Clinical Use of Hydrogen Sulfide to Protect Against Intimal Hyperplasia
 Diane Macabrey1,2†
Diane Macabrey1,2†  Florent Allagnat
Florent Allagnat- 1Department of Vascular Surgery, Lausanne University Hospital, Lausanne, Switzerland
- 2Department of Biomedical Sciences, University of Lausanne, Lausanne, Switzerland
Arterial occlusive disease is the narrowing of the arteries via atherosclerotic plaque buildup. The major risk factors for arterial occlusive disease are age, high levels of cholesterol and triglycerides, diabetes, high blood pressure, and smoking. Arterial occlusive disease is the leading cause of death in Western countries. Patients who suffer from arterial occlusive disease develop peripheral arterial disease (PAD) when the narrowing affects limbs, stroke when the narrowing affects carotid arteries, and heart disease when the narrowing affects coronary arteries. When lifestyle interventions (exercise, diet…) fail, the only solution remains surgical endovascular and open revascularization. Unfortunately, these surgeries still suffer from high failure rates due to re-occlusive vascular wall adaptations, which is largely due to intimal hyperplasia (IH). IH develops in response to vessel injury, leading to inflammation, vascular smooth muscle cells dedifferentiation, migration, proliferation and secretion of extra-cellular matrix into the vessel’s innermost layer or intima. Re-occlusive IH lesions result in costly and complex recurrent end-organ ischemia, and often lead to loss of limb, brain function, or life. Despite decades of IH research, limited therapies are currently available. Hydrogen sulfide (H2S) is an endogenous gasotransmitter derived from cysteine metabolism. Although environmental exposure to exogenous high H2S is toxic, endogenous H2S has important vasorelaxant, cytoprotective and anti-inflammatory properties. Its vasculo-protective properties have attracted a remarkable amount of attention, especially its ability to inhibit IH. This review summarizes IH pathophysiology and treatment, and provides an overview of the potential clinical role of H2S to prevent IH and restenosis.
Introduction
Prevalence of arterial occlusive disease continues to rise worldwide, largely due to the combination of aging, smoking, hypertension, and mostly diabetes mellitus (1–3).
Vascular surgery, open or endovascular, remains the only treatment for advanced arterial occlusive disease. However, the vascular trauma associated with the intervention eventually lead to secondary occlusion of the injured vessel, usually referred to as restenosis.
The overall incidence of restenosis varies greatly depending on the initial clinical presentation and the anatomic pattern of disease (e.g., coronary vs. femoro-popliteal vs. infra popliteal etc.). Overall, for open surgeries such as bypass and endarterectomy, the rate of restenosis after 1 year ranges between 20 and 30% (4). For endovascular approaches, the rate of restenosis following plain old balloon angioplasty (POBA) ranges from 30 to 60%, depending on location (5). In coronary arteries, the use of bare metal stents (BMS) lowered the rate of restenosis to 17–41% (5). In stented peripheral arteries, restenosis occurs in up to 51% of the patients 1 year after the surgery (6).
The most recent advances in the treatment of restenosis rely on the use of drug-coated balloons (DCB) and drug-eluting stents (DES), which nowadays represent a first line therapy in many endovascular approaches to treat short lesions in coronary or femoral arteries. The most used drug is the anti-tumor chemotherapy Paclitaxel (Taxol™). Several paclitaxel-coated balloons and eluting stents with various formulations and different dose of paclitaxel demonstrated superiority to POBA (7–9) or BMS (7, 10). Overall, the arrival of DES and DCB reduced the incidence of restenosis below 10% in coronary arteries (11), although restenosis has been delayed rather than suppressed (12). DES also require prolonged antiplatelet therapy and hinder future surgical revascularization. In peripheral below the knee small arteries, the use of DCB is controversial, and stents are not recommended due to the risk of thrombosis (13). In December 2018, Katsanos and colleagues reported, in a systematic review and meta-analysis, an increased risk of all-cause mortality following application of paclitaxel−coated balloons and stents in the femoropopliteal artery (14). Other groups recently confirmed these findings using the same data (15, 16). However, other meta-analyses did not find any association between paclitaxel devices and long-term survival, despite similar target populations and vessel segments (17–21). These reports questioned the widespread use of paclitaxel for the treatment of restenosis (22), and supports the need to develop other approaches or use other molecules. In coronary intervention Sirolimus is increasingly used (23), and new devices are under evaluation to validate the use of sirolimus-coated devices in below the knee peripheral arteries (24). Recent studies even report the safety and efficacy of biodegradable polymer sirolimus-eluting stent (25, 26).
Restenosis has various origins, such as secondary growth of atherosclerotic lesions or inward remodeling. However, it is due mostly to intimal hyperplasia (IH), a process whereby a “neointima” layer is formed between the internal elastic lamina (IEL) and the endothelium (Figure 1). IH is a known complication of all types of vascular procedures, including arterial bypass, angioplasty, stenting, and endarterectomy. Stenosis due to IH is also a major limitation of arteriovenous fistulas for hemodialysis patients, arteriovenous grafts, and other vascular accesses. The progressive growth of the neointima layer causes both an outward and an inward remodeling of the vessel wall, leading to a narrowing of the lumen, and eventually leads to impaired perfusion of downstream organs.
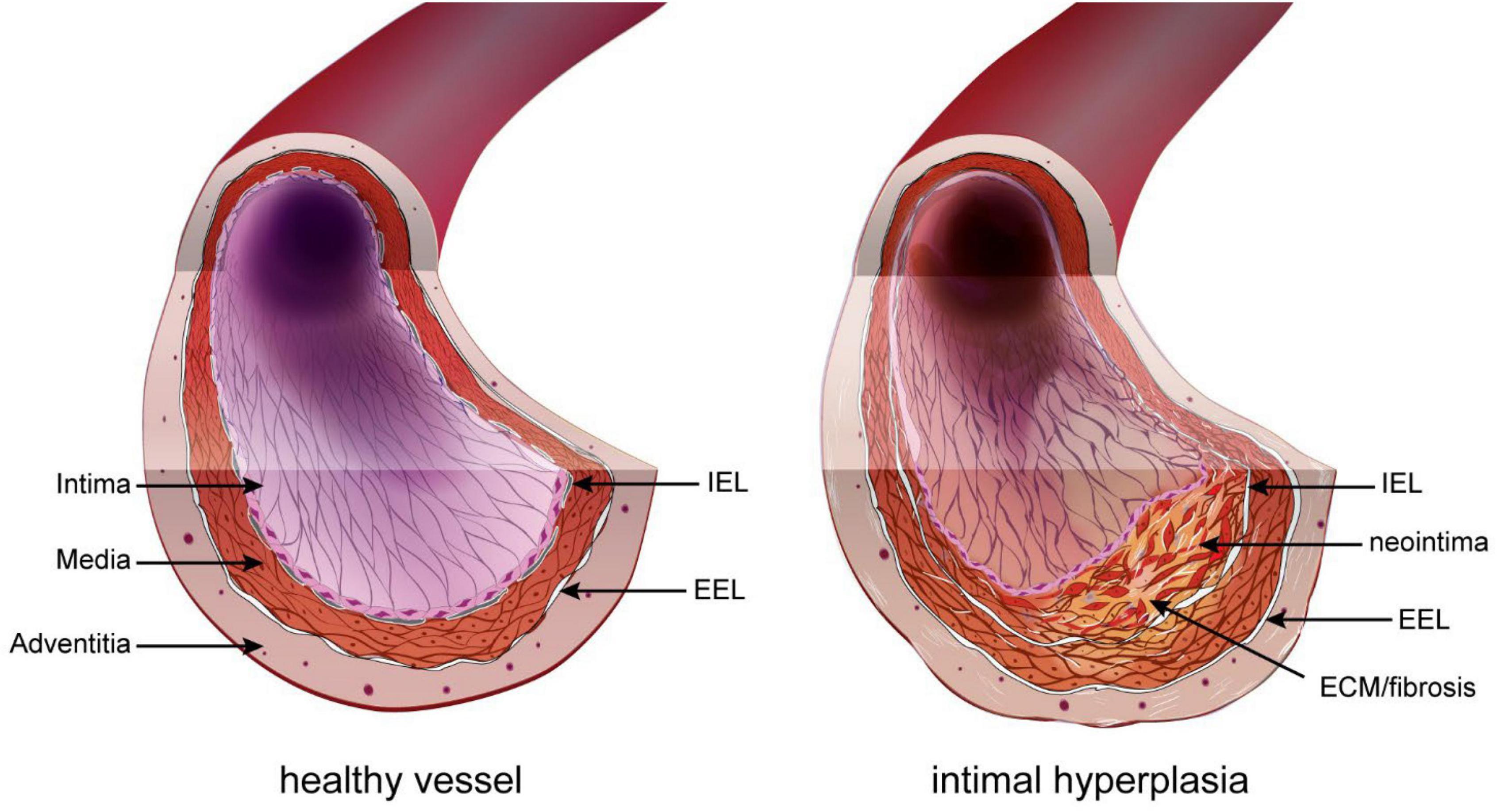
Figure 1. Intimal hyperplasia. In the healthy vessel (left), the inner intimal layer is composed only of the endothelium (pink layer). Under the endothelium sits the basement layer and internal elastic lamina (IEL). The media layer is composed of elastic fibers and smooth muscle cells (SMC). The outer part of the media and adventitia are separated by the exterior elastic lamina (EEL). Restenosis following vascular surgery is the formation of a neointima layer between the IEL and endothelium. It is composed of proliferating SMC-like cells of different origin and extracellular matrix (ECM).
All current strategies to limit IH such as Paclitaxel and Sirolimus target cell proliferation. Paclitaxel is a chemotherapeutical agent that stabilizes microtubules, thereby preventing cell division (mitosis). High dose or prolonged exposure to paclitaxel may also lead to apoptotic cell death (27). Sirolimus inhibits the mammalian target of rapamycin (mTOR), a master regulator of cell growth and metabolism (28). However, targeting cell proliferation to reduce IH also impairs re-endothelization. Endothelium repair is crucial to limit inflammation, remodeling and IH. Poor endothelial repair also prolongs the need for anti-thrombotic therapies. Therefore, there is a need for new strategies to inhibit IH while promoting endothelium recovery. In that regard, the gasotransmitter hydrogen sulfide (H2S) possesses interesting properties.
H2S is a gasotransmitter derived from cysteine metabolism (29). Circulating H2S levels are reduced in humans suffering from vascular occlusive disease (30, 31) and pre-clinical studies using water-soluble sulfide salts such as Na2S and NaHS have shown that H2S has cardiovascular protective properties [reviewed in Zhang et al. (29)], including reduction of IH in various models (32–35).
In this review, we present the pathophysiology of IH and the clinical potential of H2S against IH. The pleiotropic benefits of H2S on the cardiovascular system are described, and the interesting possibilities to target the multifactorial process leading to IH using H2S are discussed.
The Pathophysiology of Intimal Hyperplasia
The basic structure of large vessels (vein and artery) include three concentric layers: intima, media, and adventitia. The intima layer, also called endothelium, is the inner section of the vessel and is made of a single layer of endothelial cells (EC). The media is composed primarily of vascular smooth muscle cells (VSMC) and connective tissue made of collagen, elastin, and proteoglycans. The outermost adventitial layer is composed primarily of collagen and fibroblasts. In arteries, the intima and media layers are separated by a layer of elastic fibers called the internal elastic lamina (IEL), while the media and adventitia layers are separated by a second layer of elastic fibers called the external elastic lamina (EEL) (36). IH, also called neointima, develops between the intima and the IEL. The IH process is triggered in response to the injury to the blood vessel during surgery (37). This new layer is made of SMC-like cells and proteoglycan-rich extracellular matrix (ECM) (Figure 1).
Endothelial Dysfunction or Lesion
Located at the contact between the blood and the vessel wall, the EC maintain a non-thrombogenic surface and regulate the vasomotor activity (vasodilation and vasoconstriction) of vessels. In arteries, EC require high laminar shear stress to maintain proper function, i.e., secrete anti-coagulation and vasodilation agents, mainly nitric oxide (NO) and prostacyclins (36). However, the hemodynamic forces are not uniform throughout the vascular system. In straight segments of arteries, blood flow is laminar and shear stress is high. However, at bifurcations, curvatures, or other regions with complex geometry, blood flow is disturbed and turbulent. These abnormal patterns of “low” shear stress induce “endothelial dysfunction” or “endothelium activation.” Because of these disturbed arterial flow patterns, nearly all humans develop benign IH, also referred to as diffuse intimal thickening, around vessel bifurcations or in curved sections of arteries. This type of lesion serves as a precursor for the development of atherosclerosis by facilitating local inflammatory reaction and entrapment of LDL in the vessel wall (38). Inevitably, these “weak” spots of the vascular system are the sites of primary occlusion by atherosclerotic plaques that require vascular interventions. Any vascular surgery destroys the endothelial layer, furthering endothelium damage on those existing weak spots. This is the case for balloon angioplasty, stenting, or endarterectomy, which directly target the site of the atherosclerotic plaques. In the case of bypass surgery and arteriovenous fistulas, the surgery damages the endothelium while creating new regions of disturbed arterial flow patterns, which will foster IH lesions (Figure 2).
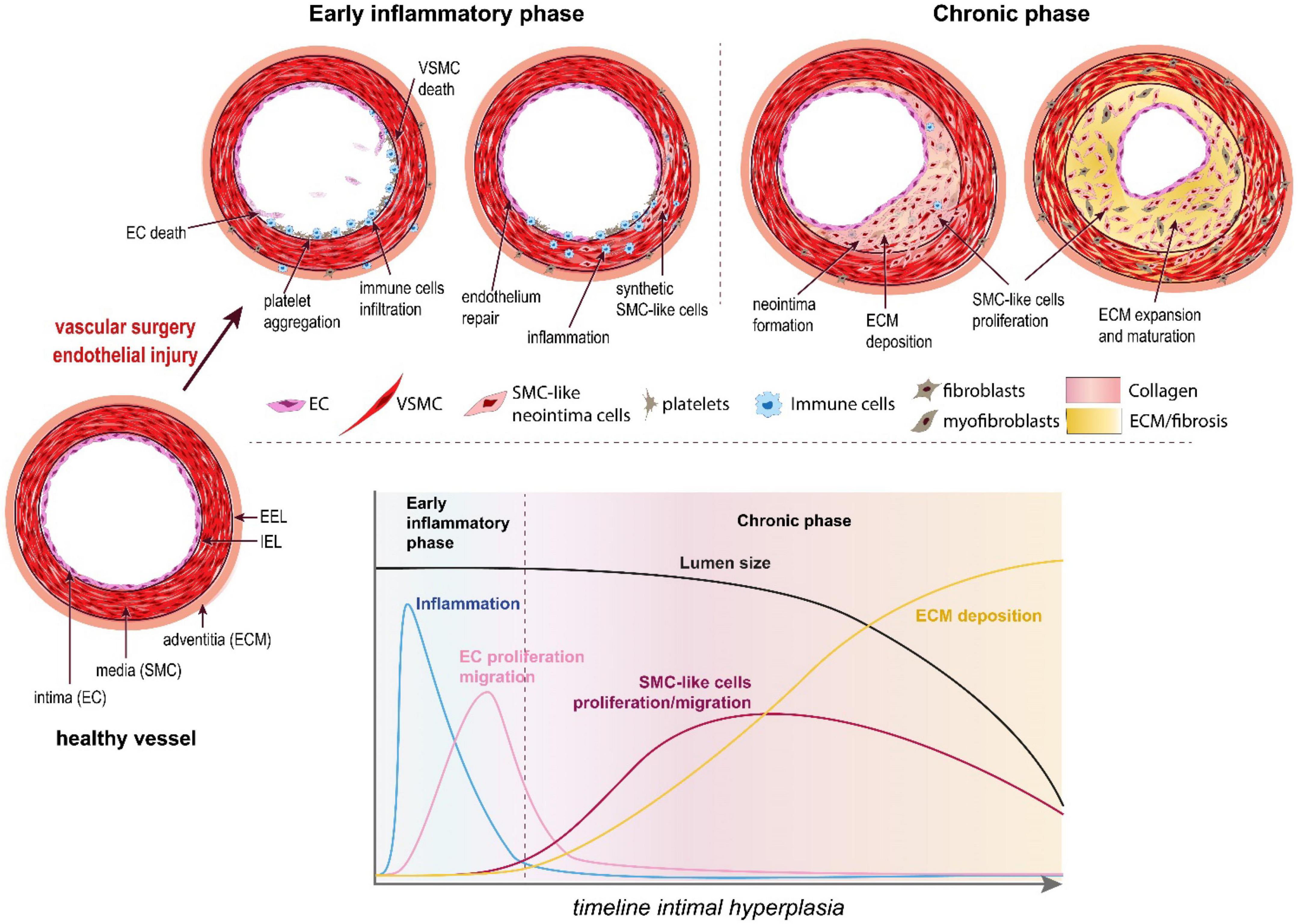
Figure 2. The pathophysiology of intimal hyperplasia. IH is triggered by endothelial injury, which activates platelets aggregation, and recruitment and activation of immune cells in the arterial wall (early inflammatory phase). The platelets and immune cells release cytokines, chemokines and growth factors, which stimulate a wound healing response mediated by SMC-like cells (mostly synthetic SMC derived from medial VSMC and myofibroblasts derived from adventitial fibroblasts). These synthetic SMC-like cells proliferate and migrate under the internal elastic lamina (IEL), forming the neointima layer. Long after inflammation is resolved and the endothelium is repaired (chronic phase), these cells continue to secrete extracellular matrix (ECM), leading to the progressive narrowing of the lumen.
Endothelial dysfunction or injury following surgery results in loss of eNOS, the enzyme producing nitric oxide (NO), a gasotransmitter maintaining healthy vessel function. Reduced NO production promotes vasoconstriction, platelet aggregation and recruitment/activation of resident and circulating inflammatory cells (mostly macrophages). The activated EC, recruited platelets and immune cells secrete cytokines and chemokines, which trigger a pro-inflammatory response (Figure 2). In addition, these cells secrete growth factors, including platelet-derived growth factor (PDGF), basic fibroblast growth factor (bFGF), transforming growth factor beta 1 (TGF-β) and thromboxane A2. Downstream of those multiple growth factors, cytokines and chemokines, the Mitogen-activated protein kinase (MAPK) pathway, including extracellular signal-regulated kinase (ERK), c-Jun-N-terminal kinase (JNK) and p38 mitogen-activated protein kinases, plays a major role in VSMC migration and proliferation in SMCs and fibroblasts. Other signals derived from oxidative stress also regulate the p38 MAPK and JNK pathways (39, 40).
Together, the secretion of these factors and the loss of NO promote vessel remodeling and reprogramming of cells composing the media and adventitia layers, leading to the formation of a neointima between the endothelium and the IEL (41) (Figures 1, 2).
The Origin of Neointimal Cells
IH is mostly formed by proliferating VSMC originating from dedifferentiated contractile medial VSMC. Unlike other terminally differentiated cells of the myogenic lineage, such as cardiac and skeletal muscle cells, adult VSMC are highly plastic and capable of phenotypic alterations in response to their environment. Modulation of VSMC from a quiescent “contractile” phenotype to a proliferative “synthetic” phenotype is important for vascular injury repair, but is also a key factor in the pathogenesis of IH (42).
Upon vascular injury, the growth factors (PDGF-BB, bFGF), chemokines (SDF-1α, MCP-1) and cytokines (TNF-α, IL-1β) secreted by activated EC, platelets and immune cells stimulate ECM production and secretion, and reduce the expression of the contractile VSMC markers smooth muscle alpha-actin (α-SMA), smooth muscle myosin heavy chain (SM-MHC), calponin, and smooth muscle 22 alpha (SM22α) (42–44). In addition, the “activated” VSMC themselves produce cytokines such as TNFα and MCP-1, leading to positive feedback cascades of enhanced VSMC migration and proliferation (42). The main signaling pathway consistently shown to play a major role in SMC-like cells reprogramming and proliferation/migration is the MAPK pathway, mainly ERK1,2, JNK, and p38 (39, 40).
However, it is now accepted that neointimal VSMCs are phenotypically heterogeneous and that their origin and identity is diverse (45). After medial VSMC, the most abundant cell involved in neointima formation are probably myofibroblasts. Myofibroblasts originate from quiescent fibroblasts in the adventitia, which have converted into proliferating cells expressing several SMC markers such as α-SMA, SM-22α and calponin (46, 47) (Figure 2). Studies also support the existence of populations of mesenchymal stem cells or multipotent progenitor cells within the vessel wall, especially the adventitia layer, which could give rise to the fibroblasts and SMC-like cells found in the neointima layer (48, 49). Further studies identified similar progenitor cells in human arterial and venous tissue (50–52), suggesting a role for these cells in arterial remodeling and IH.
Neointimal cells may also arise from circulating progenitor cells or from the bone marrow (48, 53). However, the contribution of circulating progenitor cells to IH seems to depend upon the model and the type of injury (54). Similarly, studies in the atherosclerosis field yield conflicting results as to whether neointimal SMC-like cells originate from the bone marrow [reviewed in details in Albiero et al. (55)]. Of note, in human, it has been impossible to evaluate the role of circulating progenitors and ex vivo studies using human vessels clearly demonstrated that IH forms in a vessel self-sufficient manner, independently of circulating factors (56, 57). Drawing conclusion from these studies remains challenging given the small number of studies and the variety of experimental models and methodology, especially the methods and markers employed to isolate and identify “progenitor cells.” Recent studies of VSMC lineage in the context of atherosclerosis suggest that up to 80% of SMC-derived cells in the plaques do not express the classic SMC markers α-SMA, but express macrophage markers (CD68, LGALS3), mesenchymal stem cell marker (Sca 1) or myofibroblats markers (PDGFR-β). These cells all express KLF4, a major stem cell and differentiation mediator [reviewed in Allahverdian et al. (45)]. These new evidences underscore how little is known about the identity and origin of the cells responsible for the formation of IH. Advanced techniques of single cell lineage may shed new lights on key questions in the field. Clearly, further work is required to better characterize the cells composing the neointima layer in patients.
Once the neointima starts to form, the arrival and proliferation of SMC-like cells secreting important amount of ECM progressively expand the neointima layer. At first, this expansion is compensated by an outward remodeling of the vessel under the pressure of the blood flow to maintain the lumen area. The vessel gradually thickens as fibrosis sets in. Eventually, the resistance of the arterial wall exceeds the parietal pressure and the neointima extends inward, leading to a narrowing of the lumen and impaired blood flow (Figure 2).
Hydrogen Sulfide
Endogenous Hydrogen Sulfide Production
The discovery of an endogenous pathway releasing H2S in mammalian tissues came long after the discovery of NO, in 1960 with the work of Du Vigneaud, who investigated the oxidation of sulfur-containing amino acids in tissues. He discovered a new metabolic pathway involving the inter-conversion of cysteine and homocysteine and termed this pathway “transsulfuration.” Specifically, H2S is produced by two pyridoxal 5’-phosphate (PLP) dependent enzymes, cystathionine γ−lyase (CSE) and cystathionine β−synthase (CBS). Two additional PLP-independent enzymes, 3-mercaptopyruvate sulfurtransferase (3-MST) and cysteine aminotransferase (CAT) generate sulfane sulfur that can be further processed into H2S. 3-MST and CAT are expressed ubiquitously, whereas CBS and CSE display more tissue-specific patterns of expression. Thus, CBS is the only PLP-dependent enzyme expressed in the brain, while CSE is more prominent in the cardiovascular system. In the kidney and liver, both CSE and CBS are highly expressed. Although the enzymes and pathways responsible for endogenous H2S production are well defined, little is known about their regulation and their relative contributions to H2S and sulfane sulfur levels (e.g., polysulfides, persulfides, thiosulfate) in the circulation and in tissues under normal and disease conditions.
All H2S-synthesizing enzymes have been reported to be expressed by cardiovascular cells. The study of CSE–/– mice demonstrated impaired endothelium-dependent vasorelaxation, with no apparent dysfunction at the level of VSMC (58). Furthermore, CSE seems sufficient to observe H2S-mediated vasodilation (59, 60). These observations strongly promoted the idea that CSE is the main H2S-producing enzyme in the cardiovascular system at the level of EC. However, other reports suggest a key role of 3-MST, along with CAT, in H2S production in the vascular endothelium (61). In contradiction to this early report, studies performed using CSE–/– mice generated on a pure C57BL/6J genetic background by the group of Prof. Isao Ishii failed to show impaired endothelial function and hypertension (62, 63). In addition, most studies of endogenous H2S inhibition rely of the use of high concentrations of propargylglycin (PAG) to inhibit CSE. At these concentrations, PAG may also inhibit CBS, as well as other non-specific targets. Bibli et al. recently demonstrated that CSE expression is negatively regulated by shear stress, as opposed to eNOS in the mouse aorta (64, 65). This is in line with a previous study showing that only disturbed flow regions show discernable CSE protein expression after carotid artery ligation in the mouse (66).
Cellular Effects of Hydrogen Sulfide
H2S contributes to the homeostasis of numerous systems, including the cardiovascular, neuronal, gastrointestinal, respiratory, renal, liver and reproductive systems (67).
The chemical nature of the molecules responsible for the biological activity of H2S remains elusive. HS–, polysulfides and sulfates have all been shown to affect a variety of signaling pathways and biological responses. The sulfur atom is a very potent electron acceptor/donor and H2S can undergo complex oxidation, yielding thiosulfate, sulfenic acids, persulfides, polysulfides and sulfate (68). These oxidative products are likely mediating the principal mechanism through which H2S exerts its biological actions: post-translational modification of proteins, known as persulfidation. Persulfidation is a chemical reaction whereby a persulfide group (RSSH) is formed on reactive cysteine residues of target proteins (68, 69). Since H2S has the same oxidation state as cysteine residues, a redox reaction cannot occur. Cysteine residues or H2S have to be oxidized first (for instance in the form of polysulfides H2Sn). In 2009, Mustafa et al. performed LC/MS/MS analysis on liver lysates after NaHS treatment and identified 39 proteins that were persulfidated. Amongst them, they identified GAPDH, β-tubulin, and actin. Interestingly, these proteins were not persulfidated in the liver of mice lacking CSE (CSE KO) (70). Furthermore, new high throughput techniques allowing global assessment of post-translational modification of cysteinyl thiols (-SH) to persulfides (-SSH) demonstrated extensive cysteine residues persulfidation in response to various H2S donors across various experimental designs (71–73).
Hydrogen Sulfide in Intimal Hyperplasia
Few studies directly assessed the effects of endogenous or exogenous H2S on IH. CSE expression and activity are reduced after balloon-injury in a rat model of IH (32). CSE expression and activity, as well as free circulating H2S, are also reduced in human suffering from vascular occlusive diseases (30, 74). We recently demonstrated that, in patient undergoing vascular surgery, circulating H2S levels were associated with long-term survival (75), suggesting low H2S production as a risk-factor for cardiovascular diseases. Mice lacking CSE show a significant increase in IH formation as compared to WT mice in a model of carotid artery ligation (34, 76). On the contrary, CSE overexpression decreases IH formation in a murine model of vein graft by carotid-interposition cuff technique (77). Similarly, NaHS administration limits the development of IH in in vivo models in rats (32), rabbits (33), and mice (34), and in human great saphenous vein segments ex vivo (35).
The effect of H2S against IH is probably mediated by inhibition of VSMC proliferation and migration. Indeed, it was demonstrated, using BrdU and TUNEL assays, that H2S supplementation or CSE overexpression decreases VSMCs proliferation and increases VSMCs apoptosis, respectively (33, 35, 78). VSMCs isolated from Cse–/– mice exhibit more motility than their WT counterpart, and blocking CSE activity using PAG in WT VSMCs increases cell migration (34, 79). The mechanisms whereby H2S affect VSMCs are not fully understood. In mouse VSMC, H2S has been shown to modulate the MAPK pathway, especially ERK1, 2 (32), and calcium-sensing receptors (80, 81). In addition, H2S may limit MMP2 expression and ECMs degradation, preventing migration of VSMCs from the media to the intima (34, 79). In human VSMC, we recently reported that the H2S donor Zofenopril decreases the activity of the MAPK and mTOR pathways, which correlates with reduced VSMC proliferation and migration (82). We also showed that the H2S donor salt NaHS, as well the thiol source sodium thiosulfate, inhibit microtubule polymerization, which results in cell cycle arrest and inhibition of proliferation and migration in primary human VSMC (76) (Figure 3).
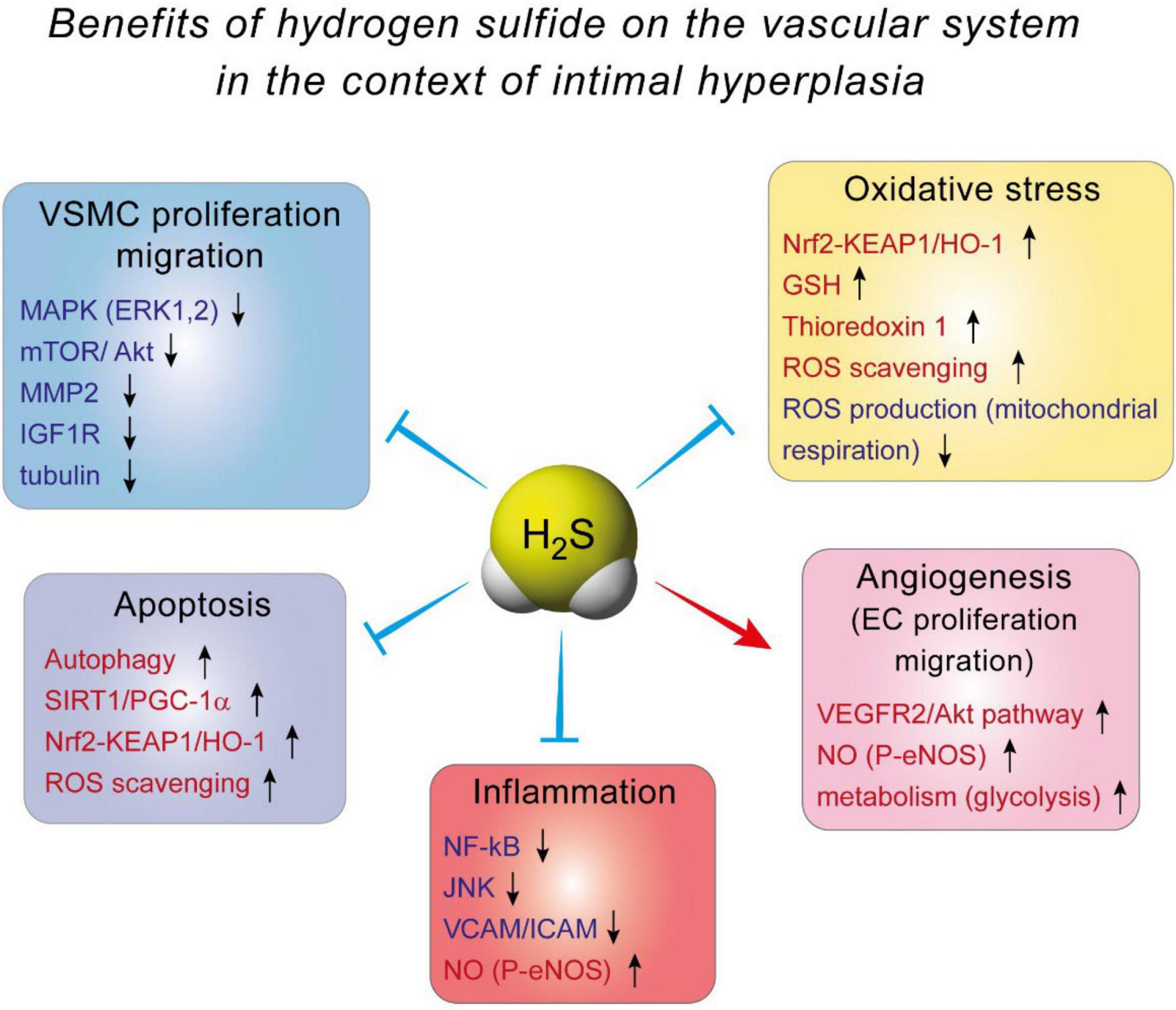
Figure 3. The benefits of hydrogen sulfide on the vascular system in the context of intimal hyperplasia.
Other studies also inform on potential mechanism of action of H2S on VSMC. The first evidence of H2S being a gasotransmitter comes from the consistent observation across species and vascular beds that H2S and other derived products induce vasodilation. H2S triggers vasodilation mostly via persulfidation of several ion channels such as KATP, voltage and Ca2+-activated K+ channels (59, 60, 83). By reducing extracellular Ca2+ entry, H2S improves VSMC relaxation. Despite the obvious fact that improved vasorelaxation may be beneficial in the context of IH, these channels may also directly regulate cell proliferation in VSMC. For instance, the anti-diabetic and KATP channel blocker glibenclamide has been shown to reduce VSMC proliferation in a recent study (84).
Other Benefits of Hydrogen Sulfide in the Cardiovascular System
Hydrogen Sulfide Stimulates Endothelial Cell Proliferation and Migration
Vascular surgery invariably leads to injury of the operated vessels. The single-cell endothelial barrier is very vulnerable and severely injured during surgery, which promotes inflammation and reprogramming of the adventitial and medial cells. The ability of endothelial cells to proliferate and migrate to restore the endothelial layer of the vessel is a key step in the resolution of post-operative inflammation to limit IH and restenosis. Unfortunately, EC are often neglected in strategies to reduce the formation of IH.
Although it has never been demonstrated in the context of IH, a large amount of studies established that H2S and polysulfides stimulate EC function and angiogenesis. Exogenous H2S treatment stimulates EC growth, motility and organization into vessel-like structure in vitro. On the contrary, inhibition of H2S biosynthesis, either via pharmacological inhibitors or via silencing of CSE, CBS or 3MST, reduces EC growth, migration and vessel-like structure formation (85, 86). Further in vivo studies of chicken chorioallantoic membranes (CAM) treated with the CSE inhibitor PAG suggest that CSE is important for vessel branching and elongation (87). Matrigel plug angiogenesis assay also confirmed the importance of CSE and H2S in vascular endothelial growth factor (VEGF)-induced angiogenesis (88, 89).
Several mechanisms have been proposed to explain H2S-induced angiogenesis. First and probably foremost, H2S stimulates the VEGF pathway in EC, through sulfhydration of the VEGF receptor VEGFR2, increasing its dimerization, autophosphorylation and activation (90). Interestingly, short term exposure of human EC to VEGF increases H2S production (87), suggesting a positive feedback loop of VEGF signaling through H2S. In addition, H2S promotes angiogenesis by inhibiting mitochondrial electron transport and oxidative phosphorylation, resulting in increased glucose uptake and glycolytic ATP production necessary to provide rapid energy for EC migration (91) (Figure 3).
H2S also promotes angiogenesis through its extensive interaction with the NO pathway. In endothelial cells, H2S may induce eNOS persulfidation at Cys433, which increases the phosphorylation of its activator site and stabilizes eNOS in its dimeric form (86, 92). H2S may also increase intracellular calcium levels, leading to increase eNOS activity and NO production (93, 94). Exogenous H2S donors have also been shown to stimulate the growth pathways Akt, p38 and ERK1/2, which all promote EC proliferation and migration (87, 89, 95). Finally, both H2S- and NO-induced angiogenesis require the other gasotransmitter (86, 92). Thus, the vascular effects of NO and H2S are interdependent and closely intertwined, with both gasotransmitter having direct and indirect effects on each other [for full review see Szabo et al. (96)]. Overall, the effect of H2S on EC may facilitate re-endothelization following vascular trauma, accelerating healing of the intima layer and limiting IH (Figure 3).
Hydrogen Sulfide Inhibits Inflammation
The surgical trauma and injury to the endothelial layer triggers inflammation, which contributes to IH. Many studies report anti-inflammatory properties of H2S, in particular in the context of atherosclerosis and cardiac failure [for full review see Pan et al. (97)]. Thus, H2S reduces adhesion and infiltration of pro-inflammatory cells and circulating levels of pro-inflammatory chemokines and cytokines in the ApoE–/– mouse model of atherosclerosis (98, 99). Similarly, several reports document that H2S donors (NaHS, DATS, SG1002, STS) or CSE overexpression decrease leukocyte and neutrophil infiltration and cytokine production following ischemic injury in various models of myocardial infarction (100–105).
Mechanistically, evidence from EC and macrophages indicate that Nuclear factor kappa B (NFkB) inhibition seems to be the key to H2S anti-inflammatory effects (98, 106–108). NF-kB is a transcription factor and a master regulator of pro-inflammatory genes, including cytokines and cell adhesion molecules. NaHS inhibits NF-kB activity probably via persulfidation/stabilization of IkB (109), which controls NF-kB (p65) translocation to the nucleus. In EC, this leads to decreased expression of adhesion molecule VCAM and ICAM, thereby limiting recruitment of leukocyte to the aortic wall (98, 106, 108). NaHS also promotes a shift in macrophages to the M2, pro-resolution state (110). Moreover, NaHS and GYY increase eNOS phosphorylation, thereby improving NO production, which reduces inflammation (106, 111). Whether or not H2S-based strategies may reduce inflammation in the context of IH remains to be tested (Figure 3).
Hydrogen Sulfide Has Anti-oxidant Properties
Several studies in various models also consistently showed that H2S holds anti-oxidant properties. First, H2S can directly scavenge reactive oxygen species (ROS), such as superoxide anions O2–, at higher rates than other classic antioxidants such as GSH. However, since H2S physiologic concentration is in the nanomolar range whereas GSH is present in millimolar quantity, it is debatable whether H2S direct contribution to anti-oxidation is significant.
Actually, the effect of H2S probably arises from stimulation of anti-oxidant pathways, rather than via direct scavenging of ROS. First, H2S increases GSH production via modulation of the transsulfuration pathway. H2S interaction with GSH has been studied in details in the central nervous system, where GSH plays a major role in maintaining the homeostasis between anti-oxidant and ROS production [reviewed in details in Shefa et al. (112)]. In the vascular system, H2S persulfidates the glutathione peroxidase 1, which promotes glutathione synthesis and results in decreased lipid peroxidation in the aortic wall in the context of atherosclerosis (113). Second, numerous studies document that H2S promotes the Nrf2 pathway [reviewed in Corsello et al. (114)]. H2S promotes the Nrf2 anti-oxidant response via persulfidation of Kelch-like ECH-associated protein 1 (Keap1), which sequesters Nrf-2 in the cytosol. Keap1 persulfidation prompts dissociation from Nrf2, which induces the expression of several proteins, among which the major antioxidant protein heme oxygenase 1 (HO-1). This mechanism reduces oxidative stress, leading to reduced atherosclerosis in diabetic low density lipoprotein receptor (LDLR) knock –out mice (115), and cardioprotection in a model of ischemia-reperfusion injury (116, 117). Finally, H2S also stimulates thioredoxin 1 (Trx) expression, via silencing the expression of inhibitory protein Trx-interacting protein (TXNIP) (118, 119). Increased Trx is instrumental in the cardioprotective effects of H2S against ischemia-induced heart failure (118) (Figure 3).
Mitochondrial respiration is a major source of ROS (120, 121). H2S has a bell-shaped effect on mitochondrial respiration. At low nanomolar concentrations, sulfide quinone oxidoreductase (SQR) transfers electrons from H2S to the coenzyme Q in the Complex II of the electron transport chain, thereby promoting mitochondrial respiration. At higher concentrations, H2S binds the copper center of cytochrome c oxidase (complex IV), thereby inhibiting respiration and limiting ROS production (122).
These anti-oxidant properties of H2S may have a beneficial impact on IH, as ROS contribute to endothelial dysfunction and VSMC dedifferentiation (123, 124).
Discussion: New Perspectives for the Treatment of Intimal Hperplasia Using Hydrogen Sulfide-Based Therapies
There is currently no clinically approved molecule exploiting the clinical potential of H2S. Most compounds available for research have poor translational potential due to their pharmacokinetic properties. Thus, the highly soluble unstable salts sodium hydrogen sulfide (NaHS) and disodium sulfide (Na2S) release H2S instantly in an uncontrollable manner, and thus have narrow clinical ranges. Other H2S-releasing molecules extracted from garlic such DATS (Diallyl trisulfide) and DADS (Diallyl disulfide), which have both been shown to possess vasoactive properties (125), are also very short lived and hard to stabilize (125). In the past years, several H2S-releasing compounds have been studied and developed for clinical purposes (Table 1).
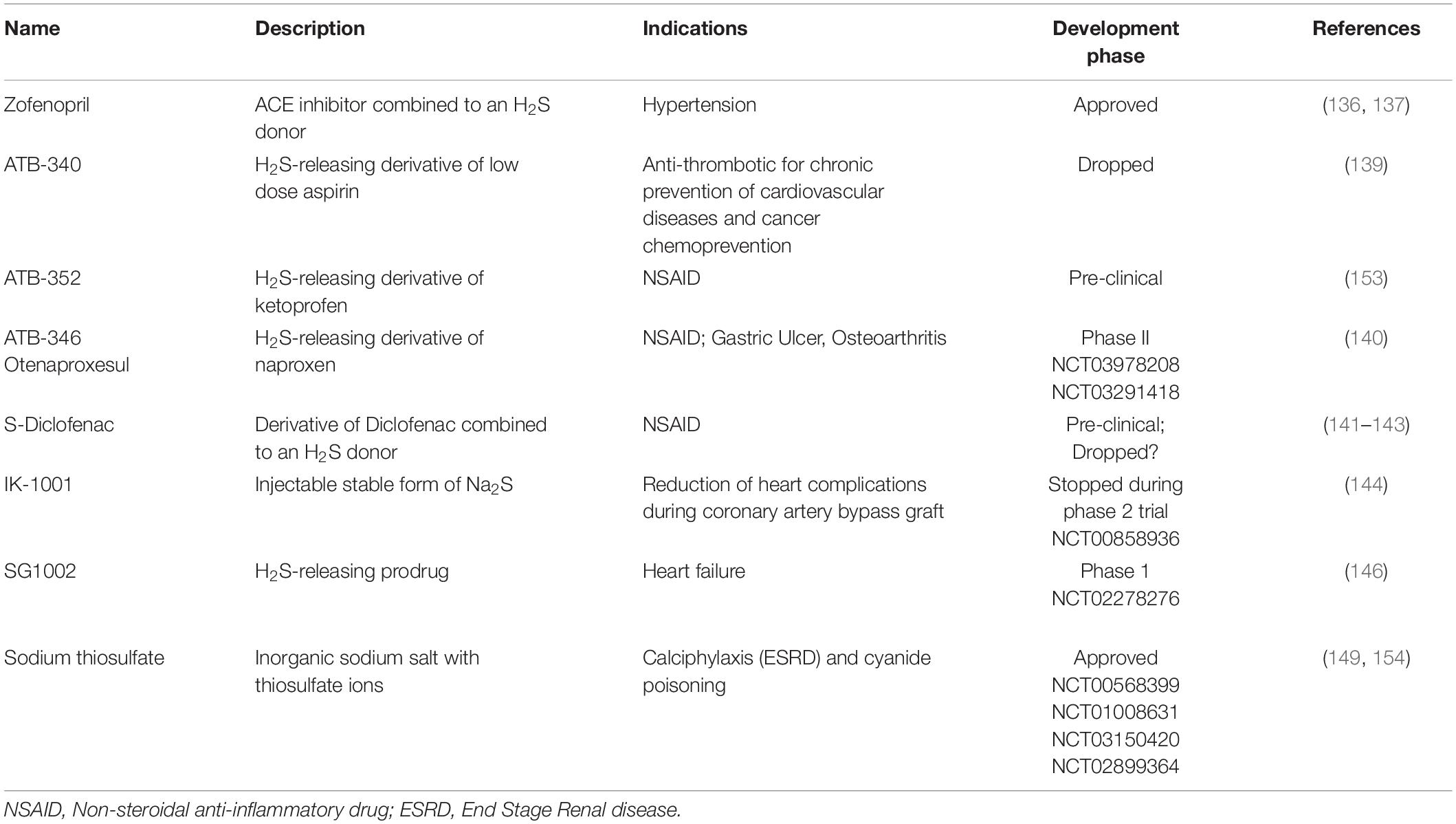
Table 1. Clinical development of H2S-releasing compound.
Hydrogen Sulfide-Based Therapies: Current Strategies
S-allylcysteine (SAC), an organosulfur compound present in abundance in garlic, has been shown to lower the mortality and reduce the infarct size in a rat model of acute myocardial infarction (126) and improves blood flow recovery after hindlimb ischemic injury in the mouse (127). S-propyl-L-cysteine and S-propargyl-L-cysteine, structural analogs of SAC found in garlic, have also been shown to slowly release H2S (128). Interestingly, S-propargyl-L-cysteine has been demonstrated to have both cardio protective (129, 130) and pro-angiogenic properties in preclinical models (131). However, despite the well-known cardiovasculo-protective properties of garlic, these active compounds are not currently used for clinical studies.
Another strategy used by pharmaceutical companies to harness the benefits of H2S has been to combine a H2S-releasing moiety with well-established parent compounds.
Zofenopril is one such product. Zofenopril is an ACE Inhibitors (ACEi) and a H2S donor combined (132). ACEi constitute one of the first-line class of antihypertensive drugs (133). Several clinical studies have shown that sulfhydrylated ACEi zofenopril has additional beneficial actions compared to non-sulfhydrylated ACEi such as enalapril or ramipril. Thus, zofenopril improves the clinical outcome of patients with different cardiovascular diseases such as acute myocardial infarction and congestive heart failure (134–137). We recently demonstrated that Zofenopril is more potent than Enalapril in reducing IH in a genetic model of hypertensive mice. In addition, it suppresses IH in normotensive condition, where other non-sulfhydrylated ACEi (Enalapril, Lisinopril and Quinapril) have no effect. Furthermore, Zofenopril prevents IH in an ex vivo model of IH in human saphenous vein. The effect of Zofenopril on IH correlates with reduced VSMC proliferation and migration and decreased activity of the MAPK and mTOR pathways (138).
Antibe Clinicals, a startup created around H2S-releasing compounds, synthesizes several H2S-releasing derivatives conjugated to NSAID for the treatments of pain and inflammation.1 ATB-340 is a H2S-releasing derivative of low-dose aspirin without the serious risk of gastrointestinal bleeding. Pre-clinical studies have demonstrated that ATB-340 caused negligible GI damage compared to low-dose aspirin (139). ATB 346, which is derived from the NSAID naproxen, was recently shown in a Phase 2B study to reduce GI toxicity compared to naproxen alone, with equivalent suppression of COX activity (140). The H2S-releasing diclofenac S-Diclofenac (ATB-337 or ACS-15), where H2S is linked to diclofenac via an ester bond, may also present advantages compared to classical Diclofenac (141, 142). S-Diclofenac has been shown to inhibit smooth muscle cell proliferation, and may play a role in restenosis in vascular injury (143). However, there was no further development of this compound.
The compound IK-1001, from the company Mallinckrodt, is an injectable stable form of Na2S (144). Despite a first phase I safety trial showing no adverse events, the development of IK-1001 was stopped by the company during a phase II efficacy trial in patients undergoing surgery for a coronary artery bypass graft (ClinicalTrials.gov ID: NCT00858936).
SG1002, initially developed by Kondo et al. (145), and further developed by the startup company Sulfagenix, is a prodrug releasing H2S. It has been tested on humans in the setting of congestive heart failure during one of the first phase 1 trial using a sulfide-based therapy to treat cardiovascular diseases. The results of this study were promising as SG1002 was able to restore sulfide and NO levels in patients with heart failure (146). However, additional studies are obviously required.
Sodium thiosulfate (STS; Na2S2O3) is an inorganic sodium salt containing thiosulfate ions in a 2:1 ratio. Pharmaceutical-grade STS is available and has been suggested to release H2S through non-enzymatic and enzymatic mechanisms (147, 148). STS is the treatment of choice for cyanide poisoning as thiosulfate is used by Rhodanese to convert cyanide to less toxic thiocyanate. Intravenous STS is also used to increase the solubility of calcium for the treatment of acute calciphylaxis, a rare vascular complication of patients with end-stage renal disease (149). Sodium thiosulfate is also under test in a number of clinical trials for the treatment of ectopic calcification (NCT03639779; NCT04251832; NCT02538939); to reduce ototoxicity in patients receiving cisplatin chemotherapy for standard risk hepatoblastoma (NCT05129748); in combination with chemotherapy to prevent low platelet count in patients with malignant brain tumors (NCT00075387). We also recently demonstrated that STS limits IH development in vivo in a model of arterial restenosis and in our ex vivo model of IH in human veins. STS treatment increases H2S bioavailability, which inhibits cell apoptosis and fibrosis, as well as VSMC proliferation and migration via microtubules depolymerization (76). Interestingly, an ongoing clinical study aims to evaluate the efficacy and safety of STS compared to placebo on myocardial infarct size in ST-segment elevation myocardial infarction (STEMI) patients treated with percutaneous coronary intervention (PCI) (NCT02899364).
The focal nature of IH lesions provide a window of opportunities for the use of local drug delivery using vascular medical devices. A number of approaches have been tested to apply treatment locally, including DCB and DES, as well as periadventitial drug delivery and targeted systemic therapies (150). Unlike current non-specific cytostatic drugs, local H2S delivery might provide a unique clinical opportunity to inhibit VSMCs proliferation while promoting ECs proliferation and endothelial repair. We recently developed and evaluated the clinical potential of an H2S-releasing biodegradable hydrogel to limit the development of IH in human veins. The thiol-triggered, controlled H2S release from peptide hydrogels provided sustained H2S concentrations over the period of hours, which inhibited VSMC proliferation and IH in human vein models more effectively than the sulfide salts (NaHS). The H2S-releasing peptide hydrogel also facilitated HUVEC proliferation and transmigration in vitro, which may promote re-endothelization, thereby supporting vascular repair (35). Recently, it was shown that a locally applicable gel containing the hydrogen sulfide releasing prodrug (GYY4137) mitigates graft failure and improve arterial remodeling in a model of vein graft surgery in the mouse (151).
Hydrogen Sulfide-Based Therapies: Advantages and Limitations
IH is a complex process, involving multiple cell types and developing over the course of several years. IH is triggered by an acute endothelial dysfunction and associated pro-inflammatory response, which triggers a cascade of event leading to the formation of the neointima layer. The neointima slowly grows over the course of months to years, long after the acute inflammation is resolved and the endothelium repaired (Figure 2). H2S is unique in the context of IH because it can have beneficial impact on both the acute pro-inflammatory response and the chronic neointima growth. Thus, on the one hand, H2S limits inflammation and oxidative damages, while promoting EC proliferation and endothelium repair. On the other hand, H2S limits the proliferation and migration of synthetic SMC-like cells forming the neointima layer. In contrast, current strategies to reduce IH aggressively target cell proliferation, which also affect re-endothelization, prolonging inflammation and the need for anti-thrombotic therapies. Moreover, recent reports suggest that paclitaxel-releasing balloons and stents may have deleterious long-term effects, which is not surprising given that it is a cytotoxic chemotherapeutic agent.
Numerous drugs have been tested over the years to limit IH, demonstrating outstanding potential in pre-clinical studies in the small animal. Yet, in most trials, the pharmacologic treatment of restenosis failed to have a positive impact (150, 152). It is probable that the lack of efficacy in humans is, at least partly, due to insufficient drug delivery at the site of injury, as much higher dosages of drugs were generally used in animal models. It will be interesting to see whether H2S-based solutions can bridge the gap between benchtop and bedside. The first challenge will be to develop stable H2S-donor molecules allowing slow and sustained H2S release over the course of months/years. Such molecules are yet to be developed and will be hard to design given the instability and short half-life of H2S. Another challenge for either systemic or local release of H2S reside in the delivery system. The development of DCB and DES releasing paclitaxel or sirolimus led to innovative delivery systems. Gels, nanoparticles, multiple-layer coatings and biodegradable scaffolds have been invented to allow sustained drug release. It will be interesting to apply this knowledge to H2S-donor molecules. Eventually, the development of H2S-releasing balloons and stents could provide much-needed device to limit VSMC proliferation while promoting EC recovery. However, combining local delivery and systemic oral drug administration will probably be necessary to prevent IH successfully.
Conclusion
Restenosis due to IH is recurrent and there is no efficient therapy. The neointima layer has a muscular and fibrotic rigid structure, which is hard to treat. Additional interventions to re-open the vessel invariably results in trauma, leading to further IH. DCB and DES improved the primary patency of vessels following endovascular surgeries but in-stent restenosis poses new challenges. Current strategies target cell proliferation to reduce IH, which also affect re-endothelization, prolonging the need for anti-thrombotic agents. Moreover, recent reports suggests that paclitaxel-releasing balloons and stents may have deleterious long-term effects.
These limitations warrant further research to better understand the molecular mechanisms of IH and develop new molecules limiting VSMC proliferation while stimulating EC proliferation and re-endothelization. Although H2S research is still in its infancy, ample evidence point to a protective role for this gaseous transmitter in the development of cardiovascular diseases. However, further animal studies are required to test the potential and safety of new H2S-based therapies. Understanding these questions will provide insightful knowledge about the biology of H2S and help design successful H2S-based therapies in the future.
Author Contributions
FA and SD made the backbone. DM, FA, AL, and SD wrote and revised the manuscript. FA made the figures. All authors contributed to the article and approved the submitted version.
Funding
The Laboratory of Unit of Vascular Surgery of the Lausanne University Hospital was supported by the Swiss National Science Foundation (grant no. FN-310030_176158 to FA and SD and PZ00P3-185927 to AL), the Novartis Foundation to FA, the Union des Sociétés Suisses des Maladies Vasculaires to SD, and the Fondation pour la recherche en chirurgie vasculaire et thoracique.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
3-MST, 3-mercaptopyruvate sulfurtransferase; AKT, Serine/Threonine Kinase 1; AOAA, aminooxyacetic acid; bFGF, Basic Fibroblast Growth Factor; CAT, cysteine aminotransferase; CBS, cystathionine β synthase; COX, cytochrome c oxidase; CSE, cystathionine γ lyase; DATS, Diacyltrisulfite; DES, Drug-Eluting Stents; DCB, Drug-Coated Balloons; EC, Endothelial Cells; ECM, Extracellular Matrix; EEL, external elastic lamina; Enos, Endothelial Nitric Oxide Synthase; ER, endoplasmic reticulum; ERK, Extracellular Signal-Regulated Kinase; ETHE1, ethylmalonic encephalopathy 1 protein; GSH, Reduced Glutathione; GSSG, oxidized GSH; ICAM1, Intercellular Adhesion Molecule 1; IEL, internal elastic lamina; IH, Intimal Hyperplasia; IL-1 β, Interleukin 1 beta; JNK, C-Jun N Terminal Kinase; KATP, ATP-sensitive K+; Keap1, kelch-like ECH-associated protein 1; LDH, lactate dehydrogenase; LDL, Low-Density Lipoprotein; MAPK, Mitogen-Activated Protein Kinase; MCP-1 (CCL-2), Monocyte Chemoattractant Protein-1; MMP, Matrix Metalloproteinase; NaHS, Sodium Hydrogen Sulfur; NF- K b, Nuclear Factor Kappa b; NO, Nitric Oxide; Nrf2, nuclear factor (erythroid-derived 2)–like 2; PCI, percutaneous coronary intervention; PCNA, Proliferating Cell Nuclear Antigen; PDGF, Platelet-Derived Growth Factor; Pdgfr- B, Beta-Type Platelet-Derived Growth Factor Receptor; PLP, pyridoxal 5’-phosphate; POBA, plain old balloon angioplasty; ROS, Reactive Oxygen Specie; SDF-1 α, Stromal Cell-Derived Factor 1; SM22 α (Tagln), Smooth Muscle 22 Alpha/Transgelin; SMA (ACTA2), Smooth Muscle Actin Alpha; SQR, sulfide quinone oxidoreductase; STEMI, ST-segment elevation myocardial infarction; STS, Sodium Thiosulfate; TNF- α, Tumor Necrosis Factor Alpha; TIMP, Tissue Inhibitors Of Metalloproteinase; Trx, thioredoxin; TXNIP, Trx-interacting protein; VCAM1, Vascular Cell Adhesion Protein 1; VSMC, Vascular Smooth Muscle Cells; WT, wild type.
Footnotes
References
1. Eraso LH, Fukaya E, Mohler ER III, Xie D, Sha D, Berger JS. Peripheral arterial disease, prevalence and cumulative risk factor profile analysis. Eur J Prev Cardiol. (2014) 21:704–11. doi: 10.1177/2047487312452968
2. Fowkes FG, Rudan D, Rudan I, Aboyans V, Denenberg JO, McDermott MM, et al. Comparison of global estimates of prevalence and risk factors for peripheral artery disease in 2000 and 2010: a systematic review and analysis. Lancet. (2013) 382:1329–40. doi: 10.1016/S0140-6736(13)61249-0
3. Song P, Rudan D, Zhu Y, Fowkes FJI, Rahimi K, Fowkes FGR, et al. Global, regional, and national prevalence and risk factors for peripheral artery disease in 2015: an updated systematic review and analysis. Lancet Glob Health. (2019) 7:e1020–30. doi: 10.1016/S2214-109X(19)30255-4
4. Simpson EL, Kearns B, Stevenson MD, Cantrell AJ, Littlewood C, Michaels JA. Enhancements to angioplasty for peripheral arterial occlusive disease: systematic review, cost-effectiveness assessment and expected value of information analysis. Health Technol Assess. (2014) 18:1–252. doi: 10.3310/hta18100
5. Buccheri D, Piraino D, Andolina G, Cortese B. Understanding and managing in-stent restenosis: a review of clinical data, from pathogenesis to treatment. J Thorac Dis. (2016) 8:E1150–62. doi: 10.21037/jtd.2016.10.93
6. Zen K, Takahara M, Iida O, Soga Y, Kawasaki D, Nanto S, et al. Drug-eluting stenting for femoropopliteal lesions, followed by cilostazol treatment, reduces stent restenosis in patients with symptomatic peripheral artery disease. J Vasc Surg. (2017) 65:720–5. doi: 10.1016/j.jvs.2016.10.098
7. Abdoli S, Mert M, Lee WM, Ochoa CJ, Katz SG. Network meta-analysis of drug-coated balloon angioplasty versus primary nitinol stenting for femoropopliteal atherosclerotic disease. J Vasc Surg. (2021) 73:1802–10.e4. doi: 10.1016/j.jvs.2020.10.075
8. Teichgraber U, Lehmann T, Aschenbach R, Scheinert D, Zeller T, Brechtel K, et al. Efficacy and safety of a novel paclitaxel-nano-coated balloon for femoropopliteal angioplasty: one-year results of the effpac trial. EuroIntervention. (2020) 15:e1633–40. doi: 10.4244/EIJ-D-19-00292
9. Caradu C, Lakhlifi E, Colacchio EC, Midy D, Berard X, Poirier M, et al. Systematic review and updated meta-analysis of the use of drug-coated balloon angioplasty versus plain old balloon angioplasty for femoropopliteal arterial disease. J Vasc Surg. (2019) 70:981–95.e10. doi: 10.1016/j.jvs.2019.01.080
10. Ding Y, Zhou M, Wang Y, Cai L, Shi Z. Comparison of drug-eluting stent with bare-metal stent implantation in femoropopliteal artery disease: a systematic review and meta-analysis. Ann Vasc Surg. (2018) 50:96–105. doi: 10.1016/j.avsg.2017.12.003
11. Fattori R, Piva T. Drug-eluting stents in vascular intervention. Lancet. (2003) 361:247–9. doi: 10.1016/S0140-6736(03)12275-1
12. Jukema JW, Verschuren JJ, Ahmed TA, Quax PH. Restenosis after PCI. Part 1: pathophysiology and risk factors. Nat Rev Cardiol. (2011) 9:53–62. doi: 10.1038/nrcardio.2011.132
13. Bjorck M, Earnshaw JJ, Acosta S, Bastos Goncalves F, Cochennec F, Debus ES, et al. Editor’s Choice – European society for vascular surgery (ESVS) 2020 clinical practice guidelines on the management of acute limb ischaemia. Eur J Vasc Endovasc Surg. (2020) 59:173–218. doi: 10.1016/j.ejvs.2019.09.006
14. Katsanos K, Spiliopoulos S, Kitrou P, Krokidis M, Karnabatidis D. Risk of death following application of paclitaxel-coated balloons and stents in the femoropopliteal artery of the leg: a systematic review and meta-analysis of randomized controlled trials. J Am Heart Assoc. (2018) 7:e011245. doi: 10.1161/JAHA.118.011245
15. Rocha-Singh KJ, Duval S, Jaff MR, Schneider PA, Ansel GM, Lyden SP, et al. Mortality and paclitaxel-coated devices: an individual patient data meta-analysis. Circulation. (2020) 141:1859–69. doi: 10.1161/CIRCULATIONAHA.119.044697
16. Royce S, Chakraborty A, Zhao Y. Us food and drug administration perspective on “mortality and paclitaxel-coated devices: an individual patient data meta-analysis”. Circulation. (2020) 141:1870–1. doi: 10.1161/CIRCULATIONAHA.120.047376
17. Dinh K, Gomes ML, Thomas SD, Paravastu SCV, Holden A, Schneider PA, et al. Mortality after paclitaxel-coated device use in patients with chronic limb-threatening ischemia: a systematic review and meta-analysis of randomized controlled trials. J Endovasc Ther. (2020) 27:175–85. doi: 10.1177/1526602820904783
18. Ipema J, Huizing E, Schreve MA, de Vries JPM, Unlu C. Editor’s choice – drug coated balloon angioplasty vs. standard percutaneous transluminal angioplasty in below the knee peripheral arterial disease: a systematic review and meta-analysis. Eur J Vasc Endovasc Surg. (2020) 59:265–75. doi: 10.1016/j.ejvs.2019.10.002
19. Katsanos K, Spiliopoulos S, Kitrou P, Krokidis M, Paraskevopoulos I, Karnabatidis D. Risk of death and amputation with use of paclitaxel-coated balloons in the infrapopliteal arteries for treatment of critical limb ischemia: a systematic review and meta-analysis of randomized controlled trials. J Vasc Interv Radiol. (2020) 31:202–12. doi: 10.1016/j.jvir.2019.11.015
20. Nordanstig J, James S, Andersson M, Andersson M, Danielsson P, Gillgren P, et al. Mortality with paclitaxel-coated devices in peripheral artery disease. N Engl J Med. (2020) 383:2538–46. doi: 10.1056/NEJMoa2005206
21. Secemsky EA, Kundi H, Weinberg I, Jaff MR, Krawisz A, Parikh SA, et al. Association of survival with femoropopliteal artery revascularization with drug-coated devices. JAMA Cardiol. (2019) 4:332–40. doi: 10.1001/jamacardio.2019.0325
22. Beckman JA, White CJ. Paclitaxel-coated balloons and eluting stents: is there a mortality risk in patients with peripheral artery disease? Circulation. (2019) 140:1342–51. doi: 10.1161/CIRCULATIONAHA.119.041099
23. Byrne RA, Stone GW, Ormiston J, Kastrati A. Coronary balloon angioplasty, stents, and scaffolds. Lancet. (2017) 390:781–92. doi: 10.1016/S0140-6736(17)31927-X
24. Teichgraber U, Ingwersen M, Platzer S, Lehmann T, Zeller T, Aschenbach R, et al. Head-to-head comparison of sirolimus- versus paclitaxel-coated balloon angioplasty in the femoropopliteal artery: study protocol for the randomized controlled sirona trial. Trials. (2021) 22:665. doi: 10.1186/s13063-021-05631-9
25. Zhu P, Zhou X, Zhang C, Li H, Zhang Z, Song Z. Safety and efficacy of ultrathin strut biodegradable polymer sirolimus-eluting stent versus durable polymer drug-eluting stents: a meta-analysis of randomized trials. BMC Cardiovasc Disord. (2018) 18:170. doi: 10.1186/s12872-018-0902-5
26. El-Hayek G, Bangalore S, Casso Dominguez A, Devireddy C, Jaber W, Kumar G, et al. Meta-analysis of randomized clinical trials comparing biodegradable polymer drug-eluting stent to second-generation durable polymer drug-eluting stents. JACC Cardiovasc Interv. (2017) 10:462–73. doi: 10.1016/j.jcin.2016.12.002
27. Yu-Wei D, Li ZS, Xiong SM, Huang G, Luo YF, Huo TY, et al. Paclitaxel induces apoptosis through the TAK1-JNK activation pathway. FEBS Open Bio. (2020) 10:1655–67. doi: 10.1002/2211-5463.12917
28. Chakraborty R, Chatterjee P, Dave JM, Ostriker AC, Greif DM, Rzucidlo EM, et al. Targeting smooth muscle cell phenotypic switching in vascular disease. JVS Vasc Sci. (2021) 2:79–94. doi: 10.1016/j.jvssci.2021.04.001
29. Zhang L, Wang Y, Li Y, Li L, Xu S, Feng X, et al. Hydrogen Sulfide (H2S)-releasing compounds: therapeutic potential in cardiovascular diseases. Front Pharmacol. (2018) 9:1066. doi: 10.3389/fphar.2018.01066
30. Islam KN, Polhemus DJ, Donnarumma E, Brewster LP, Lefer DJ. Hydrogen sulfide levels and Nuclear Factor-Erythroid 2-Related Factor 2 (NRF2) activity are attenuated in the setting of critical limb ischemia (CLI). J Am Heart Assoc. (2015) 4:e001986. doi: 10.1161/JAHA.115.001986
31. Longchamp A, MacArthur MR, Trocha K, Ganahl J, Mann CG, Kip P, et al. Plasma hydrogen sulfide production capacity is positively associated with post-operative survival in patients undergoing surgical revascularization. medRxiv. [Preprint]. (2021). doi: 10.1101/2021.02.16.21251804
32. Meng QH, Yang G, Yang W, Jiang B, Wu L, Wang R. Protective effect of hydrogen sulfide on balloon injury-induced neointima hyperplasia in rat carotid arteries. Am J Pathol. (2007) 170:1406–14. doi: 10.2353/ajpath.2007.060939
33. Ma B, Liang G, Zhang F, Chen Y, Zhang H. Effect of hydrogen sulfide on restenosis of peripheral arteries after angioplasty. Mol Med Rep. (2012) 5:1497–502. doi: 10.3892/mmr.2012.853
34. Yang G, Li H, Tang G, Wu L, Zhao K, Cao Q, et al. Increased neointimal formation in cystathionine gamma-lyase deficient mice: role of hydrogen sulfide in alpha5beta1-integrin and matrix metalloproteinase-2 expression in smooth muscle cells. J Mol Cell Cardiol. (2012) 52:677–88. doi: 10.1016/j.yjmcc.2011.12.004
35. Longchamp A, Kaur K, Macabrey D, Dubuis C, Corpataux JM, Deglise S, et al. Hydrogen sulfide-releasing peptide hydrogel limits the development of intimal hyperplasia in human vein segments. Acta Biomater. (2019) 97:374–84. doi: 10.1016/j.actbio.2019.07.042
36. Stone JR. Diseases of small and medium-sized blood vessels. In: Buja LM, Butany J editors. Cardiovascular Pathology. Cambridge: Academic Press (2016). p. 125–68. doi: 10.1016/b978-0-12-420219-1.00004-5
37. Nakano M, Otsuka F, Yahagi K, Sakakura K, Kutys R, Ladich ER, et al. Human autopsy study of drug-eluting stents restenosis: histomorphological predictors and neointimal characteristics. Eur Heart J. (2013) 34:3304–13. doi: 10.1093/eurheartj/eht241
38. Nakashima Y, Wight TN, Sueishi K. Early atherosclerosis in humans: role of diffuse intimal thickening and extracellular matrix proteoglycans. Cardiovasc Res. (2008) 79:14–23. doi: 10.1093/cvr/cvn099
39. Tong X, Khandelwal AR, Qin Z, Wu X, Chen L, Ago T, et al. Role of smooth muscle Nox4-Based NADPH oxidase in neointimal hyperplasia. J Mol Cell Cardiol. (2015) 89:185–94. doi: 10.1016/j.yjmcc.2015.11.013
40. Sterpetti AV, Cucina A, Lepidi S, Randone B, Stipa F, Aromatario C, et al. Progression and regression of myointimal hyperplasia in experimental vein grafts depends on platelet-derived growth factor and basic fibroblastic growth factor production. J Vasc Surg. (1996) 23:568–75. doi: 10.1016/s0741-5214(96)80034-6
41. Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. (2004) 84:767–801. doi: 10.1152/physrev.00041.2003
42. Liu R, Leslie KL, Martin KA. Epigenetic regulation of smooth muscle cell plasticity. Biochim Biophys Acta. (2015) 1849:448–53. doi: 10.1016/j.bbagrm.2014.06.004
43. Zhang QJ, Goddard M, Shanahan C, Shapiro L, Bennett M. Differential gene expression in vascular smooth muscle cells in primary atherosclerosis and in stent stenosis in humans. Arterioscler Thromb Vasc Biol. (2002) 22:2030–6. doi: 10.1161/01.atv.0000042206.98651.15
44. Lynch M, Barallobre-Barreiro J, Jahangiri M, Mayr M. Vascular proteomics in metabolic and cardiovascular diseases. J Intern Med. (2016) 280:325–38. doi: 10.1111/joim.12486
45. Allahverdian S, Chaabane C, Boukais K, Francis GA, Bochaton-Piallat ML. Smooth muscle cell fate and plasticity in atherosclerosis. Cardiovasc Res. (2018) 114:540–50. doi: 10.1093/cvr/cvy022
46. Sartore S, Chiavegato A, Faggin E, Franch R, Puato M, Ausoni S, et al. Contribution of adventitial fibroblasts to neointima formation and vascular remodeling: from innocent bystander to active participant. Circ Res. (2001) 89:1111–21. doi: 10.1161/hh2401.100844
47. Tinajero MG, Gotlieb AI. Recent developments in vascular adventitial pathobiology: the dynamic adventitia as a complex regulator of vascular disease. Am J Pathol. (2019) 190:520–34. doi: 10.1016/j.ajpath.2019.10.021
48. Wang G, Jacquet L, Karamariti E, Xu Q. Origin and differentiation of vascular smooth muscle cells. J Physiol. (2015) 593:3013–30. doi: 10.1113/JP270033
49. Hu Y, Zhang Z, Torsney E, Afzal AR, Davison F, Metzler B, et al. Abundant progenitor cells in the adventitia contribute to atherosclerosis of vein grafts in ApoE-deficient mice. J Clin Invest. (2004) 113:1258–65. doi: 10.1172/JCI19628
50. Torsney E, Mandal K, Halliday A, Jahangiri M, Xu Q. Characterisation of progenitor cells in human atherosclerotic vessels. Atherosclerosis. (2007) 191:259–64. doi: 10.1016/j.atherosclerosis.2006.05.033
51. Campagnolo P, Cesselli D, Al Haj Zen A, Beltrami AP, Krankel N, Katare R, et al. Human adult vena saphena contains perivascular progenitor cells endowed with clonogenic and proangiogenic potential. Circulation. (2010) 121:1735–45. doi: 10.1161/CIRCULATIONAHA.109.899252
52. Klein D, Weisshardt P, Kleff V, Jastrow H, Jakob HG, Ergun S. Vascular wall-resident Cd44+ multipotent stem cells give rise to pericytes and smooth muscle cells and contribute to new vessel maturation. PLoS One. (2011) 6:e20540. doi: 10.1371/journal.pone.0020540
53. Shimizu K, Sugiyama S, Aikawa M, Fukumoto Y, Rabkin E, Libby P, et al. Host bone-marrow cells are a source of donor intimal smooth- muscle-like cells in murine aortic transplant arteriopathy. Nat Med. (2001) 7:738–41. doi: 10.1038/89121
54. Tanaka K, Sata M, Hirata Y, Nagai R. Diverse contribution of bone marrow cells to neointimal hyperplasia after mechanical vascular injuries. Circ Res. (2003) 93:783–90. doi: 10.1161/01.RES.0000096651.13001.B4
55. Albiero M, Menegazzo L, Fadini GP. Circulating smooth muscle progenitors and atherosclerosis. Trends Cardiovasc Med. (2010) 20:133–40. doi: 10.1016/j.tcm.2010.12.001
56. Prandi F, Piola M, Soncini M, Colussi C, D’Alessandra Y, Penza E, et al. Adventitial vessel growth and progenitor cells activation in an ex vivo culture system mimicking human saphenous vein wall strain after coronary artery bypass grafting. PLoS One. (2015) 10:e0117409. doi: 10.1371/journal.pone.0117409
57. Longchamp A, Alonso F, Dubuis C, Allagnat F, Berard X, Meda P, et al. The use of external mesh reinforcement to reduce intimal hyperplasia and preserve the structure of human saphenous veins. Biomaterials. (2014) 35:2588–99. doi: 10.1016/j.biomaterials.2013.12.041
58. Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, et al. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science. (2008) 322:587–90. doi: 10.1126/science.1162667
59. Cheng Y, Ndisang JF, Tang G, Cao K, Wang R. Hydrogen sulfide-induced relaxation of resistance mesenteric artery beds of rats. Am J Physiol Heart Circ Physiol. (2004) 287:H2316–23. doi: 10.1152/ajpheart.00331.2004
60. Mustafa AK, Sikka G, Gazi SK, Steppan J, Jung SM, Bhunia AK, et al. Hydrogen sulfide as endothelium-derived hyperpolarizing factor sulfhydrates potassium channels. Circ Res. (2011) 109:1259–68. doi: 10.1161/CIRCRESAHA.111.240242
61. Shibuya N, Mikami Y, Kimura Y, Nagahara N, Kimura H. Vascular endothelium expresses 3-mercaptopyruvate sulfurtransferase and produces hydrogen sulfide. J Biochem. (2009) 146:623–6. doi: 10.1093/jb/mvp111
62. Ishii I, Akahoshi N, Yamada H, Nakano S, Izumi T, Suematsu M. Cystathionine gamma-lyase-deficient mice require dietary cysteine to protect against acute lethal myopathy and oxidative injury. J Biol Chem. (2010) 285:26358–68. doi: 10.1074/jbc.M110.147439
63. Szijarto IA, Marko L, Filipovic MR, Miljkovic JL, Tabeling C, Tsvetkov D, et al. Cystathionine gamma-lyase-produced hydrogen sulfide controls endothelial no bioavailability and blood pressure. Hypertension. (2018) 71:1210–7. doi: 10.1161/HYPERTENSIONAHA.117.10562
64. Bibli SI, Hu J, Sigala F, Wittig I, Heidler J, Zukunft S, et al. Cystathionine γ lyase sulfhydrates the RNA binding protein human antigen R to preserve endothelial cell function and delay atherogenesis. Circulation. (2019) 139:101–14. doi: 10.1161/CIRCULATIONAHA.118.034757
65. Bibli SI, Hu J, Leisegang MS, Wittig J, Zukunft S, Kapasakalidi A, et al. Shear stress regulates cystathionine gamma lyase expression to preserve endothelial redox balance and reduce membrane lipid peroxidation. Redox Biol. (2020) 28:101379. doi: 10.1016/j.redox.2019.101379
66. Yuan S, Yurdagul A Jr, Peretik JM, Alfaidi M, Al Yafeai Z, Pardue S, et al. Cystathionine γ-lyase modulates flow-dependent vascular remodeling. Arterioscler Thromb Vasc Biol. (2018) 38:2126–36. doi: 10.1161/ATVBAHA.118.311402
67. Szabo C. A timeline of hydrogen sulfide (H2S) research: from environmental toxin to biological mediator. Biochem Pharmacol. (2018) 149:5–19. doi: 10.1016/j.bcp.2017.09.010
68. Filipovic MR, Zivanovic J, Alvarez B, Banerjee R. Chemical biology of H2S signaling through persulfidation. Chem Rev. (2018) 118:1253–337. doi: 10.1021/acs.chemrev.7b00205
69. Sen N. Functional and molecular insights of hydrogen sulfide signaling and protein sulfhydration. J Mol Biol. (2017) 429:543–61. doi: 10.1016/j.jmb.2016.12.015
70. Mustafa AK, Gadalla MM, Sen N, Kim S, Mu W, Gazi SK, et al. H2S signals through protein S-Sulfhydration. Sci Signal. (2009) 2:ra72. doi: 10.1126/scisignal.2000464
71. Bibli SI, Hu J, Looso M, Weigert A, Ratiu C, Wittig J, et al. Mapping the endothelial cell S-sulfhydrome highlights the crucial role of integrin sulfhydration in vascular function. Circulation. (2021) 143:935–48. doi: 10.1161/CIRCULATIONAHA.120.051877
72. Fu L, Liu K, He J, Tian C, Yu X, Yang J. Direct proteomic mapping of cysteine persulfidation. Antioxid Redox Signal. (2020) 33:1061–76. doi: 10.1089/ars.2019.7777
73. Zivanovic J, Kouroussis E, Kohl JB, Adhikari B, Bursac B, Schott-Roux S, et al. Selective persulfide detection reveals evolutionarily conserved antiaging effects of S-Sulfhydration. Cell Metab. (2019) 30:1152–70.e13. doi: 10.1016/j.cmet.2019.10.007
74. Beard RS Jr, Bearden SE. Vascular complications of cystathionine beta-synthase deficiency: future directions for homocysteine-to-hydrogen sulfide research. Am J Physiol Heart Circ Physiol. (2011) 300:H13–26. doi: 10.1152/ajpheart.00598.2010
75. Longchamp A, MacArthur MR, Trocha K, Ganahl J, Mann CG, Kip P, et al. Plasma hydrogen sulfide is positively associated with post-operative survival in patients undergoing surgical revascularization. Front Cardiovasc Med. (2021) 8:750926. doi: 10.3389/fcvm.2021.750926
76. Macabrey D, Longchamp A, MacArthur MR, Lambelet M, Urfer S, Corpataux J-M, et al. Sodium thiosulfate acts as an H2S mimetic to prevent intimal hyperplasia via inhibition of tubulin polymerization. bioRxiv. [Preprint]. (2021). doi: 10.1101/2021.09.09.459573
77. Trocha KM, Kip P, Tao M, MacArthur MR, Trevino-Villarreal H, Longchamp A, et al. Short-term preoperative protein restriction attenuates vein graft disease via induction of cystathionine upsilon-lyase. Cardiovasc Res. (2019) 116:416–28. doi: 10.1093/cvr/cvz086
78. Yang G, Wu L, Wang R. Pro-apoptotic effect of endogenous H2S on human aorta smooth muscle cells. FASEB J. (2006) 20:553–5. doi: 10.1096/fj.05-4712fje
79. Yang G, Wu L, Bryan S, Khaper N, Mani S, Wang R. Cystathionine gamma-lyase deficiency and overproliferation of smooth muscle cells. Cardiovasc Res. (2010) 86:487–95. doi: 10.1093/cvr/cvp420
80. Wang Y, Wang X, Liang X, Wu J, Dong S, Li H, et al. Inhibition of hydrogen sulfide on the proliferation of vascular smooth muscle cells involved in the modulation of calcium sensing receptor in high homocysteine. Exp Cell Res. (2016) 347:184–91. doi: 10.1016/j.yexcr.2016.08.004
81. Zhong X, Wang Y, Wu J, Sun A, Yang F, Zheng D, et al. Calcium sensing receptor regulating smooth muscle cells proliferation through initiating cystathionine-gamma-lyase/hydrogen sulfide pathway in diabetic rat. Cell Physiol Biochem. (2015) 35:1582–98. doi: 10.1159/000373973
82. Macabrey D, Deslarzes-Dubuis C, Longchamp A, Lambelet M, Ozaki CK, Corpataux J-M, et al. Hydrogen sulfide release via the ace inhibitor zofenopril prevents intimal hyperplasia in human vein segments and in a mouse model of carotid artery stenosis. bioRxiv. [Preprint]. (2021). doi: 10.1101/2021.09.13.460108
83. Jackson-Weaver O, Osmond JM, Riddle MA, Naik JS, Gonzalez Bosc LV, Walker BR, et al. Hydrogen sulfide dilates rat mesenteric arteries by activating endothelial large-conductance Ca2+-Activated K+ channels and smooth muscle Ca2+ sparks. Am J Physiol Heart Circ Physiol. (2013) 304:H1446–54. doi: 10.1152/ajpheart.00506.2012
84. Lee KY, Kim JR, Choi HC. Gliclazide, a KATP channel blocker, inhibits vascular smooth muscle cell proliferation through the CaMKKβ-AMPK pathway. Vascul Pharmacol. (2018) 102:21–8. doi: 10.1016/j.vph.2018.01.001
85. Altaany Z, Yang G, Wang R. Crosstalk between hydrogen sulfide and nitric oxide in endothelial cells. J Cell Mol Med. (2013) 17:879–88. doi: 10.1111/jcmm.12077
86. Coletta C, Papapetropoulos A, Erdelyi K, Olah G, Modis K, Panopoulos P, et al. Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation. Proc Natl Acad Sci U S A. (2012) 109:9161–6. doi: 10.1073/pnas.1202916109
87. Papapetropoulos A, Pyriochou A, Altaany Z, Yang G, Marazioti A, Zhou Z, et al. Hydrogen sulfide is an endogenous stimulator of angiogenesis. Proc Natl Acad Sci U S A. (2009) 106:21972–7. doi: 10.1073/pnas.0908047106
88. Katsouda A, Bibli SI, Pyriochou A, Szabo C, Papapetropoulos A. Regulation and role of endogenously produced hydrogen sulfide in angiogenesis. Pharmacol Res. (2016) 113:175–85. doi: 10.1016/j.phrs.2016.08.026
89. Cai WJ, Wang MJ, Moore PK, Jin HM, Yao T, Zhu YC. The novel proangiogenic effect of hydrogen sulfide is dependent on Akt phosphorylation. Cardiovasc Res. (2007) 76:29–40. doi: 10.1016/j.cardiores.2007.05.026
90. Tao BB, Liu SY, Zhang CC, Fu W, Cai WJ, Wang Y, et al. VEGFR2 functions as an H2S-targeting receptor protein kinase with its novel Cys1045-Cys1024 disulfide bond serving as a specific molecular switch for hydrogen sulfide actions in vascular endothelial cells. Antioxid Redox Signal. (2013) 19:448–64. doi: 10.1089/ars.2012.4565
91. Longchamp A, Mirabella T, Arduini A, MacArthur MR, Das A, Trevino-Villarreal JH, et al. Amino acid restriction triggers angiogenesis via GCN2/ATF4 regulation of VEGF and H2S production. Cell. (2018) 173:117–29.e14. doi: 10.1016/j.cell.2018.03.001
92. Altaany Z, Ju Y, Yang G, Wang R. The coordination of S-Sulfhydration, S-nitrosylation, and phosphorylation of endothelial nitric oxide synthase by hydrogen sulfide. Sci Signal. (2014) 7:ra87. doi: 10.1126/scisignal.2005478
93. Szabo C, Papapetropoulos A. Hydrogen sulphide and angiogenesis: mechanisms and applications. Br J Pharmacol. (2011) 164:853–65. doi: 10.1111/j.1476-5381.2010.01191.x
94. Moccia F, Bertoni G, Pla AF, Dragoni S, Pupo E, Merlino A, et al. Hydrogen sulfide regulates intracellular Ca2+ concentration in endothelial cells from excised rat aorta. Curr Pharm Biotechnol. (2011) 12:1416–26. doi: 10.2174/138920111798281117
95. Jang H, Oh MY, Kim YJ, Choi IY, Yang HS, Ryu WS, et al. Hydrogen sulfide treatment induces angiogenesis after cerebral ischemia. J Neurosci Res. (2014) 92:1520–8. doi: 10.1002/jnr.23427
96. Szabo C. Hydrogen sulfide, an enhancer of vascular nitric oxide signaling: mechanisms and implications. Am J Physiol Cell Physiol. (2017) 312:C3–15. doi: 10.1152/ajpcell.00282.2016
97. Pan LL, Qin M, Liu XH, Zhu YZ. The role of hydrogen sulfide on cardiovascular homeostasis: an overview with update on immunomodulation. Front Pharmacol. (2017) 8:686. doi: 10.3389/fphar.2017.00686
98. Liu Z, Han Y, Li L, Lu H, Meng G, Li X, et al. The hydrogen sulfide donor, GYY4137, exhibits anti-atherosclerotic activity in high fat fed apolipoprotein E(-/-) mice. Br J Pharmacol. (2013) 169:1795–809. doi: 10.1111/bph.12246
99. Fan J, Zheng F, Li S, Cui C, Jiang S, Zhang J, et al. Hydrogen sulfide lowers hyperhomocysteinemia dependent on cystathionine γ lyase S-sulfhydration in ApoE-knockout atherosclerotic mice. Br J Pharmacol. (2019) 176:3180–92. doi: 10.1111/bph.14719
100. Elrod JW, Calvert JW, Morrison J, Doeller JE, Kraus DW, Tao L, et al. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc Natl Acad Sci U S A. (2007) 104:15560–5. doi: 10.1073/pnas.0705891104
101. Zhang Y, Li H, Zhao G, Sun A, Zong NC, Li Z, et al. Hydrogen sulfide attenuates the recruitment of CD11b+Gr-1+ myeloid cells and regulates Bax/Bcl-2 signaling in myocardial ischemia injury. Sci Rep. (2014) 4:4774. doi: 10.1038/srep04774
102. Qipshidze N, Metreveli N, Mishra PK, Lominadze D, Tyagi SC. Hydrogen sulfide mitigates cardiac remodeling during myocardial infarction via improvement of angiogenesis. Int J Biol Sci. (2012) 8:430–41. doi: 10.7150/ijbs.3632
103. Wu T, Li H, Wu B, Zhang L, Wu SW, Wang JN, et al. Hydrogen sulfide reduces recruitment of CD11b+Gr-1+ cells in mice with myocardial infarction. Cell Transplant. (2017) 26:753–64. doi: 10.3727/096368917X695029
104. Predmore BL, Kondo K, Bhushan S, Zlatopolsky MA, King AL, Aragon JP, et al. The polysulfide diallyl trisulfide protects the ischemic myocardium by preservation of endogenous hydrogen sulfide and increasing nitric oxide bioavailability. Am J Physiol Heart Circ Physiol. (2012) 302:H2410–8. doi: 10.1152/ajpheart.00044.2012
105. Ravindran S, Jahir Hussain S, Boovarahan SR, Kurian GA. Sodium thiosulfate post-conditioning protects rat hearts against ischemia reperfusion injury via reduction of apoptosis and oxidative stress. Chem Biol Interact. (2017) 274:24–34. doi: 10.1016/j.cbi.2017.07.002
106. Wang Y, Zhao X, Jin H, Wei H, Li W, Bu D, et al. Role of hydrogen sulfide in the development of atherosclerotic lesions in apolipoprotein E knockout mice. Arterioscler Thromb Vasc Biol. (2009) 29:173–9. doi: 10.1161/ATVBAHA.108.179333
107. Du J, Huang Y, Yan H, Zhang Q, Zhao M, Zhu M, et al. Hydrogen sulfide suppresses oxidized low-density lipoprotein (ox-LDL)-stimulated monocyte chemoattractant protein 1 generation from macrophages via the nuclear factor κB (NF-κB) pathway. J Biol Chem. (2014) 289:9741–53. doi: 10.1074/jbc.M113.517995
108. Wang XH, Wang F, You SJ, Cao YJ, Cao LD, Han Q, et al. Dysregulation of cystathionine gamma-lyase (CSE)/hydrogen sulfide pathway contributes to ox-LDL-induced inflammation in macrophage. Cell Signal. (2013) 25:2255–62. doi: 10.1016/j.cellsig.2013.07.010
109. Sen N, Paul BD, Gadalla MM, Mustafa AK, Sen T, Xu R, et al. Hydrogen sulfide-linked sulfhydration of NF-κB mediates its antiapoptotic actions. Mol Cell. (2012) 45:13–24. doi: 10.1016/j.molcel.2011.10.021
110. Dufton N, Natividad J, Verdu EF, Wallace JL. Hydrogen sulfide and resolution of acute inflammation: a comparative study utilizing a novel fluorescent probe. Sci Rep. (2012) 2:499. doi: 10.1038/srep00499
111. Lin Y, Chen Y, Zhu N, Zhao S, Fan J, Liu E. Hydrogen sulfide inhibits development of atherosclerosis through up-regulating protein S-nitrosylation. Biomed Pharmacother. (2016) 83:466–76. doi: 10.1016/j.biopha.2016.07.003
112. Shefa U, Kim MS, Jeong NY, Jung J. Antioxidant and cell-signaling functions of hydrogen sulfide in the central nervous system. Oxid Med Cell Longev. (2018) 2018:1873962. doi: 10.1155/2018/1873962
113. Cheung SH, Lau JYW. Hydrogen sulfide mediates athero-protection against oxidative stress via S-sulfhydration. PLoS One. (2018) 13:e0194176. doi: 10.1371/journal.pone.0194176
114. Corsello T, Komaravelli N, Casola A. Role of hydrogen sulfide in NRF2- and sirtuin-dependent maintenance of cellular redox balance. Antioxidants (Basel). (2018) 7:129. doi: 10.3390/antiox7100129
115. Xie L, Gu Y, Wen M, Zhao S, Wang W, Ma Y, et al. Hydrogen sulfide induces keap1 S-sulfhydration and suppresses diabetes-accelerated atherosclerosis via NRF2 activation. Diabetes. (2016) 65:3171–84. doi: 10.2337/db16-0020
116. Calvert JW, Jha S, Gundewar S, Elrod JW, Ramachandran A, Pattillo CB, et al. Hydrogen sulfide mediates cardioprotection through NRF2 signaling. Circ Res. (2009) 105:365–74. doi: 10.1161/CIRCRESAHA.109.199919
117. Peake BF, Nicholson CK, Lambert JP, Hood RL, Amin H, Amin S, et al. Hydrogen sulfide preconditions the db/db diabetic mouse heart against ischemia-reperfusion injury by activating Nrf2 signaling in an Erk-dependent manner. Am J Physiol Heart Circ Physiol. (2013) 304:H1215–24. doi: 10.1152/ajpheart.00796.2012
118. Nicholson CK, Lambert JP, Molkentin JD, Sadoshima J, Calvert JW. Thioredoxin 1 is essential for sodium sulfide-mediated cardioprotection in the setting of heart failure. Arterioscler Thromb Vasc Biol. (2013) 33:744–51. doi: 10.1161/ATVBAHA.112.300484
119. Tian D, Dong J, Jin S, Teng X, Wu Y. Endogenous hydrogen sulfide-mediated MAPK inhibition preserves endothelial function through TXNIP signaling. Free Radic Biol Med. (2017) 110:291–9. doi: 10.1016/j.freeradbiomed.2017.06.016
120. Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. (2005) 120:483–95. doi: 10.1016/j.cell.2005.02.001
121. Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. (2003) 552:335–44. doi: 10.1113/jphysiol.2003.049478
122. Paul BD, Snyder SH, Kashfi K. Effects of hydrogen sulfide on mitochondrial function and cellular bioenergetics. Redox Biol. (2021) 38:101772. doi: 10.1016/j.redox.2020.101772
123. Sun HJ, Zhao MX, Ren XS, Liu TY, Chen Q, Li YH, et al. Salusin-β promotes vascular smooth muscle cell migration and intimal hyperplasia after vascular injury via ROS/NFκB/MMP-9 Pathway. Antioxid Redox Signal. (2016) 24:1045–57. doi: 10.1089/ars.2015.6475
124. Wang Y, Ji L, Jiang R, Zheng L, Liu D. Oxidized high-density lipoprotein induces the proliferation and migration of vascular smooth muscle cells by promoting the production of ROS. J Atheroscler Thromb. (2014) 21:204–16. doi: 10.5551/jat.19448
125. Benavides GA, Squadrito GL, Mills RW, Patel HD, Isbell TS, Patel RP, et al. Hydrogen sulfide mediates the vasoactivity of garlic. Proc Natl Acad Sci U S A. (2007) 104:17977–82. doi: 10.1073/pnas.0705710104
126. Chuah SC, Moore PK, Zhu YZ. S-allylcysteine mediates cardioprotection in an acute myocardial infarction rat model via a hydrogen sulfide-mediated pathway. Am J Physiol Heart Circ Physiol. (2007) 293:H2693–701. doi: 10.1152/ajpheart.00853.2007
127. Syu JN, Yang MD, Tsai SY, Chiang EI, Chiu SC, Chao CY, et al. S-allylcysteine improves blood flow recovery and prevents ischemic injury by augmenting neovasculogenesis. Cell Transplant. (2017) 26:1636–47. doi: 10.1177/0963689717724792
128. Wen YD, Zhu YZ. The pharmacological effects of s-propargyl-cysteine, a novel endogenous H2S-producing compound. Handb Exp Pharmacol. (2015) 230:325–36. doi: 10.1007/978-3-319-18144-8_16
129. Wang Q, Liu HR, Mu Q, Rose P, Zhu YZ. S-Propargyl-cysteine protects both adult rat hearts and neonatal cardiomyocytes from ischemia/hypoxia injury: the contribution of the hydrogen sulfide-mediated pathway. J Cardiovasc Pharmacol. (2009) 54:139–46. doi: 10.1097/FJC.0b013e3181ac8e12
130. Huang C, Kan J, Liu X, Ma F, Tran BH, Zou Y, et al. Cardioprotective effects of a novel hydrogen sulfide agent-controlled release formulation of S-Propargyl-Cysteine on heart failure rats and molecular mechanisms. PLoS One. (2013) 8:e69205. doi: 10.1371/journal.pone.0069205
131. Kan J, Guo W, Huang C, Bao G, Zhu Y, Zhu YZ. S-propargyl-cysteine, a novel water-soluble modulator of endogenous hydrogen sulfide, promotes angiogenesis through activation of signal transducer and activator of transcription 3. Antioxid Redox Signal. (2014) 20:2303–16. doi: 10.1089/ars.2013.5449
132. Bucci M, Vellecco V, Cantalupo A, Brancaleone V, Zhou Z, Evangelista S, et al. Hydrogen sulfide accounts for the peripheral vascular effects of zofenopril independently of ace inhibition. Cardiovasc Res. (2014) 102:138–47. doi: 10.1093/cvr/cvu026
133. Christe V, Waeber G, Vollenweider P, Marques-Vidal P. Antihypertensive drug treatment changes in the general population: the colaus study. BMC Pharmacol Toxicol. (2014) 15:20. doi: 10.1186/2050-6511-15-20
135. Borghi C, Bacchelli S, Esposti DD, Bignamini A, Magnani B, Ambrosioni E. Effects of the administration of an angiotensin-converting enzyme inhibitor during the acute phase of myocardial infarction in patients with arterial hypertension. SMILE study investigators. Survival of myocardial infarction long-term evaluation. Am J Hypertens. (1999) 12:665–72. doi: 10.1016/s0895-7061(99)00042-4
136. Borghi C, Omboni S, Novo S, Vinereanu D, Ambrosio G, Ambrosioni E. Efficacy and safety of zofenopril versus ramipril in the treatment of myocardial infarction and heart failure: a review of the published and unpublished data of the randomized double-blind smile-4 study. Adv Ther. (2018) 35:604–18. doi: 10.1007/s12325-018-0697-x
137. Ambrosioni E, Borghi C, Magnani B. The effect of the angiotensin-converting-enzyme inhibitor zofenopril on mortality and morbidity after anterior myocardial infarction. The survival of myocardial infarction long-term evaluation (SMILE) study investigators. N Engl J Med. (1995) 332:80–5. doi: 10.1056/NEJM199501123320203
138. Macabrey D, Deslarzes-Dubuis C, Longchamp A, Lambelet M, Ozaki CK, Corpataux JM, et al. Hydrogen sulphide release via the angiotensin converting enzyme inhibitor zofenopril prevents intimal hyperplasia in human vein segments and in a mouse model of carotid artery stenosis. Eur J Vasc Endovasc Surg. (2021) 63:336–46. doi: 10.1016/j.ejvs.2021.09.032
139. Pavlovskiy Y, Yashchenko A, Zayachkivska O. H2S donors reverse age-related gastric malfunction impaired due to fructose-induced injury via CBS, CSE, and TST expression. Front Pharmacol. (2020) 11:1134. doi: 10.3389/fphar.2020.01134
140. Wallace JL, Nagy P, Feener TD, Allain T, Ditroi T, Vaughan DJ, et al. A proof-of-concept, phase 2 clinical trial of the gastrointestinal safety of a hydrogen sulfide-releasing anti-inflammatory drug. Br J Pharmacol. (2020) 177:769–77. doi: 10.1111/bph.14641
141. Li L, Rossoni G, Sparatore A, Lee LC, Del Soldato P, Moore PK. Anti-inflammatory and gastrointestinal effects of a novel diclofenac derivative. Free Radic Biol Med. (2007) 42:706–19. doi: 10.1016/j.freeradbiomed.2006.12.011
142. Wallace JL, Caliendo G, Santagada V, Cirino G, Fiorucci S. Gastrointestinal safety and anti-inflammatory effects of a hydrogen sulfide-releasing diclofenac derivative in the rat. Gastroenterology. (2007) 132:261–71. doi: 10.1053/j.gastro.2006.11.042
143. Baskar R, Sparatore A, Del Soldato P, Moore PK. Effect of S-diclofenac, a novel hydrogen sulfide releasing derivative inhibit rat vascular smooth muscle cell proliferation. Eur J Pharmacol. (2008) 594:1–8. doi: 10.1016/j.ejphar.2008.07.029
144. Jha S, Calvert JW, Duranski MR, Ramachandran A, Lefer DJ. Hydrogen sulfide attenuates hepatic ischemia-reperfusion injury: role of antioxidant and antiapoptotic signaling. Am J Physiol Heart Circ Physiol. (2008) 295:H801–6. doi: 10.1152/ajpheart.00377.2008
145. Kondo K, Bhushan S, King AL, Prabhu SD, Hamid T, Koenig S, et al. H2S protects against pressure overload-induced heart failure via upregulation of endothelial nitric oxide synthase. Circulation. (2013) 127:1116–27. doi: 10.1161/CIRCULATIONAHA.112.000855
146. Polhemus DJ, Li Z, Pattillo CB, Gojon G Sr, Gojon G Jr, Giordano T, et al. A Novel hydrogen sulfide prodrug, Sg1002, promotes hydrogen sulfide and nitric oxide bioavailability in heart failure patients. Cardiovasc Ther. (2015) 33:216–26. doi: 10.1111/1755-5922.12128
147. Olson KR, Deleon ER, Gao Y, Hurley K, Sadauskas V, Batz C, et al. Thiosulfate: a readily accessible source of hydrogen sulfide in oxygen sensing. Am J Physiol Regul Integr Comp Physiol. (2013) 305:R592–603. doi: 10.1152/ajpregu.00421.2012
148. Snijder PM, Frenay AR, de Boer RA, Pasch A, Hillebrands JL, Leuvenink HG, et al. Exogenous administration of thiosulfate, a donor of hydrogen sulfide, attenuates angiotensin II-induced hypertensive heart disease in rats. Br J Pharmacol. (2015) 172:1494–504. doi: 10.1111/bph.12825
149. Peng T, Zhuo L, Wang Y, Jun M, Li G, Wang L, et al. Systematic review of sodium thiosulfate in treating calciphylaxis in chronic kidney disease patients. Nephrology (Carlton). (2018) 23:669–75. doi: 10.1111/nep.13081
150. Seedial SM, Ghosh S, Saunders RS, Suwanabol PA, Shi X, Liu B, et al. Local drug delivery to prevent restenosis. J Vasc Surg. (2013) 57:1403–14. doi: 10.1016/j.jvs.2012.12.069
151. Kip P, Tao M, Trocha KM, MacArthur MR, Peters HAB, Mitchell SJ, et al. Periprocedural hydrogen sulfide therapy improves vascular remodeling and attenuates vein graft disease. J Am Heart Assoc. (2020) 9:e016391. doi: 10.1161/JAHA.120.016391
152. Sharma S, Christopoulos C, Kukreja N, Gorog DA. Local drug delivery for percutaneous coronary intervention. Pharmacol Ther. (2011) 129:260–6. doi: 10.1016/j.pharmthera.2010.11.003
153. Costa S, Muscara MN, Allain T, Dallazen J, Gonzaga L, Buret AG, et al. Enhanced analgesic effects and gastrointestinal safety of a novel, hydrogen sulfide-releasing anti-inflammatory drug (ATB-352): a role for endogenous cannabinoids. Antioxid Redox Signal. (2020) 33:1003–9. doi: 10.1089/ars.2019.7884
Keywords: restenosis, hydrogen sulfide (H2S), intimal and medial thickening, vascular SMCs, intimal hyperplasia
Citation: Macabrey D, Longchamp A, Déglise S and Allagnat F (2022) Clinical Use of Hydrogen Sulfide to Protect Against Intimal Hyperplasia. Front. Cardiovasc. Med. 9:876639. doi: 10.3389/fcvm.2022.876639
Received: 15 February 2022; Accepted: 18 March 2022;
Published: 11 April 2022.
Edited by:
Jinwei Tian, The Second Affiliated Hospital of Harbin Medical University, ChinaReviewed by:
Ming Tao, Brigham and Women’s Hospital and Harvard Medical School, United StatesHong Li, Harbin Medical University (Daqing), China
Hiroaki Neki, Hamamatsu University School of Medicine, Japan
Daniele Mancardi, University of Turin, Italy
Copyright © 2022 Macabrey, Longchamp, Déglise and Allagnat. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Florent Allagnat, Florent.allagnat@chuv.ch
†These authors have contributed equally to this work and share first authorship
‡These authors have contributed equally to this work and share senior authorship