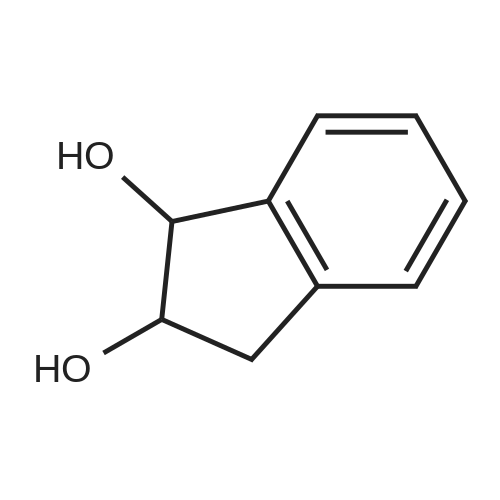
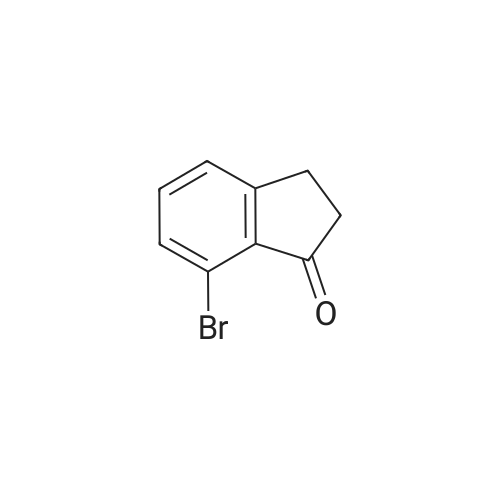
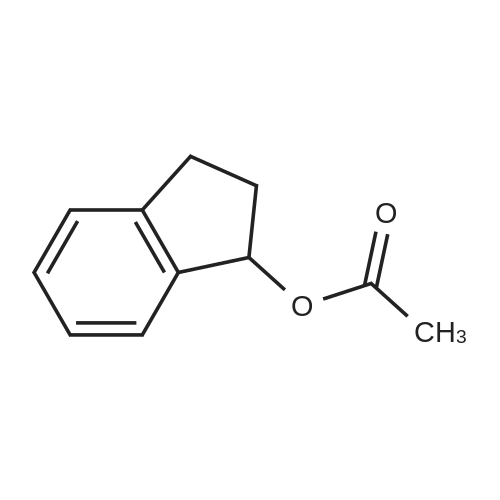
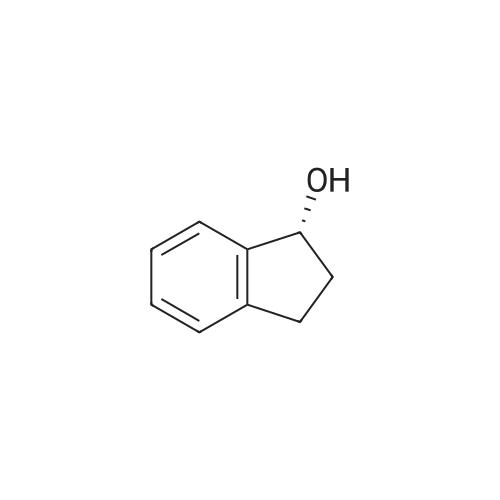
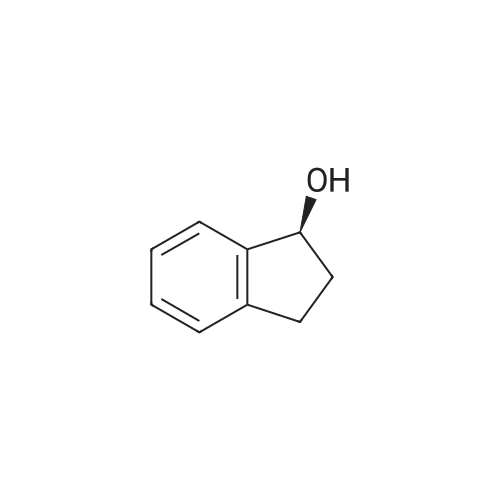
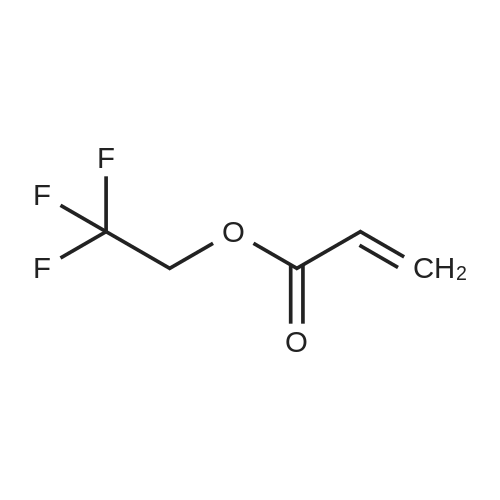
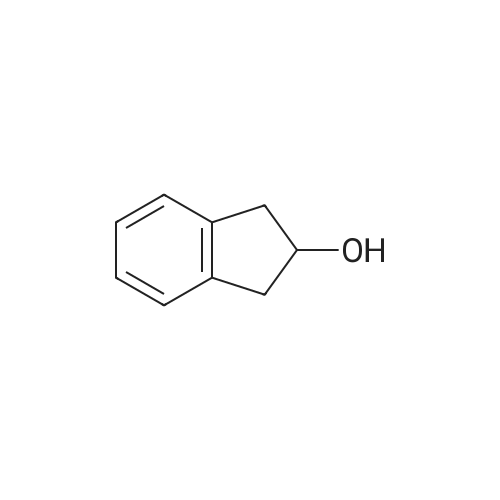
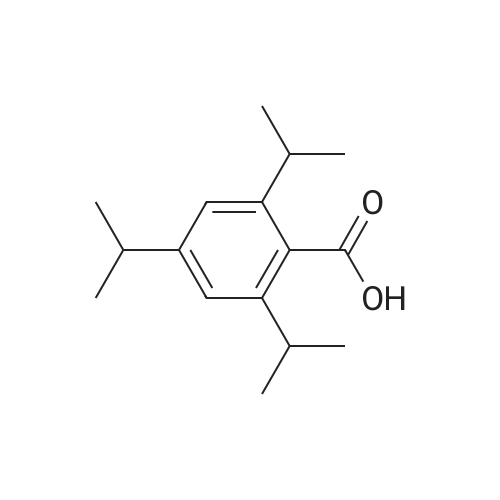
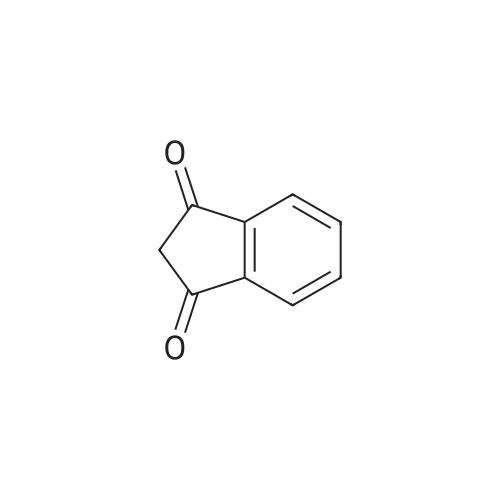
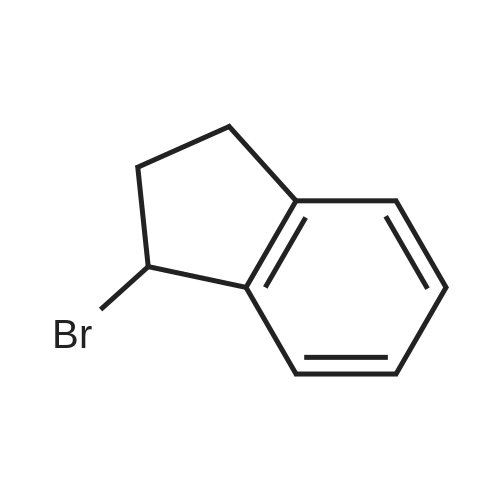
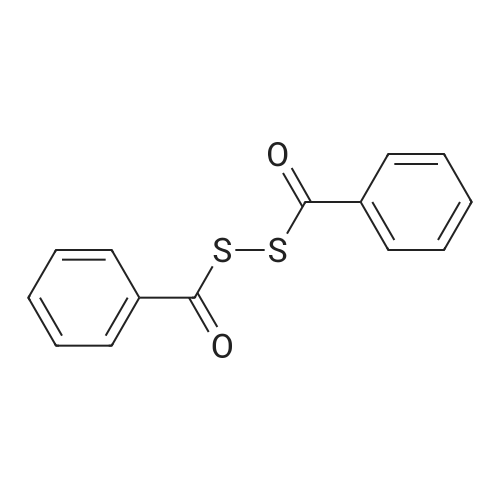
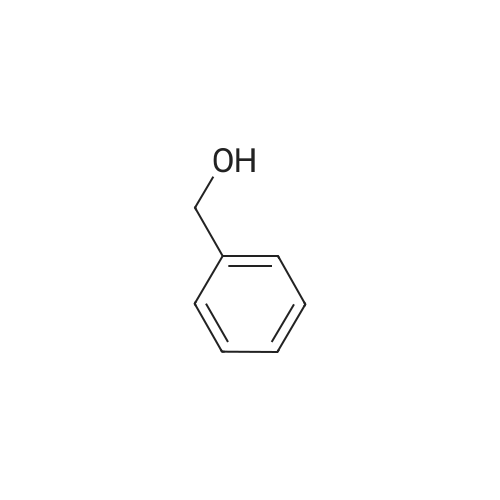
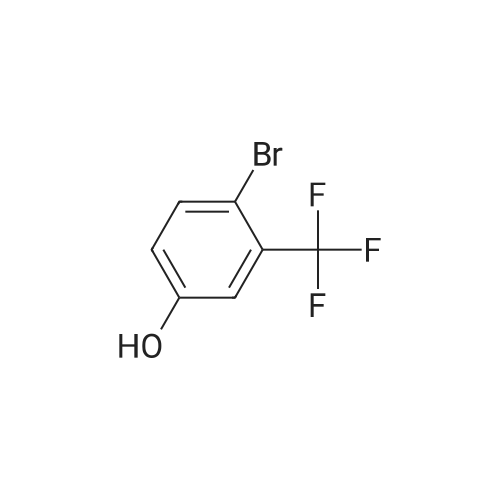
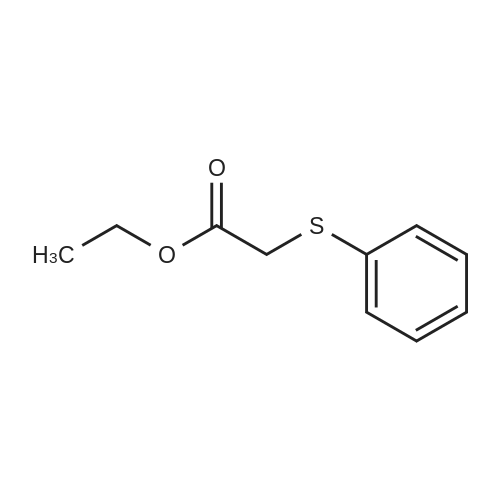
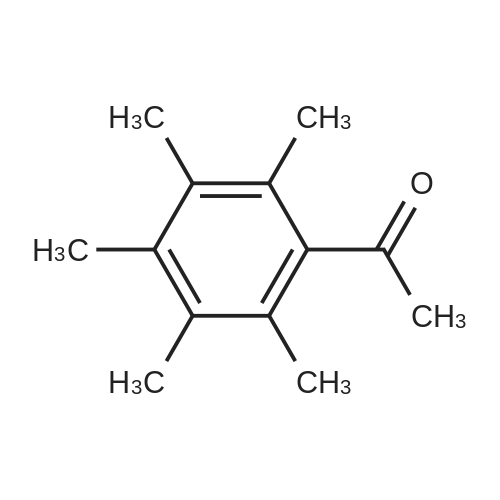
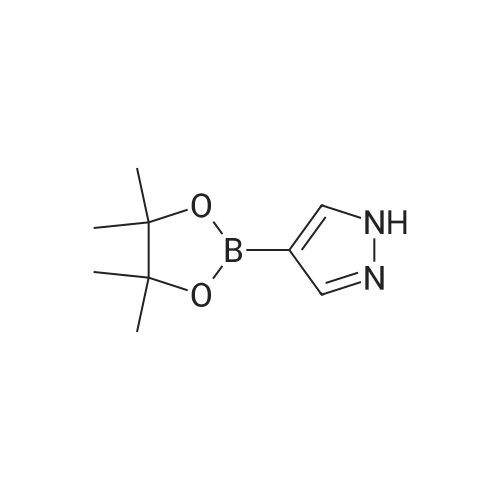
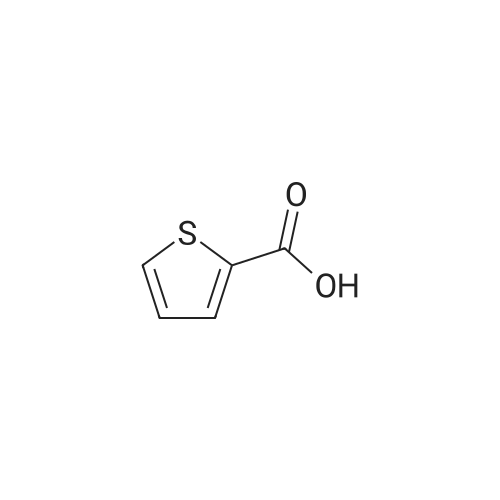
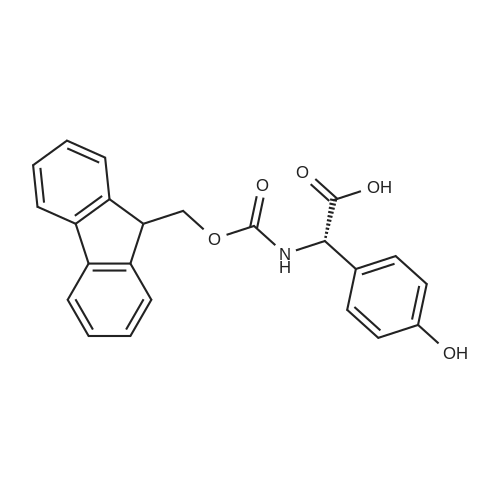
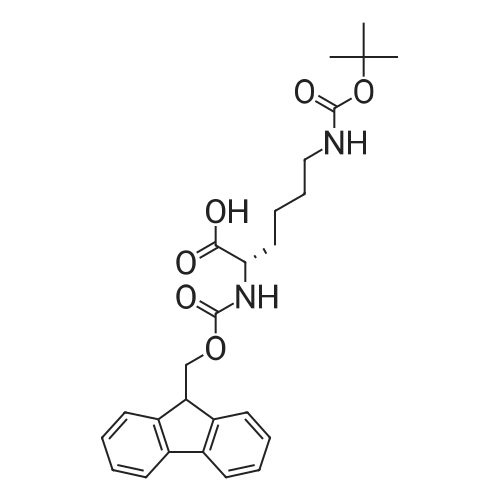
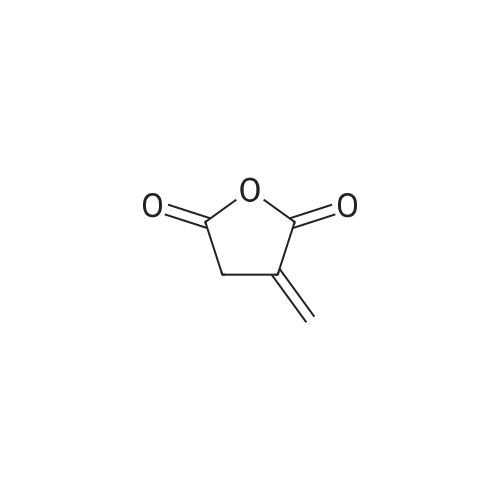
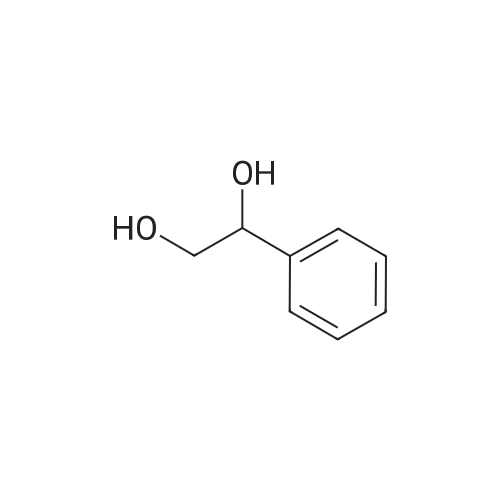
| 100% |
With iodosylbenzene; potassium bromide In lithium hydroxide monohydrate for 2h; sonication; |
|
| 100% |
With air; potassium carbonate In lithium hydroxide monohydrate at 20℃; for 2.5h; |
|
| 100% |
With C53H46ClN3P2Ru; potassium-t-butoxide; propan-2-one at 56℃; for 0.05h; |
|
| 100% |
With potassium carbonate; 4-chlorobenzoic acid In lithium hydroxide monohydrate at 27℃; for 0.5h; |
|
| 100% |
With potassium carbonate In lithium hydroxide monohydrate at 27℃; |
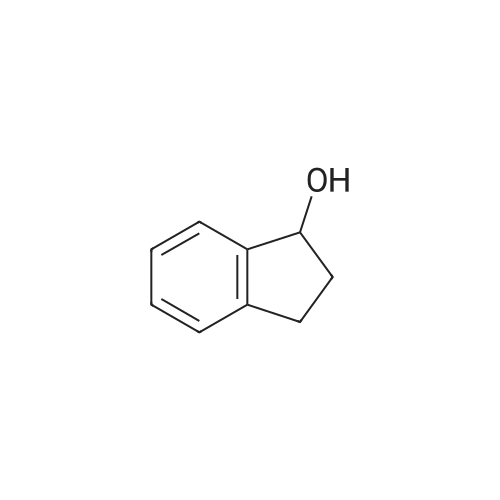
2.9. Aerobic Oxidation of 1-Indanol (1)Catalyzed by Au:HAP
The 5 atom% of Au:HAP was placed into a test tube( = 15 mm). The solution of 0.04 mmol of 1-indanol1 and 0.12 mmol (300 mol%) of K2CO3, and 2.4 mL water were added into the reaction tube. This mixture was stirred at 1,300 rpm under 27°C. The reaction mixture was quenched with 1 M HCl and filtrated with a membrane filter.The filtrate was extracted with ethyl acetate. The combined organic layer was adjusted to 50 mL in a volumetric flask. Then, quantitative analysis was performed by gas chromatography; the yield was obtained from the calibration curve. The used catalyst was pretreated at 450°Cunder vacuum for 3 h prior to use in the next cycle. |
| 99% |
With [Cp*Ru(μ-Cl)3RuCl(PPh3)2]; potassium carbonate In dichloromethane; butanone Heating; |
|
| 99% |
With air In various solvent(s) at 80℃; for 3h; |
|
| 99% |
With aluminium oxyhydroxide; ruthenium In toluene at 80℃; for 20h; |
|
| 99% |
In lithium hydroxide monohydrate; toluene at 80℃; for 36h; |
|
| 99% |
With 1,3,5,7-tetrakis[4-(diacetoxyiodo)phenyl]adamantane; Etamon In lithium hydroxide monohydrate at 20℃; for 4h; |
|
| 99% |
With oxygen; potassium hydroxide In chloroform; lithium hydroxide monohydrate at 35℃; for 1h; |
|
| 99% |
With [(2-(benzoimidazol-2-yl)-6-(3,5-dimethylpyrazol-1-yl)pyridine)RuCl2(PPh3)]; potassium-t-butoxide; propan-2-one In methanol at 56℃; for 0.166667h; Inert atmosphere; |
A typical procedure for the catalytic oxidation of alcohols
General procedure: The catalyst solutionwas prepared by dissolving complex 3(36.1 mg,0.05mmol) in methanol (5.0 mL).Under a nitrogen atmosphere, the mixture of an alcohol substrate (2.0 mmol) and1.0 mL of the catalyst solution (0.01mmol) in 20mL acetone was stirred at 56 Cfor 10 minutes. tBuOK(22.4mg, 0.2 mmol)was then added to initiate the reaction.At the stated time, 0.1 mL of the reaction mixture was sampled and immediately diluted with 0.5 mL acetone pre-cooled-to-0 C for GC or NMR analysis. After the reaction was complete, the reaction mixture was condensed under reduced pressure and subject to purification by flash silica gel column chromatography to afford the corresponding ketone product, which was identified by comparison with the authentic sample through NMR and GC analysis. |
| 99% |
With tert.-butylnitrite; oxygen; glacial acetic acid In toluene at 50℃; for 3h; |
|
| 99% |
With potassium tetrakis-μ-pyrophosphitodiplatinate(II); tetra-n-butyl-ammonium chloride In dichloromethane; lithium hydroxide monohydrate at 20℃; for 8h; Inert atmosphere; Irradiation; |
|
| 99% |
With potassium carbonate In lithium hydroxide monohydrate at 20℃; for 1.33333h; Green chemistry; |
|
| 98% |
With mesoporous silica; bis(trimethylsilyl)chromate In dichloromethane at 40℃; for 0.666667h; |
|
| 98% |
With potassium permanganate In acetonitrile at 5 - 20℃; |
|
| 98% |
With trichloroisocyanuric acid; mesoporous silica; potassium bromide In dichloromethane at 20℃; for 0.25h; |
|
| 98% |
With hydrogenchloride; 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; NaNO2 In dichloromethane; lithium hydroxide monohydrate at 20℃; for 16h; in air; |
|
| 98% |
With IBS; potassium peroxomonosulfate; N-hexadecyl-N,N,N-trimethylammonium bromide In lithium hydroxide monohydrate at 20℃; for 2h; Green chemistry; chemoselective reaction; |
IBS-catalysed alcohol oxidation in CTAB micelle; general procedure
General procedure: The alcohol (2 mmol) was added to a solution of IBS (0.02 mmol, 0.01 eq), oxone (2.2 mmol, 1.1 equiv.) and 3 wt% CTAB solution (5 mL). The mixture was stirred at room temperature. The reaction was monitored by TLC. After completion, the solution was extracted with CH2Cl2 (3 × 10 mL). The combined organic phase was then filtered through a pad of silica gel and evaporated under vacuum to afford the desired product. |
| 98% |
With ICl; Cs2CO3 In dichloromethane at 0 - 20℃; for 3h; Green chemistry; |
General Procedure for the Study of the Substrate Scope of ICl as Oxidant
General procedure: Starting alcohols (1.00 g, 1.0 eq) specified in Table 3 were dissolved in 10 mL of dried CH2Cl2 (for substrates in entries 8-20 in Table 3, 30 mL of dried CH2Cl2 was used), and the resulting mixture was stirred in an ice-water bath, followed by addition of Cs2CO3 (3.0 eq). The suspension was stirred at this temperature, and ICl (1.5 eq) dissolved in 2 mL of CH2Cl2 was added dropwise. After addition, the reaction mixture was stirred at room temperature until the reaction completed as indicated by TLC analysis, which was conducted at 0.5-h intervals.The reaction mixture was subjected to aqueous workup described previously to yield the pure carbonyl compounds specified in Table 3. |
| 98% |
With C27H42ClN2PRu; Cs2CO3 In 5,5-dimethyl-1,3-cyclohexadiene at 140℃; for 24h; Inert atmosphere; Glovebox; Sealed tube; |
|
| 97% |
In toluene for 20h; Heating; |
|
| 97% |
With Pd(0) nanoparticle supported on aminopropyl grafted silica-based mesocellular foam; air In para-xylene at 110℃; for 1h; |
|
| 97% |
With [RuCl(PPh3)2(3-phenylindenyl)]; 1,1,1,3,3,3-hexamethyldisilazane potassium In propan-2-one; toluene at 110℃; for 1h; Schlenk technique; |
|
| 97% |
Stage #1: 1-Indanol With copper(II) bromide In acetonitrile at 20℃; for 0.05h; Inert atmosphere;
Stage #2: With N,N'-di-tert-butyldiaziridin-3-one In acetonitrile at 20℃; for 6h; |
|
| 97% |
With C14H14N6O2; oxygen; anhydrous Sodium acetate; palladium diacetate at 120℃; for 48h; |
|
| 97% |
With potassium hexafluoridophosphate; tert.-butylnitrite; 9-azabicyclo<3.3.1>nonane-N-oxyl; oxygen In lithium hydroxide monohydrate at 60℃; for 2.5h; Autoclave; Green chemistry; |
|
| 97% |
With IBX; (+/-)-camphor sulfonic acid In 1,4-dioxane; dichloromethane at 20℃; Inert atmosphere; |
General procedure
General procedure: Under nitrogen atmosphere, 1.1-1.5 mmol IBX and 10 to 20 mol% (±)-CSA monohydrate was added in round bottom flask already charged with magnetic bar and 2 mL DCM:1,4-Dioxane. Stirred the mixture for 10 minutes at room temperature and added the solution of alcohol dropwise for 5 minutes. Stirred the solution at room temperature till complete consumption of alcohol. Strip off the solvent and dilute the reaction mass with DCM. Filter the suspension through sintered funnel and wash the residue properly with DCM. This residue (white powdered solid, reduced part of IBX) was successfully used for preparation of IBX. Concentrate the filtrate on rotavapor and purify the product by column chromatography. |
| 96% |
With piridinium dichromate; adogen 464; dihydrogen peroxide; anhydrous sodium carbonate In various solvent(s) for 24h; Heating; |
|
| 96% |
With potassium carbonate In lithium hydroxide monohydrate at 26.84℃; for 1.5h; |
|
| 96% |
With oxygen In lithium hydroxide monohydrate at 25℃; for 1h; |
|
| 96% |
With trifluorormethanesulfonic acid; 1-hydroxy-3H-benz[d][1,2]iodoxole-1,3-dione In 1,4-dioxane at 20℃; for 0.25h; |
General Procedure for the IBX-TfOH mediated oxidation of alcohols
General procedure: To the suspension of IBX (1.1 equiv., or 1.1 mmol) and TfOH (2-5 mol %, or 0.02-0.05 mmol) in 3 mL 1,4-dioxane, alcohol (1.0 equiv., 1.0 mmol in 2 mL 1,4-dioxane) was added at room temperature, and the reaction mixture was vigorously stirred till the complete consumption of alcohol, as indicated by TLC. The solvent was evaporated under reduced pressure, and the resulting residue was diluted with 10 mL of dichloromethane. The heterogeneous mixture was stirred for 5 minutes and filtered. The residue was washed with dichloromethane (3×3 mL) and the filtrate was evaporated to dryness to obtain the desired product in sufficiently pure form. |
| 95% |
With oxygen In lithium hydroxide monohydrate for 20h; Heating; |
|
| 95% |
