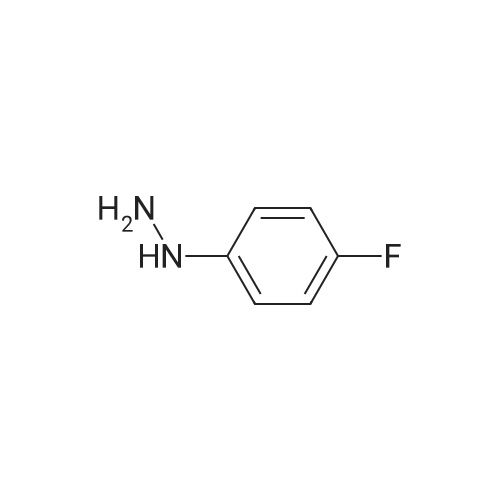
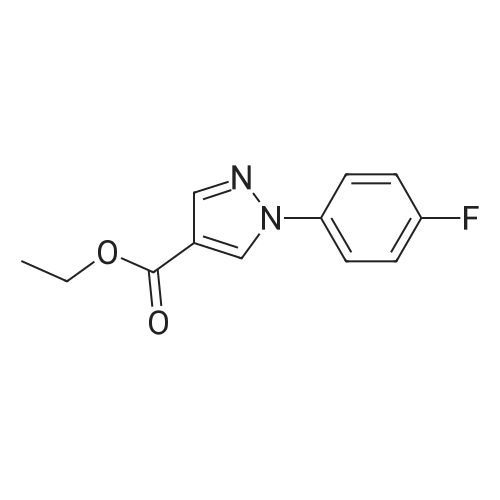
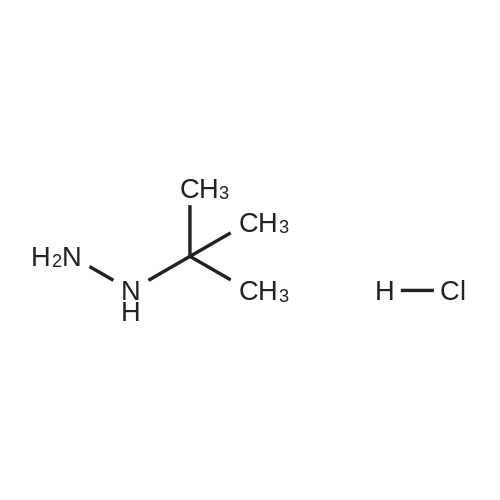
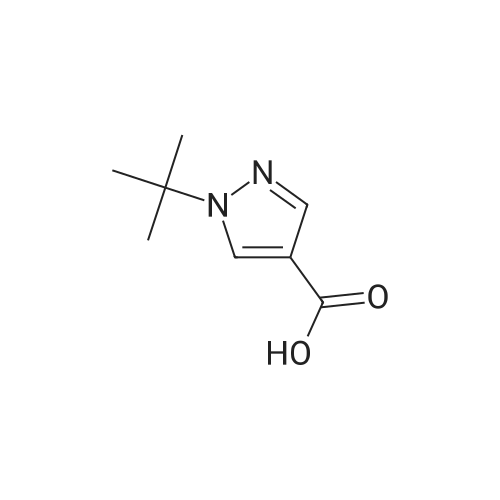
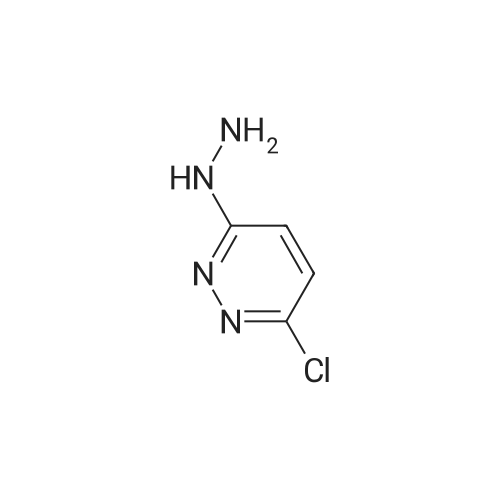
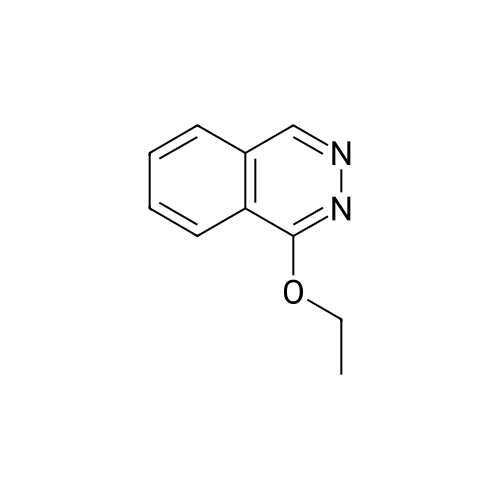
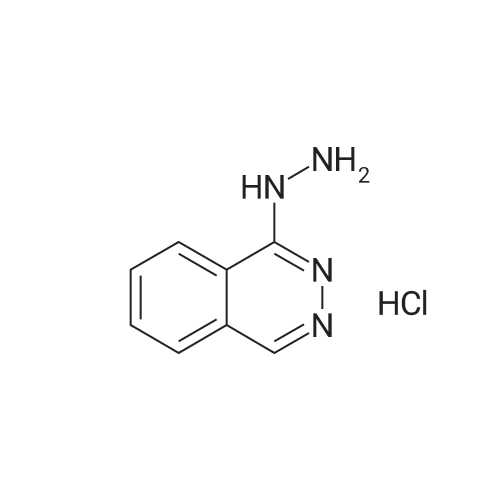
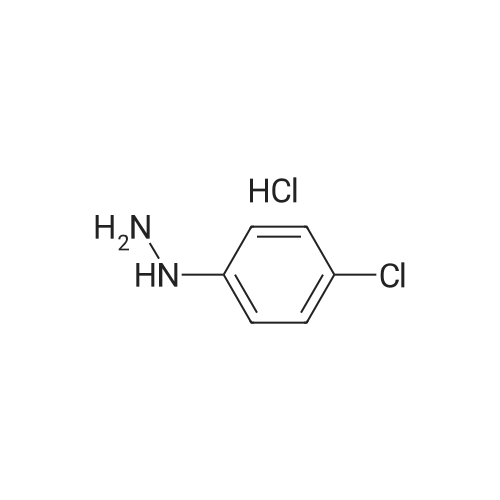
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
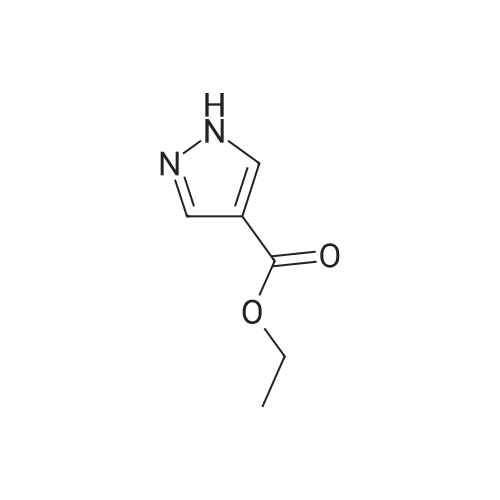
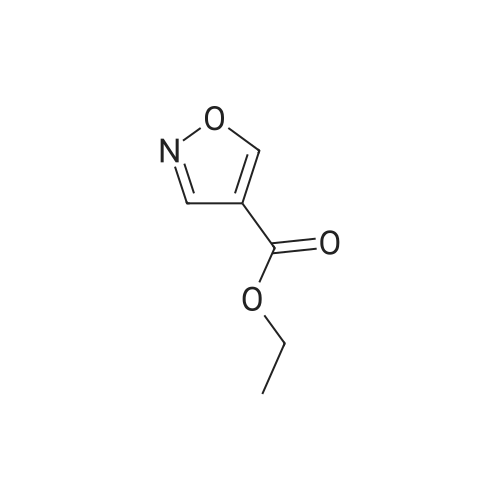
6.2 g (193 mmoles) of hydrazine was added, with ice-cooling, to a solution of 27.6 g (192 mmoles) of (ethoxycarbonyl)malondialdehyde dissolved in 150 ml of ethanol. The mixture was stirred at room temperature for 17 hours to give rise to a reaction. The reaction mixture was subjected to vacuum distillation to remove the ethanol contained therein. The residue was purified by silica gel column chromatography (developing solvent: dichloromethane-ethyl acetate mixed solvent) to obtain 19.4 g (72.4percent) of ethyl 1H-pyrazole-4-carboxylate as yellow crystals. 1H-NMR [CDCl3/TMS, δ (ppm)]:
8.08 (2H,s), 5.30 (1H,s) 4.31 (2H,q), 1.36 (3H,t)
72.4%
With hydrazine In ethanol at 20℃; for 17 h;
REFERENCE EXAMPLE 29 Production of ethyl 1H-pyrazole-4-carboxylate; 6.2 g (193 mmoles) of hydrazine was added, with ice-cooling, to a solution of 27.6 g (192 mmoles) of (ethoxycarbonyl)malondialdehyde dissolved in 150 ml of ethanol. The mixture was stirred at room temperature for 17 hours to give rise to a reaction. The reaction mixture was subjected to vacuum distillation to remove the ethanol contained therein. The residue was purified by silica gel column chromatography (developing solvent: dichloromethane-ethyl acetate mixed solvent) to obtain 19.4 g (72.4percent) of ethyl 1H-pyrazole-4-carboxylate as yellow crystals. 1H-NMR [CDCl3/TMS, δ (ppm)]: 8.08 (2H,s), 5.30 (1H,s), 4.31 (2H,q), 1.36 (3H,t)
Reference:
[1] Tetrahedron, 2008, vol. 64, # 33, p. 7745 - 7758
[2] Journal of Heterocyclic Chemistry, 1993, vol. 30, # 4, p. 865 - 872
[3] Patent: EP1364946, 2003, A1, . Location in patent: Page/Page column 196
[4] Patent: US2005/256004, 2005, A1, . Location in patent: Page/Page column 32
j00324J Step A: ethyl l-methyl-1H-pyrazole-4-carboxylate: To a 3000-mL three- necked flask was added ethyl 2-formyl-3-oxopropanoate (100 g, 694 mmol), followed by anhydrous 200-proof EtOH (694 mL) to obtain a clear yellowish solution. The reaction was cooled in an ice bath to 5 °C, and then methyihydrazine (35.8 mL, 680 mmol) was added dropwise. A vigorous exotherm was observed during hydrazine addition and the temperature was kept below 12 °C by controlling the addition rate. After the hydrazine addition was complete, the ice bath was removed, and the reaction was allowed to stir at ambient temperature overnight. The reaction was concentrated on a rotary evaporator to a crude orange oil. The crude was taken up in DCM and re-concentrated, then on high vacuum for 2 days to yield tan orange oil. LC/MS and ‘H NMR showed essentially pure ethyl 1-methyl- 1 H-pyrazole-4-carboxylate (106 g, 99.1percent).
99%
at 5 - 20℃; for 16 h;
[00352] Step A: ethyl 1 -methyl- 1 H-pyrazole-4-carboxylate: To a 3000-mL three- necked flask was added ethyl 2-formyl-3-oxopropanoate (100 g, 694 mmol), followed by anhydrous 200-proof EtOH (694 mL). The reaction was cooled in an ice bath to 5 °C, and then methylhydrazine (35.8 mL, 680 mmol) was added dropwise. A vigorous exotherm was observed during hydrazine addition and the temperature was kept below 12 °C by controlling the addition rate. After the hydrazine addition was complete, the ice bath was removed, and the reaction was allowed to stir at ambient temperature for 16 hours. The reaction was concentrated in vacuo and the residue dissolved in DCM and re-concentrated, then dried for 2 days to yield ethyl 1 -methyl- lH-pyrazole-4-carboxylate (106 g, 99percent yield) as a tan orange oil. MS (apci) m/z = 155.1 (M+H).
99.1%
at 5 - 20℃;
1006631 Step A: ethyl 1-methyl-1H-pyrazole-4-carboxylate: To a 3000-mL three- necked flask was added ethyl 2-formyl-3-oxopropanoate (100 g, 694 mmol), followed by anhydrous 200-proof EtOH (694 mE) to obtain a clear yellowish solution. The reaction was cooled in an ice bath to 5 °C, and then methyihydrazine (35.8 mL, 680 mmol) was added dropwise. A vigorous exotherm was observed during hydrazine addition and the temperature was kept below 12 °C by controlling the addition rate. After the hydrazine addition was complete, the ice bath was removed, and the reaction was allowed to stir at ambient temperature overnight. The reaction was concentrated on a rotary evaporator to a crude orange oil. The crude was taken up in DCM and re-concentrated, then on high vacuum for 2 days to yield tan orange oil. LC/MS and ‘H NMR showed essentially pure ethyl 1-methyl-1H- pyrazole-4-carboxylate (106 g, 99.1percent).
99%
at 5 - 20℃; for 16 h;
Intermediate 24 1 ',4-dimethyl- 1 -phenyl- 1 H, 1 -3 ,4'-bipyrazol-5-amine [00463] Step A: ethyl 1 -methyl- 1 H-pyrazole-4-carboxylate: To a 3000-mL three-necked flask was added ethyl 2-formyl-3-oxopropanoate (100 g, 694 mmol), followed by anhydrous 200-proof EtOH (694 mL). The reaction was cooled in an ice bath to 5 °C, and then methylhydrazine (35.8 mL, 680 mmol) was added dropwise. A vigorous exotherm was observed during hydrazine addition and the temperature was kept below 12 °C by controlling the addition rate. After the hydrazine addition was complete, the ice bath was removed, and the reaction was allowed to stir at ambient temperature for 16 hours. The reaction was concentrated under vacuum and the residue dissolved in DCM and re-concentrated, then dried for 2 days to yield ethyl 1 -methyl- lH-pyrazole-4-carboxylate (106 g, 99percent yield) as a tan orange oil. MS (apci) m/z = 155.1 (M+H).
99%
at 5 - 20℃;
To a 3000-mL three- necked flask was added ethyl 2-formyl-3-oxopropanoate (100 g, 694 mmol), followed by anhydrous 200-proof EtOH (694 mL). The reaction was cooled in an ice bath to 5 °C, and then methylhydrazine (35.8 mL, 680 mmol) was added dropwise. A vigorous exotherm was observed during hydrazine addition and the temperature was kept below 12 °C by controlling the addition rate. After the hydrazine addition was complete, the ice bath was removed, and the reaction was allowed to stir at ambient temperature for 16 hours. The reaction was concentrated under vacuum and the residue dissolved in DCM and re-concentrated, then dried for 2 days to yield ethyl 1 -methyl- lH-pyrazole-4-carboxy late (106 g, 99percent yield) as a tan orange oil. MS (apci) m/z = 155.1 (M+H).
99%
at 12 - 20℃; for 16 h;
1006621 Step A: ethyl 1-methyl-1H-pyrazole-4-carboxylate: To a 3000-mL three- necked flask was added ethyl 2-formyl-3-oxopropanoate (100 g, 694 mmol), followed by anhydrous 200-proof EtOH (694 mL). The reaction was cooled in an ice bath to 5 °C, and then methylhydrazine (35.8 mL, 680 mmol) was added dropwise. A vigorous exotherm was observed during hydrazine addition and the temperature was kept below 12 °C by controlling the addition rate. After the hydrazine addition was complete, the ice bath was removed, and the reaction was allowed to stir at ambient temperature for 16 hours. The reaction was concentrated in vacuo and the residue dissolved in DCM and re-concentrated, then dried for 2 days to yield ethyl 1-methyl-i H-pyrazole-4-carboxylate (106 g, 99percent yield) as a tan orange oil. MS (apci) m/z = 155.1 (M+H).
99.1%
at 5 - 20℃;
To a 3000-mL three-necked flask was added ethyl 2-formyl-3-oxopropanoate (100 g, 694 mmol), followed by anhydrous 200-proof EtOH (694 mL) to obtain a clear yellowish solution. The reaction was cooled in an ice bath to 5 °C, and then methylhydrazine (35.8 mL, 680 mmol) was added dropwise. A vigorous exotherm was observed during hydrazine addition and the temperature was kept below 12 °C by controlling the addition rate. After the hydrazine addition was complete, the ice bath was removed, and the reaction was allowed to stir at ambient temperature overnight. The reaction was concentrated on a rotary evaporator to a crude orange oil. The crude was taken up in DCM and re-concentrated, then on high vacuum for 2 days to yield the title compound as a tan orange oil (106 g, 99.1percent yield).
With sodium hydride In diethyl ether at 0 - 20℃; for 15 h;
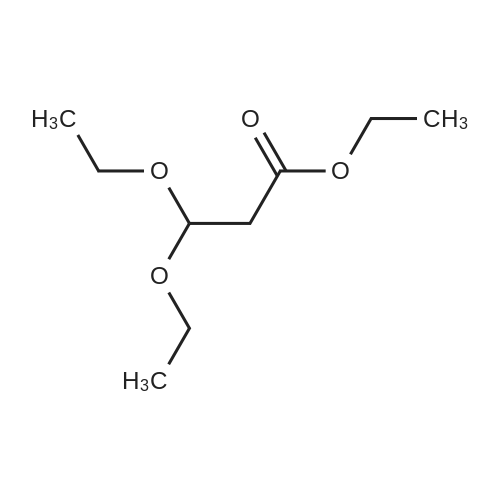
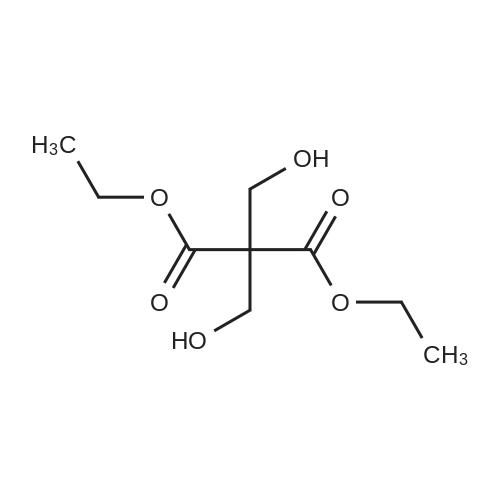
12.6 g of sodium hydride (purity: 60percent, 525.0 mmoles) was washed with diethyl ether by decantation several times and then made into a solution in 500 ml of diethyl ether. Thereto were added, in a nitrogen current at 0 to 10°C, 194 g (2.6 moles) of ethyl formate and 50 g (262.0 mmoles) of ethyl 3,3-diethoxy-propionate. The resulting mixture was stirred at room temperature for 15 hours to give rise to a reaction. After confirmation of the completion of the reaction, the reaction mixture was poured into water, followed by washing with diethyl ether. The resulting aqueous layer was allowed to have a pH of 1 with hydrochloric acid, followed by extraction with dichloromethane. The resulting organic layer was washed with an aqueous sodium chloride solution and then dried over anhydrous magnesium sulfate. The resulting solution was subjected to vacuum distillation to remove the solvent contained therein, to obtain 37.6 g (yield: 100percent) of crude (ethoxycarbonyl)malondialdehyde as a dark red oily substance. 1H-NMR [CDCl3/TMS, δ (ppm)]:
With sodium hydride In diethyl ether at 0 - 20℃; for 15 h;
REFERENCE EXAMPLE 28 Production of (ethoxycarbonyl)malondialdehyde; 12.6 g of sodium hydride (purity: 60percent, 525.0 mmoles) was washed with diethyl ether by decantation several times and then made into a solution in 500 ml of diethyl ether. Thereto were added, in a nitrogen current at 0 to 10° C., 194 g (2.6 moles) of ethyl formate and 50 g (262.0 mmoles) of ethyl 3,3-diethoxy-propionate. The resulting mixture was stirred at room temperature for 15 hours to give rise to a reaction. After confirmation of the completion of the reaction, the reaction mixture was poured into water, followed by washing with diethyl ether. The resulting aqueous layer was allowed to have a pH of 1 with hydrochloric acid, followed by extraction with dichloromethane. The resulting organic layer was washed with an aqueous sodium chloride solution and then dried over anhydrous magnesium sulfate. The resulting solution was subjected to vacuum distillation to remove the solvent contained therein, to obtain 37.6 g (yield: 100percent) of crude (ethoxycarbonyl)malondialdehyde as a dark red oily substance. 1H-NMR [CDCl3/TMS, δ (ppm)]: 9.09 (2H,s), 5.26 (1H,s), 4.27 (2H,q), 1.28 (3H,t)
99.7%
With sodium hydride In tetrahydrofuran at 0 - 20℃;
[00442] To a stirred suspension of sodium hydride (60percent, 1.68 g, 42.1 mmol) in THF (20 mL) was added ethyl formate (8.5 mL, 105.7 mmol). The solution was cooled to 0 °C and a solution of ethyl 3,3-diethoxypropanoate (4 g, 21.0 mmol) in THF (10 mL) was added dropwise over 30 mins and the reaction mixture stirred at r.t. overnight. 2M aq HCl (30 mL) was added under whilst cooling with ice and the reaction stirred at r.t. for 30 mins. The reaction mixture was extracted with diethyl ether (2 x 50 mL) and the combined organic extracts were dried over MgS04 and concentrated in vacuo to afford the title compound (3.02 g, 99.7percent) as an amber liquid. [00443] 1H NMR (250 MHz, Chloroform-d) δ 9.13 (s, 2H), 4.29 (q, J = 7.1 Hz, 2H), 1.37 - 1.28 (m, 3H).
74.2%
With sodium hydride In diethyl ether; hexane at 20℃; for 15 h; Cooling with ice
Weighed out sodium hydride (2.46 g, 61.5 mmol) in a dry 100-mL pear flask. Washed with hexanes then with diethyl ether. Suspended in ether (100 mL), cooled in ice bath then ethyl formate (24.84 ml, 308 mmol) was added then ethyl 3,3-diethoxypropanoate (5.98 ml, 30.8 mmol). The resulting mixture was stirred at room temperature for 15 hours to give rise to a reaction. After confirmation of the completion of the reaction, the reaction mixture was poured into water, followed by washing with diethyl ether. The resulting aqueous layer was allowed to have a pH of 1 with hydrochloric acid, followed by extraction with dichloromethane. The resulting organic layer was washed with an aqueous sodium chloride solution and then dried over anhydrous magnesium sulfate, filtered and concentrated to give ethyl 2-formyl-3-oxopropanoate (3.29 g, 22.83 mmol, 74.2 percent yield) as a golden syrup
70%
Stage #1: With sodium hydride In tetrahydrofuran at 0 - 20℃; Stage #2: With hydrogenchloride In water
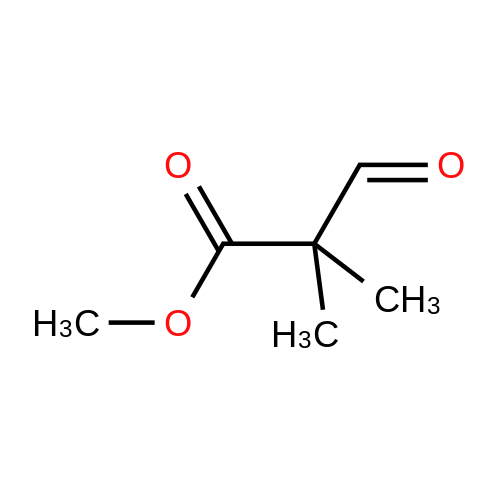
EXAMPLE 1Preparation of Ethyl-2-formyl-3-oxopropionate[0059] A three- or four-neck round bottom flask equipped with magnetic stir bar, thermocouple, digital thermometer, gas inlet and outlet and addition funnel was flushed <n="19"/>with argon. Ethyl 3,3-diethoxypropionate (64.5 g) in tetrahydrofuran were charged to the addition funnel. Sodium hydride (21.2 g of a 60percent dispersion) was charged to the reaction flask followed by tetrahydrofuran. The contents of the flask were cooled to 0- 5°C in an ice-bath, and ethyl formate (257 g) was added. The mixture was cooled to 0- 50C and the contents of the addition funnel added drop wise, maintaining an internal temperature of less than 5°C. The ice-bath was removed and the contents allowed to warm to ambient temperature. Consumption of ethyl 3,3-diethoxypropionate was monitored by TLC analysis. The reaction was quenched by addition of ice-water (10.6 vol), and extracted three times with methyl t-butyl ether (5.4 vol each), and the organic layers discarded. The aqueous phase was acidified with cone, hydrochloric acid to a pH of 1 to 1.5. The acidified aqueous layer was extracted three times with dichloromethane and the combined organic layers dried over sodium sulfate. The solvent was removed under reduced pressure, and the residue distilled under vacuum, to provide ethyl 2-formyl-3-oxopropionate, 27.92 g, 70percent yield.
