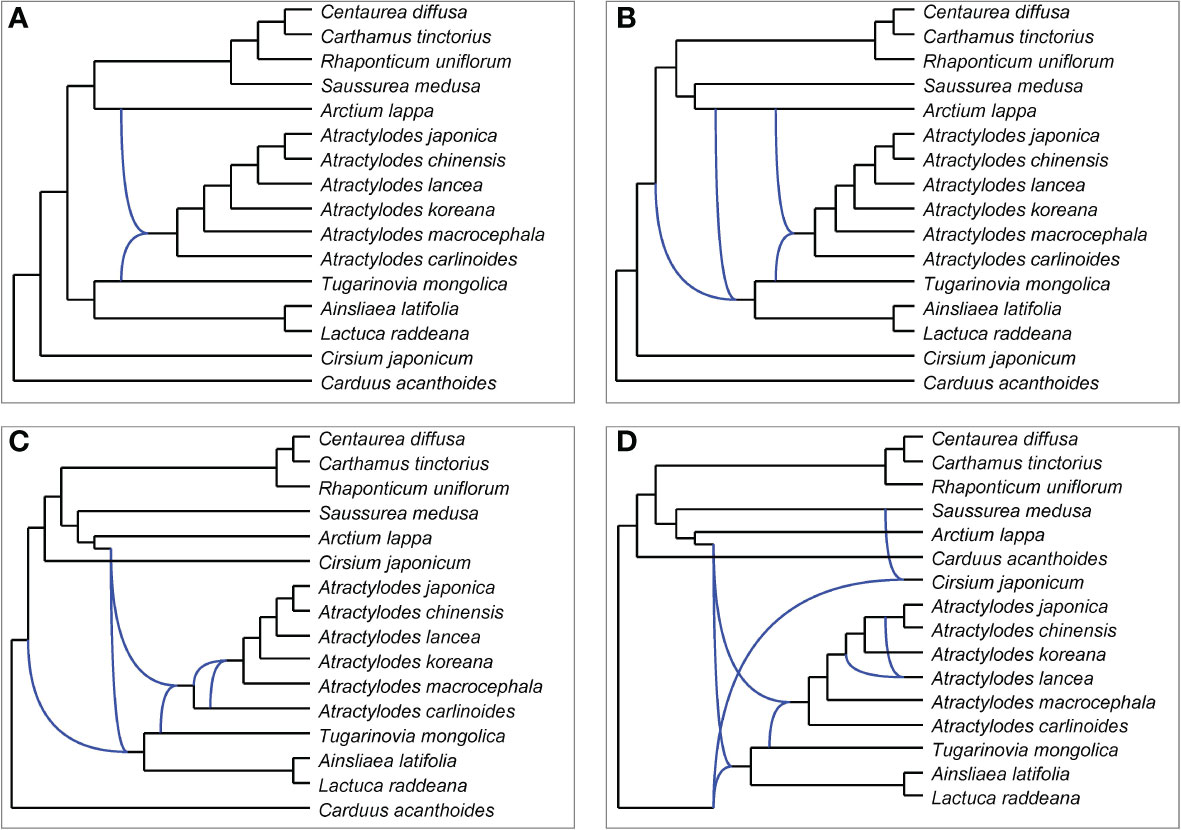
Figure 7 Networks representing plastid gene trees generated by PhyloNet MPL, allowing a maximum of 1 (A), 2 (B), 3 (C), or 4 (D) reticulations.
Discussion
Atractylodes is a small genus of the Asteraceae family mainly distributed across East Asia. It is a cross-pollination plant group, meaning that its morphological character is extremely susceptible to environmental factors (Hu et al., 2000). The phylogenetic relationships and the taxonomic treatment within the entire genus are intricate due to the continuous nature of variation in its morphological features (Peng et al., 2012). Atractylodes rhizomes distributed at low altitudes are bead-shaped and run horizontally; however, with changes in altitude and the ecological environment, rhizomes at medium altitudes appear in clumps and grow obliquely downwards (Zhu, 1981). The petioles and degree of leaf-splitting of the genus are still undergoing continuous evolution (Peng and Wang, 2007). In some instances, the lower and middle cauline leaves are petiolate, whereas basal leaves are sometimes subsessile (Hu et al., 2000). Although the leaf blades are generally divided into 3-5 pinnately arranged segments, they are occasionally undivided near the base, with a few small spiny lobes (Zhu, 1981). The 24 Atractylodes samples collected in this study also exhibited several continuously varying morphological features, especially those samples cultivated in Hubei province.
Consistent with previous studies, the phylogenetic trees presented here indicate that A. carlinoides is a basal species in the Atractylodes genus, and is a sister of the remainder of the Atractylodes species. Unlike one previous study predicting that A. macrocephala may be an A. chinensis and A. lancea hybrid (Wang et al., 2020), this study found that A. macrocephala forms an independent branch, with a bootstrap value of 100 in the case of both plastid data and nrDNA data. Regarding A. koreana, this species is mainly distributed in the Liaoning and Shandong provinces of China. Its lower and middle cauline leaves are undivided, which represents a point of distinction from the morphological characteristics of the other Atractylodes species. However, the ITS genotype of A. koreana was found here to be consistent with that of A. chinensis, and this also has been reported in a previous study (Shiba et al., 2006). A. chinensis and A. lancea are generally discriminated on the basis of differences in the shapes of their leaves. However, Shi has indicated that these morphological differences are unstable (Zhu, 1981), and past authors have been misled because the number of specimens they possessed was extremely limited, resulting in a poor understanding of the polytype of this species. Here, ten samples of A. lancea formed an independent clade in an analysis using nrDNA data from both wild and cultivated samples. A. japonica has long petioles, and the leaf blades generally divide almost to the base into 3-5 segments; these traits constitute obvious differences from other species in the A. lancea complex. This label has been considered a synonym for A. lancea, as recorded in the latest version of Flora of China. However, this study identified multiple strands of supporting evidence for a close relationship between A. japonica and other variations of the A. lancea complex in the nrDNA concatenated sequence-based phylogeny. Moreover, species trees obtained using PhyloNet confirmed that A. japonica is closely related to A. chinensis but separate from A. lancea.
Cytonuclear discordance can be caused by many factors, such as ancient hybridization and gene introgression. It is a common phenomenon in plant systematics and has been reported in many genera, such as section Galoglychia (Renoult et al., 2009) and Cotoneaster (Meng et al., 2021). Existing cpDNA and nuclear genetic evidence has revealed that sect. Galoglychia has many obvious nuclear–cytoplasmic phylogenetic tree conflicts, which are likely to be caused by ancient hybridization followed by gene introgression. In the case of Cotoneaster, sequences of the complete plastid genomes and 204 low-copy nuclear genes of 69 species were used for the phylogenetic analysis, and the results revealed there were conflicts between the plastid genome and low-copy nuclear phylogenies at both the species and clade levels. These instances of cytonuclear discordance may be caused by frequent hybridization events and incomplete lineage sorting (ILS). For species of Atractylodes, gene introgression usually results from natural hybridization among closely related species in sympatric populations (Hu et al., 2000). Shiba et al. have indicated that the continuous morphological variation in features between A. lancea and A. chinensis may be caused by the presence of such hybrids. Interspecific hybridization between A. lancea and A. chinensis has been observed in 25 samples containing nucleotide additives (Peng and Wang, 2007). Here, the phylogenetic network analysis of plastid genes revealed that A. lancea may have hybridized with the common ancestor of A. chinensis and A. japonica.
In conclusion, the results of this study tended to support treatment of A. japonica as an independent species. Although samples of A. lancea formed an independent clade, the other species in the A. lancea complex were still mingled with one another due to a complicated pattern of evolution in this genus, as shown by the phylogeny according to the plastid genomic data. In addition, this study revealed extensive discordance based on the cytonuclear genomic data, primarily involving the A. lancea complex. In future research, analysis of the A. lancea complex with sufficient single-copy nuclear genes or use of a reduced-representation genomic approach will be necessary to clarify the genetic differentiation.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
LS, JL, and XiaoZ conceived and designed the study. HX, CZ, XinZ, QW, and YL collected and identified the plant materials. JL, MS, and HX performed the experiments. JL, MS, ZZ, and WK analyzed the data. JL, LS, MS, and HX wrote the manuscript. JL, LS, and XiaoZ revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by National Key R&D Program: Intergovernmental Cooperation in International Science and Technology Innovation (2022YFE0119300), China Postdoctoral Science Foundation (2022M720504), Beijing Municipal Natural Science Foundation (7202136), the National Natural Science Foundation of China (81703659), Guangxi Science and Technology base and talent project (AD22080012).
Acknowledgments
We thank Dr Ran Wei, State Key Laboratory of Systematic and Evolutionary Botany, Institute of Botany, Chinese Academy of Sciences, for providing help with the phylogenic analysis conducted in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.1045423/full#supplementary-material
Abbreviations
IR, inverted repeat region; LSC, large single copy region; SSC, small single copy region; tRNA, transfer RNA; rRNA, ribosomal RNA; SSR, simple sequence repeats; ML, Maximum Likelihood.
References
Beier, S., Thiel, T., Münch, T., Scholz, U., Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585. doi: 10.1093/bioinformatics/btx198
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Chen, S., Yao, H., Han, J., Liu, C., Song, J., Shi, L., et al. (2010). Validation of the ITS2 region as a novel DNA barcode for identifying medicinal plant species. PloS One 5, e8613. doi: 10.1371/journal.pone.0008613
Darling, A. C., Mau, B., Blattner, F. R., Perna, N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
Deng, A.-P., Wu, Z.-T., Liu, T., Kang, L.-P., Nan, T.-G., Zhan, Z.-L., et al. (2016). Advances in studies on chemical compositions of atractylodes lancea and their biological activities. Zhongguo Zhong Yao Za Zhi 41, 3904–3913. doi: 10.4268/cjcmm20162104
Edgar, R. C. (2004). MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinf. 5, 113. doi: 10.1186/1471-2105-5-113
Galili, T. (2015). Dendextend: an r package for visualizing, adjusting and comparing trees of hierarchical clustering. Bioinformatics 31, 3718–3720. doi: 10.1093/bioinformatics/btv428
Greiner, S., Lehwark, P., Bock, R. (2019). OrganellarGenomeDRAW (OGDRAW) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 47, W59–W64. doi: 10.1093/nar/gkz238
Hu, S., Feng, X., Ji, L., Nie, S. (2000). Atractylodes lancea and its geo-varieties. Chin. Traditional Herbal Drugs 31, 781–784. Available at: https://www.tiprpress.com/zcy/article/abstract/20001033?st=search
Huson, D. H., Scornavacca, C. (2012). Dendroscope 3: an interactive tool for rooted phylogenetic trees and networks. Systematic Biol. 61, 1061–1067. doi: 10.1093/sysbio/sys062
Jin, J.-J., Yu, W.-B., Yang, J.-B., Song, Y., dePamphilis, C. W., Yi, T.-S., et al. (2020). GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21, 241. doi: 10.1186/s13059-020-02154-5
Kim, J.-H., Doh, E.-J., Lee, G. (2016). Evaluation of medicinal categorization of atractylodes japonica koidz. by using internal transcribed spacer sequencing analysis and HPLC fingerprinting combined with statistical tools. Evidence-Based Complementary Altern. Med. 2016, 2926819. doi: 10.1155/2016/2926819
Kumar, S., Stecher, G., Li, M., Knyaz, C., Tamura, K. (2018). MEGA, X Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Langmead, B., Salzberg, S. L. (2012). Fast gapped-read alignment with bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Lei, W., Ni, D., Wang, Y., Shao, J., Wang, X., Yang, D., et al. (2016). Intraspecific and heteroplasmic variations, gene losses and inversions in the chloroplast genome of astragalus membranaceus. Sci. Rep. 6, 21669–21669. doi: 10.1038/srep21669
Liu, H., He, J., Ding, C., Lyu, R., Pei, L., Cheng, J., et al. (2018). Comparative analysis of complete chloroplast genomes of anemoclema, anemone, pulsatilla, and hepatica revealing structural variations among genera in tribe anemoneae (Ranunculaceae). Front. Plant Sci. 9. doi: 10.3389/fpls.2018.01097
Liu, C., Shi, L., Zhu, Y., Chen, H., Zhang, J., Lin, X., et al. (2012). CpGAVAS, an integrated web server for the annotation, visualization, analysis, and GenBank submission of completely sequenced chloroplast genome sequences. BMC Genomics 13, 715. doi: 10.1186/1471-2164-13-715
Malinsky, M., Matschiner, M., Svardal, H. (2021). Dsuite-fast d-statistics and related admixture evidence from VCF files. Mol. Ecol. Resour. 21, 584–595. doi: 10.1111/1755-0998.13265
McPherson, H., van der Merwe, M., Delaney, S. K., Edwards, M. A., Henry, R. J., McIntosh, E., et al. (2013). Capturing chloroplast variation for molecular ecology studies: a simple next generation sequencing approach applied to a rainforest tree. BMC Ecol. 13, 8. doi: 10.1186/1472-6785-13-8
Meleshko, O., Martin, M. D., Korneliussen, T. S., Schröck, C., Lamkowski, P., Schmutz, J., et al. (2021). Extensive genome-wide phylogenetic discordance is due to incomplete lineage sorting and not ongoing introgression in a rapidly radiated bryophyte genus. Mol. Biol. Evol. 38, 2750–2766. doi: 10.1093/molbev/msab063
Meng, K.-K., Chen, S.-F., Xu, K.-W., Zhou, R.-C., Li, M.-W., Dhamala, M. K., et al. (2021). Phylogenomic analyses based on genome-skimming data reveal cyto-nuclear discordance in the evolutionary history of cotoneaster (Rosaceae). Mol. Phylogenet. Evol. 158, 107083. doi: 10.1016/j.ympev.2021.107083
Minh, B. Q., Schmidt, H. A., Chernomor, O., Schrempf, D., Woodhams, M. D., Von Haeseler, A., et al. (2020). IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534. doi: 10.1093/molbev/msaa015
Peng, H. S., Wang, D. Q. (2007). Studies on population biology of transitional types of genus atractylodes in anhui province. Zhongguo Zhong Yao Za Zhi 32, 793–797. Available at: http://www.cjcmm.com.cn/WKE3/WebPublication/paperDigest.aspx?paperID=5a670764-5069-4d94-a816-9b844c4d0625
Peng, H.-S., Yuan, Q.-J., Li, Q.-Q., Huang, L.-Q. (2012). Molecular systematics of genus atractylodes (Compositae, cardueae): Evidence from internal transcribed spacer (ITS) and trnL-f sequences. Int. J. Mol. Sci. 13, 14623–14633. doi: 10.3390/ijms131114623
Renoult, J. P., Kjellberg, F., Grout, C., Santoni, S., Khadari, B. (2009). Cyto-nuclear discordance in the phylogeny of ficus section galoglychia and host shifts in plant-pollinator associations. BMC Evolutionary Biol. 9, 248. doi: 10.1186/1471-2148-9-248
Scarcelli, N., Mariac, C., Couvreur, T. L. P., Faye, A., Richard, D., Sabot, F., et al. (2016). Intra-individual polymorphism in chloroplasts from NGS data: where does it come from and how to handle it? Mol. Ecol. Resour. 16, 434–445. doi: 10.1111/1755-0998.12462
Shiba, M., Kondo, K., Miki, E., Yamaji, H., Morota, T., Terabayashi, S., et al. (2006). Identification of medicinal atractylodes based on ITS sequences of nrDNA. Biol. Pharm. Bull. 29, 315–320. doi: 10.1248/bpb.29.315
Shi, L., Chen, H., Jiang, M., Wang, L., Wu, X., Huang, L., et al. (2019). CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 47, W65–W73. doi: 10.1093/nar/gkz345
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Sun, S.-S., Zhou, X.-J., Li, Z.-Z., Song, H.-Y., Long, Z.-C., Fu, P.-C. (2019). Intra-individual heteroplasmy in the gentiana tongolensis plastid genome (Gentianaceae). PeerJ 7, e8025. doi: 10.7717/peerj.8025
Wang, Y., Wang, S., Liu, Y., Yuan, Q., Sun, J., Guo, L. (2021). Chloroplast genome variation and phylogenetic relationships of atractylodes species. BMC Genomics 22, 103. doi: 10.1186/s12864-021-07394-8
Wang, L., Zhang, H., Wu, X., Wang, Z., Fang, W., Jiang, M., et al. (2020). Phylogenetic relationships of atractylodes lancea, a. chinensis and a. macrocephala, revealed by complete plastome and nuclear gene sequences. PloS One 15, e0227610. doi: 10.1371/journal.pone.0227610
Wen, D., Yu, Y., Zhu, J., Nakhleh, L. (2018). Inferring phylogenetic networks using PhyloNet. Systematic Biol. 67, 735–740. doi: 10.1093/sysbio/syy015
Yin, M., Xiao, C.-C., Chen, Y., Wang, M., Guan, F.-Q., Wang, Q.-z., et al. (2015). A new sesquiterpenoid glycoside from rhizomes of atractylodes lancea. Chin. Herbal Medicines 7, 371–374. doi: 10.1016/S1674-6384(15)60066-1
Zhu, S. (1981). On the nomenclature of Chinese drug “Cangzhu”. Acta Phytotaxonomica Sin. 19, 318–322. Available at: https://www.plantsystematics.com/CN/Y1981/V19/I3/318
Keywords: Atractylodes, plastid genome, nrDNA concatenated sequence, phylogeny, cyto-nuclear discordance
Citation: Liu J, Shi M, Zhang Z, Xie H, Kong W, Wang Q, Zhao X, Zhao C, Lin Y, Zhang X and Shi L (2022) Phylogenomic analyses based on the plastid genome and concatenated nrDNA sequence data reveal cytonuclear discordance in genus Atractylodes (Asteraceae: Carduoideae). Front. Plant Sci. 13:1045423. doi: 10.3389/fpls.2022.1045423
Received: 15 September 2022; Accepted: 10 November 2022;
Published: 01 December 2022.
Edited by:
Lianming Gao, Kunming Institute of Botany, Chinese Academy of Sciences (CAS), ChinaReviewed by:
Lihong Xiao, Zhejiang Agriculture and Forestry University, ChinaHanghui Kong, South China Botanical Garden, Chinese Academy of Sciences (CAS), China
Copyright © 2022 Liu, Shi, Zhang, Xie, Kong, Wang, Zhao, Zhao, Lin, Zhang and Shi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Linchun Shi, linchun_shi@163.com; Xiaoxia Zhang, zhangxiaoxia@ibcas.ac.cn
†These authors have contributed equally to this work