 Odette Leiter
Odette Leiter Tara L. Walker
Tara L. Walker- Queensland Brain Institute, The University of Queensland, Brisbane, QLD, Australia
It is now apparent that platelet function is more diverse than originally thought, shifting the view of platelets from blood cells involved in hemostasis and wound healing to major contributors to numerous regulatory processes across different tissues. Given their intriguing ability to store, produce and release distinct subsets of bioactive molecules, including intercellular signaling molecules and neurotransmitters, platelets may play an important role in orchestrating healthy brain function. Conversely, a number of neurodegenerative conditions have recently been associated with platelet dysfunction, further highlighting the tissue-independent role of these cells. In this review we summarize the requirements for platelet-neural cell communication with a focus on neurodegenerative diseases, and discuss the therapeutic potential of healthy platelets and the proteins which they release to counteract these conditions.
Introduction
Platelets are small anucleate blood cells that have been gaining recognition as important mediators of several regulatory processes. Emerging research has identified novel functions that reach well beyond the traditional role of platelets in hemostasis and wound closure, revealing them to be crucial players during immune responses and tissue remodeling processes. We have recently summarized the evidence highlighting the capacity of platelets to contribute to brain homeostasis under physiological circumstances (1). Whereas, their versatile functions make platelets important regulators of cellular processes under normal conditions, platelet dysfunction is linked to a number of pathologies, including neurodegeneration. In the following review we briefly discuss the prerequisites of intercellular communication between platelets and cells from the central nervous system and summarize the research that demonstrates the involvement of impaired platelet function in several neurodegenerative conditions, including Alzheimer's disease (AD), Huntington's disease (HD), Parkinson's disease (PD), amyotrophic lateral sclerosis (ALS), multiple sclerosis (MS), and prion diseases (Figure 1). Finally, we highlight the emerging role of platelet preparations in the development of therapeutic interventions for the treatment of neuropathologies.
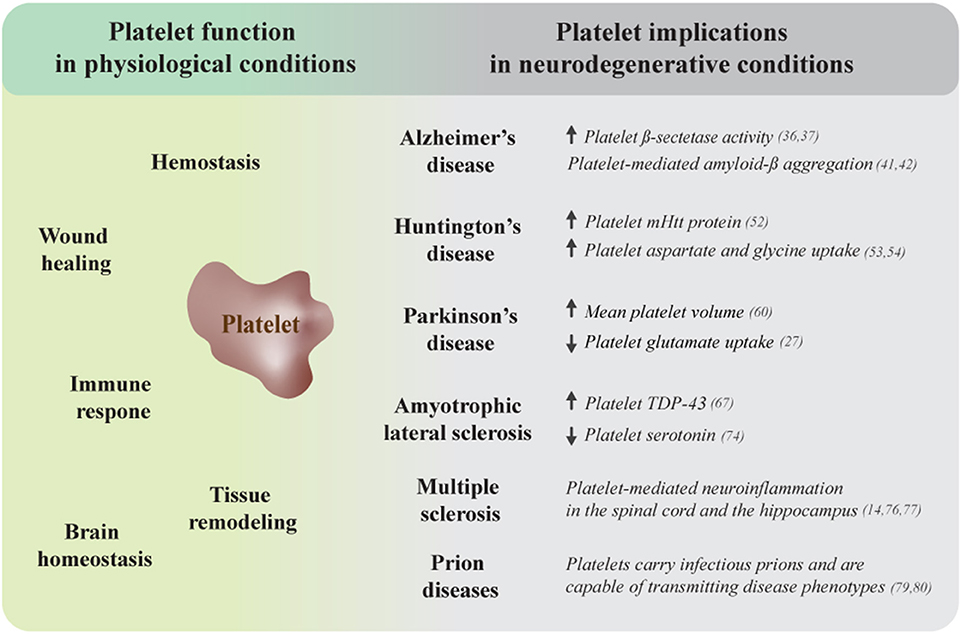
Figure 1. Platelet dysfunction is associated with several neurodegenerative disorders. Platelets are complex cells that exert numerous regulatory functions under physiological conditions, ranging from their traditional roles in hemostasis and wound healing to fundamental contributions to immune and tissue remodeling processes and brain homeostasis (left side). Platelet dysfunction, including mitochondrial abnormalities, is a common observation during neurodegeneration. The right side of this figure summarizes additional platelet-related impairments that link these cells to several neurodegenerative conditions. mHtt, mutant huntingtin protein; TDP-43, TAR DNA-binding protein of 43 kDa.
Platelets—The Diverse Properties of a Small Blood Cell
Until recently, platelets were primarily known for initiating coagulation following tissue injury and endothelial disruption. Although the platelet count in healthy humans ranges from 150,000 to 400,000 platelets per microliter of blood (2), only a small fraction of these (about 10,000 platelets per microliter) are necessary to act during hemostasis (3), supporting reports that platelets also exert other functions. Platelets are produced in the bone marrow by megakaryocytes which equip them with cytoplasm, including messenger ribonucleic acid (mRNA), mitochondria and secretory vesicles such as lysosomes, dense granules and α-granules, before they are released into the blood. Mouse and human platelets are functionally similar (4) and have short lifespans of 4–5 days and 8–12 days, respectively (5). However, a recent study found that platelets can return to the circulation following activation by thrombotic and immunological stimuli, suggesting that their lifespan could be longer than traditionally thought and that their elimination is not a direct consequence of the activation process (6). Platelet activation is required to fulfill particular functions; however, the outcome is specific to the trigger which initiates the activation. The most common platelet responses to activating stimuli include changes in shape, the upregulation of cell surface molecules, protein synthesis from mRNA, endo- and exocytosis, and the release of molecules from granule contents. In particular, the context-dependent secretion from α-granules, which provide a storage compartment for abundant bioactive molecules including growth and coagulation factors, chemokines, immune molecules and adhesion molecules, is highly regulated. Consequently, the stimulation of platelet preparations with three common agonists, adenosine diphosphate, collagen and thrombin receptor-activating peptide, results in distinct protein secretion profiles (7). In another study, it was shown that subpopulations of α-granules exist, in which proteins are stored in distinct clusters such as pro- or anti-angiogenic protein clusters (8). The selective release of these granule subtypes was triggered by the stimulation of different receptors with specific agonists, indicating that α-granule cargo is secreted in a context-dependent manner to either inhibit or promote angiogenesis as required (8). The finely tuned mechanisms whereby bioactive molecules are released from platelets represent a crucial asset in orchestrating regulatory processes across different tissues. However, disturbances in the regulation of platelet responses or hyperactivation of platelets have implications in numerous diseases, including during neurodegenerative conditions, as described in more detail below.
Platelets are Experts in Cell-Cell Communication
Platelets can communicate with other cell types in multiple ways, with their flexibility and mechanistic diversity suggesting that they likely act as inter-tissue messengers, including between blood and brain cells. Although the secretion of bioactive molecules from α- and dense granules represents a likely route of intercellular communication, additional mechanisms via which platelets may support crosstalk between the brain and the systemic environment are possible. Platelets release extracellular vesicles containing active cytoplasm components such as exosomes and microparticles (9). Both represent common ways of intercellular communication between organs and tissues in health and disease. Platelet exosomes and microparticles can also contain microRNAs, which when dysregulated are involved in various neurodegenerative disorders, including AD, PD, MS, HD, and ALS (10). Moreover, platelet-released particles, as well as platelets themselves which measure ~0.5 μm in diameter in mice (5) and from 1 to 5 μm in humans (11), are small enough to travel deep within the microcapillaries that span the brain. Thus, platelets and their released factors could interact with specific receptors in the cerebral vasculature to exert local, receptor-mediated effects. In conditions where the vascular integrity is altered or disturbed direct interactions with neural cells are possible. Platelet activity has been observed within the brain parenchyma following lesion (12) and stroke (13), as well as in the brain of experimental autoimmune encephalomyelitis (EAE)-induced mice (14). Furthermore, a direct interaction between platelets and neuronal cells has been reported, as they can bind central nervous system-specific glycolipid structures that are present in the lipid rafts of neuronal processes (15). This interaction was recently shown to stimulate the growth of new dendritic spines (16). The proposed mechanisms via which platelets communicate with neural cells have been discussed in more detail elsewhere (1); however, these mechanisms could also influence neural cell properties under neurodegenerative conditions. Moreover, as reviewed below, platelets exhibit neuron-like properties that further facilitate crosstalk between these cells and the central nervous system.
The Neuron-like Properties of Platelets—Bridging the Gap Between the Systemic Environment and Brain Pathologies?
Despite their distinct location and function, platelets and neural cells are remarkably similar, suggesting a potential path of cross-communication between the systemic environment and the brain. In particular the intercellular storage compartments in neurons, which contain neuropeptides, neurohormones and neurotransmitters, are comparable to platelet granules, including the use of similar vesicle trafficking mechanisms. Platelet dense granules resemble the small dense-core synaptic vesicles of neurons in terms of their serotonin and adenosine triphosphate contents, among other features, whereas the large dense-core vesicles of neurons are comparable to platelet α-granules. Both storage compartments carry a large variety of bioactive peptides, and stimulus-specific secretion processes are observed in both neurons (17) and platelets (8). This indicates that the strict regulation of selective exocytosis is a conserved mechanism in both cell types (18). Platelet and neuronal exocytosis are both triggered by an increase in the internal calcium concentration (19), leading to the rapid activation of the secretory machinery. Moreover, the mechanism whereby the internal vesicles fuse with the plasma membrane is highly conserved, occurring via specific docking molecules such as SNAREs, VAMPs and syntaxins (19). Other review papers have discussed the molecular similarities between platelets and neuronal cells in more detail (18–20), and have proposed that platelets could even be considered “neuronal cells” themselves, with the interaction between platelets and T cells representing a novel “neuroimmunological” synapse in the periphery (20). Likewise, platelets could act as messengers, transferring signals between the peripheral environment and brain cells. We have shown that platelet-rich plasma has direct stimulating effects on a pure population of flow cytometry-isolated hippocampal dentate gyrus-derived neural precursor cells in vitro, and that mice which have been depleted of platelets fail to show the expected exercise-induced increase in neural precursor cell proliferation in vivo (21). This work suggests that platelet-neural stem cell communication is an important regulatory mechanism in these brain cells, although the precise molecular mechanisms underlying this communication are still unclear.
Platelets carry several neurotransmitters that are essential for the intercellular communication between brain cells, including γ-aminobutyric acid (GABA), glutamate, serotonin, epinephrine, dopamine, and histamine. This suggests that platelets can send and receive signals to and from the nervous system and may act as an important relay between the brain and peripheral organs. The monoamine neurotransmitter serotonin is stored in dense granules, and peripheral serotonin release-associated regulatory functions of platelets have been described (6, 22). Although the peripheral and central nervous system serotonergic systems are thought to be separated, platelets release serotonin in response to brain-specific glycolipid structures, which are integrated into the lipid rafts of neurons and astrocytes (15). Such interactions could occur in conditions in which cerebral microvessels become leaky, including during neurodegenerative diseases (23), suggesting that platelets could act as communicators between blood and brain. This hypothesis becomes more cogent when considering the two major neurotransmitters GABA and glutamate, both of which are taken up by platelets (24). Glutamate is the most abundant excitatory neurotransmitter in the brain, and substrate-induced glutamate uptake has been demonstrated in human platelets, likely via specific glutamate receptors (25), similar to what is observed in neuronal cells (26). Platelets express various glutamate receptor subtypes and exhibit high affinity glutamate uptake activity, a process which is impaired in disorders such as PD (27), AD (28) and ALS (29). GABA, an inhibitory neurotransmitter, is crucial for healthy brain function, with perturbances in GABA receptor signaling being associated with neurodegenerative conditions [reviewed in Kim et al. (30)]. Platelets carry considerable amounts of GABA, although the concentration is 30% lower than that found in neurons (31). In both neurons and platelets GABA is metabolized by GABA transaminase (31). Moreover, similar to neurons, platelets appear to take up GABA in a substrate-induced manner, with an in vitro study reporting that the GABA concentration in platelets is negligible when the peripheral benzodiazepine receptor blocker PK11195 is present in the cell culture medium (31).
Given these similar mechanisms of neurotransmitter uptake and metabolism, platelets have been suggested as a model system of glutamate and GABA transport in patients suffering from neurodegenerative conditions (25, 31). A more recent review article has extended these concepts to other conserved mechanisms between platelets and neurons that are associated with neurodegenerative diseases, with platelet dysfunction mirroring the abnormalities observed in neurons (32). However, to date it is unclear whether platelet dysfunction occurs first or whether functional impairments in platelets arise as a consequence of other defects that occur during neurodegenerative processes.
Platelets in Neurodegenerative Conditions
It is becoming clear that neurodegenerative diseases do not solely involve cells and tissue of the central nervous system, but rather that systemic influences also play a fundamental role in the development and exacerbation of brain pathologies. As discussed above, platelets are of particular interest as important mediators of this two-way relationship. Several review papers have concluded that these blood cells can serve as potent systemic biomarkers of neurodegenerative diseases, mirroring the pathological phenotypes of neural cells (32–34). In this section we describe the studies that link platelets to neurodegenerative conditions, with a particular focus on platelet dysfunction in these disorders (summarized in Table 1).
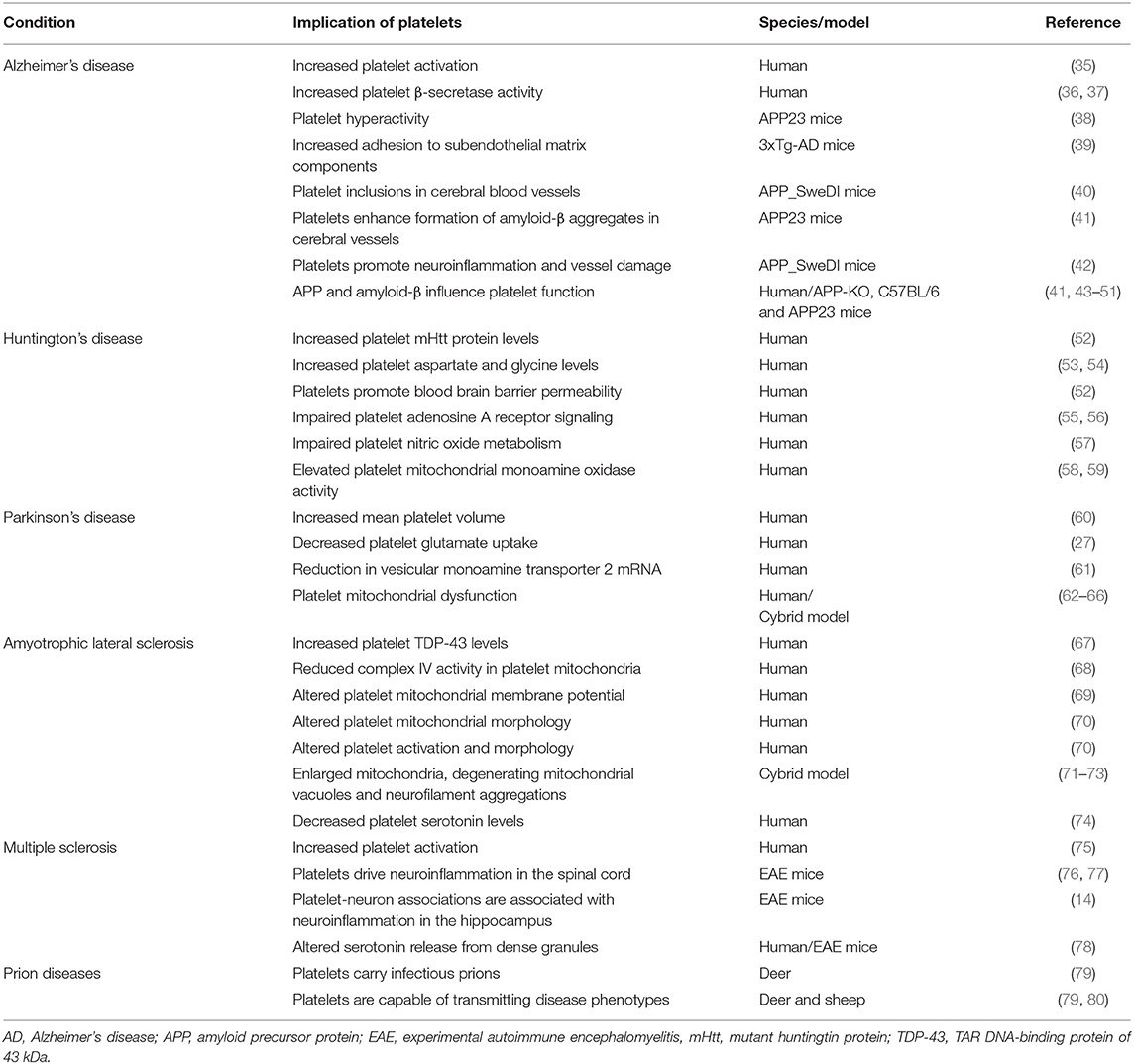
Table 1. Platelet abnormalities linked to neurodegenerative conditions.
Alzheimer's Disease
AD is a slowly developing progressive form of dementia that is accompanied by unpredictable behavior, lack of enthusiasm and memory loss. The neuropathological hallmarks of AD include neuronal and synaptic loss, neuroinflammation, the formation of intracellular neurofibrillary tangles and the deposition of amyloid-ß in brain tissue and cerebral vessels. Increasing evidence has linked platelet dysfunction to this disease, in particular in the context of amyloid-ß secretion from platelets.
Although neural cells, including astrocytes and neurons, produce and secrete amyloid-ß (81), the peptide can also be released by activated platelets (82). Platelets have been suggested to be the primary source of amyloid-ß peptide in the blood (83). The cells produce this peptide through the cleavage of its precursor protein, amyloid precursor protein (APP), which is abundantly present in platelets and is secreted following platelet activation, similar to its metabolite amyloid-ß (82, 84, 85). Both APP and amyloid-ß peptide are associated with platelet functions. Whereas, APP is involved in the regulation of thrombosis and coagulation (46–48), amyloid-ß peptide has the ability to promote platelet activation (41, 49–51), adhesion (43, 48, 50), aggregation (47, 48) and to induce reactive oxygen species generation (45, 51).
Rather than alterations in platelet count or size, changes in platelet activation appear to play a prominent role in AD, with increases in activation detected in the blood of AD patients, likely as a result of increased lipid peroxidation (35). Similarly, platelets have been shown to be hyperactive in aged APP23 transgenic mice, a model of AD (38). A subsequent study confirmed abnormalities in platelet function in a more complex mouse model of AD, 3xTg-AD mice, with increased platelet adhesion to components of the subendothelial matrix and accelerated thrombus formation, although the platelet count remained unchanged (39). In patients with mild cognitive impairment and AD, the activity of ß-secretase, one of the major enzymes required for the cleavage of APP, is significantly increased in the membranes of platelets (36, 37), suggesting further platelet-related systemic changes during the disease.
A recent parabiosis study, in which the blood circulation of APPswe/PS1dE9 transgenic AD model mice was connected with that of their wildtype counterparts demonstrated that human amyloid-ß originating from the transgenic mice accumulated in the brains of their healthy littermates, forming amyloid-ß plaques and amyloid angiopathy following 12 months of parabiosis (86). Moreover, the parabiotic wildtype mice exhibited impaired long-term potentiation in the hippocampal cornu ammonis 1 area, suggesting a reduction in synaptic plasticity, which is thought to underlie deficits in learning and memory (86). Although this study did not investigate the origin of the blood-derived amyloid-ß, the authors suggested platelets as a likely source.
Prior to amyloid-ß plaque formation, platelet inclusions in cerebral blood vessels are among the first symptoms to appear in the brains of APP_SweDI AD model mice (40). Another study demonstrated that platelets enhance the formation of amyloid-ß aggregates in the brain vasculature and that amyloid-ß itself can activate platelets (41). In the same study, the plaque burden of cerebral vessels in APP23 mice was significantly reduced following a 3-month treatment with clopidogrel, a known inhibitor of platelet activation (41). Interestingly, a trend toward reduced plaque formation was also observed within the hippocampus, a brain region which is crucial for learning and memory and is profoundly affected by AD (41). More recent work has shown that platelets isolated from APP_SweDI mice promote vessel damage and neuroinflammation in the healthy mouse brain, leading to amyloid-ß-like immunoreactivity at the damaged vessel sites (42). Together these data suggest that hyperactive AD platelets release and interact with amyloid-ß specifically at sites of vessel damage, thereby accelerating the progression of the disease (38, 39, 41, 42). This is in line with work suggesting that AD may, at least in part, be a slowly developing thrombohemorrhagic disorder (87, 88), highlighting the need to expand research beyond the brain and consider treatment of the systemic environment in AD patients. In this regard, platelets represent a potential target, with a reduction in platelet count being suggested as a means to counteract the overproduction of amyloid-β (87).
An interesting alternative theory is that amyloid-ß release represents a defense mechanism against septic agents (89, 90). Recent research indicates that amyloid-ß may be a normal component of the innate immune system, protecting individuals against microbial and viral infection (91–94). Given the emerging evidence that platelets act as fundamental immune cells, including in the brain [summarized in Leiter and Walker (1)], they could accumulate at damaged cerebral vessel sites and release amyloid-ß as a defense peptide. This is in line with a study which suggests that the release of amyloid-ß from platelets is triggered by pre-existing tissue damage and inflammation and represents a natural protective mechanism against infection during thrombosis (92). However, the platelet hyperactivity that is associated with AD may lead to the overproduction of amyloid-ß, thereby exacerbating inflammation and eventually promoting the development of plaque formation.
Although the studies described above focused on amyloid-ß, this peptide does not represent the only known link between platelets and AD, with other investigators examining the involvement of neurofibrillary tangles and impaired neurotransmitter homeostasis. These studies have been reviewed elsewhere (95).
Huntington's Disease
HD is a hereditary autosomal dominant neurodegenerative disorder caused by a CAG repeat expansion in exon 1 of the huntingtin gene, resulting in the production of a mutant huntingtin protein (mHtt). This protein accumulates in neurons, thereby leading to their eventual death and a progressive loss of motor and cognitive functions. Extensive research has shown that a number of cell subpopulations in the blood are altered in HD patients, with platelets having the highest levels of mHtt (52).
The platelets of HD patients exhibit a number of abnormalities, including aberrant amplification of adenosine A receptor (A2AR) signaling (55, 56). Given that the A2AR is expressed in GABA/enkephalin spiny neurons, it has been proposed that it may play a role in HD pathogenesis. Other studies have also reported a correlation between the density of A2AR in platelets and the rate of disease, age at onset and CAG repeat expansion (55, 96). However, whether or not A2AR activity provides a useful biomarker remains to be determined.
Dysfunction of the nitric oxide /nitric oxide synthase pathway and monoamine oxidase (MAO) have also been suggested to be critical contributors to HD pathology. Nitric oxide metabolism has been found to be dysregulated in platelets during the late stages of HD progression (57), and MAO activity has been associated with neuronal damage in a number of degenerative conditions. MAO is a mitochondrial enzyme that catalyzes the oxidative deamination of monoamines such as dopamine. MAO exists in the MAO-A and MAO-B isoforms. Whereas, some cell types express both isoforms, only MAO-B is found in platelets. Significantly elevated platelet MAO activity has been observed in HD patients during disease progression (58, 59), with the levels negatively correlating with the clinical response to drug treatment (97).
A proposed model of HD pathogenesis is the “excitatory hypothesis,” based on the observation that excitatory amino acids and N-methyl-D-aspartate receptor agonists, including aspartate and glutamate, recapitulate the striatal neuron degeneration observed in HD (98). Although early studies found no differences in glutamate and aspartate activity between normal and HD platelets (99, 100), later studies have reported significantly increased aspartate and glycine in HD platelets (53, 54).
Mitochondrial dysfunction has also been implicated in the pathogenesis of HD. A significant decrease in mitochondrial complex I activity per platelet was observed when patients were grouped according to disease severity; however, when normalized to mitochondrial DNA content, no differences were detected (101). In contrast, an earlier study found no difference in platelet mitochondrial complex activity in HD patients (102). Given the relatively small group sizes, further data are required to determine whether mitochondrial function in platelets provides a useful biomarker of HD. However, increased mitochondrial-dependent apoptosis has also been reported in HD cybrids (103).
Platelets are also important in maintaining normal vascular integrity (104). Recently, an initial study investigating the potential impact of mHtt on platelet function showed that platelets can promote blood brain barrier permeability in HD, pointing toward their potential contribution to disease pathogenesis (52).
Parkinson's Disease
PD is a degenerative disorder caused by the loss of dopaminergic neurons in the substantia nigra, thereby resulting in an impairment in motor and cognitive functions. Although the cause of sporadic PD, the most common form of the disease, is unknown, one major causal factor is mitochondrial dysfunction. This was first suggested by the finding that 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), a neurotoxin that selectively kills dopaminergic neurons, acts by inhibiting complex I of the electron transport chain (105). A plethora of studies have reported reduced complex I activity in the platelets of patients with PD (62–64), although it should be noted that other studies did not find such alterations (65, 66). Supporting the former observation, a PD cybrid model in which mitochondrial DNA from PD platelets was expressed in rho 0 human teratocarcinoma cells showed a reduction in complex I activity (106, 107). In addition, 1-methyl-4-phenyl-pyridinium ion MPP(+), the metabolite of MPTP, was shown to induce adenosine triphosphate depletion in platelets and attenuate platelet aggregation and activity, providing a potential mechanism underlying the anti-aggregation effect observed in PD patients (108).
Several studies have suggested that MAO also plays an important role in MPTP toxicity and the etiology of PD. Increased MAO-B activity has been observed in PD patients (109–111), potentially due to a G/A single nucleotide polymorphism in intron 13 which results in a splicing enhancer that stimulates intron 13 removal efficiency (110). However, the data concerning platelet MAO-B activity in PD patients are not consistent, with other studies reporting that platelet MAO-B activity is unchanged in PD patients (112, 113).
A number of other alterations in the platelets of PD patients have also been suggested as potentially useful biomarkers. These include a reduction in vesicular monoamine transporter 2 mRNA (61), an increase in mean platelet volume (60), and decreased glutamate uptake (27).
Amyotrophic Lateral Sclerosis
ALS is a fatal neurodegenerative disorder that is characterized by progressive and selective loss of motor neurons in the brain and spinal cord. Patients suffer from progressive muscle weakness and paralysis of their voluntary muscles, ultimately leading to respiratory failure and death. There is accumulating evidence that in addition to affecting motor neurons, ALS also affects platelets.
Almost all ALS cases (~97%) are characterized by pathology due to the TAR DNA-binding protein of 43 kDa (TDP-43) (114, 115). In diseased neurons, TDP-43 is relocated from its normal nuclear location to the cytoplasm, where it is phosphorylated and ubiquitinated, subsequently aggregating to form insoluble intracellular inclusions (115). A recent study found that the TDP-43 levels in platelets from patients with sporadic ALS are significantly higher than those of non-ALS age-matched controls (67). Interestingly, the TDP-43 levels in platelets tended to increase with disease progression, although a larger cohort of patients is required to confirm this observation (67).
Mitochondrial abnormalities, particularly impairments of complex IV (cytochrome c-oxidase) activity, have been implicated in ALS, although the exact role of mitochondrial dysfunction remains unclear. In addition to mitochondrial dysfunction in motor neurons of ALS patients, mitochondrial changes have also been reported in muscle, liver and blood cells, suggesting systemic involvement (116–118). Complex IV activity was found to be decreased in platelets from ALS patients in a small case-control study (68). Interestingly, the cellular mitochondrial content increased, indicating a potential compensatory mechanism (68). Further supporting the notion of mitochondrial dysfunction, a change in the mitochondrial membrane potential has been reported in platelets from ALS patients (69), as well as changes in the ultrastructure and morphology of platelets and their mitochondria (70). This is in line with an earlier study which observed platelet activation and morphological changes in ALS platelets (119). ALS cybrids (platelets fused to the rho neuronal cell lineage) also show similar cytoskeletal deformities to those found in ALS patients and transgenic superoxide dismutase 1 mice, including enlarged mitochondria, degenerating mitochondrial vacuoles and neurofilament aggregations (71–73). Despite these links between platelet mitochondrial dysfunction and ALS, larger cohort studies are required to conclusively determine whether mitochondrial function can be used as a biomarker for ALS.
Thrombospondin is a glycoprotein that is released from platelet α-granules following thrombin-induced platelet activation. Changes in blood thrombospondin levels have been detected in a number of pathological conditions, including a marked increase in thrombospondin deposition in the muscles of ALS patients (120, 121). The neurotransmitter serotonin is also decreased in the brain and spinal cord of ALS patients (122, 123). Platelets are a major source of serotonin and platelet serotonin levels have been shown to be significantly lower in ALS patients and to positively correlate with patient survival (74). However, the cause of this decrease in serotonin remains elusive. Glutamate excitotoxicity has also been implicated in the pathogenesis of the disease. Platelets contain a glutamate uptake system and express components of the glutamate-glutamine cycle, including the excitatory amino acid transporter 2 and glutamine synthetase. Increased glutamine synthetase, but normal excitatory amino acid transporter 2 expression, has been reported in the platelets of ALS patients (124). However, given that this finding is in contrast to an earlier study which reported a reduction in glutamate uptake in ALS patients (29), these data need to be confirmed.
Multiple Sclerosis
MS is an inflammatory disease, where the immune system attacks the myelin sheaths that cover nerve axons in the spinal cord and brain. The resulting nerve damage leads to communication deficits between the brain and other tissues, and depending on the affected nerves provokes a range of symptoms, including impairments in vision, deficits in motor control of the arms and legs and neuropsychological symptoms such as depression and memory loss. To date, there is no known cure for MS, as the underlying cause is still unknown.
A few studies targeting platelets and their involvement in MS and its mouse model, EAE, have shown that these conditions are associated with abnormalities in platelet function. One of these investigations found increased platelet activation in the blood of clinically stable relapsing-remitting MS patients who had not yet received treatment (116). This was evidenced by significantly larger numbers of CD62P-positive platelets and CD41-positive platelet microparticles (75). Subsequent evidence in EAE mice revealed that platelets exacerbate the development of the disease via the recruitment of leukocytes to the neural tissue (76). A more recent study cemented the involvement of platelets in EAE, demonstrating that platelets not only aggravate (76) but also drive neuroinflammation in the spinal cord (77). Possible mechanisms via which platelets could exacerbate the pathophysiology of MS are discussed in a review by Wachowicz et al. with one interesting concept being an impaired antioxidant mechanism in combination with inflammation-induced platelet activation as an additional source of reactive oxygen species to further accelerate tissue damage (125). Moreover, the secretion of serotonin from dense granules has been shown to modulate immune cell responses in a stage-depended manner. During the early stages of EAE and MS, high levels of platelet-released serotonin stimulate the proliferation and differentiation of pathogenic T cell subsets, thereby promoting proinflammatory responses (78). During later phases of the disease, however, platelets exhibit reduced serotonin levels and appear to suppress T cell activation and central nervous system inflammation (78).
Recent work investigating the brains of EAE-induced mice demonstrated that platelets were also present in the parenchyma of the hippocampus, including in the fimbria and in close proximity to neuronal cell bodies in the dentate gyrus and CA1 region (14). This phenotype was associated with the formation of a neuroinflammatory environment, supposedly due to platelet-neuron associations (14). However, this occurred in the absence of inflammatory cell infiltration, further highlighting the role of platelets in the initiation of EAE (14). In the same study, the pro-inflammatory environment in the hippocampus of EAE-induced mice, as well as their increased anxiety-like behavior, were improved following platelet depletion with polyclonal anti-platelet glycoprotein Ib α chain antibodies, suggesting that platelets could serve as a potential target for the amelioration of the symptoms of MS (14).
Prion Diseases
Prion proteins (PrPs) comprise a class of amyloid-forming proteins, with some isoforms being associated with a group of fatal neurodegenerative diseases termed transmissible spongiform encephalopathies. Once diagnosed, these conditions progress rapidly and are characterized by the chronic deterioration of physical and mental abilities, including profound memory impairments. The scrapie isoform of PrP is an abnormal, misfolded, protease-resistant isoform (126, 127) which is believed to be responsible for transmissible spongiform encephalopathies. Although considered transmissible, the paths through which prion diseases spread are unknown, with the transfusion of blood from infected donors presenting a concern.
Cellular PrP (PrPc) is carried by blood cells, including platelets, in which PrPc is present on the membranes of α-granules (128, 129). Following activation, PrPc can be released from activated platelets, mainly in the form of microparticles and exosomes (128). The function of PrPc under these circumstances is unknown, although it has been reported that the protein is unlikely to play a role in the aggregation or adhesive actions of activated platelets (128). The release of microparticles and exosomes represents a major route of intercellular communication, including crosstalk between platelets and neural cells (1). This suggests that in the course of transmissible spongiform encephalopathies, the less soluble scrapie prion isoform could be carried and released from activated platelets thereby contributing to the infection of the brain and the transmission of the disease through blood transfusion (128). Other work has confirmed that platelets and B cells in the blood of deer, infected with chronic wasting disease carry infectious prions, and are substantially involved in transmitting the disease phenotype (79). In a sheep model of variant Creutzfeldt Jakob disease, the disease could be transmitted through several blood components, such as whole blood, plasma, red blood cells, buffy coat and platelets (80). These data from animal studies suggest a high probability that spongiform encephalopathies are transmissible through blood (79, 80), even in pre-clinical stages of the disease (80). However, only a few cases suggest this possibility in humans, where the lack of a causal link between blood transfusions and the development of prion diseases makes it difficult to draw a conclusion (130–132).
Platelets—A Novel Therapeutic Avenue for the Treatment of Neurodegenerative Conditions?
Impairments in platelet function are a common observation in neurodegenerative disorders; however, healthy platelets and their secreted factors also represent a possible approach for the development of therapeutic interventions for the treatment of neurodegenerative conditions. Among the primary applications are the use of platelet lysate and platelet-rich plasma, both of which are easy to obtain from immune-compatible healthy donors. The beneficial effects of platelet-rich plasma treatment are likely to be attributable to the abundant variety of growth factors that platelets carry in their granules. Neural and glial cells express surface receptors for a range of these growth factors, including vascular endothelial growth factor, epidermal growth factor, fibroblast growth factor-2, platelet-derived growth factor, brain-derived neurotrophic factor, platelet factor 4, transforming growth factor-ß, insulin-like growth factor-1, connective tissue growth factor and bone morphogenetic protein-2,−4, and−6, suggesting a fundamental role of platelets in tissue growth and regeneration, including in the brain (133–135). Moreover, human platelet lysate comprises a plethora of growth factors, including those with neuroprotective properties. Although emerging research has shown promising results, diverse protocols for the isolation of platelet-rich plasma and platelet lysates exist, resulting in products which contain variable ranges of growth factors (136). Moreover, novel protocols are continuously being published, describing optimized preparations for specific use in different applications (137–139). These factors therefore represent an important consideration when evaluating study outcomes and planning future clinical trials across different fields.
Platelet-Rich Plasma
Platelet-rich plasma can easily be prepared from whole blood using a slow centrifugation speed and physiological washing buffers that support platelet purification. This method achieves a nearly pure population of platelets [>99.99% purity (140)], and the platelet preparation can be used immediately or stored. However, upon freezer storage and subsequent thawing of the samples, a substantial number of cells will be lyzed, leading to the release of growth factors from platelet granules. These are also present in frozen/thawed platelet-rich plasma preparations, making them a physiological cocktail of intact cells and released bioactive molecules.
Beneficial therapeutic effects of platelet-rich plasma treatment have been reported in numerous tissues, including during burn healing (141, 142), cartilage repair (143) and healing following dermal injuries (144). Other studies have demonstrated that platelet-rich plasma treatment enhances the recovery of peripheral nerves following injury, including cavernous nerve injuries (145) and damage of the facial (146) and sciatic (147) nerves. Moreover, platelet-rich plasma injections into the injured spinal cord of rats have been shown to promote locomotor recovery, local angiogenesis and neuronal regeneration (148). Another study in mice suggested the therapeutic use of platelet-rich plasma in neuroinflammatory central nervous system diseases, as platelet-rich plasma treatment considerably improved the clinical symptoms in the EAE mouse model of MS (149). This effect was accompanied by significantly lower gene expression and a decrease in the protein levels of inflammatory markers in the lumbar parts of the spinal cord, including the microglial marker Iba1 and the pro-inflammatory cytokine interleukin 1-β, as well as the reduced infiltration of inflammatory cells (149). The platelet-rich plasma injection also protected the cells from demyelination in the affected area (149). Other studies which used the plasma rich in growth factors Endoret® technology to isolate platelet-rich plasma from human blood have demonstrated that treatment with these preparations significantly reduces amyloid-β plaque density in the hippocampus and improves cognitive function in APP/PS1 AD model mice (150). Another study complemented this finding showing that the same preparations enhanced adult neurogenesis in the hippocampus of APP/PS1 mice, a process known to be affected during AD, and that this enhancement was likely due to a reduction in amyloid-β-mediated neurotoxicity (151). The same method also promoted neuronal survival and diminished the inflammatory responses in a mouse model of PD, as well as reducing the associated motor impairments (152). These data suggest that platelet-rich plasma treatment represents a promising approach which could be applied to several neurodegenerative disorders.
Platelet Lysate
Similar to platelet-rich plasma, platelet lysate can be easily obtained from whole blood samples. Platelets are first enriched by centrifugation steps, followed by freezing and thawing of the samples. An additional centrifugation step then separates the freeze/thaw-triggered secreted platelet factors, which constitutes the platelet lysate, from the remaining cell debris.
Given their essential role in wound healing and tissue repair, platelet lysates are being investigated as a therapy for a number of neurodegenerative diseases. Human platelet lysates have been investigated as a novel biotherapy for ALS and PD patients. In an NSC-34 cell-based model of ALS, human platelet lysates conferred a neuroprotective effect against staurosporine-induced apoptosis and menadione-induced oxidative stress, indicating that neuronal loss can be diminished by platelet factors in those conditions (153). In a Lund human mesencephalic cell-based model of PD, pre-treatment of the cells with human platelet lysates also protected again erastin-induced ferroptotic cell death (153). The authors further optimized the isolation protocol to produce platelet lysate preparations which are more enriched for neurotrophins and at the same time depleted of plasma proteins, thereby preventing potential adverse thrombotic effects during in vivo applications (137). Following intranasal administration of the optimized platelet lysate, obvious protective effects were observed on dopaminergic neurons in the substantia nigra and the striatum of PD model mice (137). The intranasally administered platelet factors were also found in several other regions of the brain, including the striatum, olfactory bulb, and cortex (137), making this treatment method a promising tool for application in various neurodegenerative conditions.
Although we have not addressed stroke and other brain injuries in this review, human platelet lysate treatment has also been shown to produce positive outcomes in these conditions. Following stroke, human platelet lysate injections into the lateral ventricles of rats had neuroprotective effects (154). The platelet lysate-treated rats exhibited a larger number of proliferating neural precursor cells in the subventricular zone, accompanied by increased angiogenesis (151). They also displayed lower motor function deficits (154). Another study demonstrated that administrating human platelet lysate decreased apoptosis and stimulated the survival of proliferating neural precursor cells in the same brain region after a lysolecithin-induced demyelination lesion in the corpus callosum (12), further suggesting a neuroprotective role of platelets after cerebral damage.
Platelets and Platelet Microparticles—Potential Vehicles for the Delivery of Therapeutic Drugs?
In addition to platelet-rich plasma and platelet lysate preparations, an interesting approach is emerging, whereby platelets are used as a physiological vehicle to deliver molecules to target regions that might otherwise be difficult to access. With their context-dependent and specific cell-cell communication capacity, platelets could serve as a selective and non-toxic drug delivery system in order to target specific cells and tissues. This approach has been extensively discussed previously, with a particular focus on the use of platelets to deliver chemotherapeutic agents to tumors (155). However, this novel strategy still requires additional studies to confirm its efficacy. Microparticles, which are released by platelets upon activation, have also been proposed as a natural delivery system for drugs (155, 156). The majority of all microvesicles in the blood are platelet-derived (157), indicating a vital contribution of platelets to intercellular communication. Platelet microparticles, which are 0.1–1 μm in diameter, are shed from the plasma membrane (158) and contain cytoplasm, microRNA, mRNA, lipids and proteins. These can be transferred to other cells, thereby affecting their function (159–162). Given their capacity to influence and communicate with neural cells, platelets and their secreted microparticles could also be engineered as drug carriers for the treatment of neurodegenerative disorders. However, until the exact mechanisms of the specific cell-cell communication between platelets and brain cells are fully understood, the value of this approach remains speculative. Furthermore, in order to develop human therapies with drug-loaded blood cells, extensive studies are needed to establish clinical grade protocols which standardize the varying methods of isolation and storage of platelets and platelet microparticles prior to their regulated reintroduction into individuals. Drug loading protocols for these natural vehicles, in terms of their capacity and compatibility with the drugs required to target neurodegenerative phenotypes, also need to be established. Nonetheless, in the field of regenerative medicine, considerable headway has already been made toward engineering extracellular vesicles and blood cell-inspired nanoparticles for therapeutic use (163–166).
Conclusion
As summarized in this review, data connecting platelets and the factors they secrete to neurodegeneration have accumulated over recent years. However, it remains unclear whether platelet malfunction initiates the pathophysiological events that occur in neurodegenerative conditions, or whether platelet dysfunction arises as a consequence of other unfavorable changes that occur at early stages of these disorders. More data regarding the origin of platelet dysfunction are therefore required. During the onset of neurodegenerative conditions, factors released from healthy platelets could also have a protective role, as suggested by recent studies of AD (92) and cancer, where platelets initially suppress tumor angiogenesis (167). Moreover, platelets exhibit a sophisticated endocytic machinery (168) via which they could collect products that are released into the blood from other malfunctioning cells in an attempt to clear the systemic environment of cytotoxic components in the early stages of disease.
Although platelets and their released factors are gaining recognition for their potential therapeutic value in regenerative medicine, research is still in its infancy. Furthermore, the origin of platelets, the bone marrow, should not be overlooked, as a functional predisposition may also be inherited from their parent cells, the megakaryocytes. In conclusion, it remains highly interesting, but at the same time extremely challenging, to understand how platelets exert manifold actions across different tissues in physiological as well as pathological conditions. Their functional complexity clearly demands interdisciplinary approaches in order to develop novel therapeutic interventions which benefit from the multifaceted nature of platelets, including their capacity to facilitate crosstalk between the systemic environment and the brain.
Author Contributions
OL and TW wrote the manuscript.
Funding
This work was funded by The Brazil Family Program for Neurology and The Donald and Joan Wilson Foundation Ltd.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Rowan Tweedale for her helpful comments on the paper.
Abbreviations
A2AR, adenosine A receptor; AD, Alzheimer's disease; ALS, amyotrophic lateral sclerosis; APP, amyloid precursor protein; EAE, experimental autoimmune encephalomyelitis; GABA, γ-aminobutyric acid; HD, Huntington's disease; mHtt, mutant huntingtin protein; MAO, monoamine oxidase; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; MS, multiple sclerosis; PD, Parkinson's disease; PrP, prion protein; TDP-43, TAR DNA-binding protein of 43kDa.
References
1. Leiter O, Walker TL. Platelets: The missing link between the blood and brain? Prog Neurobiol. (2019) 183:101695. doi: 10.1016/j.pneurobio.2019.101695
2. Daly ME. Determinants of platelet count in humans. Haematologica. (2011) 96:10–3. doi: 10.3324/haematol.2010.035287
3. Slichter SJ. Relationship between platelet count and bleeding risk in thrombocytopenic patients. Transfus Med Rev. (2004) 18:153–67. doi: 10.1016/j.tmrv.2004.03.003
4. Jirouskova M, Shet AS, Johnson GJ. A guide to murine platelet structure, function, assays, and genetic alterations. J Thromb Haemost. (2007) 5:661–9. doi: 10.1111/j.1538-7836.2007.02407.x
5. Schmitt A, Guichard J, Massé J-M, Debili N, Cramer EM. Of mice and men: comparison of the ultrastructure of megakaryocytes and platelets. Exp Hematol. (2001) 29:1295–302. doi: 10.1016/S0301-472X(01)00733-0
6. Cloutier N, Allaeys I, Marcoux G, Machlus KR, Mailhot B, Zufferey A, et al. Platelets release pathogenic serotonin and return to circulation after immune complex-mediated sequestration. Proc Natl Acad Sci USA. (2018) 115:e1550–e1559. doi: 10.1073/pnas.1720553115
7. Coppinger JA, O'Connor R, Wynne K, Flanagan M, Sullivan M, Maguire PB, et al. Moderation of the platelet releasate response by aspirin. Blood. (2007) 109:4786–92. doi: 10.1182/blood-2006-07-038539
8. Italiano JE, Richardson JL, Patel-Hett S, Battinelli E, Zaslavsky A, Short S, et al. Angiogenesis is regulated by a novel mechanism: pro- and antiangiogenic proteins are organized into separate platelet α-granules and differentially released. Blood. (2008) 111:1227–33. doi: 10.1182/blood-2007-09-113837
9. Heijnen HF, Schiel AE, Fijnheer R, Geuze HJ, Sixma JJ. Activated platelets release two types of membrane vesicles: microvesicles by surface shedding and exosomes derived from exocytosis of multivesicular bodies and α-granules. Blood. (1999) 94:3791–9. doi: 10.1182/blood.V94.11.3791
10. Espinosa-Parrilla Y, Gonzalez-Billault C, Fuentes E, Palomo I, Alarcón M. Decoding the role of platelets and related microRNAs in aging and neurodegenerative disorders. Front Aging Neurosci. (2019) 11:151. doi: 10.3389/fnagi.2019.00151
11. Tocantins LM. The mammalian blood platelet in health and disease. Medicine. (1938) 17:155. doi: 10.1097/00005792-193805000-00001
12. Kazanis I, Feichtner M, Lange S, Rotheneichner P, Hainzl S, Öller M, et al. Lesion-induced accumulation of platelets promotes survival of adult neural stem/progenitor cells. Exp Neurol. (2015) 269:75–89. doi: 10.1016/j.expneurol.2015.03.018
13. Schleicher RI, Reichenbach F, Kraft P, Kumar A, Lescan M, Todt F, et al. Platelets induce apoptosis via membrane-bound FasL. Blood. (2015) 126:1483–93. doi: 10.1182/blood-2013-12-544445
14. Kocovski P, Jiang X, D'Souza CS, Li Z, Dang PT, Wang X, et al. Platelet depletion is effective in ameliorating anxiety-like behavior and reducing the pro-inflammatory environment in the hippocampus in murine experimental autoimmune encephalomyelitis. J Clin Med. (2019) 8:162. doi: 10.3390/jcm8020162
15. Sotnikov I, Veremeyko T, Starossom SC, Barteneva N, Weiner HL, Ponomarev ED. Platelets recognize brain-specific glycolipid structures, respond to neurovascular damage and promote neuroinflammation. PLoS One. (2013) 8:e58979. doi: 10.1371/journal.pone.0058979
16. Dukhinova M, Kuznetsova I, Kopeikina E, Veniaminova E, Yung AWY, Veremeyko T, et al. Platelets mediate protective neuroinflammation and promote neuronal plasticity at the site of neuronal injury. Brain Behav Immun. (2018) 74:7–27. doi: 10.1016/j.bbi.2018.09.009
17. Perello M, Stuart R, Nillni EA. Prothyrotropin-releasing hormone targets its processing products to different vesicles of the secretory pathway. J Biol Chem. (2008) 283:19936–47. doi: 10.1074/jbc.M800732200
18. Goubau C, Buyse GM, Di Michele M, Van Geet C, Freson K. Regulated granule trafficking in platelets and neurons: a common molecular machinery. Eur J Paediatr Neurol. (2013) 17:117–25. doi: 10.1016/j.ejpn.2012.08.005
19. Reed GL, Fitzgerald ML, Polgár J. Molecular mechanisms of platelet exocytosis: insights into the “secrete” life of thrombocytes. Blood. (2000) 96:3334–42. doi: 10.1182/blood.V96.10.3334
20. Ponomarev ED. Fresh evidence for platelets as neuronal and innate immune cells: Their role in the activation, differentiation, and deactivation of Th1, Th17, and Tregs during tissue inflammation. Front Immunol. (2018) 9:406 doi: 10.3389/fimmu.2018.00406
21. Leiter O, Seidemann S, Overall RW, Ramasz B, Rund N, Schallenberg S, et al. Exercise-induced activated platelets increase adult hippocampal precursor proliferation and promote neuronal differentiation. Stem Cell Rep. (2019) 12:667–79. doi: 10.1016/j.stemcr.2019.02.009
22. Vanhoutte PM. Serotonin and the vascular wall. Int J Cardiol. (1987) 14:189–203. doi: 10.1016/0167-5273(87)90008-8
23. Carvey PM, Hendey B, Monahan AJ. The blood brain barrier in neurodegenerative disease: a rhetorical perspective. J Neurochem. (2009) 111:291–314. doi: 10.1111/j.1471-4159.2009.06319.x
24. Rainesalo S, Keränen T, Saransaari P, Honkaniemi J. GABA and glutamate transporters are expressed in human platelets. Brain Res Mol Brain Res. (2005) 141:161–5. doi: 10.1016/j.molbrainres.2005.08.013
25. Begni B, Tremolizzo L, D'Orlando C, Bono MS, Garofolo R, Longoni M, et al. Substrate-induced modulation of glutamate uptake in human platelets. Br J Pharmacol. (2005) 145:792–9. doi: 10.1038/sj.bjp.0706242
26. Munir M, Correale DM, Robinson MB. Substrate-induced up-regulation of Na(+)-dependent glutamate transport activity. Neurochem Int. (2000) 37:147–62. doi: 10.1016/s0197-0186(00)00018-8
27. Ferrarese C, Zoia C, Pecora N, Piolti R, Frigo M, Bianchi G, et al. Reduced platelet glutamate uptake in Parkinson's disease. J Neural Transm (Vienna). (1999) 106:685–92. doi: 10.1007/s007020050189
28. Ferrarese C, Begni B, Canevari C, Zoia C, Piolti R, Frigo M, et al. Glutamate uptake is decreased in platelets from Alzheimer's disease patients. Ann Neurol. (2000) 47:641–3. doi: 10.1002/1531-8249(200005)47:5<641::AID-ANA12>3.0.CO;2-I
29. Ferrarese C, Sala G, Riva R, Begni B, Zoia C, Tremolizzo L, et al. Decreased platelet glutamate uptake in patients with amyotrophic lateral sclerosis. Neurology. (2001) 56:270–2. doi: 10.1212/wnl.56.2.270
30. Kim J, Lee S, Kang S, Kim S-H, Kim J-C, Yang M, et al. Brain-derived neurotropic factor and GABAergic transmission in neurodegeneration and neuroregeneration. Neural Regen Res. (2017) 12:1733–41. doi: 10.4103/1673-5374.217353
31. Kaneez FS, Saeed SA. Investigating GABA and its function in platelets as compared to neurons. Platelets. (2009) 20:328–33. doi: 10.1080/09537100903047752
32. Canobbio I. Blood platelets: Circulating mirrors of neurons? Res Pract Thromb Haemost. (2019) 3:564–5. doi: 10.1002/rth2.12254
33. Behari M, Shrivastava M. Role of platelets in neurodegenerative diseases: a universal pathophysiology. Int J Neurosci. (2013) 123:287–99. doi: 10.3109/00207454.2012.751534
34. Donner L, Elvers M. Platelets and neurodegenerative diseases. In: Gresele P, Kleiman NS, Lopez JA, Page CP, editors. Platelets in Thrombotic and Non-Thrombotic Disorders: Pathophysiology, Pharmacology and Therapeutics: An Update. Cham: Springer International Publishing (2017) p. 1209–24. doi: 10.1007/978-3-319-47462-5_81
35. Ciabattoni G, Porreca E, Di Febbo C, Di Iorio A, Paganelli R, Bucciarelli T, et al. Determinants of platelet activation in Alzheimer's disease. Neurobiol Aging. (2007) 28:336–42. doi: 10.1016/j.neurobiolaging.2005.12.011
36. Johnston JA, Liu WW, Coulson DTR, Todd S, Murphy S, Brennan S, et al. Platelet β-secretase activity is increased in Alzheimer's disease. Neurobiol Aging. (2008) 29:661–68. doi: 10.1016/j.neurobiolaging.2006.11.003
37. Liu WW, Todd S, Craig D, Passmore AP, Coulson DTR, Murphy S, et al. Elevated platelet ß-secretase activity in mild cognitive impairment. Dement Geriatr Cogn Disord. (2007) 24:464–8. doi: 10.1159/000110739
38. Jarre A, Gowert NS, Donner L, Münzer P, Klier M, Borst O, et al. Pre-activated blood platelets and a pro-thrombotic phenotype in APP23 mice modeling Alzheimer's disease. Cell Signal. (2014) 26:2040–50. doi: 10.1016/j.cellsig.2014.05.019
39. Canobbio I, Visconte C, Oliviero B, Guidetti G, Zarà M, Pula G, et al. Increased platelet adhesion and thrombus formation in a mouse model of Alzheimer's disease. Cell Signal. (2016) 28:1863–71. doi: 10.1016/j.cellsig.2016.08.017
40. Kniewallner KM, Wenzel D, Humpel C. Thiazine Red(+) platelet inclusions in cerebral blood vessels are first signs in an Alzheimer's disease mouse model. Sci Rep. (2016) 6:28447. doi: 10.1038/srep28447
41. Donner L, Fälker K, Gremer L, Klinker S, Pagani G, Ljungberg LU, et al. Platelets contribute to amyloid-β aggregation in cerebral vessels through integrin αIIbβ3-induced outside-in signaling and clusterin release. Sci Signal. (2016) 9:ra52. doi: 10.1126/scisignal.aaf6240
42. Kniewallner KM, Foidl BM, Humpel C. Platelets isolated from an Alzheimer mouse damage healthy cortical vessels and cause inflammation in an organotypic ex vivo brain slice model. Sci Rep. (2018) 8:15483. doi: 10.1038/s41598-018-33768-2
43. Kowalska MA, Badellino K. ß-Amyloid protein induces platelet aggregation and supports platelet adhesion. Biochem Biophys Res Commun. (1994) 205:1829–35. doi: 10.1006/bbrc.1994.2883
44. Canobbio I, Guidetti GF, Oliviero B, Manganaro D, Vara D, Torti M, et al. Amyloid β-peptide-dependent activation of human platelets: essential role for Ca2+ and ADP in aggregation and thrombus formation. Biochem J. (2014) 462:513–23. doi: 10.1042/BJ20140307
45. Abubaker AA, Vara D, Eggleston I, Canobbio I, Pula G. A novel flow cytometry assay using dihydroethidium as redox-sensitive probe reveals NADPH oxidase-dependent generation of superoxide anion in human platelets exposed to amyloid peptide β. Platelets. (2019) 30:181–9. doi: 10.1080/09537104.2017.1392497
46. Canobbio I, Visconte C, Momi S, Guidetti GF, Zarà M, Canino J, et al. Platelet amyloid precursor protein is a modulator of venous thromboembolism in mice. Blood. (2017) 130:527–36. doi: 10.1182/blood-2017-01-764910
47. Van Nostrand WE, Schmaier AH, Farrow JS, Cunningham DD. Protease nexin-II (amyloid ß-protein precursor): a platelet α-granule protein. Science. (1990) 248:745–8. doi: 10.1126/science.2110384
48. Visconte C, Canino J, Guidetti GF, Zarà M, Seppi C, Abubaker AA, et al. Amyloid precursor protein is required for in vitro platelet adhesion to amyloid peptides and potentiation of thrombus formation. Cell Signal. (2018) 52:95–102. doi: 10.1016/j.cellsig.2018.08.017
49. Herczenik E, Bouma B, Korporaal SJA, Strangi R, Zeng Q, Gros P, et al. Activation of human platelets by misfolded proteins. Arterioscler Thromb Vasc Biol. (2007) 27:1657–65. doi: 10.1161/ATVBAHA.107.143479
50. Canobbio I, Catricalà S, Di Pasqua LG, Guidetti G, Consonni A, Manganaro D, et al. Immobilized amyloid Aβ peptides support platelet adhesion and activation. FEBS Lett. (2013) 587:2606–11. doi: 10.1016/j.febslet.2013.06.041
51. Gowert NS, Donner L, Chatterjee M, Eisele YS, Towhid ST, Münzer P, et al. Blood platelets in the progression of Alzheimer's disease. PLoS One. (2014) 9:e90523. doi: 10.1371/journal.pone.0090523
52. Denis HL, Lamontagne-Proulx J, St-Amour I, Mason SL, Rowley JW, Cloutier N, et al. Platelet abnormalities in Huntington's disease. J Neurol Neurosurg Psychiatry. (2019) 90:272–83. doi: 10.1136/jnnp-2018-318854
53. Reilmann R, Rolf LH, Lange HW. Huntington's disease: N-methyl-d-aspartate receptor coagonist glycine is increased in platelets. Exp Neurol. (1997) 144:416–9. doi: 10.1006/exnr.1997.6428
54. Reilmann R, Rolf LH, Lange HW. Huntington's disease: The neuroexcitotoxin aspartate is increased in platelets and decreased in plasma. J Neurol Sci. (1994) 127:48–53. doi: 10.1016/0022-510X(94)90134-1
55. Maglione V, Giallonardo P, Cannella M, Martino T, Frati L, Squitieri F. Adenosine A2A receptor dysfunction correlates with age at onset anticipation in blood platelets of subjects with Huntington's disease. Am J Med Genet B Neuropsychiatr Genet. (2005) 139B:101–5. doi: 10.1002/ajmg.b.30223
56. Varani K, Abbracchio MP, Cannella M, Cislaghi G, Giallonardo P, Mariotti C, et al. Aberrant A2A receptor function in peripheral blood cells in Huntington's disease. FASEB J. (2003) 17:2148–50. doi: 10.1096/fj.03-0079fje
57. Carrizzo A, Di Pardo A, Maglione V, Damato A, Amico E, Formisano L, et al. Nitric oxide dysregulation in platelets from patients with advanced Huntington Disease. PLoS One. (2014) 9:e89745. doi: 10.1371/journal.pone.0089745
58. Markianos M, Panas M, Kalfakis N, Vassilopoulos D. Platelet monoamine oxidase activity in subjects tested for Huntington's disease gene mutation. J Neural Transm (Vienna). (2004) 111:475–83. doi: 10.1007/s00702-003-0103-x
59. Norman TR, Chiu E, French MA. Platelet monoamine oxidase activity in patients with Huntington's disease. Clin Exp Pharmacol Physiol. (1987) 14:547–50. doi: 10.1111/j.1440-1681.1987.tb01511.x
60. Koçer A, Yaman A, Niftaliyev E, Dürüyen H, Eryilmaz M, Koçer E. Assessment of platelet indices in patients with neurodegenerative diseases: mean platelet volume was increased in patients with Parkinson's disease. Curr Gerontol Geriatr Res. (2013) 2013:986254. doi: 10.1155/2013/986254
61. Sala G, Brighina L, Saracchi E, Fermi S, Riva C, Carrozza V, et al. Vesicular monoamine transporter 2 mRNA levels are reduced in platelets from patients with Parkinson's disease. J Neural Transm (Vienna). (2010) 117:1093–8. doi: 10.1007/s00702-010-0446-z
62. Benecke R, Strümper P, Weiss H. Electron transfer complexes I and IV of platelets are abnormal in Parkinson's disease but normal in Parkinson-plus syndromes. Brain. (1993) 116 (Pt 6):1451–63. doi: 10.1093/brain/116.6.1451
63. Haas RH, Nasirian F, Nakano K, Ward D, Pay M, Hill R, et al. Low platelet mitochondrial complex I and complex II/III activity in early untreated Parkinson's disease. Ann Neurol. (1995) 37:714–22. doi: 10.1002/ana.410370604
64. Parker WD, Parks JK, Swerdlow RH. Complex I deficiency in Parkinson's disease frontal cortex. Brain Res. (2008) 1189:215–18. doi: 10.1016/j.brainres.2007.10.061
65. Bronstein JM, Paul K, Yang L, Haas RH, Shults CW, Le T, et al. Platelet mitochondrial activity and pesticide exposure in early Parkinson's disease. Mov Disord. (2015) 30:862–6. doi: 10.1002/mds.26164
66. Hanagasi HA, Ayribas D, Baysal K, Emre M. Mitochondrial complex I, II/III, and IV activities in familial and sporadic Parkinson's disease. Int J Neurosci. (2005) 115:479–93. doi: 10.1080/00207450590523017
67. Hishizawa M, Yamashita H, Akizuki M, Urushitani M, Takahashi R. TDP-43 levels are higher in platelets from patients with sporadic amyotrophic lateral sclerosis than in healthy controls. Neurochem Int. (2019) 124:41–5. doi: 10.1016/j.neuint.2018.12.009
68. Ehinger JK, Morota S, Hansson MJ, Paul G, Elmér E. Mitochondrial dysfunction in blood cells from amyotrophic lateral sclerosis patients. J Neurol. (2015) 262:1493–503. doi: 10.1007/s00415-015-7737-0
69. Shrivastava M, Vivekanandhan S, Pati U, Behari M, Das TK. Mitochondrial perturbance and execution of apoptosis in platelet mitochondria of patients with amyotrophic lateral sclerosis. Int J Neurosci. (2011) 121:149–58. doi: 10.3109/00207454.2010.537416
70. Shrivastava M, Das TK, Behari M, Pati U, Vivekanandhan S. Ultrastructural variations in platelets and platelet mitochondria: a novel feature in amyotrophic lateral sclerosis. Ultrastruct Pathol. (2011) 35:52–9. doi: 10.3109/01913123.2010.541985
71. Kong J, Xu Z. Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J Neurosci. (1998) 18:3241–50. doi: 10.1523/JNeurosci.18-09-03241.1998
72. Menzies FM, Cookson MR, Taylor RW, Turnbull DM, Chrzanowska-Lightowlers ZMA, Dong L, et al. Mitochondrial dysfunction in a cell culture model of familial amyotrophic lateral sclerosis. Brain. (2002) 125:1522–33. doi: 10.1093/brain/awf167
73. Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, et al. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. (1995) 14:1105–16. doi: 10.1016/0896-6273(95)90259-7
74. Dupuis L, Spreux-Varoquaux O, Bensimon G, Jullien P, Lacomblez L, Salachas F, et al. Platelet serotonin level predicts survival in amyotrophic lateral sclerosis. PLoS One. (2010) 5:e13346. doi: 10.1371/journal.pone.0013346
75. Sheremata WA, Jy W, Horstman LL, Ahn YS, Alexander JS, Minagar A. Evidence of platelet activation in multiple sclerosis. J Neuroinflammation. (2008) 5:27. doi: 10.1186/1742-2094-5-27
76. Langer HF, Choi EY, Zhou H, Schleicher R, Chung K-J, Tang Z, et al. Platelets contribute to the pathogenesis of experimental autoimmune encephalomyelitis. Circ Res. (2012) 110:1202–10. doi: 10.1161/CIRCRESAHA.111.256370
77. D'Souza Sonia C, Li Z, Luke Maxwell D, Trusler O, Murphy M, et al. Platelets drive inflammation and target gray matter and the retina in autoimmune-mediated encephalomyelitis. J Neuropathol Exp Neurol. (2018) 77:567–76. doi: 10.1093/jnen/nly032
78. Starossom SS, Veremeyko T, Yung AW, Dukhinova M, Au C, Lau AY, et al. Platelets play differential role during the initiation and progression of autoimmune neuroinflammation. Circ Res. (2015) doi: 10.1161/CIRCRESAHA.115.306847
79. Mathiason CK, Hayes-Klug J, Hays SA, Powers J, Osborn DA, Dahmes SJ, et al. B cells and platelets harbor Prion infectivity in the blood of deer infected with chronic wasting disease. J Virol. (2010) 84:5097–107. doi: 10.1128/JVI.02169-09
80. McCutcheon S, Blanco ARA, Houston EF, Wolf C de, Tan BC, Smith A, et al. All clinically-relevant blood components transmit prion disease following a single blood transfusion: a sheep model of vCJD. PLoS One. (2011) 6:e23169. doi: 10.1371/journal.pone.0023169
81. Busciglio J, Gabuzda DH, Matsudaira P, Yankner BA. Generation of ß-amyloid in the secretory pathway in neuronal and nonneuronal cells. Proc Natl Acad Sci USA. (1993) 90:2092–6. doi: 10.1073/pnas.90.5.2092
82. Li QX, Whyte S, Tanner JE, Evin G, Beyreuther K, Masters CL. Secretion of Alzheimer's disease Aß amyloid peptide by activated human platelets. Lab Invest. (1998) 78:461–9.
83. Chen M, Inestrosa NC, Ross GS, Fernandez HL. Platelets are the primary source of amyloid ß-peptide in human blood. Biochem Biophys Res Commun. (1995) 213:96–103. doi: 10.1006/bbrc.1995.2103
84. Bush AI, Martins RN, Rumble B, Moir R, Fuller S, Milward E, et al. The amyloid precursor protein of Alzheimer's disease is released by human platelets. J Biol Chem. (1990) 265:15977–83.
85. Li QX, Berndt MC, Bush AI, Rumble B, Mackenzie I, Friedhuber A, et al. Membrane-associated forms of the β A4 amyloid protein precursor of Alzheimer's disease in human platelet and brain: surface expression on the activated human platelet. Blood. (1994) 84:133–42. doi: 10.1182/blood.V84.1.133.133
86. Bu X-L, Xiang Y, Jin W-S, Wang J, Shen L-L, Huang Z-L, et al. Blood-derived amyloid-β protein induces Alzheimer's disease pathologies. Mol Psychiatry. (2018) 23:1948–56. doi: 10.1038/mp.2017.204
87. Inyushin MY, Sanabria P, Rojas L, Kucheryavykh Y, Kucheryavykh L. Aβ peptide originated from platelets promises new strategy in anti-Alzheimer's drug development. Biomed Res Int. (2017) 2017:3948360. doi: 10.1155/2017/3948360
88. Schmaier AH. Alzheimer disease is in part a thrombohemorrhagic disorder. J Thromb Haemost. (2016) 14:991–4. doi: 10.1111/jth.13277
89. Gosztyla ML, Brothers HM, Robinson SR. Alzheimer's amyloid-β is an antimicrobial peptide: a review of the evidence. J Alzheimers Dis. (2018) 62:1495–506. doi: 10.3233/JAD-171133
90. Inyushin M, Zayas-Santiago A, Rojas L, Kucheryavykh Y, Kucheryavykh L. Platelet-generated amyloid ß peptides in Alzheimer's disease and glaucoma. Histol Histopathol. (2019) 34:843–56. doi: 10.14670/HH-18-111
91. Eimer WA, Vijaya Kumar DK, Navalpur Shanmugam NK, Rodriguez AS, Mitchell T, Washicosky KJ, et al. Alzheimer's disease-associated β-amyloid is rapidly seeded by herpesviridae to protect against brain infection. Neuron. (2018) 99:56-63.e3. doi: 10.1016/j.neuron.2018.06.030
92. Kucheryavykh LY, Kucheryavykh YV, Washington AV, Inyushin MY. Amyloid ß peptide is released during thrombosis in the skin. Int J Mol Sci. (2018) 19:e1705. doi: 10.3390/ijms19061705
93. Kumar DKV, Choi SH, Washicosky KJ, Eimer WA, Tucker S, Ghofrani J, et al. Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer's disease. Sci Transl Med. (2016) 8:340ra72. doi: 10.1126/scitranslmed.aaf1059
94. Soscia SJ, Kirby JE, Washicosky KJ, Tucker SM, Ingelsson M, Hyman B, et al. The Alzheimer's disease-associated amyloid ß-protein is an antimicrobial peptide. PLoS One. (2010) 5:e9505. doi: 10.1371/journal.pone.0009505
95. Veitinger M, Varga B, Guterres SB, Zellner M. Platelets, a reliable source for peripheral Alzheimer's disease biomarkers? Acta Neuropathol Commun. (2014) 2:65. doi: 10.1186/2051-5960-2-65
96. Maglione V, Cannella M, Martino T, De Blasi A, Frati L, Squitieri F. The platelet maximum number of A2A-receptor binding sites (Bmax) linearly correlates with age at onset and CAG repeat expansion in Huntington's disease patients with predominant chorea. Neurosci Lett. (2006) 393:27–30. doi: 10.1016/j.neulet.2005.09.037
97. Mann J, Chiu E. Platelet monoamine oxidase activity in Huntington's chorea. J Neurol Neurosurg Psychiatry. (1978) 41:809–12. doi: 10.1136/jnnp.41.9.809
98. Coyle JT, Schwarcz R. Lesion of striatal neurones with kainic acid provides a model for Huntington's chorea. Nature. (1976) 263:244–6. doi: 10.1038/263244a0
99. Mangano RM, Schwarcz R. Platelet glutamate and aspartate uptake in Huntington's disease. J Neurochem. (1981) 37:1072–4. doi: 10.1111/j.1471-4159.1981.tb04502.x
100. Mangano RM, Schwarcz R. Huntington's disease. Glutamate and aspartate metabolism in blood platelets. J Neurol Sci. (1982) 53:489–500. doi: 10.1016/0022-510x(82)90245-3
101. Ehinger JK, Morota S, Hansson MJ, Paul G, Elmér E. Mitochondrial respiratory function in peripheral blood cells from Huntington's disease patients. Move Disord Clin Pract. (2016) 3:472–82. doi: 10.1002/mdc3.12308
102. Powers WJ, Haas RH, Le T, Videen TO, Hershey T, McGee-Minnich L, et al. Normal platelet mitochondrial complex I activity in Huntington's disease. Neurobiol Dis. (2007) 27:99–101. doi: 10.1016/j.nbd.2007.04.008
103. Ferreira IL, Nascimento MV, Ribeiro M, Almeida S, Cardoso SM, Grazina M, et al. Mitochondrial-dependent apoptosis in Huntington's disease human cybrids. Exp Neurol. (2010) 222:243–55. doi: 10.1016/j.expneurol.2010.01.002
104. Ho-Tin-Noé B, Demers M, Wagner DD. How platelets safeguard vascular integrity. J Thromb Haemost. (2011) 9 Suppl 1:56–65. doi: 10.1111/j.1538-7836.2011.04317.x
105. Langston JW, Ballard P, Tetrud JW, Irwin I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science. (1983) 219:979–80. doi: 10.1126/science.6823561
106. Arduíno DM, Esteves AR, Swerdlow RH, Cardoso SM. A cybrid cell model for the assessment of the link between mitochondrial deficits and sporadic Parkinson's disease. Methods Mol Biol. (2015) 1265:415–24. doi: 10.1007/978-1-4939-2288-8_31
107. Esteves AR, Arduíno DM, Swerdlow RH, Oliveira CR, Cardoso SM. Oxidative stress involvement in α-synuclein oligomerization in Parkinson's disease cybrids. Antioxid Redox Signal. (2009) 11:439–48. doi: 10.1089/ars.2008.2247
108. Lim K-M, Kim H-H, Bae O-N, Noh J-Y, Kim K-Y, Kim S-H, et al. Inhibition of platelet aggregation by 1-methyl-4-phenyl pyridinium ion (MPP+) through ATP depletion: Evidence for the reduced platelet activities in Parkinson's disease. Platelets. (2009) 20:163–70. doi: 10.1080/09537100902721746
109. Husain M, Shukla R, Dikshit M, Maheshwari PK, Nag D, Srimal RC, et al. Altered platelet monoamine oxidase-B activity in idiopathic Parkinson's disease. Neurochem Res. (2009) 34:1427–32. doi: 10.1007/s11064-009-9929-4
110. Jakubauskiene E, Janaviciute V, Peciuliene I, Söderkvist P, Kanopka A. G/A polymorphism in intronic sequence affects the processing of MAO-B gene in patients with Parkinson disease. FEBS Lett. (2012) 586:3698–704. doi: 10.1016/j.febslet.2012.08.028
111. Zhou G, Miura Y, Shoji H, Yamada S, Matsuishi T. Platelet monoamine oxidase B and plasma β-phenylethylamine in Parkinson's disease. J Neurol Neurosurg Psychiatry. (2001) 70:229–31. doi: 10.1136/jnnp.70.2.229
112. Götz ME, Gerstner A, Harth R, Dirr A, Janetzky B, Kuhn W, et al. Altered redox state of platelet coenzyme Q10 in Parkinson's disease. J Neural Transm (Vienna). (2000) 107:41–8. doi: 10.1007/s007020050003
113. Zellner M, Baureder M, Rappold E, Bugert P, Kotzailias N, Babeluk R, et al. Comparative platelet proteome analysis reveals an increase of monoamine oxidase-B protein expression in Alzheimer's disease but not in non-demented Parkinson's disease patients. J Proteomics. (2012) 75:2080–92. doi: 10.1016/j.jprot.2012.01.014
114. Ling S-C, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. (2013) 79:416–38. doi: 10.1016/j.neuron.2013.07.033
115. Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. (2006) 314:130–3. doi: 10.1126/science.1134108
116. Crugnola V, Lamperti C, Lucchini V, Ronchi D, Peverelli L, Prelle A, et al. Mitochondrial respiratory chain dysfunction in muscle from patients with amyotrophic lateral sclerosis. Arch Neurol. (2010) 67:849–54. doi: 10.1001/archneurol.2010.128
117. Curti D, Malaspina A, Facchetti G, Camana C, Mazzini L, Tosca P, et al. Amyotrophic lateral sclerosis: oxidative energy metabolism and calcium homeostasis in peripheral blood lymphocytes. Neurology. (1996) 47:1060–4. doi: 10.1212/wnl.47.4.1060
118. Nakano Y, Hirayama K, Terao K. Hepatic ultrastructural changes and liver dysfunction in amyotrophic lateral sclerosis. Arch Neurol. (1987) 44:103–6. doi: 10.1001/archneur.1987.00520130079022
119. Kiktenko AI, Zlobina GP, Brusov OS, Zakharova MN. [Structure of peripheral blood platelets surface in patients with amyotrophic lateral sclerosis and multiple sclerosis]. Zh Nevrol Psikhiatr Im S S Korsakova. (2005) 105:40–2.
120. Rao JS, Hantaï D, Festoff BW. Thrombospondin, a platelet α-granule and matrix glycoprotein, is increased in muscle basement membrane of patients with amyotrophic lateral sclerosis. J Neurol Sci. (1992) 113:99–107. doi: 10.1016/0022-510x(92)90271-l
121. Smirnova IV, Festoff BW. Alterations in serum thrombospondin in patients with amyotrophic lateral sclerosis. J Neurol Sci. (1994) 127:207–13. doi: 10.1016/0022-510x(94)90074-4
122. Bertel O, Malessa S, Sluga E, Hornykiewicz O. Amyotrophic lateral sclerosis: changes of noradrenergic and serotonergic transmitter systems in the spinal cord. Brain Res. (1991) 566:54–60. doi: 10.1016/0006-8993(91)91680-Y
123. Forrest V, Ince P, Leitch M, Marshall EF, Shaw PJ. Serotonergic neurotransmission in the spinal cord and motor cortex of patients with motor neuron disease and controls: quantitative autoradiography for 5-HT1a and 5-HT2 receptors. J Neurol Sci. (1996) 139 Suppl:83–90. doi: 10.1016/0022-510x(96)00109-8
124. Bos IWM, Hoogland G, Meine Jansen CF, Willigen G van, Spierenburg HA, van den Berg LH, et al. Increased glutamine synthetase but normal EAAT2 expression in platelets of ALS patients. Neurochem Int. (2006) 48:306–11. doi: 10.1016/j.neuint.2005.09.009
125. Wachowicz B, Morel A, Miller E, Saluk J. The physiology of blood platelets and changes of their biological activities in multiple sclerosis. Acta Neurobiol Exp. (2016) 76:269–81.
126. Collinge J, Rossor M. A new variant of prion disease. Lancet. (1996) 347:916–7. doi: 10.1016/s0140-6736(96)91407-5
127. Parchi P, Castellani R, Capellari S, Ghetti B, Young K, Chen SG, et al. Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann Neurol. (1996) 39:767–78. doi: 10.1002/ana.410390613
128. Robertson C, Booth SA, Beniac DR, Coulthart MB, Booth TF, McNicol A. Cellular prion protein is released on exosomes from activated platelets. Blood. (2006) 107:3907–11. doi: 10.1182/blood-2005-02-0802
129. Holada K, Glierova H, Simak J, Vostal JG. Expression of cellular prion protein on platelets from patients with gray platelet or Hermansky-Pudlak syndrome and the protein's association with α-granules. Haematologica. (2006) 91:1126–9.
130. Asher DM, Gregori L. Human transmissible spongiform encephalopathies: historic view. Handb Clin Neurol. (2018) 153:1–17. doi: 10.1016/B978-0-444-63945-5.00001-5
131. Ricketts MN, Cashman NR, Stratton EE, ElSaadany S. Is Creutzfeldt-Jakob disease transmitted in blood? Emerg Infect Dis. (1997) 3:155–63. doi: 10.3201/eid0302.970208
132. Urwin P, Thanigaikumar K, Ironside JW, Molesworth A, Knight RS, Hewitt PE, et al. Sporadic Creutzfeldt-Jakob disease in 2 plasma product recipients, United Kingdom. Emerg Infect Dis. (2017) 23:893–7. doi: 10.3201/eid2306.161884
133. Burnouf T, Strunk D, Koh MBC, Schallmoser K. Human platelet lysate: replacing fetal bovine serum as a gold standard for human cell propagation? Biomaterials. (2016) 76:371–87. doi: 10.1016/j.biomaterials.2015.10.065
134. Golebiewska EM, Poole AW. Platelet secretion: From haemostasis to wound healing and beyond. Blood Rev. (2015) 29:153–62. doi: 10.1016/j.blre.2014.10.003
135. Stellos K, Kopf S, Paul A, Marquardt JU, Gawaz M, Huard J, et al. Platelets in regeneration. Semin Thromb Hemost. (2010) 36:175–84. doi: 10.1055/s-0030-1251502
136. Shih DT-B, Burnouf T. Preparation, quality criteria, and properties of human blood platelet lysate supplements for ex vivo stem cell expansion. New Biotechnol. (2015) 32:199–211. doi: 10.1016/j.nbt.2014.06.001
137. Chou M-L, Wu J-W, Gouel F, Jonneaux A, Timmerman K, Renn T-Y, et al. Tailor-made purified human platelet lysate concentrated in neurotrophins for treatment of Parkinson's disease. Biomaterials. (2017) 142:77–89. doi: 10.1016/j.biomaterials.2017.07.018
138. Etulain J, Mena HA, Meiss RP, Frechtel G, Gutt S, Negrotto S, et al. An optimised protocol for platelet-rich plasma preparation to improve its angiogenic and regenerative properties. Sci Rep. (2018) 8:1–15. doi: 10.1038/s41598-018-19419-6
139. Mussbacher M, Schrottmaier WC, Salzmann M, Brostjan C, Schmid JA, Starlinger P, et al. Optimized plasma preparation is essential to monitor platelet-stored molecules in humans. PLoS One. (2017) 12:e0188921. doi: 10.1371/journal.pone.0188921
140. Burkhart JM, Vaudel M, Gambaryan S, Radau S, Walter U, Martens L, et al. The first comprehensive and quantitative analysis of human platelet protein composition allows the comparative analysis of structural and functional pathways. Blood. (2012) 120:e73-82. doi: 10.1182/blood-2012-04-416594
141. Marck RE, Middelkoop E, Breederveld RS. Considerations on the use of platelet-rich plasma, specifically for burn treatment. J Burn Care Res. (2014) 35:219–27. doi: 10.1097/bcr.0b013e31829b334e
142. Ozcelik U, Ekici Y, Bircan HY, Aydogan C, Turkoglu S, Ozen O, et al. Effect of topical platelet-rich plasma on burn healing after partial-thickness burn injury. Med Sci Monit. (2016) 22:1903–9. doi: 10.12659/MSM.895395
143. Kavadar G, Demircioglu DT, Celik MY, Emre TY. Effectiveness of platelet-rich plasma in the treatment of moderate knee osteoarthritis: a randomized prospective study. J Phys Ther Sci. (2015) 27:3863–7. doi: 10.1589/jpts.27.3863
144. Dragoo JL, Wasterlain AS, Braun HJ, Nead KT. Platelet-rich plasma as a treatment for patellar tendinopathy: a double-blind, randomized controlled trial. Am J Sports Med. (2014) 42:610–8. doi: 10.1177/0363546513518416
145. Wu Y-N, Wu C-C, Sheu M-T, Chen K-C, Ho H-O, Chiang H-S. Optimization of platelet-rich plasma and its effects on the recovery of erectile function after bilateral cavernous nerve injury in a rat model. J Tissue Eng Regen Med. (2016) 10:e294–e304. doi: 10.1002/term.1806
146. Cho H-H, Jang S, Lee S-C, Jeong H-S, Park J-S, Han J-Y, et al. Effect of neural-induced mesenchymal stem cells and platelet-rich plasma on facial nerve regeneration in an acute nerve injury model. Laryngoscope. (2010) 120:907–13. doi: 10.1002/lary.20860
147. Anjayani S, Wirohadidjojo YW, Adam AM, Suwandi D, Seweng A, Amiruddin MD. Sensory improvement of leprosy peripheral neuropathy in patients treated with perineural injection of platelet-rich plasma. Int J Dermatol. (2014) 53:109–13. doi: 10.1111/ijd.12162
148. Chen N-F, Sung C-S, Wen Z-H, Chen C-H, Feng C-W, Hung H-C, et al. Therapeutic effect of platelet-rich plasma in rat spinal cord injuries. Front Neurosci. (2018) 12: doi: 10.3389/fnins.2018.00252
149. Borhani-Haghighi M, Mohamadi Y. The therapeutic effect of platelet-rich plasma on the experimental autoimmune encephalomyelitis mice. J Neuroimmunol. (2019) 333:476958. doi: 10.1016/j.jneuroim.2019.04.018
150. Anitua E, Pascual C, Antequera D, Bolos M, Padilla S, Orive G, et al. Plasma rich in growth factors (PRGF-Endoret) reduces neuropathologic hallmarks and improves cognitive functions in an Alzheimer's disease mouse model. Neurobiol Aging. (2014) 35:1582–95. doi: 10.1016/j.neurobiolaging.2014.01.009
151. Anitua E, Pascual C, Pérez-Gonzalez R, Antequera D, Padilla S, Orive G, et al. Intranasal delivery of plasma and platelet growth factors using PRGF-Endoret system enhances neurogenesis in a mouse model of Alzheimer's disease. PLoS One. (2013) 8:e73118. doi: 10.1371/journal.pone.0073118
152. Anitua E, Pascual C, Pérez-Gonzalez R, Orive G, Carro E. Intranasal PRGF-Endoret enhances neuronal survival and attenuates NF-κB-dependent inflammation process in a mouse model of Parkinson's disease. J Control Release. (2015) 203:170–80. doi: 10.1016/j.jconrel.2015.02.030
153. Gouel F, Do Van B, Chou M-L, Jonneaux A, Moreau C, Bordet R, et al. The protective effect of human platelet lysate in models of neurodegenerative disease: involvement of the Akt and MEK pathways. J Tissue Eng Regen Med. (2017) 11:3236–40. doi: 10.1002/term.2222
154. Hayon Y, Dashevsky O, Shai E, Varon D, Leker RR. Platelet lysates stimulate angiogenesis, neurogenesis and neuroprotection after stroke. Thromb Haemost. (2013) 110:323–30. doi: 10.1160/TH12-11-0875
155. Wu Y-W, Goubran H, Seghatchian J, Burnouf T. Smart blood cell and microvesicle-based Trojan horse drug delivery: Merging expertise in blood transfusion and biomedical engineering in the field of nanomedicine. Transfus Apher Sci. (2016) 54:309–18. doi: 10.1016/j.transci.2016.04.013
156. van Dommelen SM, Vader P, Lakhal S, Kooijmans SAA, van Solinge WW, Wood MJA, et al. Microvesicles and exosomes: opportunities for cell-derived membrane vesicles in drug delivery. J Control Release. (2012) 161:635–44. doi: 10.1016/j.jconrel.2011.11.021
157. Brisson AR, Tan S, Linares R, Gounou C, Arraud N. Extracellular vesicles from activated platelets: a semiquantitative cryo-electron microscopy and immuno-gold labeling study. Platelets. (2017) 28:263–71. doi: 10.1080/09537104.2016.1268255
158. Sandberg H, Bode AP, Dombrose FA, Hoechli M, Lentz BR. Expression of coagulant activity in human platelets: release of membranous vesicles providing platelet factor 1 and platelet factor 3. Thromb Res. (1985) 39:63–79. doi: 10.1016/0049-3848(85)90122-7
159. Baj-Krzyworzeka M, Majka M, Pratico D, Ratajczak J, Vilaire G, Kijowski J, et al. Platelet-derived microparticles stimulate proliferation, survival, adhesion, and chemotaxis of hematopoietic cells. Exp Hematol. (2002) 30:450–9. doi: 10.1016/S0301-472X(02)00791-9
160. Janowska-Wieczorek A, Majka M, Kijowski J, Baj-Krzyworzeka M, Reca R, Turner AR, et al. Platelet-derived microparticles bind to hematopoietic stem/progenitor cells and enhance their engraftment. Blood. (2001) 98:3143–9. doi: 10.1182/blood.V98.10.3143
161. Janowska-Wieczorek A, Wysoczynski M, Kijowski J, Marquez-Curtis L, Machalinski B, Ratajczak J, et al. Microvesicles derived from activated platelets induce metastasis and angiogenesis in lung cancer. Int J Cancer. (2005) 113:752–60. doi: 10.1002/ijc.20657
162. Rozmyslowicz T, Majka M, Kijowski J, Murphy SL, Conover DO, Poncz M, et al. Platelet- and megakaryocyte-derived microparticles transfer CXCR4 receptor to CXCR4-null cells and make them susceptible to infection by X4-HIV. AIDS. (2003) 17:33–42. doi: 10.1097/01.aids.0000042948.95433.3d
163. Burnouf T, Agrahari V, Agrahari V. Extracellular vesicles as nanomedicine: hopes and hurdles in clinical translation. Int J Nanomed. (2019) 14:8847–59. doi: 10.2147/IJN.S225453
164. Ilahibaks NF, Lei Z, Mol EA, Deshantri AK, Jiang L, Schiffelers RM, et al. Biofabrication of cell-derived nanovesicles: a potential alternative to extracellular vesicles for regenerative medicine. Cells. (2019) 8:e1509. doi: 10.3390/cells8121509
165. de Jong OG, Kooijmans SAA, Murphy DE, Jiang L, Evers MJW, Sluijter JPG, et al. Drug delivery with extracellular vesicles: from mmagination to innovation. Acc Chem Res. (2019) 52:1761–70. doi: 10.1021/acs.accounts.9b00109
166. Panagiotou N, Neytchev O, Selman C, Shiels PG. Extracellular vesicles, ageing, and therapeutic interventions. Cells. (2018) 7:110. doi: 10.3390/cells7080110
167. Zaslavsky A, Baek K-H, Lynch RC, Short S, Grillo J, Folkman J, et al. Platelet-derived thrombospondin-1 is a critical negative regulator and potential biomarker of angiogenesis. Blood. (2010) 115:4605–13. doi: 10.1182/blood-2009-09-242065
Keywords: platelets, neurodegeneration, neuroinflammation, brain function, neuroimmune crosstalk
Citation: Leiter O and Walker TL (2020) Platelets in Neurodegenerative Conditions—Friend or Foe? Front. Immunol. 11:747. doi: 10.3389/fimmu.2020.00747
Received: 25 February 2020; Accepted: 01 April 2020;
Published: 05 May 2020.
Edited by:
Jacqueline Monique Orian, La Trobe University, AustraliaReviewed by:
Ilaria Canobbio, University of Pavia, ItalySouvarish Sarkar, Brigham and Women's Hospital and Harvard Medical School, United States
Copyright © 2020 Leiter and Walker. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tara L. Walker, t.walker1@uq.edu.au