70%
With sodium hydride In tetrahydrofuran at 0 - 20℃;
[0066] A three- or four-neck round bottom flask equipped with magnetic stir bar, thermocouple, digital thermometer, gas inlet and outlet and addition funnel was flushed with argon. Ethyl 3,3- diethoxypropionate (64.5 g) in tetrahydrofuran were charged to the addition funnel. Sodium hydride (21.2 g of a 60percent dispersion) was charged to the reaction flask followed by tetrahydrofuran. The contents of the flask were cooled to 0-50C in an ice-bath, and ethyl formate (257 g) was added. The mixture was cooled to 0-50C and the contents of the addition funnel added dropwise, maintaining an internal temperature of less than 5°C. The ice-bath was removed and the contents allowed to warm to ambient temperature. Consumption of ethyl 3,3- diethoxypropionate was monitored by TLC analysis. The reaction was quenched by addition of ice-water (10.6 vol), and extracted three times with methyl t-butyl ether (5.4 vol each), and the organic layers discarded. The aqueous phase was acidified with cone, hydrochloric acid to a pH of 1 to 1.5. The acidified aqueous layer was extracted three times with dichloromethane and the combined organic layers dried over sodium sulfate. The solvent was removed under reduced pressure, and the residue distilled under vacuum, to provide ethyl 2-formyl-3 -oxopropionate, 27.92 g, 70percent yield.
59%
With sodium hydride In tetrahydrofuran at 0 - 30℃; for 24 h;
4) In a 500 ml four-necked flask equipped with magnetic stirring, 160 ml of tetrahydrofuran and9.84g NaH, mass fraction of NaH in tetrahydrofuran solution is 60percent, the solution is cooled in an ice bath to 0 °C to 5 °C, and then 119g of ethyl formate is added to slowly increase the temperature, when the temperature stabilizes to 0 °C At 5 °C, 80 ml of a tetrahydrofuran solution containing D was continuously added dropwise into a 500 ml four-necked flask, wherein the mass fraction of D was 5percent. After the dropwise addition, the water bath was removed and heated to room temperature. At this time, a large amount of hydrogen was evolved. It will first rise to 30 °C and then gradually decrease to 25 °C. After 24 hours of reaction, the reaction solution was poured into ice water and quenched. 5) The resulting product is first extracted with 500 ml of tert-butyl methyl ether three times to remove the organic phase, and then with concentrated hydrochloric acid to adjust the pH of the aqueous phase to 1~1.5; then extract the aqueous phase with 300 ml of dichloromethane, twice The combined organic phases were combined; the combined organic phases were then dried over anhydrous sodium sulfate and evaporated to give a brown-red liquid which was evaporated in vacuo. GC yield was 99.3percent. The yield was 85.9percent. Finally, the 9 mm Hg diaphragm was distilled under reduced pressure. The colorless transparent liquid E, ethyl 2-formyl-3-oxopropanoate, was obtained in a yield of 59percent.
Reference:
[1] Patent: EP1364946, 2003, A1, . Location in patent: Page/Page column 196
[2] Patent: US2005/256004, 2005, A1, . Location in patent: Page/Page column 31-32
[3] Patent: WO2017/59191, 2017, A1, . Location in patent: Paragraph 00441-00443
[4] Journal of Organic Chemistry, 1982, vol. 47, # 11, p. 2216 - 2217
[5] Tetrahedron, 2008, vol. 64, # 33, p. 7745 - 7758
[6] Patent: WO2014/160203, 2014, A2, . Location in patent: Page/Page column 67
[7] Patent: WO2007/92372, 2007, A1, . Location in patent: Page/Page column 17-18
[8] Patent: WO2008/143667, 2008, A1, . Location in patent: Page/Page column 20
[9] Patent: CN106928060, 2017, A, . Location in patent: Paragraph 0007; 0022; 0023
[10] Patent: US2007/225280, 2007, A1, . Location in patent: Page/Page column 30-31
[11] European Journal of Medicinal Chemistry, 2017, vol. 137, p. 96 - 107
6.2 g (193 mmoles) of hydrazine was added, with ice-cooling, to a solution of 27.6 g (192 mmoles) of <strong>[80370-42-9](ethoxycarbonyl)malondialdehyde</strong> dissolved in 150 ml of ethanol. The mixture was stirred at room temperature for 17 hours to give rise to a reaction. The reaction mixture was subjected to vacuum distillation to remove the ethanol contained therein. The residue was purified by silica gel column chromatography (developing solvent: dichloromethane-ethyl acetate mixed solvent) to obtain 19.4 g (72.4percent) of ethyl 1H-pyrazole-4-carboxylate as yellow crystals. 1H-NMR [CDCl3/TMS, delta (ppm)]: 8.08 (2H,s), 5.30 (1H,s) 4.31 (2H,q), 1.36 (3H,t)
72.4%
With hydrazine; In ethanol; at 20℃; for 17h;
REFERENCE EXAMPLE 29 Production of ethyl 1H-pyrazole-4-carboxylate; 6.2 g (193 mmoles) of hydrazine was added, with ice-cooling, to a solution of 27.6 g (192 mmoles) of <strong>[80370-42-9](ethoxycarbonyl)malondialdehyde</strong> dissolved in 150 ml of ethanol. The mixture was stirred at room temperature for 17 hours to give rise to a reaction. The reaction mixture was subjected to vacuum distillation to remove the ethanol contained therein. The residue was purified by silica gel column chromatography (developing solvent: dichloromethane-ethyl acetate mixed solvent) to obtain 19.4 g (72.4percent) of ethyl 1H-pyrazole-4-carboxylate as yellow crystals. 1H-NMR [CDCl3/TMS, delta (ppm)]: 8.08 (2H,s), 5.30 (1H,s), 4.31 (2H,q), 1.36 (3H,t)
With sodium hydride; In diethyl ether; at 0 - 20℃; for 15h;
12.6 g of sodium hydride (purity: 60percent, 525.0 mmoles) was washed with diethyl ether by decantation several times and then made into a solution in 500 ml of diethyl ether. Thereto were added, in a nitrogen current at 0 to 10°C, 194 g (2.6 moles) of ethyl formate and 50 g (262.0 mmoles) of ethyl 3,3-diethoxy-propionate. The resulting mixture was stirred at room temperature for 15 hours to give rise to a reaction. After confirmation of the completion of the reaction, the reaction mixture was poured into water, followed by washing with diethyl ether. The resulting aqueous layer was allowed to have a pH of 1 with hydrochloric acid, followed by extraction with dichloromethane. The resulting organic layer was washed with an aqueous sodium chloride solution and then dried over anhydrous magnesium sulfate. The resulting solution was subjected to vacuum distillation to remove the solvent contained therein, to obtain 37.6 g (yield: 100percent) of crude (ethoxycarbonyl)malondialdehyde as a dark red oily substance. 1H-NMR [CDCl3/TMS, delta (ppm)]: 9.09 (2H,s), 5.26 (1H,s), 4.27 (2H,q), 1.28 (3H,t)
100%
With sodium hydride; In diethyl ether; at 0 - 20℃; for 15h;
REFERENCE EXAMPLE 28 Production of (ethoxycarbonyl)malondialdehyde; 12.6 g of sodium hydride (purity: 60percent, 525.0 mmoles) was washed with diethyl ether by decantation several times and then made into a solution in 500 ml of diethyl ether. Thereto were added, in a nitrogen current at 0 to 10° C., 194 g (2.6 moles) of ethyl formate and 50 g (262.0 mmoles) of ethyl 3,3-diethoxy-propionate. The resulting mixture was stirred at room temperature for 15 hours to give rise to a reaction. After confirmation of the completion of the reaction, the reaction mixture was poured into water, followed by washing with diethyl ether. The resulting aqueous layer was allowed to have a pH of 1 with hydrochloric acid, followed by extraction with dichloromethane. The resulting organic layer was washed with an aqueous sodium chloride solution and then dried over anhydrous magnesium sulfate. The resulting solution was subjected to vacuum distillation to remove the solvent contained therein, to obtain 37.6 g (yield: 100percent) of crude (ethoxycarbonyl)malondialdehyde as a dark red oily substance. 1H-NMR [CDCl3/TMS, delta (ppm)]: 9.09 (2H,s), 5.26 (1H,s), 4.27 (2H,q), 1.28 (3H,t)
99.7%
With sodium hydride; In tetrahydrofuran; at 0 - 20℃;
[00442] To a stirred suspension of sodium hydride (60percent, 1.68 g, 42.1 mmol) in THF (20 mL) was added ethyl formate (8.5 mL, 105.7 mmol). The solution was cooled to 0 °C and a solution of ethyl 3,3-diethoxypropanoate (4 g, 21.0 mmol) in THF (10 mL) was added dropwise over 30 mins and the reaction mixture stirred at r.t. overnight. 2M aq HCl (30 mL) was added under whilst cooling with ice and the reaction stirred at r.t. for 30 mins. The reaction mixture was extracted with diethyl ether (2 x 50 mL) and the combined organic extracts were dried over MgS04 and concentrated in vacuo to afford the title compound (3.02 g, 99.7percent) as an amber liquid. [00443] 1H NMR (250 MHz, Chloroform-d) delta 9.13 (s, 2H), 4.29 (q, J = 7.1 Hz, 2H), 1.37 - 1.28 (m, 3H).
74.2%
With sodium hydride; In diethyl ether; hexane; at 20℃; for 15h;Cooling with ice;
Weighed out sodium hydride (2.46 g, 61.5 mmol) in a dry 100-mL pear flask. Washed with hexanes then with diethyl ether. Suspended in ether (100 mL), cooled in ice bath then ethyl formate (24.84 ml, 308 mmol) was added then ethyl 3,3-diethoxypropanoate (5.98 ml, 30.8 mmol). The resulting mixture was stirred at room temperature for 15 hours to give rise to a reaction. After confirmation of the completion of the reaction, the reaction mixture was poured into water, followed by washing with diethyl ether. The resulting aqueous layer was allowed to have a pH of 1 with hydrochloric acid, followed by extraction with dichloromethane. The resulting organic layer was washed with an aqueous sodium chloride solution and then dried over anhydrous magnesium sulfate, filtered and concentrated to give ethyl 2-formyl-3-oxopropanoate (3.29 g, 22.83 mmol, 74.2 percent yield) as a golden syrup
70%
EXAMPLE 1Preparation of Ethyl-2-formyl-3-oxopropionate[0059] A three- or four-neck round bottom flask equipped with magnetic stir bar, thermocouple, digital thermometer, gas inlet and outlet and addition funnel was flushed <n="19"/>with argon. Ethyl 3,3-diethoxypropionate (64.5 g) in tetrahydrofuran were charged to the addition funnel. Sodium hydride (21.2 g of a 60percent dispersion) was charged to the reaction flask followed by tetrahydrofuran. The contents of the flask were cooled to 0- 5°C in an ice-bath, and ethyl formate (257 g) was added. The mixture was cooled to 0- 50C and the contents of the addition funnel added drop wise, maintaining an internal temperature of less than 5°C. The ice-bath was removed and the contents allowed to warm to ambient temperature. Consumption of ethyl 3,3-diethoxypropionate was monitored by TLC analysis. The reaction was quenched by addition of ice-water (10.6 vol), and extracted three times with methyl t-butyl ether (5.4 vol each), and the organic layers discarded. The aqueous phase was acidified with cone, hydrochloric acid to a pH of 1 to 1.5. The acidified aqueous layer was extracted three times with dichloromethane and the combined organic layers dried over sodium sulfate. The solvent was removed under reduced pressure, and the residue distilled under vacuum, to provide ethyl 2-formyl-3-oxopropionate, 27.92 g, 70percent yield.
70%
With sodium hydride; In tetrahydrofuran; at 0 - 20℃;
[0066] A three- or four-neck round bottom flask equipped with magnetic stir bar, thermocouple, digital thermometer, gas inlet and outlet and addition funnel was flushed with argon. Ethyl 3,3- diethoxypropionate (64.5 g) in tetrahydrofuran were charged to the addition funnel. Sodium hydride (21.2 g of a 60percent dispersion) was charged to the reaction flask followed by tetrahydrofuran. The contents of the flask were cooled to 0-50C in an ice-bath, and ethyl formate (257 g) was added. The mixture was cooled to 0-50C and the contents of the addition funnel added dropwise, maintaining an internal temperature of less than 5°C. The ice-bath was removed and the contents allowed to warm to ambient temperature. Consumption of ethyl 3,3- diethoxypropionate was monitored by TLC analysis. The reaction was quenched by addition of ice-water (10.6 vol), and extracted three times with methyl t-butyl ether (5.4 vol each), and the organic layers discarded. The aqueous phase was acidified with cone, hydrochloric acid to a pH of 1 to 1.5. The acidified aqueous layer was extracted three times with dichloromethane and the combined organic layers dried over sodium sulfate. The solvent was removed under reduced pressure, and the residue distilled under vacuum, to provide ethyl 2-formyl-3 -oxopropionate, 27.92 g, 70percent yield.
59%
With sodium hydride; In tetrahydrofuran; at 0 - 30℃; for 24h;
4) In a 500 ml four-necked flask equipped with magnetic stirring, 160 ml of tetrahydrofuran and9.84g NaH, mass fraction of NaH in tetrahydrofuran solution is 60percent, the solution is cooled in an ice bath to 0 °C to 5 °C, and then 119g of ethyl formate is added to slowly increase the temperature, when the temperature stabilizes to 0 °C At 5 °C, 80 ml of a tetrahydrofuran solution containing D was continuously added dropwise into a 500 ml four-necked flask, wherein the mass fraction of D was 5percent. After the dropwise addition, the water bath was removed and heated to room temperature. At this time, a large amount of hydrogen was evolved. It will first rise to 30 °C and then gradually decrease to 25 °C. After 24 hours of reaction, the reaction solution was poured into ice water and quenched. 5) The resulting product is first extracted with 500 ml of tert-butyl methyl ether three times to remove the organic phase, and then with concentrated hydrochloric acid to adjust the pH of the aqueous phase to 1~1.5; then extract the aqueous phase with 300 ml of dichloromethane, twice The combined organic phases were combined; the combined organic phases were then dried over anhydrous sodium sulfate and evaporated to give a brown-red liquid which was evaporated in vacuo. GC yield was 99.3percent. The yield was 85.9percent. Finally, the 9 mm Hg diaphragm was distilled under reduced pressure. The colorless transparent liquid E, ethyl 2-formyl-3-oxopropanoate, was obtained in a yield of 59percent.
Step 1: 2-Formyl-3-oxo-propionic acid ethyl ester Ethyl 3,3-diethoxypropionate (100 g, 525.7 mmol) was dissolved in THF (360 ml) at room temp. Ethyl formate (175.1 ml, 2.1 mol) was added at room temp. The solution was cooled in an ice-bath to 0° C. and tBuOK (1M solution in THF, 1,156 ml, 1.156 mol) was added via an addition funnel slowly over 30 min, maintaining internal temperature below 5° C. The color changed instantly from colorless to dark orange. The reaction mixture was allowed to warm up to room temp. and stirred for 2 h. The reaction was allowed to stir at room temp. for 18 h. The reaction mixture was concentrated in vacuo and 1 L of solvent was removed. The remaining brownish solution with white solid in it was cooled in an ice-bath and hydrochloric acid (6N, 200 ml) was added to adjust pH=3, maintaining internal temperature below 20° C. The resulting bright yellow suspension was then warmed up to room temp. and stirred for 1 h. Additional 700 ml solvent was removed in vacuo at room temp. Water (400 ml) was added to dissolve all the white solid and ethyl acetate (500 ml) was added and the mixture transferred to a separatory funnel. The aqueous was extracted once with ethyl acetate (200 ml). The combined organic extracts was washed once with brine (100 ml). After drying over MgSO4 and concentrating in vacuo, 2-formyl-3-oxo-propionic acid ethyl ester (75.85 g) was obtained as yellow oil.
With sodium hydride; In tetrahydrofuran; at 0 - 20℃; for 24h;
Sodium hydride (2.00 g, 50.6 mmol, 60percent) was added to a solution of ethyl formate (10.13 g, 168.7 mmol) in tetrahydrofuran while maintaining the internal temperature below 0 °C. Then, ethyl 3,3-diethoxy propionate (5.00 g, 33.75 mmol) was added dropwise to the reaction mixture and stirred for 24 h at ambient temperature. After confirming that the reaction was complete by using TLC analysis, the reaction was quenched with 20 mL water and the tetrahydrofuran was removed under reduced pressure. The mixture was extracted with ethyl acetate and then brine. The organic phase was dried over Na2SO4 overnight and the solvent was removed in vacuo to give the title compound 14.
With N-ethyl-N,N-diisopropylamine; In ethanol; for 3h;Heating / reflux;
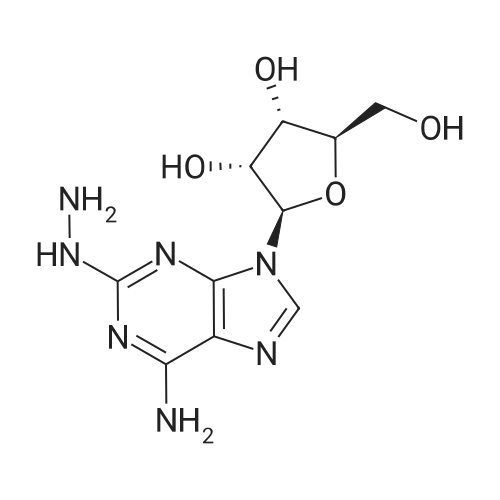
(4S,2R,3R,5R)-2-[2-hydrazino-6-(cyclopentylamino)purin-9-yl]-5- (hydroxymethyl)oxolane-3,4-diol (0.2 mmol) and ethyl 2,2-diformylacetate (0.28 mmol) were suspended in 3 mL of ethanol and to the suspension was added 5 mmol of diisopropylethylamiiie. The mixture was heated at reflux for 3 hours. Upon cooling to room temperature, the precipitate thus formed was collected by filtration, and washed with ethanol and ether to afford ethyl l-{9-[(4S,3R,5R)-3,4-dihydroxy-5- (hydroxymethyl)oxolan-2-yl]-6-(cyclopentylamino)purin-2-yl}pyrazole-4- carboxylate, a compound of Formula I.
Commercially available guanosine 5 was converted to the triacetate 6 as previously described (M. J. Robins and B. Uznanski, Can. J, Chem. (1981), 59, 2601-2607). Compound 7, prepared by following the literature procedure of Cerster et al. (J. F. Cerster, A. F. Lewis, and R.K. Robins, Org. Synthesis, 242-243), was converted to compound 9 in two steps as previously described (V. Nair et al., J. Org. Chem. , (1988), 53, 3051-3057). Compound 1 was obtained <n="30"/>by reacting hydrazine hydrate with compound 9 in ethanol at 80 C. Condensation of compound 1 with ethoxycarbonylmalondialdehyde in a mixture of AcOH and MeOH at 800C produced compound 10. Heating compound 10 in excess methylamine afforded compound11.
1-{9-[3,4-bis-(<i>tert</i>-butyl-dimethyl-silanyloxy)-5-(<i>tert</i>-butyl-dimethyl-silanyloxymethyl)-tetrahydro-furan-2-yl]-6-methylamino-9<i>H</i>-purin-2-yl}-1<i>H</i>-pyrazole-4-carboxylic acid ethyl ester[ No CAS ]
With sodium ethanolate; In ethanol; for 6h;Heating / reflux;
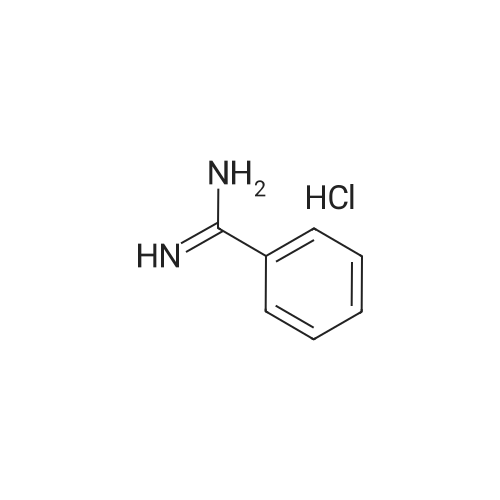
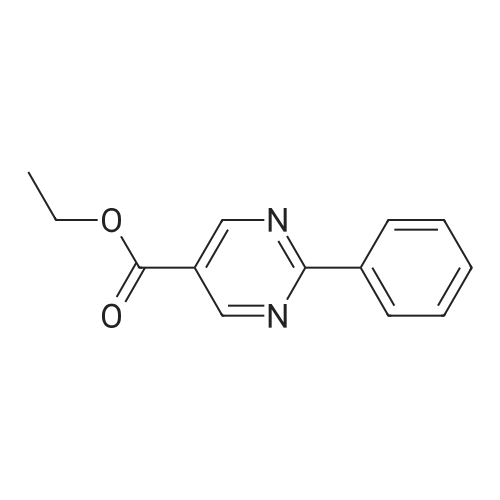
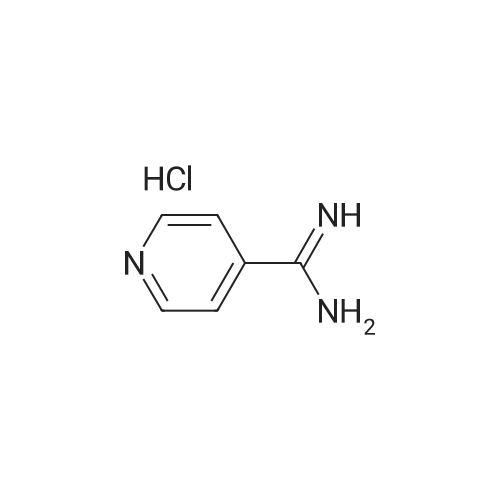
At room temperature, sodium ethoxide (590 mg) was dissolved in anhydrous ethanol (50 ml). 4-Amidinopyridine hydrochloride (1.31 g) was added to the resulting solution. After an anhydrous ethanol solution (ethanol: 50 ml) of ethyl 2,2-diformylacetate (1.20 g) was added dropwise, the resulting mixture was heated under reflux for 6 hours. Dichloromethane and water were added to the residue obtained by distilling off the solvent under reduced pressure. The organic layer thus separated was dried over anhydrous sodium sulfate. After the solvent was concentrated under reduced pressure, the residue was crystallized in ethanol, whereby the title compound (279 mg, 15percent) was obtained as colorless crystals.1H-NMR (DMSO-d6) delta: 1.46(3H,t,J=7.3Hz), 4.48 (2H, q, J=7.3Hz), 8.35 (2H, d, J=5.9Hz), 8.82(2H,d,J=5.9Hz), 9.38(2H,s). MS (FAB)m/z: 230(M+H)+.
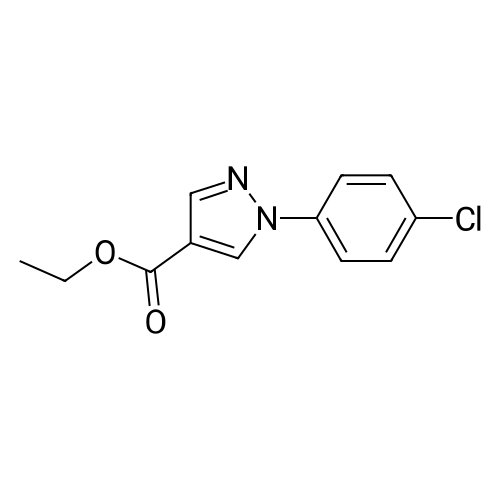
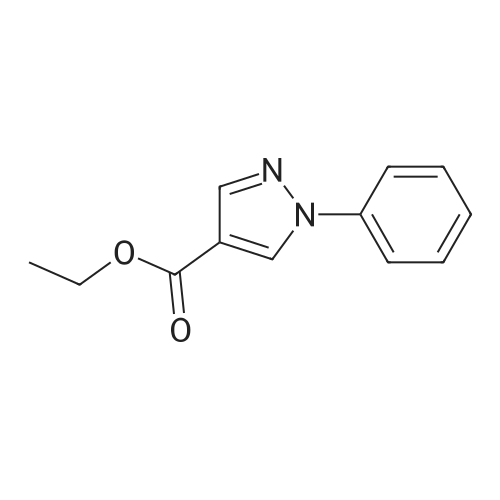
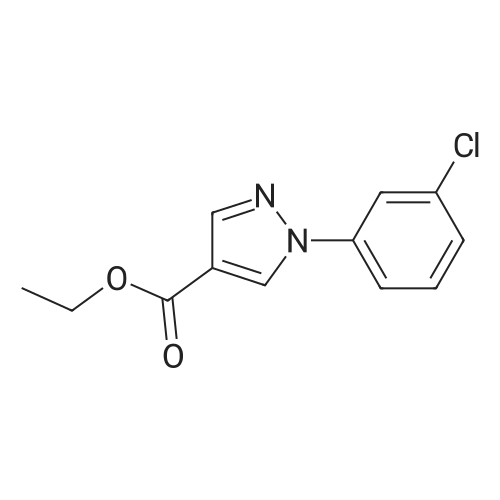
Example 16; Ethyl 1- (3-chlorophenyl)-lH-pyrazole-4-carboxylate; 3-Chlorophenylhydrazine hydrochloride (4.6 g, 25.7 mmol) in EtOH (100 mL) was added at 0 °C to a stirred solution of <strong>[80370-42-9]ethyl 2-formyl-3-oxopropanoate</strong> (3.7 g, 25.7 mmol) [J. Heterocyclic Chem. 1993,30, 865-872] in EtOH (80 mL). After addition was completed the reaction was allowed to reach rt, followed by stirring o. n. The reaction mixture was concentrated and the residue was recrystallized from EtOH to give 4.2 g (65percent) of the title compound. 1H NMR : 1.29 (t, 3H) 4.25 (q, 2H) 7.25 (d, 1H) 7.34 (t, 1H) 7.51 (d, 1H) 7.68 (s, 1H) 8.01 (s, 1H) 8.37 (s, 1H)
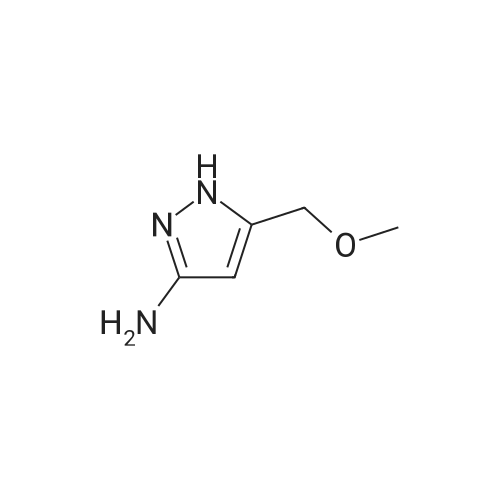
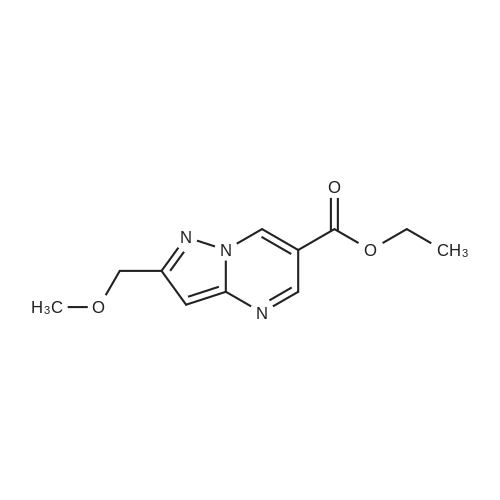
Ethyl 2-formyl-3-oxopropionate (775 mg) was added to a solution of the thus obtained 5-methoxymethyl-2H-pyrazol-3-ylamine (684 mg) in ethanol (50 ml), and stirred overnight. The reaction solution was concentrated under reduced pressure, and a saturated sodium bicarbonate solution was added to the residue which were then extracted with ethyl acetate. The organic phase was washed with a saturated saline solution and dried over sodium sulfate anhydrous. The resulting product was concentrated under reduced pressure, and the residue was purified by column chromatography (eluding solvent; n-hexane: ethyl acetate 4: 1) to give ethyl 2-methoxymethylpyrazolo[1,5-a] pyrimidine-6-carboxylate (878 mg, Y.: 69%).
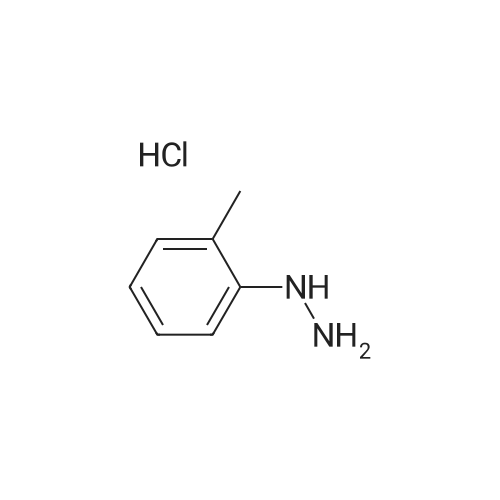
3-hydrazino-4-methyl-benzoic acid hydrochloride[ No CAS ]
[ 80370-42-9 ]
[ 869201-96-7 ]
Yield
Reaction Conditions
Operation in experiment
68%
In ethanol; at 0℃;Cooling with ice;
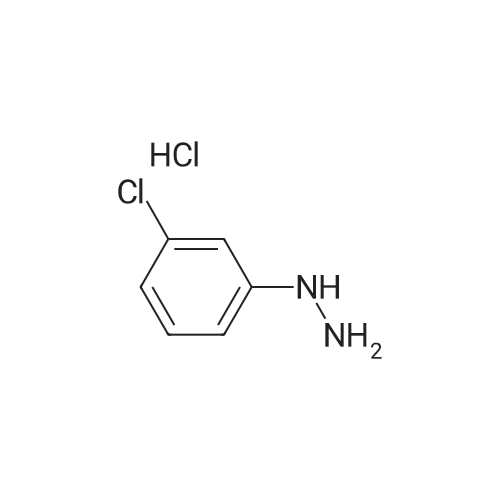
A solution of <strong>[80370-42-9]2-formyl-3-oxo-propionic acid ethyl ester</strong> (S. H. Bertz et al., J. Org. Chem., 1982, 47, 2216) (1.44 g, 10 mmol) in EtOH (10 mL) was cooled in an ice bath. A slurry of 3-hydrazino-4-methyl-benzoic acid hydrochloride (2.02 g, 10 mmol) in EtOH (50 mL) was added and the reaction was stirred overnight. The EtOH was removed under reduced pressure and the residue partitioned between water and CH2Cl2. The layers were separated and the organic layer washed with brine. The organic layer was dried over Na2SO4, filtered, and concentrated. Hexanes were added and the solution concentrated. The resulting solid was collected by vacuum filtration, washed with hexanes and dried under vacuum to provide 1-(5-carboxy-2-methyl-phenyl)-1H-pyrazole-4-carboxylic acid ethyl ester (1.86 g, 68percent) as a yellow solid: ESI MS m/z 275 [C14H14N2O4+H]+.
With sodium hydrogencarbonate; In methanol; ethyl acetate;
Example 34 Ethyl 1-(4-{2-[(acetylamino)methyl]-5oxo-2-hydroisoxazol4-yl}phenyl)pyrazole-4-carboxylate To a mixture of N-[4-(4-hydrazinylphenyl)-5-oxo-2-hydroisoxazol-2-yl]methyl}acetamide hydrochloride (150 mg, 0.5 mmol) in 3 mL of methanol was added sodium bicarbonate (50 mg, 0.6 mmol) and ethoxycarbonylmalondialdehyde (75 mg, 0.52 mmol). The mixture was stirred at room temperature overnight. The solid was collected by filtration and then washed with water, and dried to yield 140 mg of a purple solid. The crude product was subjected to silica gel chromatography (eluding with ethyl acetate followed by 5percent methanol/ethyl acetate) to yield 123 mg (66percent) of the title compound as a yellow solid. 1H NMR (300 MHz, DMSO-d6) delta9.11 (s, 1H), 9.08 (s, 1H), 8.96 (t, J=6 Hz, 1H), 8.15 (s, 1H), 7.95 (m, 4H), 5.06 (d, J=6 Hz, 2H), 4.28, (q, J=7 Hz, 2H), 1.86 (s, 3H), 1.31 (t, J=7 Hz, 3H).
With sodium ethanolate; In ethanol; for 6h;Heating / reflux; Argon atmosphere;
745mg, 5.17mmol) was dissolved in ethanol (15ml). Acetamidine hydrochloride (489mg, 5.17mmol) and sodium ethoxide in ethanol (0.41ml, 5.17mmol) were added and the mixture heated under argon at reflux for 6 hours and then overnight. The solvent was evaporated and the residue was treated with water (50ml) and extracted with diethyl EPO <DP n="34"/>ether (4 x 30ml). The diethyl ether layers were combined, dried over magnesium sulphate, filtered and evaporated. The crude product was purified using silica gel chromatography, eluting with a mixture of ethyl acetate and pentane (20-50percent) to afford the product (D33); MS (ES+) m/e 167 [M+H]+.
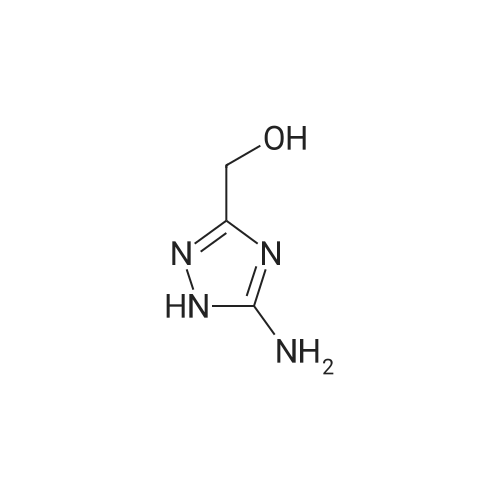
5-Amino-1H-[1,2,4]triazol-3-yl)-methanol (Bru-Magniez et al US (1995) US 5387747 A19950207) (7.9g, 41.6mmol) was mixed with <strong>[80370-42-9]2-formyl-3-oxo-propionic acid ethyl ester</strong> (Torri Sigeri et al Synthesis (1986) 5 pg 400-402), (6.0g, 41.6mmol) in acetic acid (30ml). The reaction mixture was heated to 60°C for 18hrs. After which time the mixture was cooled to room temperature and diluted with diethyl ether (200ml). Product crashes out of solution. The solids are then filtered and washed with diethyl ether (200ml), product dried in vacc oven for 5 hrs. To afford title compound as a white solid (8.0g). 1H NMR (300MHz, CDCI3): 5 1.47 (t, J=6.97, Hz, 3 H) 3.00 (bs, 1 H) 4.53 (q, J=7.16 Hz, 2 H), 5.00 (s, 2 H) 9.40 (d, J=2.07, Hz, 1 H) 9.50 (d, J=2.07, Hz, 1 H)
2-amino-3-pyridin-2-yl-pyrazolo[1,5-a]pyrimidine-6-carboxylic acid ethyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
61%
acetic acid; at 180℃; for 0.25h;Microwave irradiation;
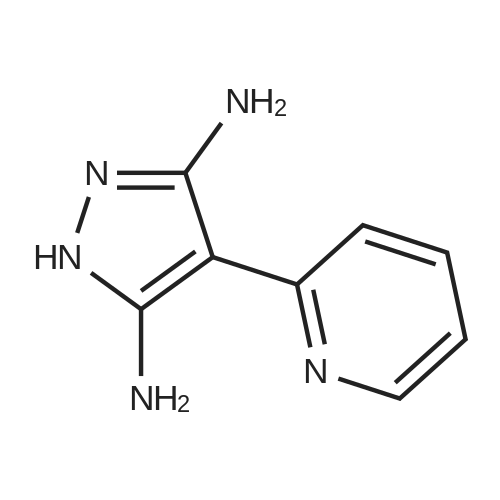
Example 8 [0286] 2-Amino-3-pyridm-2-yl-pyrazolo[1,5-a]pyrimidine-6-carboxyIic acid ethyl ester IV-1A mixture of 4-(pyridin-2-yl)-lH-pyrazole-3,5-diamine (0.10 g, 0.57 mmol), <strong>[80370-42-9](ethoxycarbonyl)malondialdehyde</strong> ( 0.084 g, 0.57 mmol) and 2 drops acetic acid in 1- propanol (3 mL) was heated in the microwave for 15 min at 180°C with stirring. The reaction was cooled down to room temperature and filtered, washing with ethanol and vacuum dried at 60°C for 3 days to provide the title compound as a brown solid (0.098 g, 61 percent yield). MS (ES+) 284. deltaEta (500 Mhz, DMSO-d6) 1.35 (3Eta, t), 4.36 (2H, q), 7.19 (1H, m), 7.36 (2H, s), 7.87 (1H, dt), 8.58 (2H, m), 8.82 (1H, d), 9.18 (1H, d)
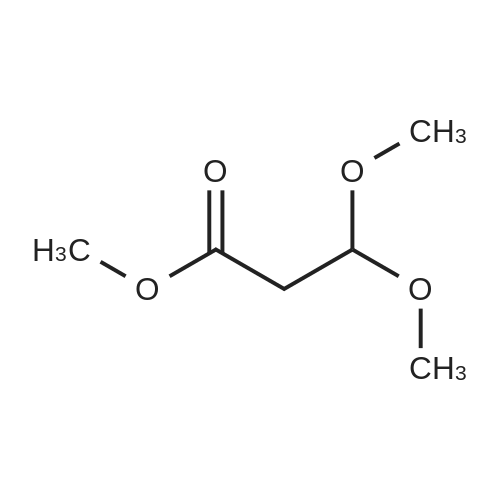
To a dry 250ml 3-necked flask, sodium hydride (60percent in oil) (778mg, 32.4mmol) was added in diethyl ether (35ml). A condenser was attached and the flask placed under an atmosphere of argon. The mixture was stirred and cooled to 0 0C. Ethyl formate (22ml, 270mmol) was added dropwise, followed by a solution of methyl 3,3- bis(methyloxy)propanoate (3.83ml, 27.0mmol) in diethyl ether (25ml) added dropwise over 10 minutes. The mixture was stirred at 0 0C for 1.5 hours and then warmed to room temperature overnight. Poured into 100ml of ice-water and extracted with diethyl ether (3 x 50 ml) which was discarded. The aqueous phase was acidified (pH 3) with concentrated HCI (3ml) and extracted with dichloromethane (5 x 50ml), dried, filtered and evaporated. The residue was purified using silica gel chromatography, eluting with a mixture of methanol and dichloromethane (0-25percent) to afford the product (D32); MS (ES+) m/e 143 [M-H]".
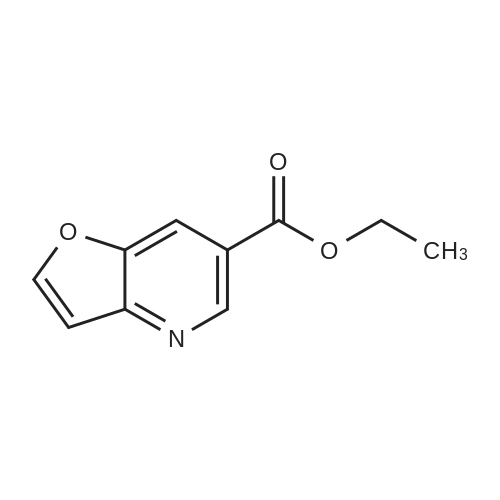
Step A: Preparation of ethyl furo[3,2-b]pyridine-6-carboxylate To a solution of conc. hydrogen chloride (100 mL) containing zinc chloride (11.6 g, 83 mmoL) was added 3-(t-butoxycarbamoyl)furan (13.8 g, 75.4 mmol). After stirring for 5 minutes, the reaction mixture was diluted with ethanol (60 mL) and a solution of <strong>[80370-42-9]ethoxycarbonylmalonaldehyde</strong> (11.6 g, 80.6 mmol) in ethanol (40 mL) was added. This mixture was warmed at 80° C. for two hours and then cooled and poured into ice/water and made basic with ammonium hydroxide. The product was extracted into methylene chloride, dried over anhydrous sodium sulfate, filtered through a pad of charcoal and evaporated. This residue was chromatographed on silica gel eluding with chloroform to give 5.92 g of purified product. Crystallization from hexane provided product with, mp: 63°-65° C. Analysis calculated for C10 H9 NO3: N, 7.33; C, 62.82; H, 4.74. Found: N, 7.23; C, 63.09; H, 4.87.
ethyl-2-(pyridin-4-yl)-5-pyrimidinecarboxylic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
15%
With sodium ethanolate; In ethanol; dichloromethane; water;
[Referential Example 142] Ethyl-2-(4-pyridyl)-5-pyrimidinecarboxylic acid Sodium ethoxide (590 mg) was dissolved in anhydrous ethanol (50 ml) at room temperature. To the resulting solution, 4-amidinopyridine hydrochloride (1.31 g) was added, followed by the dropwise addition of a solution of ethyl 2,2-diformylacetate (1.20 g) in anhydrous ethanol (50 ml). The resulting mixture was heated under reflux for 6 hours. To the residue obtained by distilling off the solvent under reduced pressure, dichloromethane and water were added. The organic layer thus separated was dried over anhydrous sodium sulfate. After the solvent wasconcentrated under reduced pressure, the residue was crystallized in ethanol, whereby the title compound (279 mg, 15percent) was obtained as colorless crystals. 1H-NMR (DMSO-d6) delta: 1.46(3H,t,J=7.3Hz), 4.48(2H,q,J=7.3Hz), 8.35(2H,d,J=5.9Hz), 8.82(2H,d,J=5.9Hz), 9.38(2H,s). MS (FAB) m/z: 230 (M+H)+. Elementary analysis for C12H11N3O2 Calculated: C, 62.87; H, 4.84; N, 18.33. Found: C, 62.80; H, 4.78; N, 18.25.
1-[2-(2-hydroxy-4-methylphenoxy)phenyl]-1H-pyrazole-4-carboxylic acid ethyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
77%
With sodium acetate; In ethanol; for 3h;Heating / reflux;
1- [2-(2-Hydroxy-4-methyl-phenoxy)-phenyl]-IH- pyrazolbeta-4-carboxylic acid ethyl esterA solution of 2- (2-hydrazino-phenoxy) -5-methyl-phenol (Example 66) (100 mg, 0.434 mmol) , <strong>[80370-42-9]2-formyl-3-oxo-propionic acid ethyl ester</strong> (Bertz et al, J. Org. Chem. , 1982, 47, 2216) (63 mg, 0.434 mmol) and sodium acetate (36 mg, 0.434 mmol) in ethanol (1 mL) was refluxed for 3 hrs. The reaction was evaporated under reduced pressure and the residue was purified by flash chromatography on silica column eluted with ethyl acetate/hexane (30percent) . The Example title compound (113 mg, 77percent) was obtained asa white powder: M.P.: 118 - 119 0C; Ci9Hi8N2O4 (338.13): GC-MS (EI+) m/e: 338. This product was analyzed by 1H-NMR. The EPO <DP n="101"/>corresponding 1H-NMR spectrum was consistent with the structure of the anticipated product.
ethyl (2Z)-3-({9-[(4S,2R,3R,5R)-3,4-dihydroxy-5-(hydroxymethyl)-oxolan-2-yl]-2-[4-(ethoxycarbonyl)pyrazolyl]purin-6-yl}amino)-2-formylprop-2-enoate[ No CAS ]
[ 946390-52-9 ]
Yield
Reaction Conditions
Operation in experiment
In ethanol; for 2h;Heating / reflux;
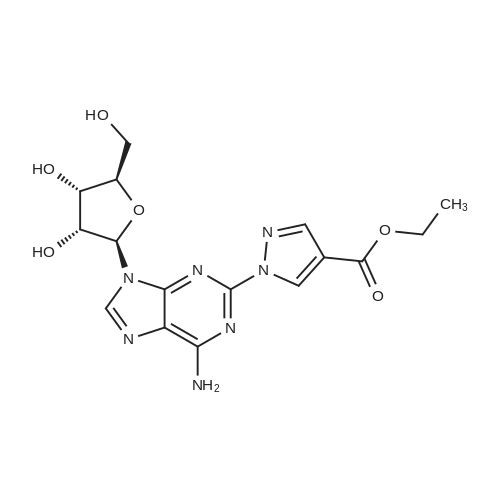
EXAMPLE 5Preparation of Ethyl f 2EV3 -f i9- IT4S.2R,3R.5IO-3 ,4-dihvdroxy-5-rhvdroxymethylV oxolan-2-yl1-2-r4-(ethoxycarborivpipyrazolyllpurin-6-yl>aminoV2-forrnylprorho-2-enoate[0066] A mixture of 2-hydrazinoadenosine (100 g, 0.34 mol), ethyl 2-formyl-3- oxopropionate (242g, 1.7 mol) and absolute ethanl were charged to a reactor, and the mixture heated to reflux for 2 hours. When the reaction was judged complete, the heat source was removed and the mixture gradually cooled to 5-100C over a period of 3 hours. The slurry was stirred for 30 minutes at this temperature, and the mixture filtered. The solid material was washed with cold (5-100C) absolute ethanol, and then dried under vacuum at a temperature that did not exceed 400C, to provide ethyl (2E)-3- ({9-[(4S,2R,3R,5R)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-2-[4- (ethoxycarbonyl)-pyrazolyl]purin-6-yl}amino)-2-formylprop-2-enoate.[0067] An elemental analysis gave the following results: C, 48.75percent; H, 4.86percent; N, 18.05percent; O, 27.57. Theoretical: C, 49.72percent; H, 4.74percent; N, 18.45percent; O, 27.09. The analysis corresponds within experimental error limits to the hemihydrate of the desired product. (C, 48.89percent; H, 4.81percent; N, 18.1percent; O5 28.12)[0068] 1H and 13C NMR spectra were obtained in the following manner. 20.2mg of the compound of formula (4) was dissolved in ~0.75 ml of DMSO-d6, and the spectra obtained at ambient temperature on a JEOL ECX-400 NMR spectrometer operating at 400 MHz for 1H and 100 MHz for 13C. The chemical shifts were referenced to the DMSO solvent, 2.50 ppm for 1H and 39.5 ppm for 13C.
In ethanol; for 2h;Heating / reflux;
[0073] A mixture of 2-hydrazinoadenosine (100 g, 0.34 mol), <strong>[80370-42-9]ethyl 2-formyl-3-oxopropionate</strong> (242g, 1.7 mol) and absolute ethanol were charged to a reactor, and the mixture heated to reflux for 2 hours. When the reaction was judged complete, the heat source was removed and the mixture gradually cooled to 5-100C over a period of 3 hours. The slurry was stirred for 30 minutes at this temperature, and the mixture filtered. The solid material was washed with cold (5-100C) absolute ethanol, and then dried under vacuum at a temperature that did not exceed 400C, to provide ethyl (2E)-3-({9-[(4S,2R,3R,5R)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2- yl]-2-[4-(ethoxycarbonyl)-pyrazolyl]purin-6-yl}amino)-2-formylprop-2-enoate.
ethyl (2Z)-3-({9-[(4S,2R,3R,5R)-3,4-dihydroxy-5-(hydroxymethyl)-oxolan-2-yl]-2-[4-(ethoxycarbonyl)pyrazolyl]purin-6-yl}amino)-2-formylprop-2-enoate[ No CAS ]
[ 946390-52-9 ]
[ 313348-16-2 ]
Yield
Reaction Conditions
Operation in experiment
With hydrogenchloride; In ethanol;Heating / reflux;
[0074] A reaction vessel is charged with 2-hydrazinoadenosine (450 g) and absolute ethanol (11376 g). HCl (7.47 g) and <strong>[80370-42-9]ethyl 2-formyl-3-oxopropionate</strong> (1557 g) are added. The mixture is heated to reflux and sampled until the level of residual 2-hydrazinoadenosine in the mixture is not more than 0.50percent and the level of the compound of formula (3) is not more than 2.5percent. The mixture is slowly cooled to approximately 100C. The product, the compound of formula (4) is isolated by filtration and washed with absolute ethanol (5121 g). The product is dried under vacuum at up to 400C until residual ethanol is not more than 5000 ppm to give ethyl (2E)-3-({9- [(4S,2R,3R,5R)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-2-[4-(ethoxycarbonyl)- pyrazolyl]purin-6-yl}amino)-2-formylprop-2-enoate.[0075] An elemental analysis of the product of Example 5 A gave the following results: C, 48.75percent; H, 4.86percent; N, 18.05percent; O, 27.57. Theoretical: C, 49.72percent; H, 4.74percent; N, 18.45percent; O, 27.09. The analysis corresponds within experimental error limits to the hemihydrate of the desired product. (C, 48.89percent; H, 4.81percent; N, 18.1percent; O, 28.12)[0076] 1H and 13C NMR spectra were obtained in the following manner. 20.2mg of the compound of formula (4) was dissolved in -0.75 ml of DMSO-d6, and the spectra obtained at ambient temperature on a JEOL ECX-400 NMR spectrometer operating at 400 MHz for 1H and 100 MHz for 13C. The chemical shifts were referenced to the DMSO solvent, 2.50 ppm for 1H and 39.5 ppm for 13C. <n="27"/>RESULTS[0077] The 1H and 13C chemical shifts are listed in Table 1. Two isomers in a ratio of -60/30 were observed in both the 1H and the 13C spectra, labeled as major and minor in the table. <n="28"/>The compound of formula (4) was confirmed to be a mixture of the following two isomers:Major Minor
Step 3: 1-r6-rtetrahydro-2H-pyran-4-aminol-9-((1 R,2S,3R,4S)-2,3-dihvdroxy-4-propionylamino- cvclopentyl)-9H-purin-2-yll-1 H-pyrazole-4-carboxylic acid ethyl ester:To a solution of N-[(1 S, 2R, 3S, 4R)-4-(6-[tetrahydro-2H-pyran-4-amino]-2-hydrazino-purin-9-yl)- 2,3-dihydroxy-cyclopentyl]-propionamide in dry ethyl alcohol is added <strong>[80370-42-9]2-formyl-3-oxo-propionic acid ethyl ester</strong>, prepared according to the process illustrated at page 2217, Intermediate 1 of Bertz <n="35"/>52289A 34S. H., Dabbagh G. and Cotte P.; J. Org. Chem. 1982, 47, 2216-2217. The reaction mixture is heated at reflux for 8 hours then concentrated in vacuo. The crude residue is purified by chromatography on silica eluting with MeOH in chloroform to afford the title compound.
e)190 mg of <strong>[80370-42-9]ethyl 2-formyl-3-oxopropionate</strong> (1.32 mmol) are dissolved in 5 ml of absolute pyridine, and 252 mg of 3-hydroxymethylbenzamidinium acetate (1.2 mmol) are added. This suspension is heated at 90° for 2 hours in a heating block, during which everything dissolves. The reaction mixture is stirred into 30 ml of water. The deposited crystals are filtered off with suction, washed well with water and dried overnight at 80° in vacuo in a drying cabinet; yield: 279 mg of beige crystals=90percent of theory; ESI 301 (M+H), HPLC: Rt=3.06 min (method B).
Step 3: 1-[6-[Bis-(4-methoxy-phenyl)-methyl]-amino}-9-((1R,2S,3R,4S)-2,3-dihydroxy-4-propionylamino-cyclopentyl)-9H-purin-2-yl]-1H-pyrazole-4-carboxylic acid ethyl ester To a solution of N-[(1S,2R,3S,4R)-4-(6-[Bis-(4-methoxy-phenyl)-methyl]-amino}-2-hydrazino-purin-9-yl)-2,3-dihydroxy-cyclopentyl]-propionamide (0.1 g, 0.177 mmol) in dry ethyl alcohol (5 ml) is added <strong>[80370-42-9]2-formyl-3-oxo-propionic acid ethyl ester</strong> (synthesised from ethyl-3,3-diethoxy-propionate, as described in: Bertz S. H., Dabbagh G. and Cotte P.; J. Org. Chem. (1982) 47, pp 2216-2217) (0.033 g, 0.231 mmol). The reaction mixture is heated at reflux for 8 hours then concentrated in vacuo. The crude residue is purified by chromatography on silica (60-120 mesh) eluding with 3percent methanol in chloroform to afford the title compound. LC-MS (0.1percent formic acid, acetonitrile): (MH+ 671).
ethyl 1-[4-(3,4-dichlorophenyl)-1,3-thiazol-2-yl]-1H-pyrazole-4-carboxylate To a solution of <strong>[80370-42-9]ethyl 2-formyl-3-oxopropanoate</strong> (0.57 g, 3.0 mmol) in ethanol (5 mL) was added thiosemicarbazide (1.37 g, 15 mmol), and the reaction mixture was stirred at room temperature for 1 hr. 2-Bromo-1-(3,4-dichlorophenyl)ethanone (0.54 g, 2.0 mmol) was added, and the mixture was stirred at 80°C for 30 min, and cooled to room temperature. The precipitated crystals were collected by filtration to give the title compound (545 mg, 74percent) as orange crystals. 1H NMR (400 MHz, CHLOROFORM-d) delta ppm 1.40 (3 H, t, J=7.2 Hz), 4.37 (2 H, q, J=7.2 Hz), 7.35 (1 H, s), 7.51 (1 H, d, J=8.6 Hz), 7.72 (1 H, dd, J=8.4, 2.1 Hz), 8.03 (1 H, d, J=2.2 Hz), 8.11 (1 H, d, J=0.5 Hz), 8.89 (1 H, d, J=0.5 Hz) LCMS(ESI+)M+H: 368.
(3-(Difluoromethoxy)phenyl)hydrazine hydrochloride (4.2 g, 19 9 mmol; CAS 479581-64-1) was suspended in ethanol (80 ml) and cooled to 0° C. A solution of <strong>[80370-42-9]ethyl 2-formyl-3-oxopropanoate</strong> (2.87 g, 19 9 mmol; CAS 80370-42-9) in ethanol (40 ml) was added, and the reaction was stirred overnight. The solvent was removed under reduced pressure and the residue partitioned between sodium bicarbonate solution and ethyl acetate. The organic layers were combined, dried (MgSO4) and evaporated to yield an orange solid. The solid was suspended in pentane (50 ml) and stirred at 35° C. for 90 min. The suspension was cooled in an ice bath for one hour and the solid was filtered and washed with pentane. After drying 5.12 g (91percent) of a yellow solid was obtained MS (ISP): 283.1 ([M+H]+).
91%
(3-(Difluoromethoxy)phenyl)hydrazine hydrochloride (4.2 g, 19.9 mmol; CAS 479581-64-1) was suspended in ethanol (80 ml) and cooled to 0 °C. A solution of ethyl 2-formyl-3- oxopropanoate (2.87 g, 19.9 mmol; CAS 80370-42-9) in ethanol (40 ml) was added, and the reaction was stirred overnight. The solvent was removed under reduced pressure and the residue partitioned between sodium bicarbonate solution and ethyl acetate. The organic layers were combined, dried (MgS04) and evaporated to yield an orange solid. The solid was suspended in pentane (50 ml) and stirred at 35 °C for 90 min. The suspension was cooled in an ice bath for one hour and the solid was filtered and washed with pentane. After drying 5.12 g (91percent) of a yellow solid was obtained MS (ISP): 283.1 ([M+H]+).
(3-(difluoromethoxy)phenyl)hydrazine hydrochloride (4.2 g, 19.9 mmol; CAS 479581-64-1) was suspended in ethanol (80 ml) and cooled to 0 °C. A solution of <strong>[80370-42-9]ethyl 2-formyl-3-oxopropanoate</strong> (2.87 g, 19.9 mmol; CAS 80370-42-9) in ethanol (40 ml) was added, and the reaction was stirred overnight. The solvent was removed under reduced pressure and the residue partitioned between sodium bicarbonate solution and ethyl acetate. The organic layers were combined, dried (MgS04) and evaporated to yield an orange solid. The solid was suspended in pentane (50 ml) and stirred at 35 °C for 90 min. The suspension was cooled in an ice bath for one hour and the solid was filtered and washed with pentane. After drying 5.12 g (91 percent) of a yellow solid was obtained MS (ISP): 283.1 ([M+H]+).
4-(trifluoromethoxy)phenylhydrazine hydrochloride[ No CAS ]
[ 1202942-22-0 ]
Yield
Reaction Conditions
Operation in experiment
241 mg
In ethanol; at 0℃;
(4- (Trifluoromethoxy)phenyl)hydrazine hydrochloride (229 mg, 1.0 mmol; CAS 133115-72-7) was suspended in ethanol (5 ml) and cooled to 0 °C. A solution of ethyl 2-formyl-3- oxopropanoate (2.87 g, 19.9 mmol; CAS 80370-42-9) in ethanol (1 ml) was added, and the reaction was stirred overnight. The solvent was removed under reduced pressure and the residue partitioned between sodium bicarbonate solution and ethyl acetate. The organic layers were combined, dried (MgS04) and evaporated to yield yellow crystals (241 mg, 80 percent). The product was used directly for the next step
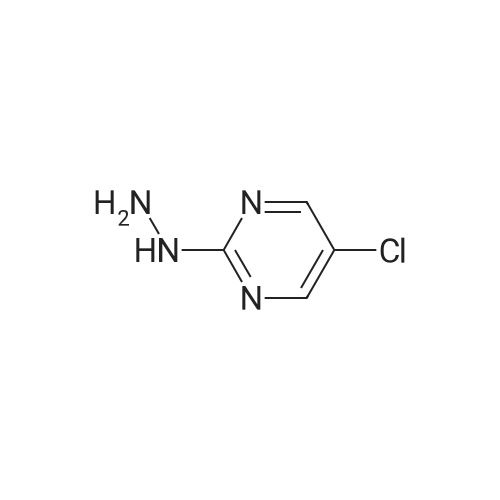
Under N2, 5-chloro-2-hydrazinylpyrimidine (200 mg, 1.38 mmol, Eq: 1.00) was combined with ethanol (15 ml). 4 M HCl in dioxane (346 mu, 1.38 mmol, Eq: 1.00) (the reaction mixture became a colourless solution) was added and the reaction mixture was cooled to 0 °C (the reaction mixture became a white suspension). A solution of <strong>[80370-42-9]ethyl 2-formyl-3-oxopropanoate</strong> (CAS 80370-42-9) (199 mg, 1.38 mmol, Eq: 1.00) in ethanol (2.6 ml) was added (the reaction mixture became a dark yellow suspension), and the reaction was stirred at RT over 1 hour. The solvent was removed and the reaction mixture was suspended in CH2CI2, filtered-off and concentrated in vacuo to yield 230 mg white solid. MS (ISP): 253.0 ([M+H]+).
1-amino-2-((3R,5R,6S)-1-((S)-2-(tert-butylsulfonyl)-1-cyclopropylethyl)-6-(4-chloro-3-fluorophenyl)-5-(3-chlorophenyl)-3-methyl-2-oxopiperidin-3-yl)ethaniminium[ No CAS ]
ethyl 3-bromopyrazolo[1,5-a]pyrimidine-6-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
100%
In ethanol; for 1.33333h;Reflux;
Ethyl 2-formyl-3-oxopropanoate (3.29 g, 22.84 mmol) and 4-bromo-lH-pyrazol-5 -amine (3.7 g, 22.84 mmol) were combined in ethanol (30 mL) and heated for 80 min at reflux. The mixture was cooled and the solid product was collected by filtration to give ethyl 3- bromopyrazolo[l,5-a]pyrimidine-6-carboxylate (6.17 g, 22.84 mmol, 100 percent yield) as a brown solid
In a sealed tube, (4-bromo-3-fluorophenyl)-hydrazine (931 mg; 4.54 mmol) was added at 0°C to a solution of <strong>[80370-42-9]ethyl 2-formyl-3-oxopropanoate</strong> (654 mg; 4.54 mmol) in EtOH (3.8 mL). The reaction mixture was stirred at rt for 18 hours. The solid was filtered off, washed with EtOH and dried on frit to give 1.21 g (85percent) of intermediate (U5) as a pale orange solid.
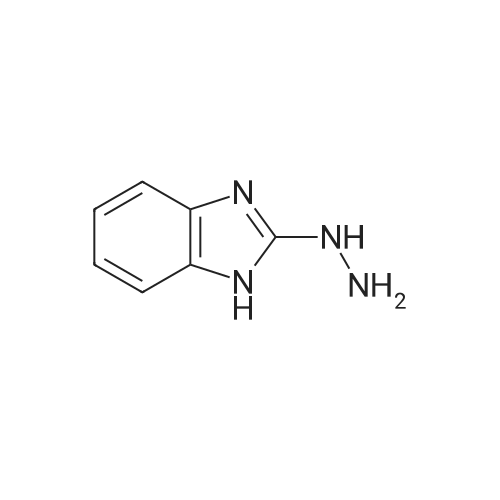
2-Hydrazinyl-1H-benzimidazole (20.0 g, 128 mmol) was provided in a mixture of THF/ethanol (600 mL, 2:1). Ethyl 2-formyl-3-oxopropanoate (16 mL, 130 mmol) was added, and the mixture was stirred at 80 °C for 1.5 h. After cooling down to room temperature, the precipitate was collected by filtration, was washed with hexane and was dried under reduced pressure. 27.2 g (83percent of theory) of the title compound were obtained. LC-MS (method 2): Rt = 1.00 min; MS (ESIpos): m/z = 257 [M+H]+ 1H-NMR (400 MHz, DMSO-d6) delta [ppm]: 1.300 (7.55), 1.318 (16.00), 1.335 (7.71), 2.523 (0.99), 4.266 (2.38), 4.284 (7.27), 4.302 (7.16), 4.319 (2.28), 7.220 (0.90), 7.230 (5.07), 7.238 (4.79), 7.245 (4.99), 7.253 (5.56), 7.264 (1.01), 7.549 (2.98), 7.558 (3.14), 7.565 (3.02), 7.573 (2.54), 8.345 (8.73), 8.973 (9.08).
83%
In tetrahydrofuran; ethanol; at 80℃; for 1.5h;
2-Hydrazinyl-lH-benzimidazole (20.0 g, 128 mmol) was provided in a mixture of THF/ethanol (600 mL, 2:1). Ethyl 2-formyl-3-oxopropanoate (16 mL, 130 mmol) was added, and the mixture was stirred at 80 °C for 1.5 h. After cooling down to room temperature, the precipitate was collected by filtration, was washed with hexane and was dried under reduced pressure. 27.2 g (83percent of theory) of the title compound were obtained. LC-MS (method 2): t = 1.00 min; MS (ESIpos): m/z = 257 [M+H]+ (0425) 1H-NMR (400 MHz, DMSO-d6) delta [ppm]: 1.300 (7.55), 1.318 (16.00), 1.335 (7.71), 2.523 (0.99), 4.266 (2.38), 4.284 (7.27), 4.302 (7.16), 4.319 (2.28), 7.220 (0.90), 7.230 (5.07), 7.238 (4.79), 7.245 (4.99), 7.253 (5.56), 7.264 (1.01), 7.549 (2.98), 7.558 (3.14), 7.565 (3.02), 7.573 (2.54), 8.345 (8.73), 8.973 (9.08).
ethyl 1-(1,3-benzothiazol-2-yl)-1H-pyrazole-4-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
82%
In tetrahydrofuran; ethanol; at 80℃; for 2h;
2-Hydrazinyl-l,3-benzothiazole (20.0 g, 121 mmol) was provided in a mixture of THF/ethanol (600 mL, 2: 1). Ethyl 2-formyl-3-oxopropanoate (15 mL, 120 mmol) was added, and the mixture was stirred at 80 °C for 2 h. After concentration, the remaining material was stirred in a mixture of hexane/ethanol (600 mL, 5: 1). The precipitate was collected by filtration, was washed with hexane and was dried under reduced pressure at 50 °C. 27.1 g (82percent of theory) of the title compound were obtained. LC-MS (method 2): R, = 1.34 min; MS (ESIpos): m/z = 274 [M+H]+ 'H-NMR (400 MHz DMSO-d6) delta [ppm] : 1.304 (7.16), 1.322 (16.00), 1.340 (7.38), 2.523 (0.82), 4.274 (2.09), 4.292 (6.78), 4.310 (6.79), 4.327 (2.10), 7.462 (1.06), 7.466 (1.09), 7.481 (2.15), 7.483 (2.38), 7.501 (1.93), 7.504 (1.83), 7.555 (1.70), 7.558 (1.60), 7.573 (1.58), 7.575 (2.39), 7.579 (1.88), 7.593 (1.32), 7.597 (1.09), 7.960 (2.23), 7.963 (2.21), 7.980 (2.10), 7.984 (1.83), 8.144 (2.13), 8.148 (2.08), 8.164 (2.00), 8.168 (1.84), 8.334 (6.43), 9.066 (7.07).
6-(ethoxycarbonyl)pyrazolo[1,5-a]pyrimidine-3-carboxylic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
48%
With acetic acid; at 110℃; for 1h;Microwave irradiation;
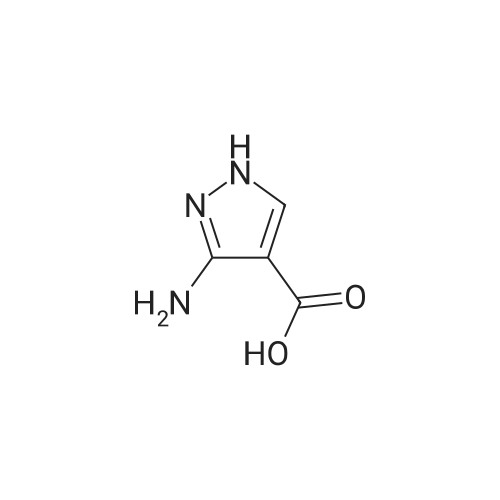
A mixture of 5-amino-i H-pyrazole-4-carboxylic acid (1 .00 g, 7.87 mmol) and ethyl 2-formyl-3- oxopropanoate (2.0 ml, 16 mmol) in acetic acid (6.0 ml) was heated for 1 h at 1100 in a microwave reactor (Biotage Initator). Upon cooling to room temperature, the precipitate wascollected by filtration, washed with water and dried under high vacuum at SOC to give the title compound (900 mg, 48percent yield).[C-MS (Method 1): R = 0.68 mm; MS (ESIpos): m/z = 236.1 [M+i]iH-NMR (400 MHz, DMSO-d6): 6 [ppm] = 12.59 (5, iH), 9.72 (d, iH), 9.13 (d, iH), 8.76 (5, 1 H), 4.41 (q, 2H), 1.38 (t, 3H).
2-methyl-pyrazolo[1,5-a]pyrimidine-6-carboxylic acid ethyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
86.3%
With triethylamine; diisopropylamine; N-ethyl-N,N-diisopropylamine; In tetrahydrofuran; at 0℃; for 0.5h;
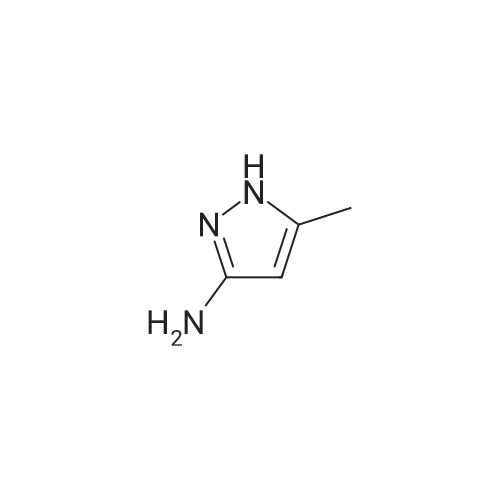
0.97 g of 3-amino-5-methylpyrazole and the acid binding agent were dissolved in tetrahydrofuran in 100 ml of a three-necked flask, and 2-formyl-3-oxo Ethyl propionate was added, and the mixture was stirred at 0 ° C for 0.5 h.After the reaction was completed, the temperature was raised to room temperature. After evaporation of the solvent, 100 ml of distilled water was added and the aqueous phase was extracted with ethyl acetate (100 x 3 ml). The organic phase was dried over anhydrous sodium sulfate for 20 minutes and concentrated to 1.95 g of intermediate I, The crude product was purified by silica gel column chromatography (eluent was a mixed solution of ethyl acetate and petroleum ether in a volume ratio of 1: 2) to give 1.77 g of intermediate I extract in a yield of 86.3percent; wherein the 2-formyl -3-oxo-propionic acid ethyl ester, 3-amino-5-methylpyrazole, acid binding agent, tetrahydrofuran in a weight ratio of 1: 1: 1.2: 4; a fluoric acid agent is triethylamine, diisopropylamine, Diisopropylethylamine, and the weight ratio is 3: 4: 1;
Ethyl-2-formyl-3-oxopropionate (131 mg, 0.91 mmol) is added to a stirred suspension of crude {1 -[6-(3-Aza-bicyclo[3. 1 .0]hex-3-yl)-pyridin-3-yl]-cyclopropyl}-hydrazine hydrochloride (Intermediate 56, 200 mg) in 15 mL of ethanol and the reactionmixture is stirred 7 days at room temperature. The solvent is removed under reduced pressure and the residue is partitioned between EtOAc and 1 N HCI aqueous solution. The organic layer is separated and concentrated under reduced pressure. The residue is purified by flash chromatography (20 to 100percent EtOAc in cyclohexane) and the impure title product (yield 60 mg) is used without further purification for thenext step.LC (Method 1): tR = 1 .03 mm; Mass spectrum (ES+): m/z = 292 [M+H].
ethyl 1-(6-amino-9H-purin-2-yl)-1H-pyrazole-4-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
80%
In ethanol; for 2h;Reflux;
2-hydrazinadenine (V) (15 g, 90.8 mmol),Add 300 ml of anhydrous ethanol,2-formyl-3-oxopropionate (15.7 g, 109.0 mmol) was added and the mixture was heated under reflux for 2 hours to complete the reaction, ethyl 1-(6-amino-9H-purin-2-yl)-1H-pyrazole-4-carboxylate (I) (19.8 g, molar yield 80%) was obtained by cooling to room temperature, ).
ethyl 1-(2-chloro-5-nitrophenyl)-1H-pyrazole-4-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In ethanol; at 80℃; for 2h;Microwave irradiation;
Into a 100-mL round-bottom flask, was placed (2-chloro-5-nitrophenyl)hydrazine (1 g, 5.33 mmol, 1.00 equiv), ethanol (40 mL), m<strong>[80370-42-9]ethyl 2-formyl-3-oxopropanoate</strong> (760 mg, 5.34 mmol, 1.00 equiv). The final reaction mixture was irradiated with microwave radiation for 2 h at 80 °C. The solids were filtered out. The resulting mixture was concentrated under vacuum. This resulted in 600 mg (crude) of the title compound that was used without further purification Analytical Data; 1 H NMR (300 MHz, Methanol-d4) delta 8.69 (d, J = 0.6 Hz, 1 H), 8.52 (d, J = 2.6 Hz, 1H), 8.39 (q, J = 2.7 Hz, 1H), 8.20 (d, J = 0.6 Hz, 1H), 7.95 (d, J = 8.7 Hz, 1H), 4.37 (q, J = 7,2 Hz, 2H).






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping