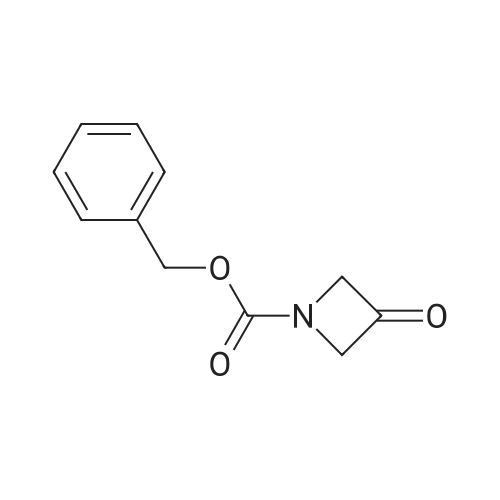
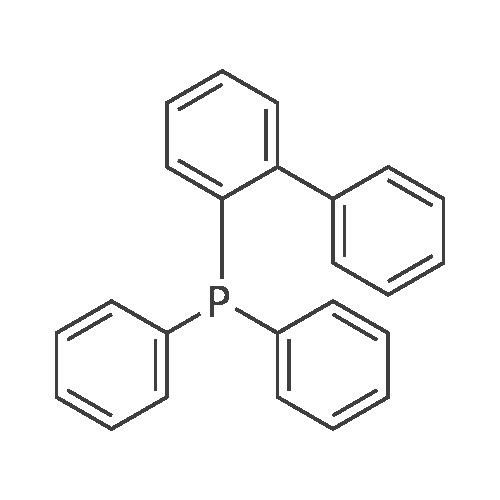
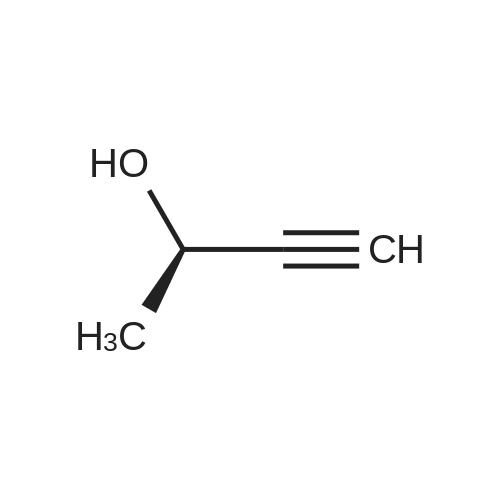
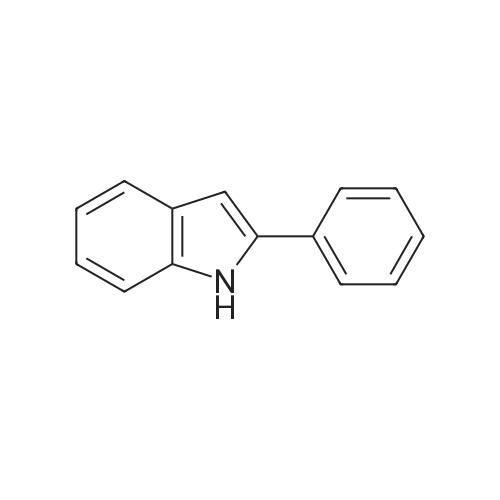
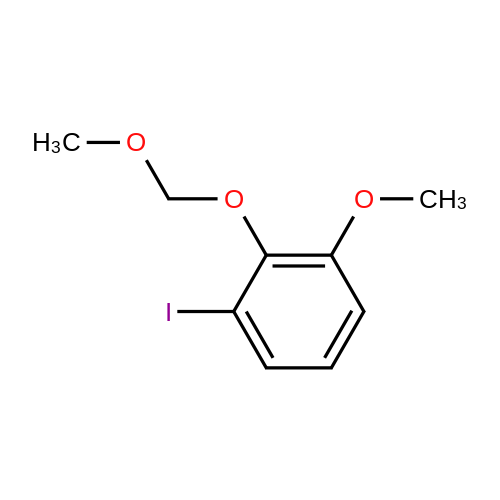
| 76%Chromat. |
With N-methylcyclohexylamine;(1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; In 1,4-dioxane; at 80℃; for 17h; |
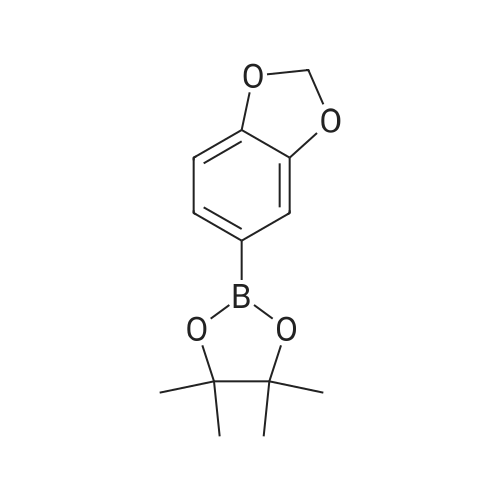
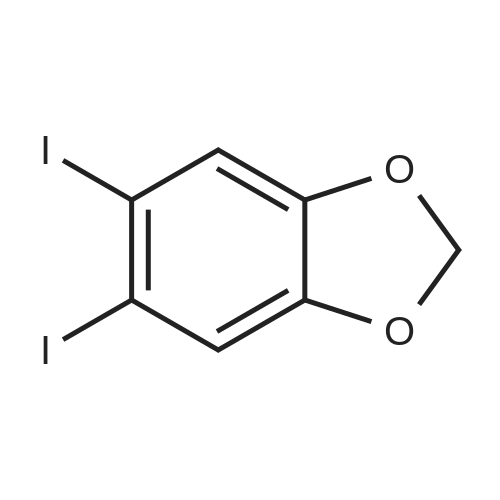
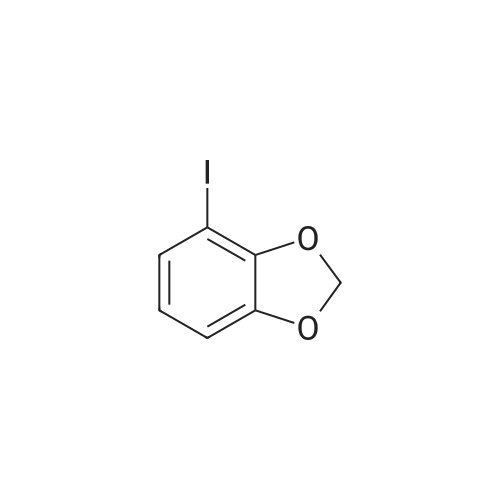
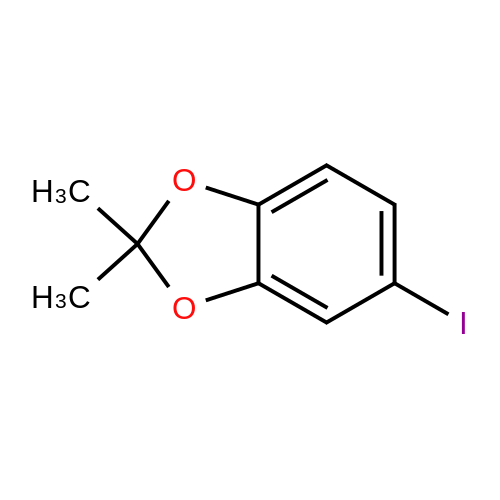
[00228] To 26.1 mg PdCl2(dppf).CH2Cl2 in a reaction tube under nitrogen were added 4 ml dioxane, 0.12 ml (1.0 mmol) of N-methypiperidine, 0.16 ml (1.1 mmol) pinacolborane and 253 mg (1.02 mmol) 1-iodo-3,4-methylenedioxybenzene. The red reaction solution was warmed to 80 C. with stirring for 17 h in an oil bath. Analysis of the dark green reaction solution by gc as described above gave the reaction product distribution: 1,3-benzodioxole (16% of gc peak area); the pinacol ester of phenylboronic acid (7%) and the desired arylboronic acid pinacol ester (peak area 76%). Increasing the N-methypiperidine concentration to 3 mmol had little effect on product distribution. Found: 1,3-benzodioxole (17% of gc peak area); the pinacol ester of phenylboronic acid (8%) and the pinacol ester of 3,4-methylenedioxyphenylboronic acid (peak area 75%). |
| 13%Chromat. |
With triethylamine;palladium diacetate; In 1,4-dioxane; at 80℃; for 1h; |
[00209] To 22 mg of Pd(II)acetate in a reaction tube under nitrogen were added 4 ml dioxane, 260 mg (1.05 mmol) 1-iodo-3,4-methylenedioxybenzene, 0.44 ml (3.1 mmol) triethylamine and 0.22 ml (1.5 mmol) pinacolborane. The solution became black on addition of the pinacolborane to the other reaction ingredients. The reaction solution was warmed to 80 C. with stirring in an oil bath. An aliquot (ca. 0.3 ml) of the reaction solution was removed after 1 h and extracted into diethyl ether and washed several times with water and analysed by gc (fid detector, SGE HT5 capillary column). Besides some 1,3-benzodioxole, the only other product peak in the gc (13% of total peak area) was that due to the desired arylboronic acid pinacol ester. |
| 81%Chromat. |
With triethylamine;(1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; In 1,4-dioxane; at 80℃; for 16h; |
[00221] This example demonstrates that there is an inverse relationship between the amount of base (triethylamine) used in the reaction and the extent of dehalogenation of 1-iodo-3,4-methylenedioxybenzene. It also demonstrates that the amount of pinacolborane required for the complete reaction of the aryl halide can be less than 1.5 equivalents. Unreacted pinacolborane was found at the completion of the reaction when 1.1 equivalents were used. Formation of 5-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3-benzodioxole [00222] [C00045] [00223] To 25.1 mg PdCl2(dppf).CH2Cl2 in a reaction tube under nitrogen were added 4 ml dioxane, 0.42 ml (3 mmol) triethylamine, 0.16 ml (1.1 mmol) pinacolborane and 256 mg (1.03 mmol) 1-iodo-3,4-methylenedioxybenzene. The reaction solution was warmed to 80 C., with stirring, for 16 h in an oil bath. An aliquot (ca. 0.25 ml) of the reaction solution was removed, extracted into ethyl acetate and washed several times with water and brine solution and analysed by gc (fid detector, SGE HT5 capillary column). Hydrogen evolution was observed on the initial contact of the reaction sample with water, indicative that excess pinacolborane was present at the completion of the reaction even though only 1.1 equivalents had been used. Besides the 1,3-benzodioxole (10% of gc peak area) and pinacol ester of phenylboronic acid (7%), the only other product peak in the gc (area of 81%) was that due to the desired arylboronic acid pinacol ester. In a parallel reaction in which the only change was a reduction in the amount of triethylamine used, from 3.0 equivalents to 1.0 equivalents, the product distribution found was 1,3-benzodioxole (23% of gc peak area), the pinacol ester of phenylboronic acid (6%) and the desired arylboronic acid pinacol ester (peak area 69%). |
|
With N,N-dimethyl acetamide; triethylamine;(1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; In 1,4-dioxane; at 80℃; for 24h; |
[00347] In a reaction tube under nitrogen, a mixture of PdCl2(dppf)CH2Cl2 (23 mg; 0.028 mmol), triethylamine (0.36 ml; 2.58 mmol), HB(pin) (0.19 ml; 1.31 mmol), 1-iodo-3,4-methylenedioxybenzene (216 mg; 0.871 mmol) and N,N-dimethylacetamide (41 mg; 0.47 mmol) in dioxane (5 ml; dried over 4 ? sieves) was sealed and stirred at 80 C. GC analysis after 24 hours showed the reaction was complete and the desired arylborate compound had formed (new peak at 11.2 minutes). |
| 80%Chromat. |
With 2.6-dimethylpiperidine;(1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; In 1,4-dioxane; at 80℃; for 4h; |
[00230] To 27.7 mg PdCl2(dppf).CH2Cl2 in a reaction tube under nitrogen were added 4 ml dioxane, 0.14 ml (1.0 mmol) of 2,6-dimethylpiperidine, 0.23 ml (1.5 mmol) pinacolborane (crimson solution) and 261 mg (1.05 mmol) 1-iodo-3,4-methylenedioxybenzene. The crimson reaction solution was warmed to 80 C. with stirring in an oil bath. Analysis of the reaction solution by gc, as described above, was carried out at intervals (see Table 19.1). The reaction is fast and is complete after 4 h reaction time. The initial reaction rate with 1 mmol of 2,6-dimethylpiperidine as base exceeds that found using 3 mmol of triethylamine (see Table 17.3; catalyst not activated prior to reaction) and the final product distribution is essentially the same. This is not the case if one compares the reactions in which either 1 mmol of 2,6-dimethylpiperidine or 1 mmol of triethylamine is used as base. As noted above, dehalogenation in reactions using 1 mmol of triethylamine is considerable (23% of peak area in the gc is due to 1,3-benzodioxole) and the desired product, the pinacol ester of 3,4-methylenedioxyphenylboronic acid is only 69% of peak area. [00231] The results are different when 3 mmol of 2,6-dimethylpiperidine is used in the reaction. The reaction rate is reduced, especially after the first 1 to 2 hours (see Table 19.2). The results suggest that in situ catalyst activation may be initially enhanced using 2,6-dimethylpiperidine but excess of this base also retards the reaction. Dehalogenation occurs predominantly during the earlier part of the reaction. [TABLE-US-00007] TABLE 19.1 Rate of product formation on reaction of 1-iodo-3,4-methylenedioxybenzene with pinacolborane at 80 C., catalyst PdCl2(dppf).CH2Cl2, base 2,6-dimethylpiperidine (1.0 mmol). The concentrations are expressed in area % (uncorrected for response factors) determined by gc analysis of aliquots of the reaction solution taken at selected reaction times*. Reaction Time (h) [C00047] [C00048] [C00049] [C00050] 1 8.7 2 70 20 2 8.4 4.3 42 44 3 8.5 6.2 16 67 4 11.5 6.5 0.3 80 5 11.6 6.4 0 80 *The reaction was quenched at the selected reaction time by addition of the aliquot of reaction solution to a water/ethyl acetate mixture. [TABLE-US-00008] TABLE 19.2 Rate of product formation on reaction of 1-iodo-3,4-methylenedioxybenzene with pinacolborane at 80 C., catalyst PdCl2(dppf).CH2Cl2, base 2,6-dimethylpiperidine (3.0 mmol). The concentrations are expressed in area % (uncorrected for response factors) determined by gc analysis of aliquots of the reaction solution taken at selected reaction times*. Reaction Time (h) [C00051] [C00052] [C00053] [C00054] 1 8.9 1.3 69 20 2 9.2 2.8 49 38 3 9.6 3.5 33 53 4 11 5.1 25 58 5 10 5.6 18 66 6 10 5.9 13 71 25.5 12 6.1 2.2 78 *The reaction was quenched at the selected reaction time by addition of the aliquot of reaction solution to a water/ethyl acetate mixture. |
| 92%Chromat. |
|
[00237] 6.8 mg of Pd(OAc)2 (0.03 mmol) and 13 mg (0.03 mmol) of bis(1,2-diphenylphosphino)ethane were placed in a reaction tube under nitrogen together with 4 ml of dioxane and 0.45 ml of triethylamine. The tube was heated in an oil bath at 80 C. for 15.5 h and the solution became reddish in colour. Then added at room temperature 267 mg (1.08 mmol) of 1-iodo-3,4-methylenedioxybenzene and 0.23 ml (1.5 mmol) of pinacolborane. The reaction mixture was then warmed to 80 C. After 3 h, the reaction was complete with 92% of the total peak areas due to the desired product. |
| 89%Chromat. |
|
[00235] To 24.4 mg PdCl2[(PCy3)2] in a reaction tube under nitrogen was added 4 ml dioxane and 0.43 ml (3.0 mmol) of triethylamine. The mixture was heated at 80 C. to give an lime-green coloured solution which still contained solids, presumably PdCl2[(PCy3)2]. To this mixture, at room temperature, were added 0.23 ml (1.5 mmol) pinacolborane and 247 mg (1.0 mmol) 1-iodo-3,4-methylenedioxybenzene. The reaction solution was warmed to 80 C. with stirring in an oil bath. Analysis of the reaction solution by gc, as described above, was carried out at intervals (see Table 20.2). After the initial hour reaction period, the reaction rate increased sharply and the reaction was essentially complete after 3.5 hours. Solubilisation and activation of the catalyst appears to occur over the first hour of heating the reaction mixture. The reaction solution remains virtually colourless over the course of the reaction and the catalyst is all dissolved at the end of the reaction. Importantly, the absence of phenyl groups on the catalyst gives a product which is not contaminated by the pinacol ester of that particular phenylboronic acid. This eliminates the major difficulty in the purification of the product arylboronic acid ester or, in the case of one-pot coupling reactions, it eliminates the need to separate a biaryl mixture. [TABLE-US-00010] TABLE 20.2 Rate of product formation on reaction of 1-iodo-3,4-methylenedioxybenzene with pinacolborane at 80 C. The catalyst PdCl2[PCy3)2] was heated with the base triethylamine (3.0 mmol) prior to reaction. The concentrations are expressed in area % (uncorrected for response factors) determined by gc analysis of aliquots of the reaction solution taken at selected reaction times*. Reaction Time (mins) [C00060] [C00061] [C00062] [C00063] 6 0 0 98 1 16 0 0 98 2 31 0 0 97 3 60 0 0 92 8.3 150 (2.5 h) 3.9 0 29 67 210 (3.5 h) 9.6 0 1.7 89 300 (5 h) 11 0 0 89 *The reaction was quenched at the selected reaction time by addition of the aliquot of reaction solution to a water/ethyl acetate mixture. |
| 88%Chromat. |
|
[00233] To 24.7 mg PdCl2[(PPh2(CH2)5PPh2] in a reaction tube under nitrogen was added 4 ml dioxane and 0.43 ml (3.0 mmol) of triethylamine. The mixture was heated at 80 C. to give an orange coloured solution which still contained solids, presumably PdCl2[(PPh2(CH2)5PPh2]. To this mixture, at room temperature, were added 0.23 ml (1.5 mmol) pinacolborane (solution became brown but still contained insolubles) and 255 mg (1.03 mmol) 1-iodo-3,4-methylenedioxybenzene. The reaction solution was warmed to 80 C. with stirring in an oil bath. Analysis of the reaction solution by gc, as described above, was carried out at intervals (see Table 20.1). The reaction was complete after 5 h reaction time. The reaction solution was a bright crimson colour at the completion of the reaction and the only solids present on cooling to room temp. appeared to be the triethylamine salt. The amount of pinacol ester of phenylboronic acid formed is low with PdCl2[(PPh2(CH2)5PPh2] as catalyst and forms only later in the reaction. [TABLE-US-00009] TABLE 20.1 Rate of product formation on reaction of 1-iodo-3,4-methylenedioxybenzene with pinacolborane at 80 C. The catalyst PdCl2[(PPh2(CH2)5PPh2] was heated with the base triethylamine (3.0 mmol) prior to reaction. The concentrations are expressed in area % (uncorrected for response factors) determined by gc analysis of aliquots of the reaction solution taken at selected reaction times*. Reaction Time (mins) [C00056] [C00057] [C00058] [C00059] 5 0 0 99 1 15 0 0 90 10 30 4.0 0 60 36 60 5.2 0 40 54 150 7.1 1.8 13 78 210 7.9 2.1 3.5 86 300 9.3 2.4 0 88 *The reaction was quenched at the selected reaction time by addition of the aliquot of reaction solution to a water/ethyl acetate mixture. |
| 39.5%Chromat. |
|
[00447] In a reaction tube under nitrogen, a mixture of PdCl2(dppf)CH2Cl2 (22 mg; 0.027 mmol) and triethylamine (0.36 ml; 2.58 mmol) in toluene (4 ml; dried over 4 ? sieves) was sealed and stirred at 80 C. overnight (18 h). After cooling to room temperature, HB(pin) (0.19 ml; 1.31 mmol) and 1-iodo-3,4-methylenedioxybenzene (214 mg; 0.863 mmol) were added and the reaction mixture was stirred at 40 C. [00448] GC analysis after 1.25 h showed a peak at 9.59 mins (39.5%) which was identified by GC/MS as the desired compound. |
| 70 - 94%Chromat. |
|
[00216] This example demonstrates that palladium catalysts can be activated by treatment with a base prior to their use in promoting the reaction of an organic halide with a dialkoxyborane. In particular, the catalytic activity of PdCl2(dppf).CH2Cl2 can be increased significantly, especially the initial activity, by treatment, in the reaction solvent, with triethylamine prior to the addition of the pinacolborane and substrate. Besides the rate enhancement observed in the formation of the required product boronic acid ester (e.g. pinacol ester of 3,4-methylenedioxyphenylboronic acid) there is a further advantage in the prior activation of the catalyst in that the amount of bi-product formed in the reaction (viz. 1,3-benzodioxole through dehalogenation of the substrate and the pinacol ester of phenylboronic acid in which the phenyl groups are from the catalyst ligand) is significantly reduced. Formation of 5-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3-benzodioxole [00217] [C00024] [00218] To 24.6 mg PdCl2(dppf).CH2Cl2 in a reaction tube under nitrogen was added 4 ml dioxane and 0.42 ml (3 mmol) triethylamine. The mixture was heated at 80 C. for ca 17 h. The red-orange suspension of PdCl2(dppf).CH2Cl2 dissolved to give a dark red-brown solution. To this solution, at room temperature, was added 0.23 ml (1.5 mmol) pinacolborane and 253 mg (1.02 mmol) 1-iodo-3,4-methylenedioxybenzene. The reaction solution was warmed to 80 C. with stirring for 1 h in an oil bath. The solution remained a dark red-brown in colour. An aliquot (ca. 0.25 ml) of the reaction solution was removed, extracted into ethyl acetate and washed several times with water and brine solution and analysed by gc (fid detector, SGE HT5 capillary column). Apart from a small amount of 1,3-benzodioxole (5% of uncorrected gc peak area) and pinacol ester of phenylboronic acid (3%), the only other product peak in the gc (area of 92%, uncorrected) was that due to the desired arylboronic acid pinacol ester. There was no evidence of biaryl formation. The rate of reaction of 1-iodo-3,4-methylenedioxybenzene with pinacolborane at 80 C. with activated catalyst is indicated also in Table 17.1. Table 17.2 shows that side product formation can be reduced still further by carrying out the reaction at 30 C. [TABLE-US-00002] TABLE 17.1 Rate of product formation on reaction* of 1-iodo-3,4-methylenedioxybenzene with pinacolborane at 80 C. in which the catalyst, PdCl2(dppf).CH2Cl2, was activated, prior to employment in the reaction, with triethylamine. The concentrations are expressed in area % (uncorrected for response factors) determined by gc analysis of aliquots of the reaction solution taken at selected reaction times. Reaction Time (mins) [C00025] [C00026] [C00027] [C00028] 6 4.4 0.74 54 40 10 4.4 1.0 40 55 15 5.2 2.1 26 66 20 5.9 2.9 16.3 75 25 5.7 2.9 9.2 82 30 6.0 3.2 3.6 87 35 5.9 3.4 1.2 89 40 5.7 3.4 0.7 90 50 5.6 3.4 0 91 180 5.7 3.5 0 91 *Used 25.5 mg of PdCl2(dppf).CH2Cl2. 4 ml dioxane, 0.43 ml (3.0 mmol) triethylamine and warmed to 80 C. for 16 h. Then added 0.23 ml (1.5 mmol) pinacolborane and 247 mg (1.0 mmol) 1-iodo-3,4-methylenedioxybenzene at room temp. before warming the reaction to 80 C. The reaction was quenched at the selected reaction time by addition of the aliquot of reaction solution to a water/ethyl acetate mixture. [TABLE-US-00003] TABLE 17.2 Rate of product formation on reaction* of 1-iodo-3,4-methylenedioxybenzene with pinacolborane at 30 C. in which the catalyst, PdCl2(dppf).CH2Cl2, was activated, prior to employment in the reaction, with triethylamine. The concentrations are expressed in area % (uncorrected for response factors) determined by gc analysis of aliquots of the reaction solution taken at selected reaction times Reaction Time (h) [C00029] [C00030] [C00031] [C00032] 1 1.6 0 94 4.8 2 1.7 0 89 9.7 3 2 0 84 13.8 4 2 0 81 17 7 2.3 0 71 26 28 4 0.6 27 68 71.5 4.2 1.9 0 94 *Used 25 mg of PdCl2(dppf).CH2Cl2, 4 ml dioxane, 0.43 ml (3.0 mmol) triethylamine and warmed to 80 C. for 16 h. Then added 0.23 ml (1.5 mmol) pinacolborane and 262 mg (1.05 mmol) 1-iodo-3,4-methylenedioxybenzene at room temp. before warming the reaction to 80 C. The reaction was quenched at the selected reaction time by addition of the aliquot of reaction solution to a water/ethyl acetate mixture. [00219] When the catalyst PdCl2(dppf).CH2Cl2 is treated with the amine together with the borane ester prior to use in the reaction, the initial reaction rate is enhanced, indicating that some catalyst has been activated. The overall reaction, however, is slower than that when the catalyst receives no pretreatment. Catalyst presumably unactivated by the pretreatment with triethylamine and pinacolborane appears to be more resistant to activation during the progress of the boronation reaction. This can be seen by comparison of Tables 17.3 and 17.4. In Table 17.3, the catalyst was not activated prior to use and the reaction rate over the first 1 to 2 hours is slow. In T... |
| 51%Chromat. |
|
[00341] In a reaction tube under nitrogen, a mixture of PdCl2(dppf)CH2Cl2 (22 mg; 0.027 mmol) and triethylamine (0.36 ml; 2.58 mmol) in dioxane (4 ml; dried over 4 ? sieves) was sealed and stirred at 80 C. overnight (18 h). After cooling to room temperature, HB(pin) (0.18 ml; 1.24 mmol), 1-iodo-3,4-methylenedioxybenzene (216 mg; 0.871 mmol) and N-methylacetamide (39 mg; 0.56 mmol) were added and the reaction mixture was stirred at 80 C. GC analysis after 30 minutes showed a new peak at 11.1 minutes (51%) which was identified by GC/MS as the desired arylborate compound. |
| 46.1%Chromat. |
|
[00450] In a reaction tube under nitrogen, a mixture of PdCl2(dppf)CH2Cl2 (22 mg; 0.027 mmol) and triethylamine (0.36 ml; 2.58 mmol) in dioxane (4 ml; dried over 4 ? sieves) was sealed and stirred at 80 C. overnight (18 h). After cooling to room temperature, HB(pin) (0.19 ml; 1.31 mmol) and 1-iodo-3,4-methylenedioxybenzene (214 mg; 0.863 mmol) were added and the reaction mixture was stirred at 40 C. [00451] GC analysis after 1.25 h showed a peak at 9.53 mins (46.1%) which was identified by GC/MS as the desired compound. |
| 57.8%Chromat. |
|
[00444] In a reaction tube under nitrogen, a mixture of PdCl2(dppf)CH2Cl2 (22 mg; 0.027 mmol) and triethylamine (0.36 ml; 2.58 mmol) in distilled dimethoxyethane (4 ml) was sealed and stirred at 80 C. overnight (18 h). After cooling to room temperature, HB(pin) (0.19 ml; 1.31 mmol) and 1-iodo-3,4-methylenedioxybenzene (216 mg; 0.870 mmol) were added and the reaction mixture was stirred at 40 C. [00445] GC analysis after 1.25 h showed a peak at 9.61 mins (57.8%) which was identified by GC/MS as the desired compound. |
| 7.2%Chromat. |
|
[00441] In a reaction tube under nitrogen, a mixture of PdCl2(dppf)CH2Cl2 (22 mg; 0.027 mmol) and triethylamine (0.36 ml; 2.58 mmol) in distilled 1,2-dichloroethane (4 ml) was sealed and stirred at 80 C. overnight (18 h). After cooling to room temperature, HB(pin) (0.19 ml; 1.31 mmol) and 1-iodo-3,4-methylenedioxybenzene (215 mg; 0.867 mmol) were added and the reaction mixture was stirred at 40 C. [00442] GC analysis after 1.25 h showed a peak at 9.54 mins (7.2%) which was identified by GC/MS as the desired compound. |
|
|
[00411] In a reaction tube under nitrogen, a mixture of PdCl2(dppf)CH2Cl2 (24 mg; 0.029 mmol) and triethylamine (0.36 ml; 2.58 mmol) in dimethylsulphoxide (4 ml; dried over 4 A sieves) was sealed and stirred at 80 C. for 18 hours. After cooling to room temperature HB(pin) (0.19 ml; 1.31 mmol) was added followed by 1-iodo-3,4-methylenedioxybenzene (211. mg; 0.851 mmol). The reaction mixture was stirred at 80 C. GC analysis after 18 hours showed that the desired arylborate compound had formed. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping