In silico mapping of the dynamic interactions and structure-activity relationship of flavonoid compounds against the immune checkpoint programmed-cell death 1 pathway
 Geraldo Rodrigues Sartori1,2
Geraldo Rodrigues Sartori1,2  Aline de Oliveira Albuquerque1,3
Aline de Oliveira Albuquerque1,3  Andrielly Henriques Santos-Costa1,2
Andrielly Henriques Santos-Costa1,2  Luca Milério Andrade1,2
Luca Milério Andrade1,2  Diego da Silva Almeida1,2
Diego da Silva Almeida1,2  Eduardo Menezes Gaieta1,2
Eduardo Menezes Gaieta1,2  Jean Vieira Sampaio1,2
Jean Vieira Sampaio1,2  Vitória Taiana de Melo Lima Albuquerque1,2
Vitória Taiana de Melo Lima Albuquerque1,2  João Hermínio Martins Da Silva1,2,3*
João Hermínio Martins Da Silva1,2,3*- 1Laboratory of Structural and Functional Biology Applied to Biopharmaceuticals, Oswaldo Cruz Foundation (Fiocruz), Eusébio, Ceará, Brazil
- 2Postgraduate Program in Computational and Systems Biology, Oswaldo Cruz Foundation (Fiocruz), Rio de Janeiro, Rio de Janeiro, Brazil
- 3Postgraduate Program in Natural Resource Biotechnology, Federal University of Ceará, Fortaleza, Ceará, Brazil
Flavonoids are a class of natural products widely available in medicinal and dietary plants. Their pharmacological use has shown the potential to reduce the risk of different types of cancer, among other prevalent diseases. Their molecular scaffold inhibits the PD-1/PD-L1 axis, an important pathway related to the adaptive immune resistance of cancer cells already targeted for developing new cancer immunotherapy. However, despite the availability of kinetic and thermodynamic experimental data on the flavonoid–PD-1/PD-L1 interaction, there is still a lack of reliable information about their binding mode at the atomic level. Thus, we aimed to computationally predict the binding mode of flavonoid molecules with PD-1 and/or PD-L1 proteins using unbiased computational methodologies such as blind docking and supervised molecular dynamics simulation. The molecular interactions and dynamics of these predicted poses of protein-flavonoid complexes were further analyzed through multiple molecular dynamics simulations. This information, corroborated with the IC50 and KD values from available literature, was used to perform molecular matched-pair analysis to comprehensively describe the main interactions governing the inhibition of the complex PD-1/PD-L1 by the flavonoid scaffold. By analyzing the effect of substitutions in such a scaffold, we observed a clear correspondence with literature binding assays. Thus, we propose, for dimeric PD-L1, that the 7-O-glucoside forces the molecule displacement in the dimer interface. Furthermore, the 3-OH plays an essential role in stabilizing the buried binding mode by water-bridged hydrogen bonds with Asp122 and Gln66 in both extremities of the pocket. In PD-1, we suggest that flavonoids could bind through the BC loop by inducing a flip of Phe56 after a conformational change of the Asn58 glycosylation. Hence, our results introduced unprecedented information on flavonoid interaction and dynamics when complexed with PD-1 checkpoint pathway proteins and can pave the road for developing new flavonoid derivatives with selective anticancer activity.
1 Introduction
Cancer is a complex disease involving a diverse set of molecular and cellular changes, being considered one of the leading causes of mortality worldwide. Therefore, understanding the molecular changes in signaling pathways may provide helpful insights for the development of new therapies (Jiang et al., 2020; Xue et al., 2022). In this regard, cancer treatment research has changed the understanding of the biology of the disease (Sun et al., 2021; Xue et al., 2022). Particularly, therapies based on blocking immune checkpoints have drawn attention by showing promising results against various malignities (Sun et al., 2021).
The checkpoint protein PD-1 (CD279) is a co-inhibitory transmembrane receptor expressed primarily on the surface of T cells and is a member of the CD28 family of costimulatory receptors (Dermani et al., 2019; Liu et al., 2021a). Its primary function is to suppress T cell activity by regulating the TCR signaling cascade through binding to any of its two ligands, PD-L1 or PD-L2 transmembrane proteins (Buchbinder and Desai, 2016; Lin et al., 2020). PD-1 and PD-L1 interact with each other along their IgV domains. The CC′, CC″, and FG loops and GFCC′ strands of the front β-face of PD-1 bind to the GFCC' strands of the front β-face of PD-L1 (Freeman, 2008) (Supplementary Figure S1). Both PD-1 and PD-L1 are highly glycosylated proteins, with the Asn58 glycosylation site of PD-1 being particularly important for the PD-1/PD-L1 interaction (Sun et al., 2020). Several cancer types have an increased expression of the PD-1 ligands, making the inhibition of the PD-1 checkpoint pathway a promising strategy for tumor treatment (Dermani et al., 2019; Liu et al., 2021a).
More specifically, blocking PD-1/PD-L1 interactions can shut down PD-1-mediated signaling pathways and reactivate T cell-mediated antitumor immune responses (Kumar et al., 2021). Thus far, only monoclonal antibodies (mAbs) have been approved as PD-1 pathway inhibitors by the Food and Drug Administration (FDA) (Krueger et al., 2017; Kumar et al., 2021). However, there are several drawbacks to using mAbs: high cost, immunogenicity, low oral bioavailability, and low tissue penetration. Such shortcomings make alternative drug types, such as small molecules, attractive (O’Sullivan Coyne et al., 2014; Naidoo et al., 2015; Sidaway, 2016; Inman et al., 2017; Akinleye and Rasool, 2019; Lin et al., 2020). Although there are still no approved compounds from this class of inhibitors, there is a great potential for their use as a way to overcome the lack of response of many patients to mAbs therapy, (Patsoukis et al., 2020; Liu et al., 2021a; Wang et al., 2021).
The search for small-molecule inhibitors to target the PD-1 pathway has been primarily focused on two main classes of molecules: biphenyl derivatives and peptide mimetics (Wu et al., 2021; Sasikumar and Ramachandra, 2022). The biphenyl molecules have demonstrated the capacity to stabilize the PD-L1 dimerization in solution (Zak et al., 2016). Although PD-L1 oligomerization naturally occurs in living cells to regulate the protein complex glycosylation (Zhou et al., 2022), the interaction with PD-1 only happens in the monomeric state. Examples from both classes were able to inhibit the PD-1/PD-L1 interaction competitively and suppress tumor growth in preclinical tests, with some currently in clinical trials, such as INCB-086550 and CA-170 (Piha-Paul et al., 2020; Sasikumar et al., 2021; Koblish et al., 2022; Sasikumar and Ramachandra, 2022). Another class of molecules whose derivatives have shown potential as PD-1/PD-L1 competitive inhibitors are flavonoids.
Flavonoids are polyphenolic compounds produced as secondary metabolites of several plant species (Rice-Evans et al., 1996). They have been increasingly studied because of their reported activity against various diseases, especially cancer (Kopustinskiene et al., 2020; Liu et al., 2021a; Lee et al., 2021). In vivo and in vitro studies indicate that some of these molecules are capable of interfering with biochemical functions related to PD-1 and PD-L1 through the reduction of IFN-γ-induced PD-L1 expression or the inhibition of PD-1/PD-L1 interaction by competitive antagonism (Coombs et al., 2016; Li et al., 2019a; Ke et al., 2019; Mazewski et al., 2019; Liu et al., 2021a).
Despite growing evidence about the antineoplastic utility of the flavonoid scaffold and its ability to modulate the PD-1/PD-L1 interaction, the molecular mechanism through which the inhibition of complex formation happens is still understudied. Nonetheless, computational tools can be used to elucidate such mechanisms, as they have already been used to gain insight into the finer details of the PD-1/PD-L1 interaction and the interactions between PD-L1 and many of its known small molecule antagonists (Wang et al., 2019b; Mejías and Guirola, 2019; Shi et al., 2019; Konieczny et al., 2020; Lin et al., 2020; Liu et al., 2021b; Guo et al., 2021; Urban et al., 2022).
As such, we sought to characterize the binding mode of several different flavonoids with reported inhibitory activity against PD-1/PD-L1. Furthermore, we searched for possible new interaction hotspots in PD-1/PD-L1. These investigations were made using in silico techniques, including docking and conventional and supervised Molecular Dynamics (MD), applied to a set of conformationally diverse protein structures. The obtained results allowed us to propose the binding mode and Structure-Activity Relationship (SAR) of the flavonoid scaffold in the PD-1/PD-L1 pathway. This unprecedented information regarding the flavonoid-protein interaction and dynamics might be helpful in the molecular optimization and development of new drug-like molecules (Figure 1).
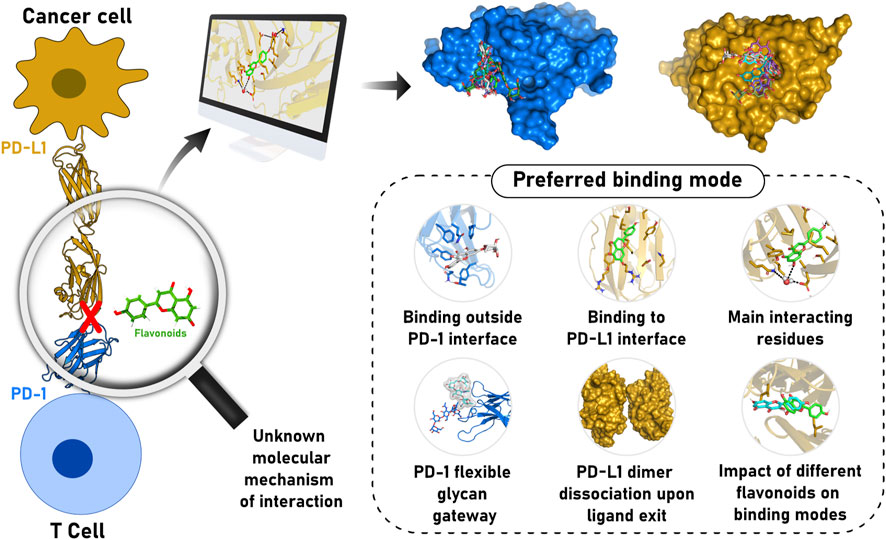
FIGURE 1. Schematic representation of flavonoid relevance to cancer treatment through PD-1/PD-L1 axis modulation and our major findings regarding the structural and dynamics determinants for molecule recognition. Flavonoids can inhibit the PD-1/PD-L1 complex, allowing lymphocytes to recognize cancer cells and promote controlled cell death. However, the mode of interaction of the molecules with PD-1 and/or PD-L1 remains to be understood. In this context, computational methods were applied to propose an accurate model representing the binding mode of different flavonoid derivatives. We suggested flavonoids could interact with PD-1 in a transiently occluded pocket outside the GFCC’ strands interface. Furthermore, we described how the flavonoid modifications influence the interaction with the main residues of the PD-L1 dimer interface cavity and the effect on the molecule activity.
2 Materials and methods
2.1 Small molecule and proteins’ structure preparation
Initially, the flavonoid compounds were chosen based on experimental data and structure similarity. Nine selected ligands, namely apigenin, cosmosiin, eriodictyol, fisetin, kaempferol, kaempferol-7-O-rhamnoside, kaempferol-3,7-dirhamnoside, luteolin, and quercetin, were identified through literature (Supplementary Table S1) and then obtained from the PubChem database (Kim et al., 2016). From this flavonoid pool, only luteolin and kaempferol-3,7-dirhamnoside are unable to inhibit the PD-1/PD-L1 interaction. The conversion from smiles to pdb format used the Open Babel program (O’Boyle et al., 2011), and the MarvinSketch program determined the molecules’ protonation state.
For the proteins’ structure selection, five conformations of PD-1, monomeric PD-L1 (PD-L1m), and dimeric PD-L1 (PD-L1d) were chosen from the Protein Data Bank (PDB) following the criteria: form (free or complexed with antibody or ligand), absence of mutation and glycosylation, high coverage of amino acid sequence, and high resolution (Supplementary Table S2). Thus, for PD-1, we chose PDBs 2M2D, 4ZQK, 5GGS, and 7E9B. In addition, a fifth structure was considered based on the structural rearrangement of the C'D loop observed through MD simulations with organic probes (Andrade et al., 2022). The PDBs 3FN3, 4ZQK, 5JDS, 5J89, and 7C88 were representative of PD-L1m. Similarly, the structures 5N2F, 6NM7, 6VQN, chains A and B of 7NLD, and chains C and D of 7NLD were selected for the following computational experiments with the PD-L1d.
Initial identification of the proteins’ cavities was accomplished by analyzing their PDB files with the DoGSiteScorer web server (Volkamer et al., 2012). Then, as a structure pre-treatment for the following steps, we removed existing hydrogen atoms, ions, water, and small molecules from the original PDB files. The protonation profiles of PD-1 and the monomeric and dimeric forms of PD-L1 at pH 7.4 were generated using the PropKa software (Dolinsky et al., 2007; Olsson et al., 2011). The default protonation state was kept for all residues. As such, all histidine residues were defined as histidine epsilon (HIE). Finally, prior to the parametrization for molecular dynamics, the proteins’ N-terminal and C-terminal regions were capped with acetyl (ACE) and methylamine (NME) groups, respectively.
The effect of glycosylation on PD-1 cavity formation was evaluated by simulating Asn58-glycosylated PD-1 obtained from the PDB ID: 6JJP (Wang et al., 2019). Glycosylation of structure 6JJP was converted to Amber format via CHARMM-GUI (Jo et al., 2008).
2.2 Molecular docking protocol
Blind docking assays using the Quick Vina 2 program (Alhossary et al., 2015) were performed to identify binding sites and modes of interaction of flavonoids with PD-1, PD-L1m, and PD-L1d. The pdbqt files were prepared using the AutoDockTools program (Morris et al., 2009) by merging non-polar hydrogen atoms and adding Gasteiger charges. Calculations were performed using a box positioned on the center of mass of the proteins, with dimensions sufficient to encompass the entire protein structure (Supplementary Table S3). The exhaustiveness was set to 15, and a maximum of 10 models were generated per ligand. To validate the docking calculations, we performed re-docking and cross-docking assays involving the structures of the chosen PDBs. The same grid configurations previously used were adopted. The results were visually inspected to assess the ability of blind docking to correctly reproduce the site of interaction of ligands in PD-1 and PD-L1.
2.3 Selection of docking poses for MD simulations
The resulting docking structures were clustered based on their relative all-against-all RMSD using the Complete Link Hierarchical clustering algorithm in the RDKit software. Complexes were chosen to prioritize the diversity of the proposed binding mode for each flavonoid against PD-1, PD-L1m, or PD-L1d. A visual inspection was performed to ensure pose diversity from the lowest branch level of the clustering tree. Thus, we reduced the number of poses necessary to describe most of the conformational space explored by the docking algorithm. This contributed to reducing the simulated time and, therefore, the computational cost. From the initial 1,350 docking poses, we selected 865 complexes to submit to molecular dynamics simulations. The hierarchical clustering tree was also used to distinguish the different cavities of PD-1, PD-L1m, and PD-L1d.
2.4 Molecular dynamics simulation
2.4.1 Protein-ligand setup and parametrization
First, to obtain the parameter files of the flavonoid molecules, ligands’ Restrained Electrostatic Potential (RESP) charges were derived using R.E.D. Server (Vanquelef et al., 2011) with its default parameters, except for CHR_TYP = RESP-X1. Then, Antechamber and tLeap, included on AmberTools21, were used to build a suitable ligand library using gaff2 force field for complex preparation. Subsequently, protein, ligands, and glycan were parametrized in the tLeap module from AmberTools21 using ff19sb (Tian et al., 2020), gaff2, and GLYCAM-06J (Kirschner et al., 2008) forcefields, respectively.
Each system containing a protein-ligand complex or a free protein was solvated using OPC water in a truncated octahedral box with dimensions defined to ensure a minimum distance of 12 Å from solute atoms to the box wall. All complexes were neutralized, and Na+ and Cl- ions were added to achieve ionic strength of 120 mM. The hydrogen atoms had their mass changed using Hydrogen Mass Repartitioning (Feenstra et al., 1999; Hopkins et al., 2015) implemented on Parmed (Shirts et al., 2017) to support a timestep of 4 fs during simulation.
2.4.2 Molecular dynamics simulation protocol
After generating the topology and input coordinates, we minimized and equilibrated all 865 complexes. First, the solvent was minimized for 2000 cycles in the CPU version of pmemd. All following steps of minimization and simulation were carried out in GPUs, starting with a subsequent round of minimization, keeping ligand and protein constrained, was performed. A subsequent minimization round constraining only the protein followed (7,000 steps in each of the two previous rounds). Finally, a whole system minimization was run until convergence. Each minimization step employed the steepest descent and conjugate gradient methods.
Next, the minimized systems were gently heated to 310 K for 200 ps at constant volume, followed by water equilibration at constant pressure, constraining the protein and ligand with 10 kcal mol−1 force. The final equilibration step consisted of a 5 ns unconstrained simulation in the NPT ensemble. After the equilibration, 25 ns of conventional Molecular Dynamics simulation was performed for each complex, totaling 21.6 μs (865 systems, 25 ns each) of simulation. The cut-off for non-bounded interactions was set to 10 Å, and the SHAKE algorithm was used to constrain bonds involving hydrogen atoms. We used an integration step of 4 fs. As needed, the Monte Carlo barostat and Langevin thermostat were used. Detailed parameters are described in the Supplementary Information S1. From the 865 docking outputs, we selected 67 complexes that remained stable during the 25 ns simulations in the priority binding sites. A second round of MD simulations, each 50 ns long, was performed in quintuplicate using the same preparation steps and input files as the previous round, adding 18 μs to the simulation time. Finally, additional 450 ns of simulations were run continuing from at least one stable pose per flavonoid in the priority binding site identified from previous simulations for PD-1, PD-L1m, and PD-L1d, totaling 34 systems and 17 us. All simulations were executed using pmemd.cuda from the Amber20 package (Case et al., 2005). MM/GBSA calculations using the default parameters of MMPBSA.py script implemented in AmberTools20 suite were performed over the last 10% of simulation frames of 50 and 500 ns simulations, considering one frame each 100 ps.
2.5 Supervised molecular dynamics
Supervised molecular dynamics (SuMD) is an adaptative method that improves the studying of the time-dependent evolution of ligand-protein molecular recognition using little computational effort. In this implementation, the geometrical supervision of MD trajectories is written in a bash programming language, according to the algorithm published by Sabbadin and Moro (2014).
The protein center of mass was defined considering all the residues. The ligand was placed at 40 Å so that the initial coordinate varied according to the x, y, and z axes for each ligand, taking the Cartesian plane of the protein as a reference. The sampling corresponded to six replicates for each ligand, totalling an amount of 270 SuMD for each protein. In this case, only PD-1 and PD-L1m were considered. Pre-treatment of the systems was performed similarly to the cMD. Position restraints were applied on the protein backbone, and gradually released throughout the 1 ns of equilibration (ensemble NPT). The SuMD trajectory was composed of 1 ns MD simulations, carried out at 310 K using the NPT ensemble.
3 Results
Herein, we sought to compare the potential binding modes of several flavonoid-derivate molecules to the PD-1 and PD-L1 proteins (Figure 2). First, the flavonoid compounds were carefully selected to enable a Matched-molecular pair analysis, which would integrate the available experimental binding/inhibitory activity and our in silico protocol (Figures 2A, 3). Seven of the nine selected compounds are reportedly active, while the other two are inactive (luteolin and kaempferol-3,7-dirhamnoside) as inhibitors of the PD-1/PD-L1 interaction (Figure 3). Then, knowing that protein-ligand interaction is highly dependent on the target conformation, we selected five distinct crystallographic structures for the three evaluated targets: PD-1 and the monomeric and dimeric states of PD-L1 (PD-L1m and PD-L1d, respectively) (Figure 2A). The list and reasoning for selecting each protein structure are described in Supplementary Table S2. Next, we identified the putative binding sites in the proteins through orthogonal methods, initially detecting and characterizing the pockets in the chosen pdb structures and then scanning its surface for flavonoid interaction sites using unbiased approaches, such as SuMD and blind docking (Figures 2B,C). The identified flavonoid interaction poses were simulated and clustered to select the most probable interaction cavity (Figure 2D). Subsequently, we extended our simulations to refine the predicted poses and present a reliable flavonoid binding mode (Figure 2E). Finally, these binding modes were used to propose how each variation in the flavonoid scaffold affected the interaction dynamics, correlating with the known activity of the different compounds (Figure 2E). In the following topics, we described the putative pockets using three orthogonal methodologies.
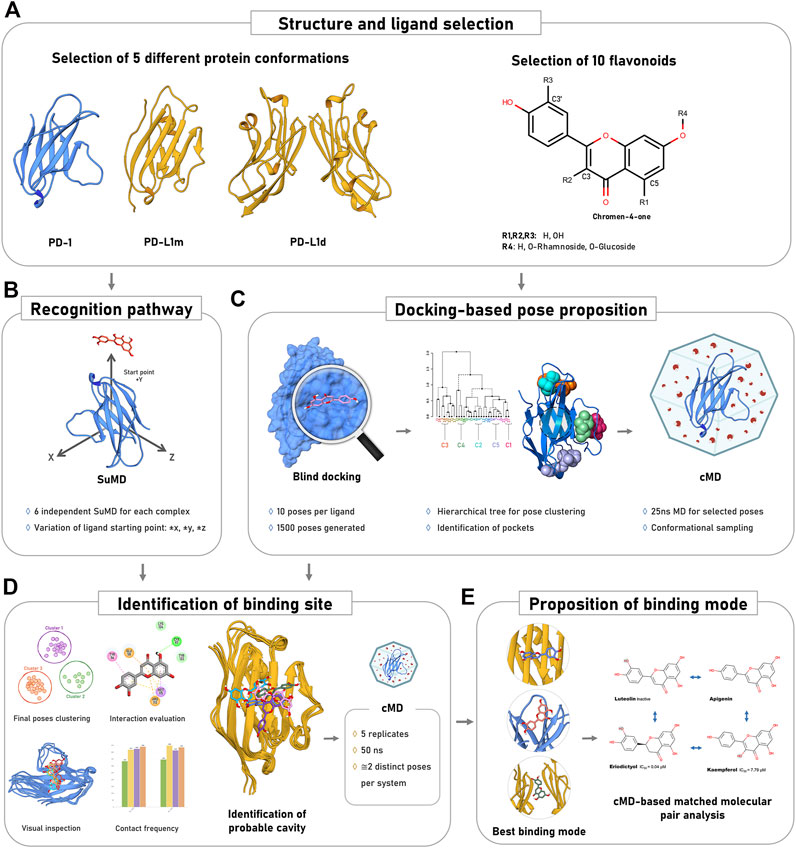
FIGURE 2. Overview of the methodological approach. (A) The inputs for simulations and docking encompassed different protein states and diverse flavonoid structural constructs. R1 to R4 represent substitution positions in the flavonoid scaffold. (B) Construction of 540 complexes to search for new favorable cavities through unbiased Supervised Molecular Dynamics. (C) Prediction and simulation of flavonoid binding modes prior to complex stability evaluation. (D) Proposition of the best interaction cavities on the PD-1 and PD-L1. (E) Correlation between the flavonoid-protein interaction and experimental data.
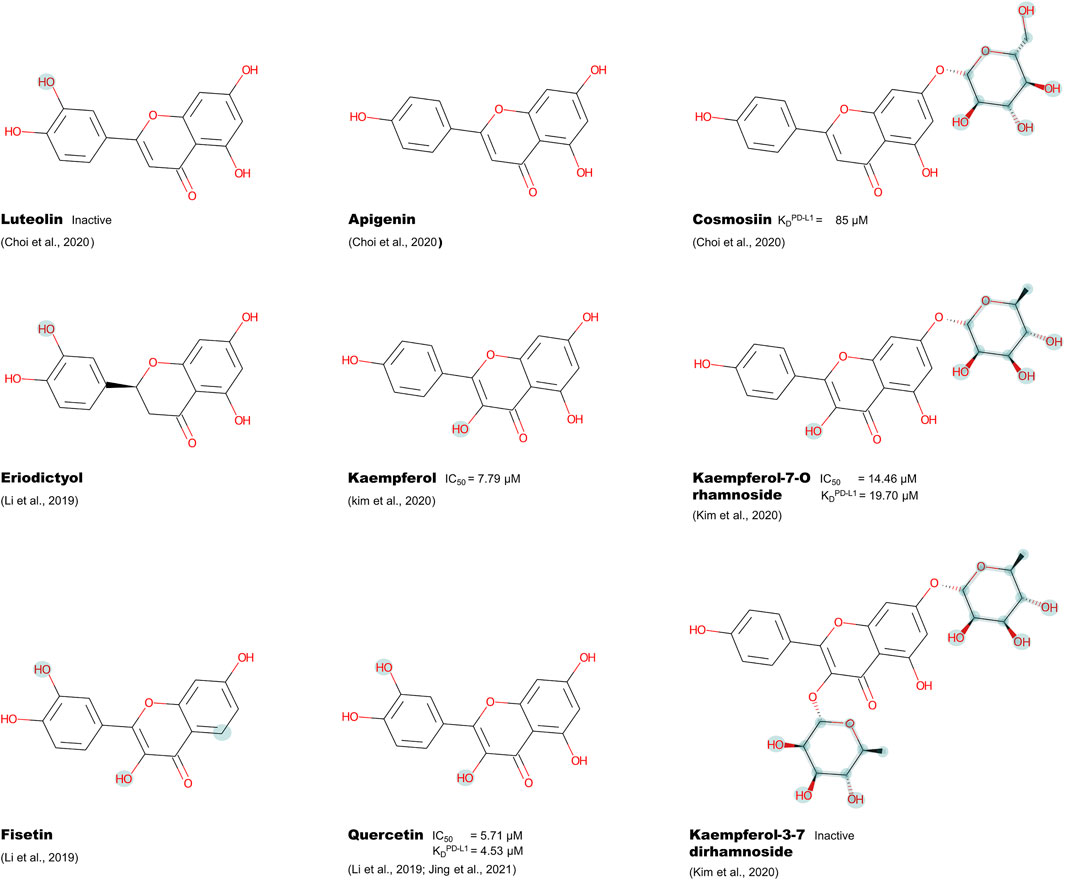
FIGURE 3. Chemical structure of selected flavonoids with the determined KD or IC50 against PD-1 or PD-L1, highlighting the different functional groups compared to apigenin.
3.1 Exposed pocket site is dependent on the protein base structure
Initially, we evaluated the currently exposed cavities in the PDB files of each of the fifteen selected structures for the three proteins using DoGSiteScorer (Supplementary Table S11). We found two cavities in all considered PD-L1d structures, with the highest druggability score among the five dimer conformations of 0.83. The first pocket, C1PD−L1d, is the well-established hydrophobic tunnel formed by PD-L1 dimerization and centered at Met115, currently used to develop small molecule inhibitors. The C2PD−L1d, located at the tunnel entrance near A:Asp61 and B:Arg125, shows a more hydrophilic profile and is already under research as a target for new-generation PD-L1-dimerization molecules (Figure 4C).
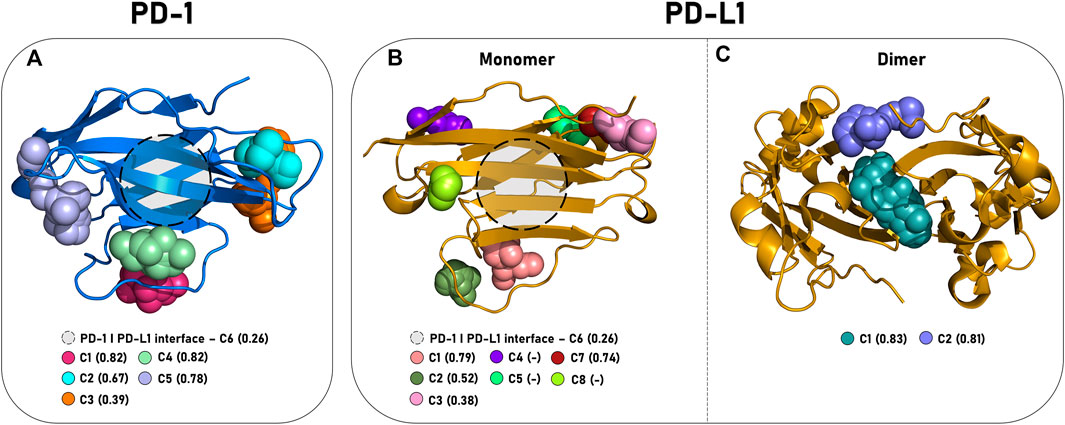
FIGURE 4. Positioning of the interaction pockets in PD-1 and PD-L1. The main cavities of (A) PD-1 (cartoon, blue), (B) PD-L1m (cartoon, yellow), and (C) PD-L1d (cartoon, yellow) are represented by colored spheres, and the druggability score descriptor obtained from the DoGSiteScorer web server is shown in parenthesis. (-) define cavities not identified by the server.
In contrast to its dimeric form, PD-L1m showed a structure-dependent behavior (Figure 4B). From the five identified cavities by DogSiteScorer, only the C1PD−L1m, located at the C″D loop and centered at the residue Gln77, was detected in all structures. The cavity with the second highest druggability score among the evaluated conformations was the C7PD−L1m, located in the BC loop and centered at residue Leu92. Notably, the PD-L1 interface of dimerization/interaction with PD-1, defined as C6PD−L1m, was identified in only three structures, with a maximum druggability score of 0.26, consistent with the difficulty of targeting the front face of PD-L1.
PD-1 had six pockets recognized despite being considered a hard-to-target protein (Figure 4A). Unsurprisingly, the C6PD−1 pocket at the front sheet GFCC′ had the lowest druggability score value (maximum of 0.26). The other detected cavities were C1PD−1 and C4PD−1, located inside and outside of C’D loops, and C2PD−1, C3PD−1, and C5PD−1, located at the BC, DE, and CC′ loops, respectively.
In summary, we identified two, five, and six putative pockets along PD-L1d, PD-L1m, and PD-1 static surfaces, some of which showed conformational-dependent behavior. Subsequently, we investigated whether the flavonoids preferentially recognize a specific cavity from the previously described ones or could access additional regions of the proteins since new sites might become accessible during MD.
3.2 Flavonoids are mainly anchoring at the PD-L1 interface with PD-1
Considering the description of new cavities in PD-1 and PD-L1, elucidating the potential interactions between the ligand and all protein sites is fundamental for understanding the most probable flavonoid anchoring region in the PD-1 pathway proteins. Supervised Molecular Dynamics simulation (SuMD) favors this understanding by enabling the compound to scan the entire surface of the protein. This way, it is possible to observe structural rearrangements and specific mechanisms of the binding event and interpret which cavities can attract and maintain the flavonoids’ scaffold.
The ligand must first be capable of scanning the entire protein space. Since the target binding site, i.e., the reference region to calculate the distance from the ligand, is located at the center of mass of the protein, ligands tend to arrange themselves in the strand areas, as they assume smaller distances from the reference than the loop regions. To circumvent this difficulty, we specified six starting positions for each ligand-protein pair. We also analyzed the trajectories with true approximation to the protein (also known as productive trajectories) and filtered residues with contact values above 300 consecutive frames, comprising 1.5 ns.
The SuMD performed with the PD-1 systems revealed the main regions with prevalent contact with all flavonoids selected in this study. These regions are described as cavities C5PD−1 and C1PD−1/C4PD−1 (Supplementary Table S4/Figure 4A). In C5PD−1, Arg69, Ser71, and Asp77 of the CC′ loop accommodated the chromen-4-one moiety through polar interactions. Asn116 flanked this region and stabilized the ligands. In the cavities C1PD−1 and C4PD−1, Lys78, Arg86, Gln91, and Arg96 were the most accessed residues. Both regions are closely related to the C’D loop, differing only in being positioned at the entrance (C1PD−1) or inside the loop (C4PD−1). Hence, while Thr98 anchored the ligands to C1PD−1, Arg96 coordinated with the hydroxyl group of the molecules and stabilized binding with C4PD−1. Moreover, polar residues on the loop can stabilize the flavonoid compounds in either cavity through hydrogen bonding and other polar interactions.
It is essential to highlight that C5PD−1 and C1PD−1/C4PD−1 are sites distant from the interface region. Unexpectedly, SuMD indicated that the interaction between PD-1 and flavonoids could not primarily occur in the protein-protein interface with PD-L1. Moreover, kaempferol-3,7-dirhamnoside was the only molecule from our pool of flavonoids that did not stably interact with any residues in cavity C1PD−1. Secondary pockets, such as C6PD−1 (CC’FG and BED sheet), C3PD−1 (located between Phe56 and Asn58), and C2PD−1, showed only marginal or molecule-dependent stability.
In the PD-L1m systems, all the flavonoids preferentially accessed the C6PD−L1m pocket located at the interface region of PD-L1 (Supplementary Table S5). The orthosteric pocket C6PD−L1m, formed by Tyr56, Arg113, Met115, Tyr123, and Arg125, encompasses the chromen-4-one group in a manner that mimics the binding mode of crystallographic ligands to PD-L1. Regarding the prevalence of contacts, other exploited cavities were C1PD−L1m, C2PD−L1m, C7PD−L1m, and the ABED sheet. Compared to the PD-L1 interface with PD-1, those cavities had a smaller number of residues recruited for the interaction in fewer reported replicates. Still, the identical residues were prevalent for all ligands, demonstrating the flavonoids’ ability to scan different regions in the protein and find favorable areas for interaction. Kaempferol was the only ligand stabilized in cavity C3PD−L1m, in contrast to kaempferol-3,7-dirhamnoside and apigenin, which exclusively interacted with cavity C8PD−L1m. Inactive ligands did not behave differently from active ones.
Overall, we observed that SuMD favored the analysis of binding events in PD-1 and PD-L1. Despite the tendency of SuMD to guide the ligands to the centroid of proteins, we were able to identify alternative regions that allow stable interaction with the flavonoids’ scaffold. Briefly, the flavonoids tend to move toward the CC’ and C'D loops of PD-1, away from the interface with PD-L1. For PD-L1m, the most populated cavity was the C6PD−L1m, responsible for interfacing with PD-1. Moreover, we recovered similar binding modes for flavonoids to the ones from co-crystallized ligands in PD-L1, supporting our findings.
3.3 Diverse cavities along the PD-1 and PD-L1 are suitable for accommodating flavonoids
Simultaneously to the hotspot determination by SuMD, we applied an orthogonal approach to determine the suitable binding sites for flavonoids (Figure 2C). First, we sought to determine whether the blind docking procedure could identify a similar pose to the native one, obtained from PD-L1 complexes crystal structures. This validation step proved that our docking methodology could recover at least one pose with RMSD lower than 5 Å from each crystallographic ligand (Supplementary Figures S2, S3; Supplementary Table S6). The validation also included the 1,4-benzodioxane as a negative control. However, as expected, we could not distinguish between active/inactive molecules or correct/incorrect poses from docking outputs based on the energy score. Nevertheless, we blind-docked each selected ligand against five different structures of PD-1, PD-L1m, and PD-L1d, and clustered the outputs according to the ligands’ flavonoid scaffold to identify the main interacting cavities (Figure 2C, Supplementary Figure S3).
We generally observed more favorable interaction energies involving PD-L1d (Supplementary Table S7–S9). In this system, we identified two prominent interaction cavities, C1PD−L1d and C2PD−L1d, with most of the predicted poses located inside or around the tunnel at the dimer interface (Figure 4C; Supplementary Table S10), following the DogSiteScorer prediction (Supplementary Table S11).
In addition, the PD-L1m docking detected eight putative pockets capable of recognizing flavonoid scaffolds (Figure 4B; Supplementary Table S12), namely cavities C1PD−L1m to C8PD−L1m. Noteworthy, cavity C6PD−L1m, located at the interface of PD-L1 with PD-1, was only accessed by docking to the structures with PDB IDs 5J89 and 7C88, co-crystallized with a small molecule and an antibody, respectively. Moreover, DogSiteScorer prediction showed a low druggability score for this pocket. Altogether, these results imply that flavonoids can bind only to specific conformations of the PD-L1 interface. Furthermore, DogSiteScorer was unable to identify cavities C4PD−L1m, C5PD−L1m, and C8PD−L1m located at AB, DE, and CC’ loops, respectively. Still, they were accessed by some SuMD replicates, validating these three cavities as possible binding spots.
Lastly, for the PD-1 protein, the predicted binding modes were clustered in six cavities (Figure 4A). Most of the identified pockets in this protein were located inside or near loops. It is worth noting that the cavity C4 in the C’D loop is only accessible when the E10 structure, previously obtained using cosolvent molecular dynamics simulation, was used as the target (Andrade et al., 2022). Similarly, the cavity C1PD−L1m in the C″D loop of PD-L1m is only accessible in 5JDS, a conformationally-distinct PD-L1 structure co-crystallized with a nanobody.
Although our docking methodology recognized the GFCC’ sheet - the interface of PD-1 with PD-L1 - as the putative pocket C6PD−1 for the flavonoid scaffold, it accommodated only 4.5% of the predicted poses of all structures (Supplementary Table S13), indicating a possible binding mechanism of these molecules in regions yet unexplored. Together, the low druggability of C6PD−1, reinforces the difficulty in designing new molecules against this pocket.
The docking protocol suggested the possibility of flavonoids binding to pockets far from the PD-1 or PD-L1 interface. Specifically for PD-1, we could observe that the predicted poses are diverse on the proteins’ surface. In brief, we prospected the possible cavities accessed by distinct flavonoids in PD-1, PD-L1m, and PD-L1d. As expected, the buried cavity C1PD−L1d, already explored for PD-1/PD-L1 modulation, showed the most promising results. While the interface cavity C6PD−L1m had a low druggability score, we showed that such a score drastically improved on the dimer. Furthermore, the analysis of docking and SuMD results also indicated that flavonoids could interact with the PD-L1 interface (C6PD−L1m) before the dimer assembly but are not restricted to this site. Lastly, several putative pockets were proposed for PD-1, although compounds did not have a clear tendency to interact with the interface cavity (C6PD−1), as conjectured in the existing literature.
3.4 Flavonoids showed stability and convergence in both PD-1 and PD-L1 proteins
The detailed scanning of protein surfaces and cavities generated several possible flavonoid binding modes. Next, we evaluated the stability of the proposed conformations by performing extensive molecular dynamics simulations starting from the selected docking output to distinguish a true predicted pose in a close-to-native conformation from a false positive one. After a visual inspection of the predicted poses and evaluation of the clustering results, we chose the inputs for MD, prioritizing one pose from each lowest branch level of the clustering tree to ensure pose diversity for each ligand. Roughly half of the 50 predicted poses for each ligand were selected, resulting in more than 20 μs of simulation (Figure 2C, Supplementary Figure S4). One would anticipate that the flavonoid scaffold’s almost ideal pose would hold steady around the docking site. In contrast, the others would leave their original cluster or even dissociate from the protein.
After the simulations, a new clustering considering only the flavonoid scaffold atoms using both the docking output and the last frame of each simulation gave us an insight into the most reliable cavity for each studied protein. Each system’s clustering parameters were fine-tuned independently to distinguish the previously identified cavities into different clusters (Figures 2C, 4). In the first instance, we evaluated the permanence ratio, which describes the percentage of molecules that remained in the same cluster compared to the amount that started. Then, a second round of clustering was performed for each identified group to identify distinct scaffold conformations inside each cavity and detect possible convergence. For clarity, PD-1, PD-L1m, and PD-L1d will be separately discussed in the three subsequent subtopics.
3.4.1 Low convergence of flavonoids in the PD-1 interface indicates the existence of an undescribed cavity
Starting with PD-1, clusters 0 and 2, representing the C1PD−1 and C5PD−1 cavities, retained the ligands in 57% (47/82) and 61% (17/28) of the simulations, respectively. Even though these two pockets were considered hotspots by SuMD, they appear to have limited capacity to retain the molecules in more extended simulations compared to the other evaluated sites. Cluster 5, equivalent to cavity C6PD−1, also showed poor molecule retention, with a permanence ratio of only 54%. Contrastingly, cluster 6, encompassing the C4PD−1 pocket in the C’D loop, showed a 75% permanence ratio during the simulations (6/8), but pose convergence was not observed. Finally, clusters 3 and 1, corresponding to cavities C2PD−1 and C3PD−1, respectively, retained the ligand in 58% (11/19) and 77% (33/43) of the simulations (Supplementary Table S14).
Reclustering within the previously identified conformational clusters was used to evaluate pose convergence further. This second clustering round indicated that the flavonoid scaffold recognizes cavity C3PD−1 preferentially. This preferential interaction is supported by the higher number of stable simulations grouped within the same subcluster. Additionally, this cavity interacted with the ligands in about 60% of the trajectories from the 25 ns simulations. We must highlight that an interacting trajectory was defined as one in which the ligand stayed in contact with any cavity residues for at least 80% of the frames from the last 5 ns of the trajectory.
The C3PD−1 pocket hydrophobic core comprises Phe56, Phe63, Phe82, and Phe106. The latter is at the base of the pocket and interacts in 65% of the analyzed MD trajectories, emerging as the main anchor for flavonoids. However, at first analysis, the molecules remained partially exposed to the solvent without adequately interacting with PD-1. Such an interaction pattern could be explained by ligand rigidity and the apparent small cavity depth. Although the results point to the C3PD−1 cavity as the one that could recognize the flavonoids more efficiently, it is still a frail inconclusive observation.
As kaempferol-7-O-rhamnoside has an experimental KD of 31 μM against PD-1 (Kim et al., 2020), we used it as a positive control for running new extended 50-ns simulations to identify the most reliable cavity for binding of flavonoids (Supplementary Figure S5). Unfortunately, none of the extended molecular dynamics starting with this known binder in the cavities C1PD−1, C5PD−1, or C6PD−1 showed convergence between replicates, except for one that pointed the glycoside to the base of cavity C1PD−1. Since this group is a particular feature of the rhamnose-derived kaempferol molecule, such a pose cannot represent the overall binding mode of flavonoids in PD-1. We thus discarded these cavities as possible for kaempferol-7-O-rhamnoside recognition.
3.4.2 Initially occluded Asn-58-glycosilation cavity emerges as a potential binding site in PD-1
The last possible cavity, C3PD−1, is supposed to be blocked by the glycosylation of Asn58. It has been reported that glycans can function as a gateway to binding pockets, obstructing or allowing access depending on the assumed conformation (Seitz et al., 2020; Vuorio et al., 2021). To assess if C3PD−1 could have its exposure regulated by the saccharide, five MD replicates of 50 ns with the Asn58-glycosylated PD-1 were run. We showed that while in most frames, the polysaccharide hinders the accessibility to the pocket, in a few others (Figure 5B), its flexible nature allows enough distance to expose the cavity, accommodate a ligand and possibly cooperate with the protein in forming the binding groove (Figure 5A).
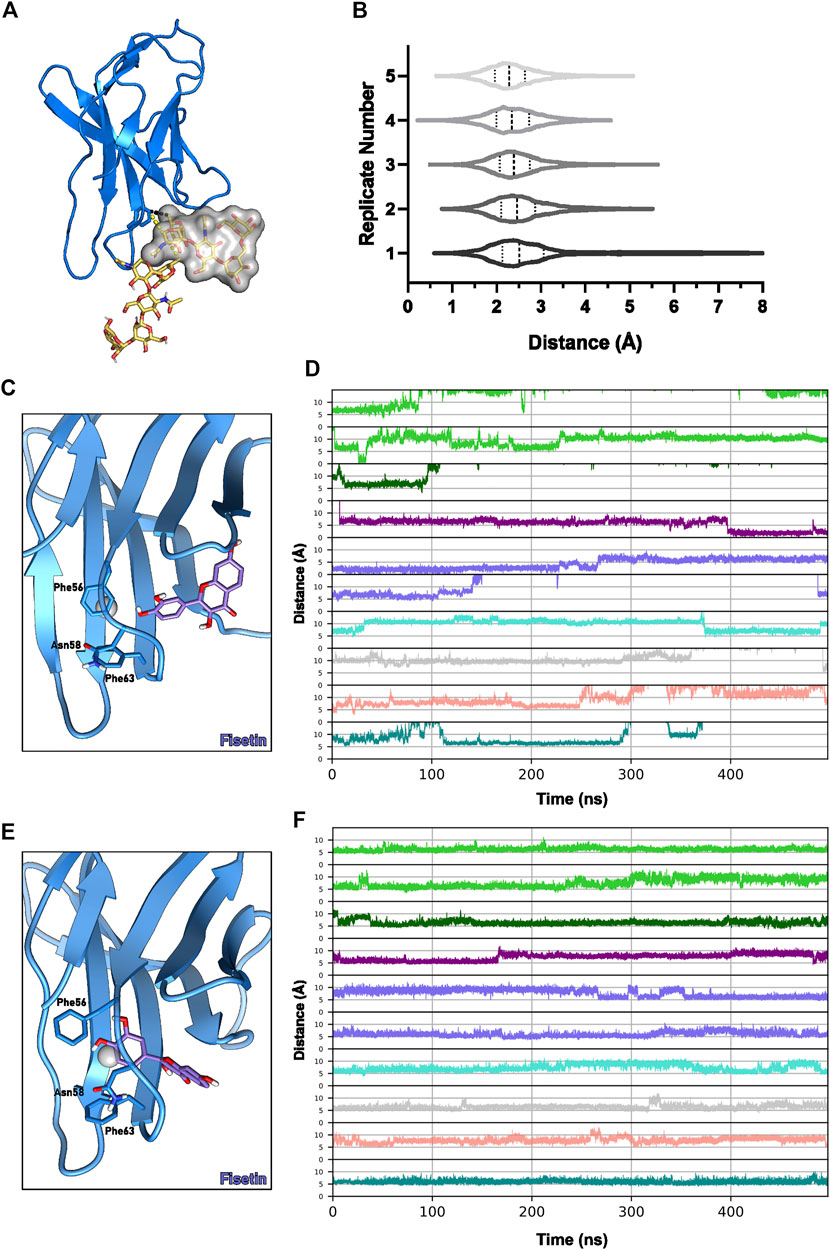
FIGURE 5. C3PD−1 cavity dynamics. (A) Conformations of the glycoside attached to Asn58 before (yellow sticks and surface) and after (yellow sticks) a MD simulation of Asn58-glycosilated PD-1 (cartoon, blue). (B) Distances between Cζ-Phe63 and the glycan in five MD replicates. Example of a flavonoid pose (C) outside and (E) inside the pocket formed by the Phe56/Phe63 dihedral flip in the BC loop. Blue sticks represent the fisetin molecule, and the pocket centroid is a gray sphere. (D) Molecules’ distance to the centroid of the pocket defined between the Cα-Phe56 and Cα-Phe63 throughout 500-ns simulations. (F) Distances between the Cα-Phe56 and Cα-Phe63 during the 500-ns simulations. Plot lines are colored according to the molecule complexed with PD-1: apigenin, green; cosmosiin, dark green; eriodictyol, purple; fisetin, blue; kaempferol, cyan; kaempferol-7-O-rhamnoside, gray; luteolin, salmon; quercetin, teal.
We next simulated the docking poses of kaempferol-7-O-rhamnoside inside this putative C3PD−1 binding site in a non-glycosylated PD-1 and demonstrated it fits the pocket adequately (Figure 6B). Its chrome-4-one moiety was stabilized by a T-shaped π-π stacking interaction with Phe82 or Phe106. Moreover, its 3-OH group formed a polar interaction with the backbone of Phe56. The other moieties remained solvent-exposed. The rhamnoside group was located between Pro83 and Phe82, and the hydroxyphenyl was placed near Gly103, without interacting with PD-1.
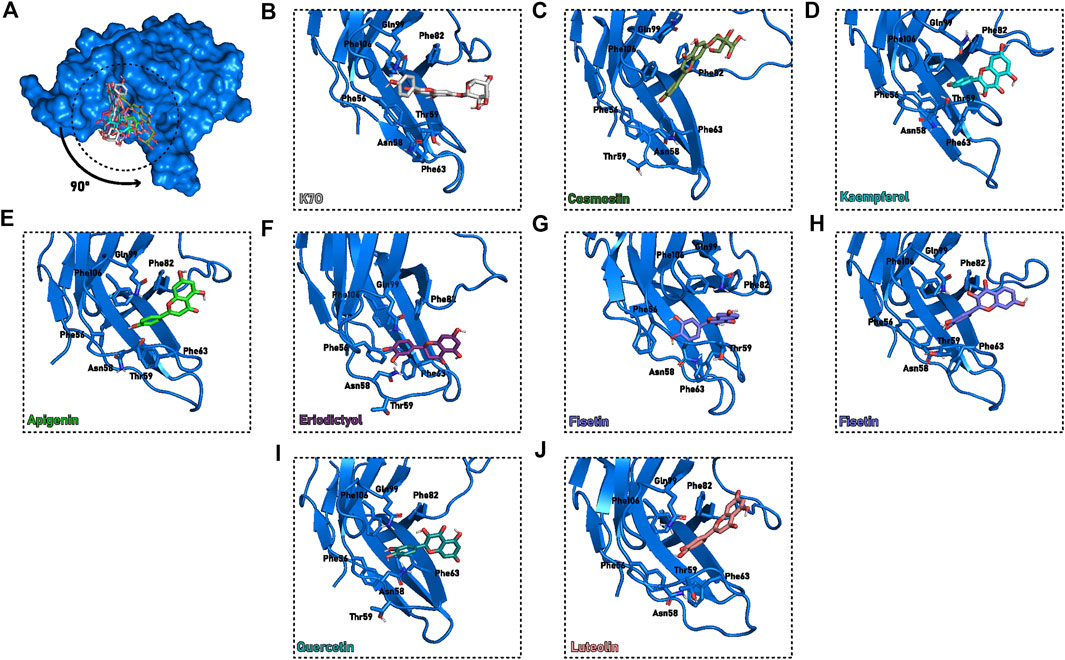
FIGURE 6. Proposed flavonoids binding modes in cavity C3PD−1 at the end of the 50 ns cMD. (A) PD-1 (blue surface) with the molecules (sticks) superimposed to cavity C3PD−1. (B–J) Detailed description of C3PD−1 binding cavity (blue cartoon) with the interacting residues and molecules highlighted (sticks). The protein orientation was rotated 90° compared to (A). Different colors were assigned for each flavonoid: (B) Kaempferol-7-O-rhamnoside, or K7O, grey; (C) Cosmosiin, dark green; (D) Kaempferol, cyan; (E) Apigenin, green; (F) Eriodictyol, purple; (G,H) Fisetin, blue; (I) Quercetin, teal; (J) Luteolin, salmon.
PD-1 complexes with other flavonoids in a similar orientation to kaempferol-7-O-rhamnoside were also simulated. However, most simulations diverged, suggesting it is not the prevailing flavonoid binding mode. Moreover, C3PD−1 could not accommodate ligands with bulkier radicals, such as kaempferol-3-7-dirhamnoside. In this case, the two rhamnoside moieties, which must remain solvent-exposed, would clash with the native glycoside. The inability of kaempferol-3-7-dirhamnoside to fit into C3PD−1 could explain the inactivity of the molecule against PD-1.
From the remaining stable poses of the 25 ns simulations, we ran new 50 ns MD replicates. For all molecules except kaempferol-3-7-dirhamnoside, the converging pose is described by the (di)hydroxyphenyl group pointed toward the Asn58, inducing the protein to form a narrow pocket enclosing the ligand with residues Ser60, Phe63, Leu65, Phe82, Phe105, Thr59, Phe56, Pro83, Gln99 (Figures 6A,C–J).
The pair Phe56-Phe63 initially performs T-shaped π-stacking with each other (distance of ∼6 Å between their centers of mass). Interestingly, in one of the fisetin and apigenin MD replicates, a flip of Phe56 caused the opening of a tunnel through the BC loop (Figures 6G,H), enabling the ligand access. While the apigenin quickly returned to its original state (5 ns of permanence), fisetin remained inside the tunnel for more than 40 ns until the end of the simulation. Following the Phe56 sidechain flip and ligand entrance, the hydroxyphenyl group is sandwiched between Phe63 and Phe56 in a potential triple-stacking interaction, and the hydroxyl group interacts with N-terminal residues. These results suggest that both hydroxyphenyl and dihydroxyphenyl have enough size and chemical features to induce the conformational change in this loop. Furthermore, no other substitution appears to influence this binding mode during simulations.
To better understand the mechanism of the pocket opening caused by the flip of Phe56 χ1 dihedral, we ran an additional 450 ns of simulation from one selected replicate for each flavonoid. The chosen replicates had the flavonoid stable at the C3PD−1 cavity in the end of the previous 50 ns MD. The two replicates where Phe56 χ1 flipping was detected (one with a fisetin complex and one with apigenin) were also extended to 500 ns. Cavity opening was primarily characterized by the flips of Phe56 and/or Phe63 χ1 dihedral, which allowed the distances between Phe56 and Phe63 centers of mass to reach values higher than 10 Å (Figures 5C,E,F). Fisetin evaded the cavity after 250 ns of simulation (Figures 5C,D,E). Interestingly, eriodictyol also entered the pocket between Phe56 and Phe63 after 400 ns of simulation and remained stably inside until the end of the simulation (Figure 5D). Following ligand exit, the T-shaped π-stacking between the phenylalanine residues was promptly restored (Figures 5C,E,F).
Only in four of the ten 500 ns MDs (buried apigenin, buried fisetin, eriodictyol, and kaempferol, respectively the lime green lower, medium slate blue upper, purple, and turquoise lines of Figure 5D) the flavonoid remained in the C3PD−1 pocket, which could be explained by the lack of polar interactions and the very exposed pocket entrance (Supplementary Figure S6). These four stable flavonoids showed similar ΔG values of roughly –23 kcal mol−1 (Supplementary Table S18). It is worth highlighting that cooperative interactions with the glycoside may provide additional stability to the interaction in this cavity.
In summary, we suggest flavonoid ligands could not interact with the C6PD−1 cavity at the PD-1 interface, given the lack of pose convergence during simulations. Instead, we propose that the Asn58 glycosylation is flexible enough to unveil the C3PD−1 cavity, and that the presence of flavonoids’ hydroxyphenyl moiety could induce a flip change in Phe56/Phe63 χ1 dihedral. This flip naturally occurs in the PD-1 structure, although maintaining the Phe56-Phe63 stacking, as observed in NMR structures (PDB ID 2M2D). In our systems, flavonoids exposed a pocket through PD-1’s BC loop and enabled the ligand entry. Due to the importance of Asn58-glycosylation to the interaction with PD-L1, modulation of this pocket using a small molecule could impair the PD-1/PD-L1 complex formation.
3.4.3 Flavonoids interact with interface sheets in monomeric PD-L1
After describing a possible binding mode for PD-1, we turned our attention to the PD-L1 protein. Initially considering the monomeric state of this protein, PD-L1m, the simulations indicated the flavonoid ligands showed attractive stability in clusters 3 (permanence ratio of 68%, 21/31) and 10 (permanence ratio of 67%, 2/3), both describing the C6PD−L1m in the GFCC’ sheet of the PD-L1, responsible for PD-1 binding (Figure 4B; Supplementary Table S15).
After the reclustering step, subclusters 0 and 3 from cluster 3 contained more final than initial simulation poses (seven and three, respectively), which could suggest that simulations converged from the docking conformation to a predicted near-native pose. Their binding mode was characterized by anchoring one of the flavonoid aromatic rings to Met115, while the second ring is stacking with either Tyr123 or Tyr56. The Met115 anchoring is also present in the molecules of cluster 10, suggesting it is the major hotspot for flavonoid interaction in the C6PD−L1m cavity. In agreement with the cMD hotspots, the most accessed residues during SuMD were also Tyr123 and Met115.
We further identified the cavity C6PD−L1m as the only one with multiple residues contacting the ligand in more than 80% of the last 5 ns of simulation in at least 50% of trajectories. Specifically, Met115, Ala121, Tyr123, Tyr56, and Ser117 stably contacted the ligand in 75, 50, 55, 57.5, e 52.5% of the simulations starting in C6PD−L1m. Despite the high contact ratio, no stable hydrogen bond interactions were observed, reinforcing the hydrophobic pattern of binding.
It is essential to highlight that these hotspot residues have already been explored to propose ligands that induce PD-L1 dimerization. The superimposition of the ligand UGZ, co-crystallized with PD-L1 and available at PDB:ID 7NLD, with the identified clusters revealed that cluster 10 describes the expected orientation of the molecule in the dimeric PD-L1 (Supplementary Figure S7). Albeit the cluster 3 conformations are slightly displaced from those observed in structure 7ND7, one could not discard it as the native one in PD-L1m since some reorientation could occur due to dimer formation.
To gain better insight into the pose convergence and determine the most probable binding mode of flavonoids in the C6PD−L1m at the interface of PD-L1, we carried out extended MD simulations of selected ligands docking poses (Figure 2D). This approach highlighted a clear binding mode for apigenin and kaempferol in the C6PD−L1m pocket at the PD-L1 interface (Figures 7A–C). Chromen-4-one and hydroxyphenyl groups are generally perpendicular to each other. In the most prevalent binding mode, the first is usually stacking with Tyr123, while the second is placed in a cleft described by residues Met115, Ser117, Ile54, and Tyr56. We also identified a possible hydrogen bond between chromen-4-one and Arg125 or Arg113 and between (di)hydroxyphenyl and Tyr56. The 3-OH and 3′-OH groups are exposed to the solvent and do not seem to affect the binding mode with PD-L1m (Figures 2E, 7). A similar binding mode was identified luteolin. Although this compound remained stable in the specific orientation in the pocket of the monomeric PD-L1, the unexplored molecule’s behavior throughout the dimerization process required for cavity assembly might explain its inactivity against PD-L1 in cell-based assays (Choi et al., 2020).
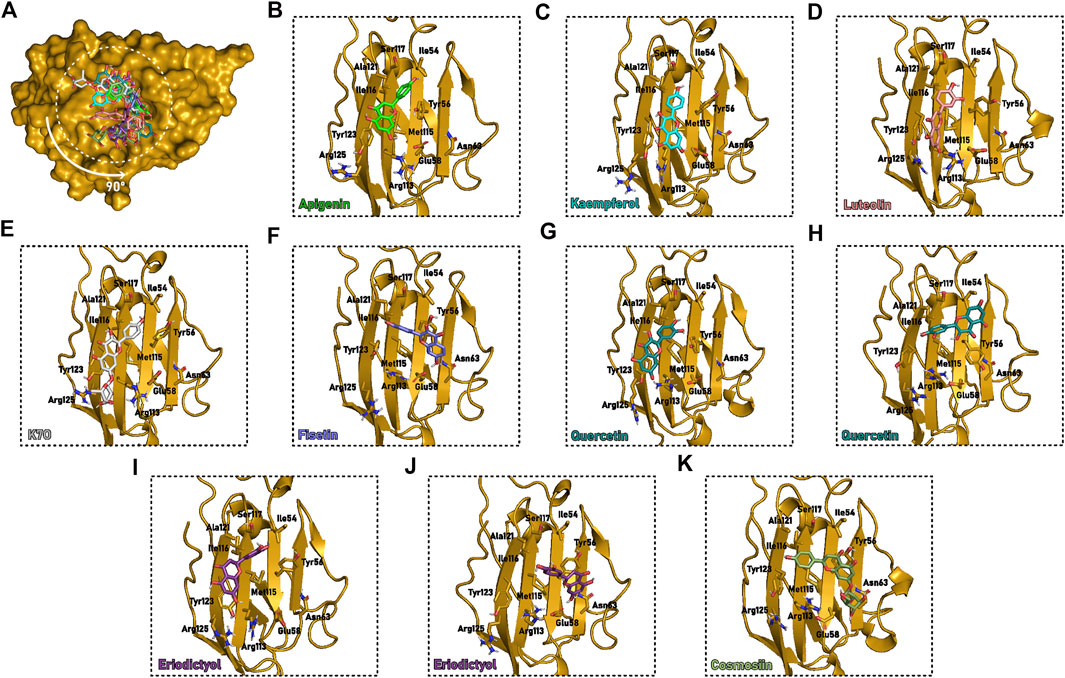
FIGURE 7. Flavonoids selected binding mode in the interface cavity of PD-L1m at the end of the 50 ns cMD. (A) PD-L1m (yellow surface) with the molecules (sticks) superimposed to cavity C6PD−L1m. (B–L) Detailed description of binding cavity C6PD−L1m (yellow cartoon) with the interacting residues and molecules highlighted (sticks). The protein orientation was rotated 90° compared to (A). Different colors describe the various flavonoid: (B) Apigenin, green; (C) Kaempferol, cyan; (D) Luteolin, salmon; (E) Kaempferol-7-O-rhamnoside, or K7O, grey; (F) Fisetin, blue; (G,H) Quercetin, teal; (I,J) Eriodictyol, purple; (K) Cosmosiin, dark green.
The additional glucoside group in kaempferol-7-O-rhamnoside was placed between Arg125 and Arg113, causing a slight displacement of the flavonoid scaffold toward Tyr56 (Figure 7E). The absence of hydroxyl in position C7 of fisetin enabled an inversion in the molecule orientation. In this case, convergence was achieved when chromen-4-one was stacked with Tyr56, and the dihydroxyphenyl was placed between Met115, Ala121, and Tyr123. Interestingly, the PD-L1m/fisetin simulations starting from a similar orientation to the stable binding mode detected for apigenin, with chromen-4-one near Tyr123, diverged and dissociated from the protein (Figure 7F).
Although quercetin has no substituents at position C3, it converged towards two binding modes during the 50 ns MD, both anchoring. Both anchored the dihydroxyphenyl portion within the vicinity of Ile116. At the same time, the first pose resembled the binding mode of apigenin, luteolin, kaempferol, and kaempferol-7-O-rhamnoside. Contrastingly, the second one projected the chromen-4-one in the direction of Ile54 (Figures 7G,H). In consonance with quercetin’s behavior, eriodictyol also accessed two different poses despite the change of chromen-4-one to its reduced analog, dihydrochrome-4-one (Figures 7I,J). While the first one is similar to one of the already described binding modes of quercetin (Figure 7I), the second adopts the exact positioning of fisetin (Figure 7J). We hypothesize that the least aromaticity of dihydrochromen-4-one disfavors the π-π stacking interaction with Tyr123. Moreover, the second pose provides extra hydrogen bonding with Glu61 and Asn63 and stacking with Tyr56, the latter directly affecting the orientation of the flavonoid hydroxyphenyl. Furthermore, the break in the molecule’s planarity led to a more adjusted molecule positioning at the pocket.
No stable rearrangement was found for cosmosiin, which could be explained by the lack of a similar docking pose to the one of the converged kaempferol-7-O-rhamnoside simulation (Figure 7K, Supplementary Figures S8, S9). Lastly, kaempferol-3-7-dirhamnoside did not show pose convergence, which was expected for an inactive molecule. This instability was related to the required exposure to the solvent of the rhamnoside and hydroxyphenyl groups, as explored in the results of the shorter cMD. Interestingly, all the converging simulations for the different flavonoids started from poses generated with PD-L1 structure 7C88, obtained from a complex structure with a monoclonal antibody, instead of the 5J89 structure, co-crystallized with a small molecule in the interface. No significant difference in residues rotamer near the predicted binding site was identified between these two structures.
A simulation time of 450 ns was added to one of the 50-ns replicates of each flavonoid to evaluate the molecules’ retention time in the interface sheet. Despite the lack of polar interactions, the non-polar feature of flavonoids and the C6PD−L1 cavity appears sufficient to maintain the interaction in nine out of ten simulations. Furthermore, despite each flavonoid interacting distinctly with the pocket, all were anchored between residues Ile54, Tyr56, Met115, and Ser117, confirming them as the most relevant hotspot of the PD-L1 interface. MM/GBSA analysis was inconclusive, as all molecules had predicted ΔG between –15 and –18 kcal mol−1, except for kaempferol-7-O-rhamnoside with a slightly better interaction of –20 kcal mol−1 (Supplementary Table S19), presumable due to the glucoside moiety.
In summary, we observed a convergence toward the probable binding mode from different flavonoids. Chromen-4-one generally stacks with Tyr123, and hydroxyphenyl is accommodated in a hydrophobic cleft near Met115. When present, the saccharides slightly displace the scaffold to promote hydrogen bonding with Arg125 and Arg113. No clear correlation with the literature experimental binding data was noted, presumably due to the presence of aromatic compounds in the interface, which would favor PD-L1 dimerization. We emphasize that the stabilization of the dimer by small molecules is a relevant aspect that was not addressed until now.
3.4.4 Substitutions in flavonoids clearly affect the binding mode and dynamics of compounds in PD-L1d
We then clustered the resulting MD conformations of the complexes containing the dimeric state PD-L1d, which would be favored by the presence of a hydrophobic molecule in the interface cavity C6PD−L1m. We found that cluster 1, equivalent to C1PDL1d, had the highest permanence ratio among the conformational clusters evaluated (92%, 12/13) and showed evidence of pose convergence. After reclustering this conformational group, we detected a high concentration of poses in subcluster 0. Still, two poses predicted by docking did not remain in the cavity after the 25 ns simulation. Otherwise, subclusters 1 and 3 had 100% permanence during simulations and are very similar conformationally. They are located in the middle of the PD-L1d tunnel and overlap with cluster 10 from PD-L1m and the 7LND ligand (Supplementary Figure S10). The contact analysis for PD-L1d simulations agreed with the observed for the PD-L1m simulations. Only the tunnel cavity has shown residues with a high contact ratio among the PD-L1d simulations, specifically the Ile54, Tyr56, Met115, Ile116, Ser117, Ala121, Asp122, and Tyr123, all present in more than 60% of simulations. Except for residues Ile54, Ile116 and Asp122, all others were also identified as frequent contacting residues in PD-L1m simulations.
3.4.4.1 Glucoside is stabilized by hydrogen bond interaction and displaces the flavonoid scaffold toward the Tyr56 residue
Particularly for cosmosiin, from the 20 docking poses submitted to the single-replicate 25 ns of MD, six ended with the ligand inside the tunnel cavity of PDL1d. It is worth noting that only five simulations started with the hydroxyphenyl moiety pointed to the center of C1PD−L1d, indicating that a short simulation is sufficient for a cosmosiin molecule, initially located at the outer surface of the pocket, to enter the cavity fully. The hydrogen bonding of the hydroxyphenyl group with A:Ile116 backbone and the π-π stacking between A:Tyr56 and chromen-4-one ring are the main interactions responsible for stabilizing the flavonoid core of cosmosiin. The glucoside moiety performs hydrogen bonding with A:Glu58, B:Lys124, and the backbone of B:Tyr123. Moreover, the A:Glu61 had a salt bridge with B:Arg125 disrupted by the ligand.
Further 50 ns simulation replicates for cosmosiin starting from Models 16 and 19 confirmed that the glucoside moiety guided the binding mode through hydrogen bond interactions with A:Glu58, A:Asp122, A:Lys124, B:Asp61, B:Glu58, B:Gln66, B:Tyr56, Tyr118, B:Asp122 and/or B:Lys124. This interaction network stabilized the chromen-4-one group outside the β-sheet in one of the dimer chains, reinforced by stacking with B:Tyr56 or A:Tyr56. The hydroxyphenyl group also contributed to ligand stability by interacting with B:Ile116. The saccharide further interacts via water-bridge hydrogen bond interaction with the residues A/B:Asp61 and A/B:Glu58 depending on the replicate. However, only a few replicates showed a complete entry of cosmosiin into the PD-L1d tunnel, characterized by a fully stretched molecule and the absence of most interactions.
The absence of glucoside in apigenin led to starting poses with either the hydroxyphenyl or chromen-4-one groups pointed to the tunnel’s center in PD-L1d simulations. However, despite the pocket having enough volume to accommodate the bulky group, burying of the chromen-4-one induces a severe perturbation in the PD-L1d interface (Supplementary Figure S9), suggesting destabilization of the complex. In contrast, none of the 11 poses of apigenin with the hydroxyphenyl moiety inserted in the pocket show significant rearrangements in PD-L1 chains. Hence, these poses could be grouped into two binding modes: the first one is almost identical to the observed for cosmosiin, while the second displaced the hydroxyphenyl toward A:Gln66, putting chromen-4-one in the hydrophobic cleft comprised by A/B:Met115, A:Ala121 and B:Ile54. In this pose, the ligand is completely buried between the PD-L1 chains.
Extended MD confirmed apigenin’s ability to bury itself into the interface even when starting from a partially exposed position. Almost all 15 replicates of Models 1, 4, and 5 simulations converged to a similar, buried pose (Figure 8C1). The interaction is stabilized by hydrogen bonding of 4′-OH with A:Gln66, 7-OH, or 5-OH with B:Gln66 and/or B:Ser117. Both hydroxyphenyl and chromen-4-one are further stabilized by Hydrogen-water-bridge involving A/B:Gln66 and A/B:Asp122. We also observed the possibility of water-bridging with B:Ala121. Moreover, a possible stacking between B:Tyr56 and chromen-4-one could also be formed. Our findings suggest that, when fully buried in the PD-L1d tunnel, the flavonoid is sealed inside the pocket by water-bridged interactions.
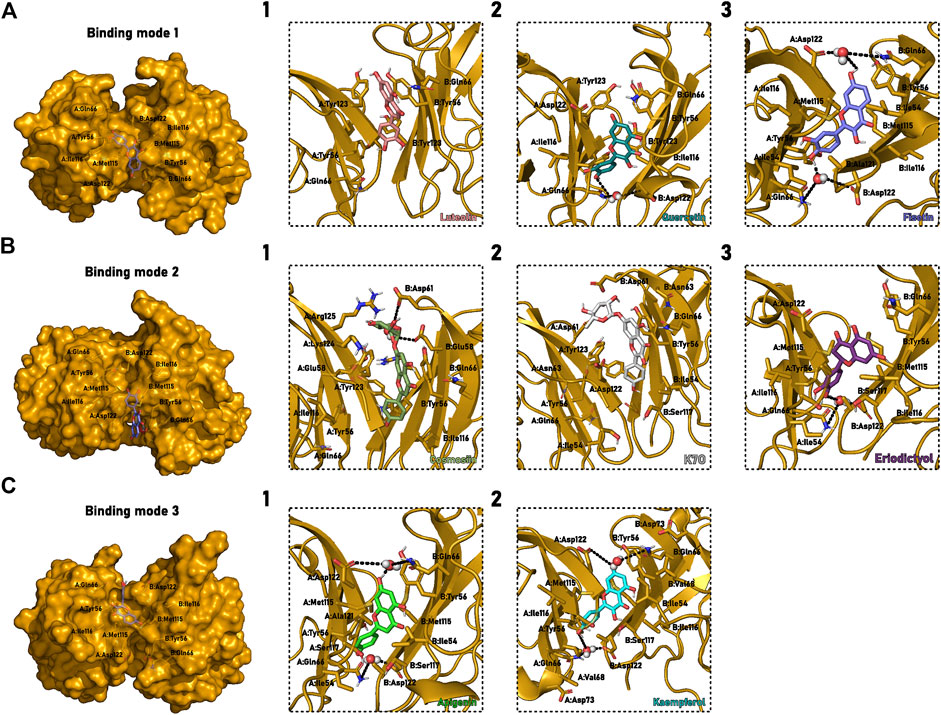
FIGURE 8. Flavonoids selected binding mode in the cavity between the monomers that form the PD-L1 dimer. (A) Binding mode 1, characterized by hydroxyphenyl and chromen-4-one buried between the dimer β-sheets. (B) Binding mode 2, described by the buried hydroxyphenyl. (C) Binding mode 3, marked by the buried chromen-4-one group. (1–3) Detailed description of C1PD−L1d binding cavity (yellow cartoon) with the interacting residues and molecules highlighted (sticks). Different colors describe the various flavonoid: (A1) Luteolin, salmon; (A2) Quercetin, teal; (A3) Fisetin, blue; (B1) Cosmosiin, dark green; (B2) Kaempferol-7-O-rhamnoside, or K7O, grey; (B3) Eriodictyol, purple; (C1) Apigenin, green; (C2) Kaempferol, cyan. Water molecules forming Hydrogen-water-bridges are represented as spheres, and the interactions are black dash lines. The representation used the same perspective for all molecules, although PD-L1d is a symmetric homodimer and the interactions can occur in two different orientations.
3.4.4.2 3-OH group is important for hydrogen bond with Ile116 and for stabilizing of the buried binding mode of flavonoids
The 3-OH substitution is also widespread among flavonoids. The kaempferol was thus evaluated to check whether the inclusion of this group affects the interaction with PD-L1d. The overall behavior of this compound is quite similar to the first binding mode above described for apigenin. Chromen-4-one moiety is stacked with B:Tyr56, and hydroxyphenyl remains buried around A:Ile116, performing hydrogen bond interaction with this residue. Interestingly, kaempferol also adopted a binding mode like the second pose identified for apigenin in one of the simulations. In this case, 3-OH played a vital role in stabilizing binding by forming a hydrogen bond with the same B:Ile116, which usually interacts with hydroxyphenyl moiety.
A new round of simulations indicated that kaempferol could effectively enter the cavity depending on the starting point of the simulation. A selected pose on 5N2F structure, where the molecule was almost entirely exposed to solvent, converged to an apigenin-like buried conformation in four of five replicates (Figure 8C2). These simulations reinforced the importance of 3-OH in performing hydrogen bonding with B:Ile116/B:Ala121. As expected, the trajectories that finished with buried kaempferol also showed the water-bridge interaction with the residues A/B:Gln66 and A/B:Asp122. Hydrophobic interactions with B:Tyr56, B:Val68, and B:Ile54 and a water-mediated hydrogen bond with B:Asp73 stabilized the chromen-4-one-exposed final pose, which was observed in three of the replicates for the Model 23. A further set of extended simulations based on a 6VQN-kaempferol pose led to a PD-L1 dimer dissociation upon the ligand exit from the cavity in one replicate, suggesting the dimer stability could be dependent on the presence of small molecule within the dimerization interface. As all PD-L1d simulations without complexed ligands showed only slight variation in the distance between the chains’ centroid and low RMSDinterface (Supplementary Figure S8), we reasoned the dissociation of kaempferol forced the disruption of essential interactions responsible for maintaining the dimer stability.
The inclusion of the rhamnoside group in position 7 has a similar effect to that of cosmosiin compared to apigenin. Again, the binding is mediated by glucoside, responsible for stabilizing the molecule only partially buried in the cavity. As observed for cosmosiin, the rhamnoside moiety interacts with B:Tyr56, B:Glu58, B:Asn63, B:Asp61, and B:Tyr123 through Hydrogen bonds. That enables the formation of a π-π stacking interaction between chromen-4-one and A:Tyr56 or B:Tyr56. No direct effect of 3-OH was observed in this binding mode. Additionally, the insertion of another bulky group, such as the O-rhamnoside at 3-OH made the molecule unable to bind to the tunnel, confirming the inactivity of kaempferol-3,7-O-dirhamnoside.
3.4.4.3 Apparently disfavoured 3′-OH group seems to only be tolerated in the presence of a 3-OH substitution in the flavonoid scaffold
The inclusion of hydroxyl in position 3′ was also evaluated. Luteolin remained inside the cavity during the simulations but without an apparent convergence of pose or interaction. We could infer that luteolin destabilizes the dimer based on the observed change in the relative orientation of PD-L1 chains (Supplementary Figure S11). The volume increase caused by 3′-OH occurs in the tunnel’s most profound, narrow, and hydrophobic point, which contains primarily aliphatic side chains. In contrast with the kaempferol and apigenin 50 ns simulation replicates, we did not observe the entry of luteolin in the tunnel, despite a similar starting point between these molecules (Supplementary Figure S10). Instead, luteolin predominantly interacts in the cavity border, forming π-stacking interactions with residues A:Tyr56, B:Tyr56, or B:Tyr123, and scarce hydrogen bonding with Gln66. Thus, our simulations could support the observation that luteolin cannot inhibit the PD-1/PD-L1 complex formation.
The influence of the 3′-OH substituent was further evaluated by simulating the quercetin-PD-L1d complex. The short simulations starting from the docking complexes showed pose convergence to the buried state of ligand in two MD simulations. Otherwise, the simulation starting at the buried pose moved the ligand toward the tunnel’s extremity to expose the 3′,4′-dihydroxyphenyl group to the solvent. The expected interaction with B:Ile116 was formed with the 3-OH instead of 4′-OH. Such an interaction reinforces the hypothesis that dihydroxyphenyl group is unlikely to be in the middle of the pocket. An increase in the simulation time supported the previous observation. Quercetin entered the cavity in all five replicates starting from Model 1. The new hydroxyl 3-OH consistently interacted through hydrogen bonds with B:Ile116, suggesting a compensatory effect of 3-OH to tolerate the presence of the dihydroxyphenyl group. Despite the lack of hydrogen bonding between quercetin and PD-L1d, a water-bridge bond between A:Gln66, B:Asp122, and 4′-OH of the molecule occurred during ∼30% of the simulation time summing all four stable replicates of Model 1.
3.4.4.4 Flavonoid interaction with dimeric PD-L1 was not affected by 5-OH substitution
We next evaluated the importance of 5-OH substitution in the flavonoid. As all previously tested molecules had this group, we chose fisetin to understand this substituent’s role in interaction with the PD-L1d system. The simulations started with the ligand at least somewhat buried in the tunnel and converged to either a partially buried - with the hydroxyphenyl or the chromen-4-one moiety located between the dimer β-sheets - or a fully buried binding mode. The hydroxyphenyl-buried pose is described by the dihydroxyphenyl located near B:Ile116 and chromen-4-one stacking with B:Tyr56 (Figure 8B). The chromen-4-one-buried pose could be identified by a stacking between dihydroxyphenyl and A:Tyr56 and the chromen-4-one located near A:Met115 and B:Met115 (Figure 8C). Finally, the fully buried binding mode has a unique hydrogen bond interaction between 3-OH and B:Ile116, and the dihydroxyphenyl is placed between A:Gln66 and B:Asp122 (Figure 8A). The absence of 5-OH substituents had little effect on the binding mode of fisetin in comparison with quercetin. The only detected difference was the change in the rotamer of Ile54, one of the residues responsible for packing the chromen-4-one moiety. The fully buried pose was fisetin’s most probable binding mode, as a complete convergence to the this pose was observed for ten extended simulations replicates generated from Models 1 and 32. This convergence occurs despite five replicates having started in the hydroxyphenyl-exposed pose.
Moreover, 3-OH substituent performs Hydrogen bonding with B:Ile116 or B:Asp122 in all simulations. Again, the water-bridged interactions with residues A:Gln66, A:Asp122, B:Asp122, B:Ala121 and B:Gln66 were frequent, as we have shown for all fully buried binding modes (Figure 8A3).
3.4.4.5 Aromaticity of 5-chromen-4-one could play an important role in stabilizing the flavonoid binding mode through a water-bridge hydrogen bond
The last modification tested was the aromaticity of the chromen-4-one group. In eriodyctiol, the chromen-4-one of luteolin was replaced by dihydrochromen-4-one. The molecule was docked in the centered binding mode (Supplementary Figure S10), and remained in this position throughout MD. The binding mode is guided by hydrophobic interactions, with the dihydrochromen-4-one surrounded by B:Met115, B:Ile54, A:Ala121, and A:Tyr123. The dihydroxyphenyl was located between A:Met115, A:Ser117, and B:Ala121 residues. No apparent hydrogen bonding or stacking was observed. However, such a binding mode was not maintained during the 50 ns simulation replicates starting from Models 6 and 34. Only two replicates converged to the above pose, with hydrogen bonds between 6-OH and B:Ile116, suggesting instability of the PD-L1d-eriodictyol complex.
This dihydroxyphenyl orientation had not been observed yet in previous simulations of other molecules. In the two replicates ending with eriodictyol in an entirely buried pose, we observed water-bridge interaction only with hydroxyphenyl moiety (residues A:Gln66 and B:Asp122), instead of with both groups as seen for the buried pose of the previous molecules. Another possible converged pose exposed the dihydrochromen-4-one to the solvent without stacking with B:Tyr56, as observed for luteolin. We hypothesize that this group’s partial loss of aromaticity is related to the loss of the interaction with B:Tyr56. The remaining replicates diverged into distinct poses. We observed the dimer dissociation in a single trajectory, but unlike the kaempferol simulation, the eriodyctiol was still bound to the PD-L1 interface. These observations reinforce the hypothesis that the aromaticity of chromen-4-one group is important to the interaction and that the dihydroxyphenyl is only tolerated in flavonoid scaffold by PD-L1d when 3-OH substituent is also present.
We extended the 50 ns cMD to 500 ns for two selected replicates of all complexes with our flavonoid pool. The replicate selection was based on the flavonoid pose at the end of the 50 ns MDs comprehending one in the buried state and the other in the chromen-4-one-exposed state. Only one replicate was selected for apigenin and fisetin since both fully converged to the buried position. Fisetin and apigenin remained stable in a fully buried state throughout the 500 ns simulations, supporting our previous conclusion that this is likely the near-native flavonoid pose.
Furthermore, all trajectories based on buried poses showed similar behavior except for the eriodictyol one, which displaced the dihidroxyphenyl moiety to the solvent (Figure 8C, and Supplementary Figure S12). Meanwhile, three simulations presenting an exposed chromen-4-one pose moved the flavonoid toward the buried pose, pointing to the robustness of this pose as the near-native one. However, for luteolin and kaempferol-7-O-rhamnoside, the flavonoid motion was followed by a relative displacement between the dimer chains. The chain movement is characterized by increasing the distance between A:Gly119. A and B:Gly119 to ∼15 Å compared to the 7–10 Å range observed in other simulations and crystallographic structures, suggesting such flavonoids could destabilize the PD-L1 dimer (Supplementary Tables S16, S17). In contrast, quercetin adopted the buried pose without disturbing the dimer interface.
Lastly, simulations with kaempferol-7-o-rhamnoside, cosmosiin, or eriodictyol - initially in a chromen-4-one exposed pose - did not show ligand entrance into the pocket. As observed in 50 ns simulations, the glucoside moiety of the two former flavonoids favors the establishment of multiple hydrogen bond interactions with polar residues in the tunnel entrance, stabilizing this binding mode. The most favorable binding free energy value calculated by MM/GBSA for cosmosiin supports the stability of this pose (Supplementary Tables S20, S21). Yet our simulations have described that eriodictyol could not enter the tunnel without altering the dimer interface.
Altogether, we have described the binding mode of a series of flavonoids in the dimeric form of PD-L1. The glucoside moiety in position 7 of cosmosiin is expected to increase the molecule’s affinity when compared with apigenin due to the high number of hydrogen bond interactions occurring at the border of the cavity, consistent with the literature. Additionally, such moiety modifies the binding mode favoring the stacking of chromen-4-one with B:Tyr123, B:Tyr56, A:Tyr56. The 7-O-glucosylated form of kaempferol did not show this hydrogen bond interaction. As kaempferol has a better IC50, we hypothesize the 3-OH substitution improves the interaction of flavonoid molecules. The displacement caused by rhamnoside is detrimental to the binding, but it is balanced by a cluster of polar interactions in the PD-L1d pocket entrance. Contrastingly, the second glucoside of kaempferol-3-7-dirhamnoside prevents the molecule from stabilizing the PD-L1d interface.
PD-L1 readily recognizes the 3-OH hydroxyphenyl group as a favorable modification when we compare the dynamics of apigenin and kaempferol. The 3-OH in the latter promotes an extra hydrogen bond with B:Ile116. Such a beneficial effect is also observed comparing luteolin, fisetin, and quercetin, where the 3-OH reverted the harmful impact caused by a 3′-OH of the dihydroxyphenyl in the luteolin (inactive compound). Like luteolin, fisetin and quercetin contain the 3′-OH substituent, but owing to the presence of 3-OH, both showed good stability during simulations. The Kd and IC50 data for quercetin are possibly reflected in these computational results. Otherwise, luteolin and eriodyctiol disturbed the PD-L1 dimer and did not converge to the fully buried pose during simulations. Furthermore, the aliphatic ring of eriodyctiol cannot perform stacking interactions with Tyr56 residues of the PD-L1d interface, enhancing the detrimental effect of 3′-OH substituent. Additionally, it causes a change in the binding mode, adopting a distinct conformation pointing the 3′-OH toward the backbone of Asp104 and 6-OH to Ile214.
We successfully identified multiple possible binding sites in PD-1, PD-L1m, and PD-L1d surfaces. We observed concurrent indications from molecular docking and SuMD suggesting the PD-L1 interface as the most likely binding site for flavonoids in the monomeric and dimeric states of the protein. Extensive molecular dynamics simulations supported interaction at the interface by demonstrating the pose convergence and stability of different flavonoids in this pocket. We could also propose the interaction determinants from the MD simulations through molecular matched-pair analysis. Although the observations for PD-1 were generally inconclusive, we identified a putative mechanism of glycosylation displacement to unveil a BC loop pocket. The conformational change of a gate glycoside in the presence of a ligand suggests a cryptic site-like mechanism that was still unexplored.
4 Discussion
Proteins of the PD-1 pathway are well-established targets for the development of cancer treatments (Han et al., 2020). Examples of tumors applying PD-1 pathway blockade as therapeutic strategy are urothelial carcinoma or metastatic urothelial carcinoma (Nakanishi et al., 2007), non-small cell lung (Takada et al., 2017), and prostate cancer (Gevensleben et al., 2016). Seven commercially available antibodies against PD-1 and PD-L1 are currently approved (Upadhaya et al., 2022). Nonetheless, due to the high mutation rate associated with these cancer forms, some individuals do not respond to treatment with highly specific immunoglobulins (Chamoto et al., 2020; Kim et al., 2021; Machiraju et al., 2021; Hwang et al., 2022). Small molecules represent the conventional alternative. Due to the intrinsic difficulty of modulating the protein-protein interaction interface (PPI) (Lu et al., 2020; Martino et al., 2021), there is no currently available pharmaceutical chemical targeting this pathway (Wu et al., 2021). As is typical for PPI, the PD-1 and PD-L1 interfaces are flat, hydrophobic, and do not form a deep pocket capable of accommodating such compounds. However, small molecules can induce PD-L1 dimerization, creating a druggable pocket (Zak et al., 2016). CA-170 (Clinical Trial Phase 3) from Curis/Aurigene is currently the most promising drug candidate against the PD-1 pathway, as it can modulate the PD-1 pathway by binding to PD-L1 without effectively blocking the PD-1/PD-L1 complex formation (Sasikumar et al., 2021). Other compounds, including Incyte INCB-086550 (Clinical trial Phase 1) (Piha-Paul et al., 2020; Koblish et al., 2022), also promoted PD-L1 dimerization and were based on the pioneer biphenyl core proposed by Bristol Myers Squibb researchers (Zak et al., 2016). Recently, PD-1/PD-L1 inhibitory flavonoids have been identified through cell-based and in vitro studies (Li et al., 2019b; Choi et al., 2020; Kim et al., 2020; Jing et al., 2021). These natural product fragments bind with high affinity to both proteins (Choi et al., 2020) and represent an alternative to the biphenyl core and to previously discovered fragments. Due to their structural similarity with biphenyl compounds, flavonoid scaffold molecules are expected to bind primarily to the PD-L1 interface (Cheng et al., 2020; Choi et al., 2020; Kim et al., 2020). Still, no exhaustive binding mode studies have yet been conducted for these substances.
In this study, PD-1, PD-L1m, and PD-L1d were individually analyzed using unbiased computational methods such as SuMD and blind ensemble docking followed by brief cMD. We did not observe flavonoids’ propensity to interact with the PD-1 interface. Instead, we identified five other cavities in this protein that could accommodate small molecules. Combining the results from SuMD, docking, and cMD, we narrow that to Cavities 1, 3, and 5, with only Cavity 3 exhibiting ligand stability throughout extended replicates for one of the know PD-1 binders, Kaempferol-7-O-rhamnoside. However, it is known that this protein is heavily glycosylated, and the Asn58 was shown to significantly impact the PD-1/PD-L1 interaction (Sun et al., 2020). According to the crystal structure, the glucoside occludes cavity 3 (BC loop), which is characterized by residues Phe106, Phe82, Thr59, and Gln99. In an effort to determine if small molecules could modulate this pocket, we performed a simulation with Asn58-glycosylated PD-1, concluding that the glucoside is sufficiently flexible to reach a fully exposed conformation and reveal the pocket for small molecule interaction. The presence of flavonoids in this cavity was primarily stabilized by a hydrogen bond between the hydroxyphenyl group and Thr59 backbone, which can induce a flip of Phe56, thereby opening the cavity in the direction of the N-terminal residue Pro34. Thus, we propose that the flavonoid ligands could bind to cavity 3 and disturb the Asn58 glycosylation conformation, weakening the PD-1/PD-L1 complex.
PD-L1m was evaluated using a comparable procedure. Despite the identification of multiple potential cavities surrounding the protein, there was an evident preference for flavonoids to bind to the interface. Indeed, the only successfully explored pocket for designing small molecules for this pathway thus far is located at the PD-L1 dimerization interface. It is hypothesized that ligand interaction triggers the dimerization at the interface of monomeric PD-L1 (Zak et al., 2016; Shi et al., 2019). In spite of the strong interaction between the PD-L1 monomers in the absence of a ligand, which was also corroborated here by the maintenance of dimer during our 25 replicates of 50 ns MD using five PDB structures as the starting point, we observed for the first time the dimer dissociation event following ligand unbinding. Hence, we propose that the ligand exit disrupts essential interaction between dimer chains, resulting in the dissociation of the complex. Moreover, flavonoids appear to have a well-defined binding mode at the interface. The chromen-4-one moiety is stabilized via π-π stacking with Tyr23 residue and a hydrogen bond with Arg125 or Arg113.
The glucoside of cosmosiin and kaempferol-7-O-rhamnoside are located in close proximity to the mentioned arginine residues, causing a slight displacement of these molecules. Nevertheless, during simulations, kaempferol-3-7-dirhamnoside diverged, as expected for an inactive compound. Notably, PD-L1 dimerization generates a symmetric cavity (Yang and Hu, 2019), implying that ligands can interact in two possible orientations. Our simulations reveal a distinct orientation preference of flavonoids in PD-L1m, possibly explained by the difference in the chemical microenvironment. While in the dimer, there is a hydrophobic environment where the ligand can stay fully buried, in the monomer, the ligand strives to maximize its interactions, particularly the aromatic ones. The bulky chromen-4-one moiety stacks with Tyr123, while the hydroxyphenyl is accommodated by hydrophobic side chains. In the inverse orientation of ligand, chromen-4-one is not well stabilized by the hydrophobic cleft and diverges in most simulations.
The binding mechanism of the different flavonoids to PD-L1d is susceptible to substitutions in the flavonoid core, which directly drives the reported activity of these molecules. We suggest that dihydroxyphenyl (3′-OH) is only tolerated in the presence of 3-OH, which is consistent with the inactivity of luteolin (Choi et al., 2020). The findings of Choi et al. also support a similar interaction pattern for eriodyctiol and luteolin, except for the inability of the dihydrochroman-4-one to perform stable π-stacking with Tyr56. They further suggested that 7-O-glucosylation improves the binding of cosmosiin and homoplantagnin via polar interactions with residues Tyr56, Glu58, Asp61, and Asn63. Our simulation supported their hypothesis, showing that this group consistently interacts with Glu58, Asp122, Lys124, and Arg125.
In addition, we demonstrated that these interactions shifted the flavonoid toward the solvent, an effect that is mitigated by the stacking between chromen-4-one and Tyr56 in a binding mode remarkably similar to that reported in PD-L1m simulations. We also demonstrated that a fully buried flavonoid scaffold in PD-L1d simulations engages in simultaneous water-mediated interactions with Gln66 and Asp122 from both chains at the entrances of the dimer tunnel using the chromen-4-one and (di)hydroxyphenyl groups, as seen in the apigenin, fisetin, and quercetin simulations. Intriguingly, the reduction of the chromen-4-one in eriodictyol not only alters its conformation, but also hinders the occurrence of water-mediated bonds with dihydrochromen-4-one. Our simulations suggested it is more favorable for the flavonoid to remain fully buried in the dimer interface than to engage in stacking contact with the Tyr56, increasing the compound’s solvent exposure. Thus, a linker in position C7 between the glicoside moiety and the flavonoid scaffold could enhance the binding affinity of the compounds.
5 Conclusion
Here, we have described the possible binding mode of nine flavonoid molecules against the PD-1/PD-L1 pathway proteins. Our computational approach reinforced that these molecules could promote PD-L1 dimerization and extensively described their determinants for binding to the interface. Different substituents in the flavonoid scaffold do not seem to affect the binding to monomeric PD-L1 but could impair the formation of PD-L1d. Specifically, 5′-OH appears to have a deleterious effect on the interaction in the absence of 3-OH but is well-tolerated when such a group is present. The 3-OH group proved essential for stablishing an extra hydrogen bond interaction with the backbone of Ile116.
Additionally, the 7-O-glucoside significantly changes the proposed binding mode. In that case, the glucoside interacts with residues Glu58, Asp122, Lys124, and Arg125 and displaces the flavonoid scaffold, stabilized by stacking with Tyr56. Our simulations also showed the loss of π-stacking interaction when the chromen-4-one is replaced by dihydrochromen-4-one. Contrastingly, position 5 appears not to have a significant effect on binding. Nevertheless, we showed that despite flavonoids having a lower affinity toward the PD-1 interface, they could interact with residues Phe63 and Phe56 in the BC loop. Although Asn58 is a known PD-1 glycosylation point, we demonstrated that the glucoside has enough flexibility to expose the cavity, providing sufficient space for ligand binding. Flavonoids appear to have interesting stability in this cavity and could induce Phe56 flip to form a tunnel through the loop, enabling interactions with N-terminal residues. The detailed description of how flavonoids inhibit the formation of the PD-1/PD-L1 complex and the definition of a novel possible cavity for modulation of interaction in PD-1 protein could shed light on the development of novel flavonoid-based molecules to advance in the chemotherapy of cancer.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Materials, further inquiries can be directed to the corresponding author.
Author contributions
GRS and JHMS designed experiments. GRS, AHSC, AOA, LMA, EMG, JVS, DSA, VTMLA performed the experiments and analysed the results. JHMS, GRS, AOA, AHSC, LMA wrote the manuscript with contributions from EMG, JVS, DSA and VTMLA. All authors approved the final manuscript.
Acknowledgments
The authors gratefully acknowledge Dr. Ivan Rosa e Silva and Dr. Igor Muccilo Prokopczyk for the helpful discussion during manuscript writing. We also acknowledge the Amber developers for making the software available.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This project is supported by National Council for Scientific and Technological Development - CNPq under project Universal (grant number 431296/2018-9) and Cearense Foundation for the Support of Scientific and Technological Development (grant number FIO016700007010020).
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fddsv.2022.1032587/full#supplementary-material
References
Akinleye, A., and Rasool, Z. (2019). Immune checkpoint inhibitors of PD-L1 as cancer therapeutics. J. Hematol. Oncol. 12, 92. doi:10.1186/s13045-019-0779-5
Alhossary, A., Handoko, S. D., Mu, Y., and Kwoh, C. K. (2015). Fast, accurate, and reliable molecular docking with QuickVina 2. Bioinformatics 31, 2214–2216. doi:10.1093/BIOINFORMATICS/BTV082
Andrade, L., Albuquerque, A., Santos-Costa, A., Vasconcelos, D., Savino, W., Sartori, G. R., et al. (2022). Investigation of unprecedented sites and proposition of new ligands for programmed cell death protein i through molecular dynamics with probes and virtual screening. J. Chem. Inf. Model. 62, 1236–1248. doi:10.1021/ACS.JCIM.1C01122/SUPPL_FILE/CI1C01122_SI_001.PDF
Buchbinder, E. I., and Desai, A. (2016). CTLA-4 and PD-1 pathways: Similarities, differences, and implications of their inhibition. Am. J. Clin. Oncol. 39, 98–106. doi:10.1097/COC.0000000000000239
Case, D., Cheatham, T., Merz, K., Luo, R., Roitberg, A., and Walker, R. (2005). The amber molecular dynamics package.
Chamoto, K., Hatae, R., and Honjo, T. (2020). Current issues and perspectives in PD-1 blockade cancer immunotherapy. Int. J. Clin. Oncol. 25, 790–800. doi:10.1007/s10147-019-01588-7
Cheng, B., Ren, Y., Niu, X., Wang, W., Wang, S., Tu, Y., et al. (2020). Discovery of novel resorcinol dibenzyl ethers targeting the programmed cell death-1/programmed cell death-ligand 1 interaction as potential anticancer agents. J. Med. Chem. 63, 8338–8358. doi:10.1021/ACS.JMEDCHEM.0C00574/SUPPL_FILE/JM0C00574_SI_003.CSV
Choi, J. G., Kim, Y. S., Kim, J. H., Kim, T. I., Li, W., Oh, T. W., et al. (2020). Anticancer effect of salvia plebeia and its active compound by improving T-cell activity via blockade of PD-1/PD-L1 interaction in humanized PD-1 mouse model. Front. Immunol. 11, 598556. doi:10.3389/fimmu.2020.598556
Coombs, M. R. P., Harrison, M. E., and Hoskin, D. W. (2016). Apigenin inhibits the inducible expression of programmed death ligand 1 by human and mouse mammary carcinoma cells. Cancer Lett. 380, 424–433. doi:10.1016/j.canlet.2016.06.023
Dermani, F. K., Samadi, P., Rahmani, G., Kohlan, A. K., and Najafi, R. (2019). PD-1/PD-L1 immune checkpoint: Potential target for cancer therapy. J. Cell. Physiol. 234, 1313–1325. doi:10.1002/jcp.27172
Dolinsky, T. J., Czodrowski, P., Li, H., Nielsen, J. E., Jensen, J. H., Klebe, G., et al. (2007). PDB2PQR: Expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 35, W522–W525. doi:10.1093/nar/gkm276
Feenstra, K. A., Hess, B., and Berendsen, H. J. C. (1999). Improving efficiency of large time-scale molecular dynamics simulations of hydrogen-rich systems. J. Comput. Chem. 20, 786–798. doi:10.1002/(SICI)1096-987X(199906)20:8<786:AID-JCC5>3.0.CO;2-B
Freeman, G. J. (2008). Structures of PD-1 with its ligands: Sideways and dancing cheek to cheek. Proc. Natl. Acad. Sci. U. S. A. 105, 10275–10276. doi:10.1073/PNAS.0805459105/ASSET/28D27112-4D16-4E9A-86D2-F67C9FA4DD84/ASSETS/GRAPHIC/ZPQ9990844480001.JPEG
{kind=link}
Gevensleben, H., Dietrich, D., Golletz, C., Steiner, S., Jung, M., Thiesler, T., et al. (2016). The immune checkpoint regulator PD-L1 is highly expressed in aggressive primary prostate cancer. Clin. Cancer Res. 22, 1969–1977. –. doi:10.1158/1078-0432.CCR-15-2042
Guo, Y., Liang, J., Liu, B., and Jin, Y. (2021). Molecular mechanism of food-derived polyphenols on PD-L1 dimerization: A molecular dynamics simulation study. Int. J. Mol. Sci. 22, 10924. doi:10.3390/ijms222010924
Han, Y., Liu, D., and Li, L. (2020). PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 10, 727–742. Available at: /pmc/articles/PMC7136921/(Accessed August 29, 2022).
Hopkins, C. W., Le Grand, S., Walker, R. C., and Roitberg, A. E. (2015). Long-time-step molecular dynamics through hydrogen mass repartitioning. J. Chem. Theory Comput. 11, 1864–1874. doi:10.1021/CT5010406/ASSET/IMAGES/LARGE/CT-2014-010406_0017.JPEG
{kind=link}
Hwang, B. J., Tsao, L. C., Acharya, C. R., Trotter, T., Agarwal, P., Wei, J., et al. (2022). Sensitizing immune unresponsive colorectal cancers to immune checkpoint inhibitors through MAVS overexpression. J. Immunother. Cancer 10, e003721. doi:10.1136/JITC-2021-003721
Inman, B. A., Longo, T. A., Ramalingam, S., and Harrison, M. R. (2017). Atezolizumab: A PD-L1-blocking antibody for bladder cancer. Clin. Cancer Res. 23, 1886–1890. doi:10.1158/1078-0432.CCR-16-1417
Jiang, N., Dai, Q., Su, X., Fu, J., Feng, X., and Peng, J. (2020). Role of PI3K/AKT pathway in cancer: The framework of malignant behavior. Mol. Biol. Rep. 47, 4587–4629. doi:10.1007/s11033-020-05435-1
Jing, L., Lin, J., Yang, Y., Tao, L., Li, Y., Liu, Z., et al. (2021). Quercetin inhibiting the PD-1/PD-L1 interaction for immune-enhancing cancer chemopreventive agent. Phytother. Res. 35, 6441–6451. doi:10.1002/PTR.7297
Jo, S., Kim, T., Iyer, V. G., and Im, W. (2008). CHARMM-GUI: A web-based graphical user interface for charmm. J. Comput. Chem. 29, 1859–1865. doi:10.1002/JCC.20945
Ke, M., Zhang, Z., Xu, B., Zhao, S., Ding, Y., Wu, X., et al. (2019). Baicalein and baicalin promote antitumor immunity by suppressing PD-L1 expression in hepatocellular carcinoma cells. Int. Immunopharmacol. 75, 105824. doi:10.1016/j.intimp.2019.105824
Kim, J. H., Kim, Y. S., Choi, J. G., Li, W., Lee, E. J., Park, J. W., et al. (2020). Kaempferol and its glycoside, kaempferol 7-O-rhamnoside, inhibit PD-1/PD-L1 interaction in vitro. Int. J. Mol. Sci. 21, 3239. doi:10.3390/IJMS21093239
Kim, K. H., Kim, H. K., Kim, H. D., Kim, C. G., Lee, H., Han, J. W., et al. (2021). PD-1 blockade-unresponsive human tumor-infiltrating CD8 + T cells are marked by loss of CD28 expression and rescued by IL-15. Cell. Mol. Immunol. 18, 385–397. doi:10.1038/S41423-020-0427-6
Kim, S., Thiessen, P. A., Bolton, E. E., Chen, J., Fu, G., Gindulyte, A., et al. (2016). PubChem substance and compound databases. Nucleic Acids Res. 44, D1202–D1213. doi:10.1093/NAR/GKV951
Kirschner, K. N., Yongye, A. B., Tschampel, S. M., González-Outeiriño, J., Daniels, C. R., Foley, B. L., et al. (2008). GLYCAM06: A generalizable biomolecular force field. Carbohydrates. J. Comput. Chem. 29, 622–655. doi:10.1002/JCC.20820
Koblish, H. K., Wu, L., Wang, L. C. S., Liu, P. C. C., Wynn, R., Rios-Doria, J., et al. (2022). Characterization of INCB086550: A potent and novel small-molecule PD-L1 inhibitor. Cancer Discov. A-POTENT-AND-NOVEL 12, 1482–1499. doi:10.1158/2159-8290.CD-21-1156
Konieczny, M., Musielak, B., Kocik, J., Skalniak, L., Sala, D., Czub, M., et al. (2020). Di-bromo-Based small-molecule inhibitors of the PD-1/PD-L1 immune checkpoint. J. Med. Chem. 63, 11271–11285. doi:10.1021/acs.jmedchem.0c01260
Kopustinskiene, D. M., Jakstas, V., Savickas, A., and Bernatoniene, J. (2020). Flavonoids as anticancer agents. Nutrients 12, 457. doi:10.3390/nu12020457
Krueger, J., Jules, F., Rieder, S. A., and Rudd, C. E. (2017). CD28 family of receptors inter-connect in the regulation of T-cells. Recept. Clin. Investig. 4, e1581. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6753945/.
Kumar, S., Ghosh, S., Sharma, G., Wang, Z., Kehry, M. R., Marino, M. H., et al. (2021). Preclinical characterization of dostarlimab, a therapeutic anti-PD-1 antibody with potent activity to enhance immune function in in vitro cellular assays and in vivo animal models. MAbs 13, 1954136. doi:10.1080/19420862.2021.1954136
Lee, J., Han, Y., Wang, W., Jo, H., Kim, H., Kim, S., et al. (2021). Phytochemicals in cancer immune checkpoint inhibitor therapy. Biomolecules 11, 1107. doi:10.3390/biom11081107
Li, W., Kim, T. I., Kim, J. H., and Chung, H.-S. (2019a). Immune checkpoint PD-1/PD-L1 CTLA-4/CD80 are blocked by rhus verniciflua Stokes and its active compounds. Molecules 24, E4062. doi:10.3390/molecules24224062
Li, W., Kim, T. I., Kim, J. H., and Chung, H. S. (2019b). Immune checkpoint PD-1/PD-L1 CTLA-4/CD80 are blocked by rhus verniciflua Stokes and its active compounds. Molecules 24, 4062. doi:10.3390/MOLECULES24224062
Lin, X., Lu, X., Luo, G., and Xiang, H. (2020). Progress in PD-1/PD-L1 pathway inhibitors: From biomacromolecules to small molecules. Eur. J. Med. Chem. 186, 111876. doi:10.1016/j.ejmech.2019.111876
Liu, C., Seeram, N. P., and Ma, H. (2021a). Small molecule inhibitors against PD-1/PD-L1 immune checkpoints and current methodologies for their development: A review. Cancer Cell. Int. 21, 239. doi:10.1186/s12935-021-01946-4
Liu, Y., Zhou, X., Luo, L., Zhong, A., Wang, Q., and Zheng, T. (2021b2022). Structure-based pharmacophore modeling, virtual screening, molecular docking, ADMET, and molecular dynamics (MD) simulation of potential inhibitors of PD-L1 from the library of marine natural products. Mar. Drugs 20, 29. doi:10.3390/MD20010029
Lu, H., Zhou, Q., He, J., Jiang, Z., Peng, C., Tong, R., et al. (2020). Recent advances in the development of protein–protein interactions modulators: Mechanisms and clinical trials. Signal Transduct. Target. Ther. 51 (5), 213–223. doi:10.1038/s41392-020-00315-3
Machiraju, D., Schäfer, S., and Hassel, J. C. (2021). Potential reasons for unresponsiveness to anti-PD-1 immunotherapy in young patients with advanced melanoma. Life 11, 1318. doi:10.3390/LIFE11121318
Martino, E., Chiarugi, S., Margheriti, F., and Garau, G. (2021). Mapping, structure and modulation of PPI. Front. Chem. 9, 843. doi:10.3389/fchem.2021.718405
Mazewski, C., Kim, M. S., and Gonzalez de Mejia, E. (2019). Anthocyanins, delphinidin-3-O-glucoside and cyanidin-3-O-glucoside, inhibit immune checkpoints in human colorectal cancer cells in vitro and in silico. Sci. Rep. 9, 11560. doi:10.1038/s41598-019-47903-0
Mejías, C., and Guirola, O. (2019). Pharmacophore model of immunocheckpoint protein PD-L1 by cosolvent molecular dynamics simulations. J. Mol. Graph. Model. 91, 105–111. doi:10.1016/j.jmgm.2019.06.001
Morris, G. M., Ruth, H., Lindstrom, W., Sanner, M. F., Belew, R. K., Goodsell, D. S., et al. (2009). AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791. doi:10.1002/JCC.21256
Naidoo, J., Page, D. B., Li, B. T., Connell, L. C., Schindler, K., Lacouture, M. E., et al. (2015). Toxicities of the anti-PD-1 and anti-PD-L1 immune checkpoint antibodies. Ann. Oncol. 26, 2375–2391. doi:10.1093/annonc/mdv383
Nakanishi, J., Wada, Y., Matsumoto, K., Azuma, M., Kikuchi, K., and Ueda, S. (2007). Overexpression of B7-H1 (PD-L1) significantly associates with tumor grade and postoperative prognosis in human urothelial cancers. Cancer Immunol. Immunother. 56, 1173–1182. doi:10.1007/S00262-006-0266-Z
O’Boyle, N. M., Banck, M., James, C. A., Morley, C., Vandermeersch, T., and Hutchison, G. R. (2011). Open Babel: An open chemical toolbox. J. Cheminform. 3, 33. doi:10.1186/1758-2946-3-33
Olsson, M. H. M., Søndergaard, C. R., Rostkowski, M., and Jensen, J. H. (2011). PROPKA3: Consistent treatment of internal and surface residues in empirical p K a predictions. J. Chem. Theory Comput. 7, 525–537. doi:10.1021/ct100578z
O’Sullivan Coyne, G., Madan, R. A., and Gulley, J. L. (2014). Nivolumab: Promising survival signal coupled with limited toxicity raises expectations. J. Clin. Oncol. 32, 986–988. doi:10.1200/JCO.2013.54.5996
Patsoukis, N., Wang, Q., Strauss, L., and Boussiotis, V. A. (2020). Revisiting the PD-1 pathway. Sci. Adv. 6, eabd2712. doi:10.1126/sciadv.abd2712
Piha-Paul, S., Mitchell, T., Sahebjam, S., Mehnert, J., Karasic, T., O’Hayer, K., et al. (2020). 419 Pharmacodynamic biomarkers demonstrate T-cell activation in patients treated with the oral PD-L1 inhibitor INCB086550 in a phase 1 clinical trial. J. Immunother. Cancer 8. doi:10.1136/jitc-2020-SITC2020.0419
Rice-Evans, C. A., Miller, N. J., and Paganga, G. (1996). Structure-antioxidant activity relationships of flavonoids and phenolic acids. Free Radic. Biol. Med. 20, 933–956. doi:10.1016/0891-5849(95)02227-9
Sabbadin, D., and Moro, S. (2014). Supervised molecular dynamics (SuMD) as a helpful tool to depict GPCR-ligand recognition pathway in a nanosecond time scale. J. Chem. Inf. Model. 54, 372–376. doi:10.1021/CI400766B/SUPPL_FILE/CI400766B_SI_010.MPG
Sasikumar, P. G., and Ramachandra, M. (2022). Small molecule agents targeting PD-1 checkpoint pathway for cancer immunotherapy: Mechanisms of action and other considerations for their advanced development. Front. Immunol. 13, 752065. doi:10.3389/fimmu.2022.752065
Sasikumar, P. G., Sudarshan, N. S., Adurthi, S., Ramachandra, R. K., Samiulla, D. S., Lakshminarasimhan, A., et al. (2021). PD-1 derived CA-170 is an oral immune checkpoint inhibitor that exhibits preclinical anti-tumor efficacy. Commun. Biol. 4, 699. doi:10.1038/s42003-021-02191-1
Seitz, C., Casalino, L., Konecny, R., Huber, G., Amaro, R. E., and McCammon, J. A. (2020). Multiscale simulations examining glycan shield effects on drug binding to influenza neuraminidase. Biophys. J. 119, 2275–2289. doi:10.1016/J.BPJ.2020.10.024/ATTACHMENT/368B2112-0B8E-4864-B1FB-BFFFA1ACE6A7/MMC1.PDF
Shi, D., An, X., Bai, Q., Bing, Z., Zhou, S., Liu, H., et al. (2019). Computational insight into the small molecule intervening PD-L1 dimerization and the potential structure-activity relationship. Front. Chem. 7, 764. doi:10.3389/fchem.2019.00764
Shirts, M. R., Klein, C., Swails, J. M., Yin, J., Gilson, M. K., Mobley, D. L., et al. (2017). Lessons learned from comparing molecular dynamics engines on the SAMPL5 dataset. J. Comput. Aided. Mol. Des. 31, 147–161. doi:10.1007/s10822-016-9977-1
Sidaway, P. (2016). Skin cancer: Avelumab effective against Merkel-cell carcinoma. Nat. Rev. Clin. Oncol. 13, 652. doi:10.1038/nrclinonc.2016.156
Sun, L., Li, C. W., Chung, E. M., Yang, R., Kim, Y. S., Park, A. H., et al. (2020). PD-1-INDUCES-POTENT-ANTI, 80, 2298–2310. doi:10.1158/0008-5472.CAN-19-3133/653933/AM/TARGETING-GLYCOSYLATED-Targeting glycosylated PD-1 induces potent antitumor immunityCancer Res.
Sun, Y., Jiang, L., Wen, T., Guo, X., Shao, X., Qu, H., et al. (2021). Trends in the research into immune checkpoint blockade by anti-PD-1/PDL1 antibodies in cancer immunotherapy: A bibliometric study. Front. Pharmacol. 12, 670900. doi:10.3389/fphar.2021.670900
Takada, K., Toyokawa, G., Okamoto, T., Shimokawa, M., Kozuma, Y., Matsubara, T., et al. (2017). A comprehensive analysis of programmed cell death ligand-1 expression with the clone SP142 antibody in non-small-cell lung cancer patients. Clin. Lung Cancer 18, 572–582. doi:10.1016/J.CLLC.2017.02.004
Tian, C., Kasavajhala, K., Belfon, K. A. A., Raguette, L., Huang, H., Migues, A. N., et al. (2020). Ff19SB: Amino-Acid-Specific protein backbone parameters trained against quantum mechanics energy surfaces in solution. J. Chem. Theory Comput. 16, 528–552. doi:10.1021/ACS.JCTC.9B00591/SUPPL_FILE/CT9B00591_SI_002.ZIP
Upadhaya, S., Neftelinov, S. T., Hodge, J., and Campbell, J. (2022). Challenges and opportunities in the PD-1/PDL1 inhibitor clinical trial landscape. Nat. Rev. Drug Discov. 21, 482–483. doi:10.1038/D41573-022-00030-4
Urban, V. A., Nazarenko, P. S., Perepechko, S. A., and Veresov, V. G. (2022). Using PD-L1 full-length structure, enhanced induced fit docking and molecular dynamics simulations for structural insights into inhibition of PD-1/PD-L1 interaction by small-molecule ligands. Mol. Simul. 0, 1269–1283. doi:10.1080/08927022.2022.2080824
Vanquelef, E., Simon, S., Marquant, G., Garcia, E., Klimerak, G., Delepine, J. C., et al. (2011). R.E.D. Server: A web service for deriving RESP and ESP charges and building force field libraries for new molecules and molecular fragments. Nucleic Acids Res. 39, W511–W517. doi:10.1093/nar/gkr288
Volkamer, A., Kuhn, D., Grombacher, T., Rippmann, F., and Rarey, M. (2012). Combining global and local measures for structure-based druggability predictions. J. Chem. Inf. Model. 52, 360–372. doi:10.1021/ci200454v
Vuorio, J., Škerlová, J., Fábry, M., Veverka, V., Vattulainen, I., Řezáčová, P., et al. (2021). N-Glycosylation can selectively block or foster different receptor–ligand binding modes. Sci. Rep. 111 (11), 5239. doi:10.1038/s41598-021-84569-z
Wang, M., Wang, J., Wang, R., Jiao, S., Wang, S., Zhang, J., et al. (2019a). Identification of a monoclonal antibody that targets PD-1 in a manner requiring PD-1 Asn58 glycosylation. Commun. Biol. 21 2, 392. doi:10.1038/s42003-019-0642-9
Wang, Y., Gu, T., Tian, X., Li, W., Zhao, R., Yang, W., et al. (2021). A small molecule antagonist of PD-1/PD-L1 interactions acts as an immune checkpoint inhibitor for NSCLC and melanoma immunotherapy. Front. Immunol. 12, 654463. doi:10.3389/fimmu.2021.654463
Wang, Y., Guo, H., Feng, Z., Wang, S., Wang, Y., He, Q., et al. (2019b). PD-1-Targeted Discovery of peptide inhibitors by virtual screening, molecular dynamics simulation, and surface plasmon resonance. Molecules 24, 3784. doi:10.3390/molecules24203784
Wu, Q., Jiang, L., Li, S., He, Q., Yang, B., and Cao, J. (2021). Small molecule inhibitors targeting the PD-1/PD-L1 signaling pathway. Acta Pharmacol. Sin. 42, 1–9. doi:10.1038/s41401-020-0366-x
Xue, C., Li, G., Zheng, Q., Gu, X., Bao, Z., Lu, J., et al. (2022). The functional roles of the circRNA/Wnt axis in cancer. Mol. Cancer 21, 108. doi:10.1186/s12943-022-01582-0
Yang, J., and Hu, L. (2019). Immunomodulators targeting the PD-1/PD-L1 protein-protein interaction: From antibodies to small molecules. Med. Res. Rev. 39, 265–301. doi:10.1002/med.21530
Zak, K. M., Grudnik, P., Guzik, K., Zieba, B. J., Musielak, B., Dömling, A., et al. (2016). Structural basis for small molecule targeting of the programmed death ligand 1 (PD-L1). Oncotarget 7, 30323–30335. doi:10.18632/ONCOTARGET.8730
Keywords: flavonoids, PD-1, PD-L1, structure-activity relationship, binding mode, molecular dynamics simulation, glycosylation
Citation: Sartori GR, Albuquerque AdO, Santos-Costa AH, Andrade LM, Almeida DdS, Gaieta EM, Sampaio JV, Albuquerque VTdML and Martins Da Silva JH (2022) In silico mapping of the dynamic interactions and structure-activity relationship of flavonoid compounds against the immune checkpoint programmed-cell death 1 pathway. Front. Drug. Discov. 2:1032587. doi: 10.3389/fddsv.2022.1032587
Received: 31 August 2022; Accepted: 25 November 2022;
Published: 12 December 2022.
Edited by:
Carmen Cerchia, University of Naples Federico II, ItalyReviewed by:
Sangwook Wu, Pukyong National University, South KoreaJulien Diharce, Université de Paris, France
Copyright © 2022 Sartori, Albuquerque, Santos-Costa, Andrade, Almeida, Gaieta, Sampaio, Albuquerque and Martins da Silva. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: João Hermínio Martins Da Silva, joao.martins@fiocruz.br