With 3 A molecular sieve; oxygen; triethylamine In tetrahydrofuran; toluene at 25℃; for 12h; |
|
| 95% |
With iodosylbenzene In dichloromethane at 20℃; for 1h; |
|
| 95% |
With palladium diacetate; oxygen; triethylamine In tetrahydrofuran; toluene at 20℃; for 12h; |
|
| 95% |
With sodium chlorine monoxide; N-oxyl-immobilized silica gel; Sodium hydrogenocarbonate In propan-2-one at 0℃; for 0.5h; |
|
| 95% |
With iodine; oxygen In acetonitrile at 20℃; for 2.5h; Irradiation; |
|
| 95% |
With oxygen In lithium hydroxide monohydrate for 20h; Heating; |
|
| 95% |
With C32H16Cl2Cr2N2O10Ru2; propan-2-one at 80℃; for 15h; |
|
| 95% |
With sodium chlorine monoxide; C186H204N12O36; Sodium hydrogenocarbonate; potassium bromide In dichloromethane; lithium hydroxide monohydrate at 0 - 15℃; for 1h; |
|
| 95% |
With (diacetoxyiodo)benzene In dichloromethane at 20℃; for 1h; |
|
| 95% |
With C32H25Cl2N6O2Rh2(1+)*Cl(1-); sodium hydroxide In lithium hydroxide monohydrate at 100℃; for 16h; Sealed tube; Green chemistry; |
|
| 95% |
With C29H35Cl2IrN2O2; anhydrous Sodium acetate In 2,2,2-trifluoroethanol for 20h; Inert atmosphere; Reflux; Schlenk technique; |
|
| 95% |
With cerium(III) bromide; dihydrogen peroxide In 1,4-dioxane; lithium hydroxide monohydrate at 20℃; |
|
| 94% |
With potassium peroxomonosulfate; (o-C6H4-CO2CH2)2CO; (ethylenedinitrilo)tetraacetic acid disodium salt; Sodium hydrogenocarbonate In acetonitrile for 5h; Ambient temperature; |
|
| 94% |
With poly[4-(diacetoxyiodo)styrene]; 2,2,6,6-tetramethyl-1-piperidinyloxy free radical In propan-2-one at 20℃; for 6h; |
|
| 94% |
With 2,2,6,6-tetramethyl-1-piperidinyloxy free radical; CAN; oxygen In acetonitrile at 82℃; for 0.5h; |
|
| 94% |
With manganese(IV) oxide; potassium permanganate at 20℃; for 0.833333h; |
|
| 94% |
With aluminium(III) chloride; 1-decyl-4-aza-1-azoniabicyclo[2.2.2]octane chlorochromate In acetonitrile for 2.2h; Heating; |
|
| 94% |
With doubly supported catalyst Pd(at)MIL-88B-NH2(at)nano-SiO2; air In para-xylene at 150℃; for 12h; |
|
| 94% |
With tert.-butylhydroperoxide In hexane; lithium hydroxide monohydrate at 50℃; for 5.5h; |
|
| 94% |
With triethylamine In o-dimethylbenzene at 140℃; for 36h; Inert atmosphere; Sealed tube; |
|
| 93% |
With 4,4'-bis(dichloroiodo)biphenyl; Etamon In chloroform at 20℃; for 0.5h; |
|
| 93% |
With sodium tetrahydridoborate; oxygen; potassium carbonate In ethanol; lithium hydroxide monohydrate at 20℃; for 0.333333h; |
|
| 93% |
With sodium tetrahydridoborate; 1% Pd/C; lithium hydroxide monohydrate; oxygen; potassium carbonate In ethanol at 20℃; for 0.33h; |
|
| 93% |
With Cu/AlO(OH); orthoperiodic acid In lithium hydroxide monohydrate at 27℃; for 3h; chemoselective reaction; |
|
| 93% |
With 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; potassium-t-butoxide; copper trifluoromethanesulfonate; (S)-(-)-5-(2-pyrrolidinyl)-1H-tetrazole In N,N-dimethyl-formamide at 25℃; for 3h; |
4 4.1.1. The oxidation of secondary alcohols
General procedure: A round-bottom flask was charged with alcohol (2 mmol), CuOTf (0.1 mmol, 0.05 equiv) (S)-5-(pyrrolidin-2-yl)-1H-tetrazole (0.1 mmol, 0.05 equiv), TEMPO (0.1 mmol, 0.05 equiv), t-BuOK (2 mmol, 1 equiv) and DMF (5 ml). The reaction mixture was stirred at 25 °C open to air until the completion of the reaction, as monitored by TLC. The mixture was then diluted with CH2Cl2 (20 ml), washed with water, dried over Na2SO4, and evaporated under vacuum to give the crude product, which was purified by column chromatography to give the pure product. |
| 93% |
With methyl 3,5-bis((1H-1,2,4-triazol-1-yl)methyl)benzoate; oxygen; anhydrous Sodium acetate; nickel(II) bromide at 120℃; for 48h; |
|
| 93% |
With tert.-butylhydroperoxide at 80℃; for 7.25h; Green chemistry; |
General procedure for the oxidation of alcohols tocarbonyl compounds
General procedure: The alcohol (1 mmol) was added to a mixture of TBHP(1 mmol) and VO(ephedrine)2MNPs (50 mg) in PEG(1 mL), and then the mixture was refluxed at 80 C for thetime specified. The progress was monitored by TLC (EtOAc/n-hexane, 1/2). After completion of the reaction, the catalystwas separated from the product by an external magnet(within 5 s), and the mixture was washed with EtOAc(25 mL) and decanted. The decanted mixturewas washedwith 30% NaOH (5 mL) and the organic layer was dried overanhydrous Na2SO4. The evaporation of EtOAc underreduced pressure gave the pure products in 85e98% yields. |
| 93% |
With iron (ΙΙΙ) nitrate nonahydrate; oxygen; 2,3-dicyano-5,6-dichloro-p-benzoquinone In 1,2-dichloro-ethane at 60℃; for 3h; Schlenk technique; Green chemistry; |
|
| 92% |
With [bis(acetoxy)iodo]benzene; 2,6-bis(2-oxazolinyl)pyr Ru(II)-pyr-2,6-dicarboxylate In dichloromethane at 20℃; for 5h; |
|
| 92% |
With silica chromate; mesoporous silica at 20℃; for 0.75h; |
|
| 92% |
With Shvo's catalyst; C33H33CoN3O6; oxygen In acetonitrile at 75℃; for 17h; |
|
| 92% |
With potassium peroxomonosulfate In lithium hydroxide monohydrate; acetonitrile at 20℃; for 3.5h; |
Typical procedure of the SiO2-supported iodoarene-RuCl3 catalyzed oxidation of alcohols and aromatic hydrocarbons:
General procedure: Oxone (0.374 g; 0.6 mmol) was added to a mixture of 1-phenylethanol (25 mg, 0.2 mmol), catalyst 5 or 6 (15 mg) in acetonitrile (1 mL) and water (1 mL) in one portion under stirring at room temperature. The reaction was monitored by TLC by the disappearance of 1-phenylethanol. Then ethyl acetate (3 mL) and water (5 mL) were added and the mixture was stirred for 5 min. The catalyst was filtered, washed with water (2x1 mL), ethyl acetate (2 x 1 mL) and collected, thereby directly being used for next run under the same conditions. The organic solution was separated and the aqueous phase was extracted with ethyl acetate (2 x 5 mL). The organic phases were combined, washed with brine (5 mL), and dried over anhydrous Na2SO4. Removal of the solvent under vacuum afforded acetophenone. The oxidation of the other alcohols and hydrocarbons was performed using a similar procedure. In all cases, conversions were measured by GC-MS with a prior column calibration using authentic samples of reactants and products. The reaction products were isolated by removal of the solid resin followed by aqueous work-up of organic solution; products 10 and 14 were identified by comparison of the retention times and MS data with those obtained for authentic samples or by 1H NMR. Representative spectra are provided below. In the oxidation protocol, the bifunctional SiO2-supported iodoarene-RuCl3 catalysts 5 and 6 were easily separated by filtration and directly reused without noticeable loss of their activity. |
| 92% |
With manganese bis(trifluoromethanesulfonate); adamantane-1-carboxylic acid; C32H38N4O2; dihydrogen peroxide In lithium hydroxide monohydrate; acetonitrile at 0℃; for 2h; chemoselective reaction; |
|
| 92% |
With potassium carbonate In lithium hydroxide monohydrate; dimethyl sulfoxide at 60℃; for 0.833333h; |
|
| 92% |
With potassium carbonate In toluene at 90℃; for 20h; |
2.2.11 General procedure for the oxidation of benzyl alcohols
General procedure: A mixture of K2CO3 (1 mmol) and 2mg of Fe3O4N-CPd Y-S (B) (1 mol% of Pd) in PhCH3 (5 mL) was prepared in a two-necked rounded bottom flask. A solution of the benzyl alcohol (1 mmol) in PhCH3 (5 mL) was injected into the solution, and the resulting mixture was stirred at 90°C under air. The progress of the reaction was monitored by thin-layer chromatography (TLC). After the completion of the reaction, the catalyst was separated from the reaction mixture by external magnetic field and washed with ethanol and ethyl acetate. Then the reaction mixture was concentrated, and then the residue was purified by using thin layer chromatography over SiO2 (n-Hexane:Ethyl acetate 9:1 as v:v%). The solvent was removed under vacuum to yield pure product. |
| 92% |
With encapsulated manganese dioxide nanoparticles in mesoporous silica hollow spheres; air In acetonitrile at 70℃; |
|
| 92% |
With oxygen In dimethyl sulfoxide at 20℃; for 24h; Irradiation; |
|
| 91% |
With potassium permanganate; Rexyn 101 H ion exchange resin In dichloromethane for 4h; Heating; |
|
| 91% |
With 2,2,6,6-tetramethyl-1-piperidinyloxy free radical; oxygen; copper chloride (I) In various solvent(s) at 65℃; for 30h; |
|
| 91% |
With tert-butyl 1-hydroxy-2-methyl-6-trifluoromethyl-1H-indole-3-carboxylate; oxygen; copper chloride (I) In N,N-dimethyl-formamide at 50℃; for 16h; chemoselective reaction; |
2.2 General procedure for aerobic oxidation of allylic and benzylic alcohols
General procedure: To a 10 mL Schlenk tube, NHI-1 (0.2 mmol, 63 mg) and CuCl (0.2 mmol, 19.6 mg) and DMF (2 mL) were added and stirred at 50 °C for about 30 min to form a dark red solution. Alcohol 11 (2 mmol) was added, the mixture was left to stir at 50 °C under an oxygen balloon (1 atm). The reaction progress was monitored by TLC or GC. After completion, the mixture was allowed to cool to room temperature, quenched with 1M HCl and diluted with H2O (50 mL), extracted with EtOAc (EA) (10 mL × 3), the combined organic layer was washed with brine and dried over MgSO4, the crude was purified by flash column chromatography (EtOAc : hexane = 1 : 10 to 1: 3) to afford ketone or aldehyde 12. |
| 91% |
With 9H-fluoren-9-one In dimethyl sulfoxide at 20℃; Irradiation; |
|
| 91% |
With oxygen at 120℃; for 8h; Green chemistry; |
|
| 91% |
With ruthenium(III) trichloride hydrate; oxygen; C25H44NO2PS In 1,2-dichloro-ethane at 60℃; for 17h; |
|
| 90.8% |
With tris(triphenylphosphine)ruthenium(II) chloride; potassium carbonate; propan-2-one at 56℃; for 1h; |
|
| 90% |
With di(pentafluorophenyl)hydroxyborane; magnesium(II) sulfate; 2,2-dimethypropanal In toluene for 5h; Ambient temperature; |
|
| 90% |
With tetrabutylammonium bromide; palladium (II) chloride at 120℃; for 22h; |
|
| 90% |
With potassium peroxodisulfate; molybdenum trioxide In lithium hydroxide monohydrate; acetonitrile for 1.8h; Reflux; |
|
| 90% |
With [bis(acetoxy)iodo]benzene; Ru2(bbpmp)(OAc)2 acetate In tetrahydrofuran; acetonitrile at 40℃; for 4h; Inert atmosphere; |
|
| 90% |
With tert.-butylhydroperoxide; (Ph2PRuCl2(η6-p-cymene))(ferrocene-1,1'-diyl)C(O)NHCH2COOCH3 In lithium hydroxide monohydrate at 20℃; for 24h; |
|
| 90% |
With ammonium nitrate; hydrogenchloride; 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; oxygen In lithium hydroxide monohydrate; acetonitrile at 60℃; for 6h; Green chemistry; |
|
| 90% |
With 6-((cobalt(II) 4,9,16,23-tetraaminephthalocyanin-4-yl))cellulose; oxygen; potassium hydroxide In o-dimethylbenzene at 20℃; for 2.5h; Green chemistry; |
Typical procedure for the oxidation of 1-phenyl-ethanol
General procedure: 1-Phenyl-ethanol (0.14 g, 1.00 mmol) was added to a two-necked flask equipped with a gas bubbling tube containing colloidal of CoPcCell (0.05 g) and KOH (0.25 mmol) in o-xylene (5 mL) at room temperature. The mixture was stirred at room temperature under O2 atmosphere provided with a balloon. The progress of the reaction was followed by thin layer chromatography (TLC). Upon completion, CoPcCell was separated by filtration and washed with acetone (5 mL). Acetophenone was isolated from the mixture using column chromatography with n-hexane in 90% yield. |
| 90% |
With ammonium nitrate; hydrogenchloride; 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical In acetonitrile at 60℃; Green chemistry; chemoselective reaction; |
|
| 90.1% |
With tert.-butylhydroperoxide at 60℃; for 5h; |
Catalytic oxidation of alcohols
General procedure: In a typical process, into a 5-ml two-necked round-bottomflask equipped with a magnetic stirrer were addedRu(pbbp)(pydic) (0.002 mmol) and alcohol (2 mmol)successively at room temperature. The mixture washeated to 60 C under stirring, and then TBHP (70%aqueous solution) was slowly dropped in 0.5 h. Thereaction was monitored by GC equipped with a SE 54column (30 m 9 0.5 lm). After reaction, the product waspurified by column chromatography over silica gel (eluent:n-hexane/ethyl acetate) and characterized by 1HNMR. |
| 90% |
With Potassium bicarbonate; 9-oxo-9-azabicyclo<3.3.1>nonanium tetrafluoroborate In acetonitrile at 25℃; for 18h; Inert atmosphere; |
4. General procedures of ABNO+BF4- mediated alcohol oxidation
General procedure: A 15 mm flame-dried test tube, which was equipped with a magnetic stir bar and charged with alcohol (0.3 mmol, in case of solid), ABNO+BF4- (2.0 equiv, 0.6 mmol), and KHCO3 (1.0 equiv, 0.3 mmol), was evacuated and backfilled with nitrogen (this process was repeated 3 times). After 0.3 mL of CH3CN was added, alcohol (0.3 mmol, in case of liquid), and CH3CN (0.3 mL) were added in sequence. The reaction mixture was stirred for 18 h at 25 oC under N2 balloon. The reaction was diluted by adding EtOAc and washed 4 M HCl aqueous solution. Two layers were separated, and the aqueous layer was extracted with EtOAc. The organic layer was dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography to give the desired carbonyl products. |
| 90% |
With oxygen at 20℃; for 1.5h; Sealed tube; Irradiation; |
2.5. General procedure for the photooxidation of alcohols
General procedure: RuXN-C (0.2 mol% of Ru) was added to a test tube containingbenzyl alcohol stock solution (1 mmol, in benzotrifluoride (1 mL)) andbenzotrifluoride (4 mL). The mixture was left to stir for 30 min to dispersethe catalyst under a dioxygen atmosphere and sealed with arubber septum. The mixture was then irradiated with a 5W Xenon HIDlamp with an irradiance of 12.5mW cm-2 at room temperature. Afterthe photoreaction, the photocatalyst could be efficiently and simplyrecovered by external magnetic field and washed with acetonitrile, anddried in air and reused for the next cycle. |
| 89% |
With dimethyl sulfoxide; triphenylphosphine dibromide 1:1 addition complex; triethylamine In dichloromethane at -78 - 20℃; for 3.25h; |
|
| 89% |
With aluminium(III) chloride; 1-butyl-4-aza-1-azoniabicyclo[2.2.2]octane chlorochromate In acetonitrile for 3h; Heating; |
|
| 89% |
With HNO3; diphosphorus pentoxide; mesoporous silica for 0.05h; |
|
| 89% |
With 4,5,6,7-F4-1-OH-1-oxo-1H-1λ5-benzo[d][1,2]iodoxol-3-one In lithium hydroxide monohydrate; acetonitrile at 20℃; for 4h; |
|
| 89% |
With 1,3,5,7-tetrakis[4-(diacetoxyiodo)phenyl]adamantane; 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical In dichloromethane at 20℃; |
|
| 89% |
With potassium hydroxide In o-dimethylbenzene at 20℃; for 0.5h; |
|
| 89% |
With potassium hydroxide In o-dimethylbenzene at 20℃; for 0.5h; |
2.4 Typical procedure for the oxidation of benzylalcohol
General procedure: Benzylalcohol (0.1 g, 1.0 mmol) was added to a two-necked flask equipped with a gas bubbling tube containing colloidal of Co(II)-EDANC (0.2 g) and KOH (0.5 mmol) in o-xylene (5mL). Air was bubbled at arate of 5mL/min into the reaction mixture and the progress of the reaction was followed by thin-layer chromatography (TLC). Upon completion, Co(II)-EDANC was separated by filtration and washed with CH3CN (2 × 5mL). The filtrate solvent was evaporated under vacuum and benzylalcohol was purified with column chromatography with n-hexane:ethylacetate (2:1) in 95% yield. |
| 89.6% |
With tert.-butylhydroperoxide at 70℃; for 4.3h; |
|






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping