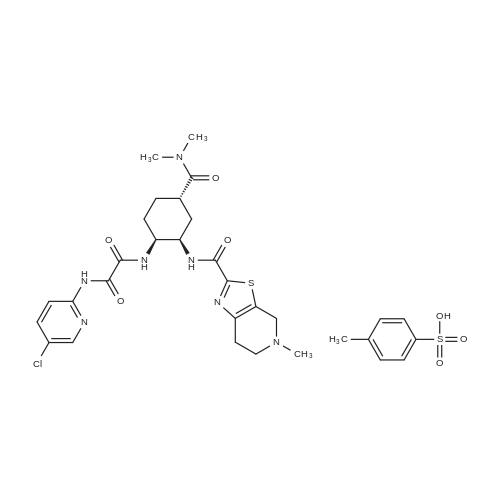
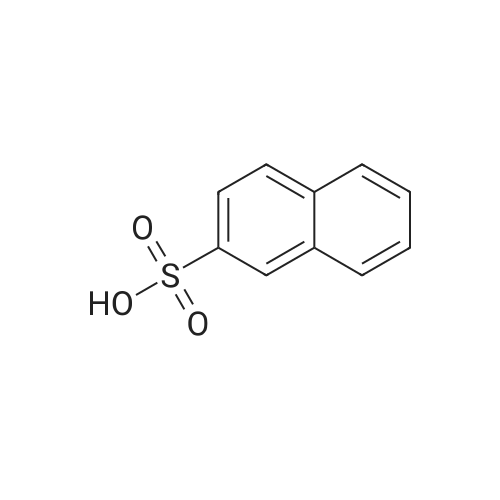
| 94.2% |
In ethanol; water at 10 - 70℃; |
3
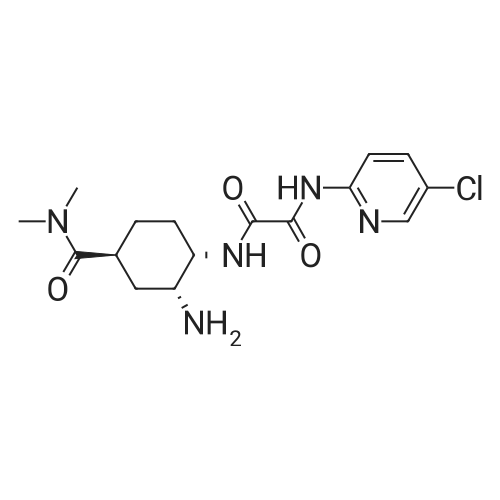
(Test Example 3) Based on the results of Test Examples 1 and 2, the present inventor had an idea that the excess of p-toluenesulfonic acid that promotes the decomposition of compound B be avoided for dissolving compound B under high temperatures, and the excess of p-toluenesulfonic acid under low temperatures be created for crystallization to reduce the solubility of compound A, resulting in improvement in the yield of step (a). An attempt was made to perform a p-toluenesulfonic acid division method reflecting this idea. Specifically, 21 ml of water, 49 ml of ethanol, and 3.30 g of TsOH·H2O (0.95 molar equivalent with respect to compound B) were added to 10.0 g of compound B, followed by dissolution at 70°C. The dissolved solution was filtered through a filter, and the filter was washed with a mixed solution of 3 ml of water and 17 ml of ethanol. Subsequently, a mixed solution of the filtered mother liquor and the washes was slowly cooled, and 521 mg of TsOH·H2O (0.15 molar equivalent with respect to compound B) and 150 ml of ethanol were added thereto. After stirring at 10°C, crystals of compound A were collected by filtration, and its yield was determined. Moreover, two lots of compound B were used to evaluate reproducibility. To prepare a control, 21 ml of water, 49 ml of ethanol, and 3.47 g of TsOH·H2O (1.0 molar equivalent with respect to compound B) were added to 10.0 g of compound B, followed by dissolution at 70°C. The dissolved solution was filtered through a filter, and the filter was washed with a mixed solution of 3 ml of water and 17 ml of ethanol. Subsequently, a mixed solution of the filtered mother liquor and the washes was slowly cooled, and 150 ml of ethanol was added thereto. After stirring at 10°C, crystals of compound A were collected by filtration, and its yield was determined. A loss into the mother liquor means the compound A remaining in the mother liquor without being deposited as crystals. Loss into mother liquor (%) described in Table 1 was calculated by converting the weight of the compound A remaining in the mother liquor into the weight of the compound B and indicating this weight as a ratio (%) to the weight of the compound B before the start of the reaction. The results are shown in Table 3. |
| 92% |
Stage #1: edoxaban; toluene-4-sulfonic acid In ethanol; 1,2-dichloro-ethane at 20℃;
Stage #2: In ethanol; water at 20 - 60℃; for 1h; |
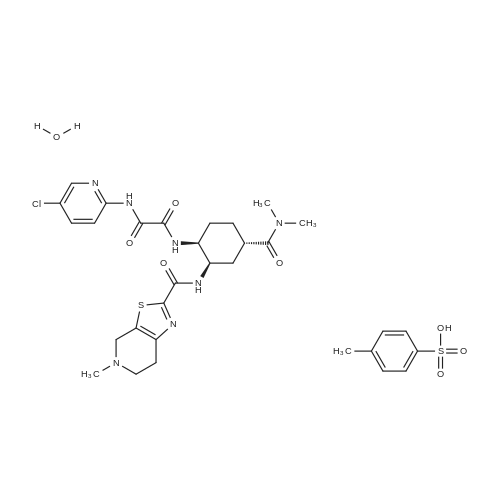
2 N1-(5-chloropyridin-2-yl)-N2-((1S,2R,4S)-4-[(dimethylamino)carbonyl]-2-[(5-methyl-4,5,6,7-tetrahydrothiazole[5,4-c]pyridin-2-yl)carbonyl]amino}cyclohexyl)ethanediamide 4-toluene sulfonic acid monohydrate
0.25 g of the compound synthesized in Experimental Example 1 was usedN1- (5-chloropyridin-2-yl) -N2- ((lS, 2R, 4S)4 - [(dimethylamino) carbonyl] -2 - [(5-methyl-4,5,6,7-tetrahydrothiazole [5,4-c]Pyridin-2-yl) carbonyl] amino} cyclohexyl) ethanediamide was dissolved in 5 ml of dichloroethane, 0.52 ml of 1 mol per liter of p-toluenesulfonic acid ethanol solution was added,The mixture was stirred at room temperature. The solvent was then distilled off.Add 3.9 ml of 15% aqueous ethanol to the residue,And the mixture was dissolved with stirring at 60 ° C.The mixture was then allowed to cool to room temperature and stirred for 1 hour.The precipitated crystals were collected by filtration and rinsed with ethanol,And then dried under reduced pressure at room temperature for 2 hours to obtain0.29 g of the title compound (yield 92%). |
| 92% |
In ethanol; water at 70℃; for 1h; |
4; 5; 9 Preparation of edoxaban p-toluenesulfonic acid monohydrate
To a 1 L three-necked flask, 68 g of DXB-ABC and 332 ml of absolute ethanol, 147 ml of purified water were added, stirred, and 22.4 g of p-toluenesulfonic acid monohydrate was added at room temperature, heated to 70 ° C, all solids were dissolved, filtered while hot, and used. The filter cake was rinsed with 128 ml of 85% ethanol, transferred to a 1 L three-necked flask, and 3.6 g of p-toluenesulfonic acid monohydrate and 1020 ml of absolute ethanol were added thereto, and the mixture was cooled to 5-10 ° C, and stirred for lh. After suction filtration, the cake was dried under vacuum at 30-40 ° C to give a white solid. The yield was 95% and the purity was 99.82%. 5. Purification of edoxaban p-toluenesulfonic acid monohydrate80 g of edoxaban p-toluenesulfonic acid monohydrate and 34 ml of absolute ethanol, 6 ml of purified water were added to a 250 ml three-necked flask, stirred, stirred at room temperature for 20 h, cooled to 5-10 ° C and stirred for 1 h. Filter by suction and the filter cake was dried under vacuum. Yield: 92%, purity: 99.94%. |
| 88.3% |
With water In ethanol at 75 - 80℃; |
11 Example 11 N1-(5-Chloro-2-pyridyl)-N2-[(1S,2R,4S)-4-[(dimethylamino)carbonyl]-2-[[(4,5,6,7-Tetrahydro-5-methylthiazolo[5,4-c]pyridin-2-yl)carbonyl]amino]cyclohexyl]oxalamide p-toluenesulfonateSynthesis of compound (Edoxaban tosylate hydrate)
Add absolute ethanol (2700g) to the reaction flask, add purified water (1350g), then add 109TM-01 (540g, 0.9853mol) obtained in Example 10, stir and disperse evenly, add p-toluenesulfonic acid monohydrate ( 200g, 1.051mol), heated to 7580 to completely dissolve, add activated carbon, keep warm and stir to decolorize for about 3040min; filter while hot, rinse the filter residue with absolute ethanol, collect the filtrate and cool to 05, keep warm Crystallize for about 2 to 4 hours; filter, rinse the filter cake with 75% ethanol, and collect the solids; blow drying at about 40° C. to obtain about 642 g (theoretical amount: 727.4 g) of dry 109TM-11 with a purity of 99.93%. Yield: 88.3%. |
| 82% |
In water; acetone at 0.5 - 0.6℃; for 0.01h; |
4E Example 4E: Preparation of Ethanediamide, /Vi-(5-chloro-2-pyridinyl)-/V2-[(lS, 2R,4S) -4- [(dimethylamino)carbonyl] -2- [ [(4, 5, 6, 7 -tetrahydro-5-methy lthiazolo[5, 4, c]-pyridin-2-yl)carbonyl]amino]cylohexyl]-, 4-methyl benzene sulfonate, hydrate (1:1:1) (compound of formula I)
The compound of formula I-A (20.0g) as obtained in Example 3E -3F was added to acetone (125 mL), water (125 mL) mixture and p-toluene sulfonic acid 8.30g) was added to it and flushed with 50% aq. (50 mL). The reaction mass was then heated to about 50°C to 60°C to get clear solution. The obtained clear solution was stirred for about 60 min and hot filtered and washed with acetone and water mixture. The filtrate was then cooled to about 5°C to 10°C and maintained at same temperature for about 240 minutes. The reaction mass was then filtered and dried at about 45°C to 50°C for about 12 hours to get compound of formula I. Yield: 22.0g (82.0%), HPLC purity 99.94% |
| 75% |
In ethanol; dichloromethane |
12 Example 12: Preparation of Edoxaban Tosylate (Formula 1):
P-toluenesulfonic acid (1.73g) was added to a stirred solution of Edoxaban(5.0g), methylene dichloride(5ml),and ethanol(85ml). The mixture was distilled under atmospheric pressure and water(12.5 ml) was addedand the mixturewas heated to 70-75 °C to obtain a clear solution. The reaction mixture was cooled to room temperature and stirred for 16 hours. The solution was filtered to obtain a white precipitate, which was washed with ethanol (5ml) then dried under reduced pressure at room temperature to obtain Formula 1 (5.0g, yield: 75%, HPLC Purity: 99.7%). |
| 75% |
In ethanol; water at 65℃; |
4: Preparation of compound (VII)
In a 5000mL three-necked flask, add 625mL of absolute ethanol, 250mL of purified water, 125g of compound (VI), and 50g of p-toluenesulfonic acid monohydrate, heat up to 65°C until the system is completely dissolved, add 6.3g of activated carbon and stir for 40min, Heat rate, the filtrate was transferred to a 5000mL three-necked flask, cooled to 50°C, 2250mL of ethanol was added dropwise to the system, and the temperature was lowered to 0°C for crystallization for 3h after the dropwise addition. Suction filtration, the filter cake was rinsed with 300 mL of water 10% anhydrous ethanol for 3 times, dried at 30 °C,An off-white solid was obtained, namely edoxaban tosylate monohydrate, and the molar yield was 75%. |
|
|
311 N⊃1⊆-(5-Chloropyridin-2-yl)-N⊃2⊆-((1S,2R,4S)-4-[(dimethylamino)carbonyl]-2-[(5-methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-yl)carbonyl]amino}cyclohexyl)ethanediamide p-toluenesulfonate monohydrate:
[Example 311] N1-(5-Chloropyridin-2-yl)-N2-((1S,2R,4S)-4-[(dimethylamino)carbonyl]-2-[(5-methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-yl)carbonyl]amino}cyclohexyl)ethanediamide p-toluenesulfonate monohydrate: The compound (6.2 g) obtained in Example 310 is dissolved in methylene chloride (120 ml), a 1 mol/L ethanol solution (11.28 ml) of p-toluenesulfonic acid was added to the solution, and the solvent was distilled off.. ethanol (95 ml) containing 15% water was added to the residue, and the mixture was stirred at 60°C to dissolve it.. The solution was then cooled to room temperature and stirred for a day.. Crystals deposited were collected by filtration, washed with ethanol and dried at room temperature for 2 hours under reduced pressure to obtain the title compound (7.4 g).1H-NMR (DMSO-d6) δ: 1.45-1.54(1H,m),1.66-1.78(3H,m),2.03-2.10(2H,m),2.28(3H,s),2.79(3H,s),2.91-3.02(1H,m), 2.93(3H,s),2.99(3H,s),3.13-3.24(2H,m),3.46-3.82(2H,m), 3.98-4.04(1H,m),4.43-4.80(3H,m),7.11(2H,d,J=7.8Hz), 7.46(2H,d,J=8.2Hz),8.01(2H,d,J=1.8Hz),8.46(1H,t,J=1.8Hz), 8.75(1H,d,J=6.9Hz),9.10-9.28(1H,br),10.18(1H,br), 10.29(1H,s). MS (ESI) m/z: 548(M+H)+. Elemental analysis: C24H30ClN7O4S·C7H8O3S·H2O. Calculated: C;50.43,H;5.46,N;13.28,Cl;4.80,S;8.69. Found: C;50.25,H;5.36,N;13.32,Cl;4.93,S;8.79. mp (decomposed): 245∼248°C. |
|
In ethanol; dichloromethane |
7
(Reference Example 7) N1-(5-Chloropyridin-2-yl)-N2-((1S,2R,4S)-4-[(dimethylamino)carbonyl]-2-[(5-methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-yl)carbonyl]amino}cyclohexyl)ethanediamide mono-p-toluenesulfonate monohydrate (A-a) N1-(5-Chloropyridin-2-yl)-N2-((1S,2R,4S)-4-[(dimethylamino)carbonyl]-2-[(5-methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-yl)carbonyl]amino}cyclohexyl)ethanediamide (A) (6.2 g) was dissolved in methylene chloride (120 ml). To the solution, a 1 mol/L solution of p-toluenesulfonic acid in ethanol (11.28 ml) was added, and the solvent was distilled off. To the residue, 15% hydrous ethanol (95 ml) was added, and the mixture was dissolved by stirring at 60°C. Then, the mixture was cooled to room temperature and stirred for 1 day. The precipitated crystals were collected by filtration, washed with ethanol, and then dried under reduced pressure at room temperature for 2 hours to obtain the title compound (A-a) (7.4 g). 1H-NMR (DMSO-d6) δ: 1.45-1.54 (1H, m), 1.66-1.78 (3H, m), 2.03-2.10 (2H, m), 2.28 (3H, s), 2.79 (3H, s), 2.91-3.02 (1H, m), 2.93 (3H, s), 2.99 (3H, s), 3.13-3.24 (2H, m), 3.46-3.82 (2H, m), 3.98-4.04 (1H, m), 4.43-4.80 (3H, m), 7.11 (2H, d, J=7.8 Hz), 7.46 (2H, d, J=8.2 Hz), 8.01 (2H, d, J=1.8 Hz), 8.46 (1H, t, J=1.8 Hz), 8.75 (1H, d, J=6.9 Hz), 9.10-9.28 (1H, br), 10.18 (1H, br), 10.29 (1H, s). MS (ESI) m/z: 548 (M+H)+. Anal.: C24H30ClN7O4S·C7H8O3S·H2O Theoretical: C; 50.43, H; 5.46, N; 13.28, Cl; 4.80, S; 8.69. Found: C; 50.25, H; 5.36, N; 13.32, Cl; 4.93, S; 8.79. mp (dec.) : 245-248°C. (Example 1) Tert-butyl {(1R,2R,5S)-2-azido-5-[(dimethylamino)carbonyl]cyclohexyl}carbamate (1) wherein Boc is as defined above. |
|
In ethanol; dichloromethane at 20℃; |
5
(Reference Example 5) N1-(5-Chloropyridin-2-yl)-N2-((1S,2R,4S)-4-[(dimethylamino)carbonyl]-2-[(5-methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-yl)carbonyl]amino}cyclohexyl)ethanediamide mono-p-toluenesulfonate monohydrate (A-a) N1-(5-Chloropyridin-2-yl)-N2-((1S,2R,4S)-4-[(dimethylamino)carbonyl]-2-[(5-methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-yl)carbonyl]amino}cyclohexyl)ethanediamide (A) (6.2 g) was dissolved in methylene chloride (120 ml). To the solution, a 1 mol/L solution of p-toluenesulfonic acid in ethanol (11.28 ml) was added, and the mixture was stirred at room temperature. Then, the solvent was distilled off. To the residue, 15% hydrous ethanol (95 ml) was added, and the mixture was dissolved by stirring at 60°C. Then, the mixture was cooled to room temperature and stirred for 1 day. The precipitated crystals were collected by filtration, washed with ethanol, and then dried under reduced pressure at room temperature for 2 hours to obtain the title compound (7.4 g). 1H-NMR (DMSO-d6) δ: 1.45-1.54 (1H, m), 1.66-1.78 (3H, m), 2.03-2.10 (2H, m), 2.28 (3H, s), 2.79 (3H, s), 2.91-3.02 (1H, m), 2.93 (3H, s), 2.99 (3H, s), 3.13-3.24 (2H, m), 3.46-3.82 (2H, m), 3.98-4.04 (1H, m), 4.43-4.80 (3H, m), 7.11 (2H, d, J=7.8 Hz), 7.46 (2H, d, J=8.2 Hz), 8.01 (2H, d, J=1.8 Hz), 8.46 (1H, t, J=1.8 Hz), 8.75 (1H, d, J=6.9 Hz), 9.10-9.28 (1H, br), 10.18 (1H, br), 10.29 (1H, s). MS (ESI) m/z: 548 (M+H)+. Anal.: C24H30ClN7O4S·C7H8O3S·H2O Theoretical: C;50.43, H;5.46, N;13.28, Cl;4.80, S;8.69. Found: C;50.25, H;5.36, N;13.32, Cl;4.93, S;8.79. mp (dec.): 245-248°C. |
| 7.4 g |
Stage #1: edoxaban; toluene-4-sulfonic acid In ethanol; dichloromethane
Stage #2: In ethanol; water at 20 - 60℃; for 24h; |
7 (Reference Example 7) N1-(5-Chloropyridin-2-yl)-N2-((1S,2R,4S)-4-[(dimethylamino)carbonyl]-2-[(5-methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-yl)carbonyl]amino}cyclohexyl)ethanediamide mono-p-toluenesulfonate monohydrate (X-a) (production method described in the pamphlet of International Publication No. WO 2007/032498)
N1-(5-Chloropyridin-2-yl)-N2-((1S,2R,4S)-4-[(dimethylamino)carbonyl]-2-[(5-methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-yl)carbonyl]amino}cyclohexyl)ethanediamide (X) (6.2 g) was dissolved in methylene chloride (120 ml). To the solution, a 1 mol/L solution of p-toluenesulfonic acid in ethanol (11.28 ml) was added, and the solvent was distilled off. To the residue, 15% hydrous ethanol (95 ml) was added, and the mixture was dissolved by stirring at 60°C. Then, the mixture was cooled to room temperature and stirred for 1 day. The precipitated crystals were collected by filtration, washed with ethanol, and then dried under reduced pressure at room temperature for 2 hours to obtain the title compound (X-a) (7.4 g). 1H-NMR (DMSO-d6) δ : 1. 45-1. 54 (1H, m), 1.66-1.78 (3H, m), 2.03-2.10 (2H, m), 2.28 (3H, s), 2.79 (3H, s), 2.91-3.02 (1H, m), 2.93 (3H, s), 2.99 (3H, s), 3.13-3.24 (2H, m), 3.46-3.82 (2H, m), 3.98-4.04 (1H, m), 4.43-4.80 (3H, m), 7.11 (2H, d, J = 7.8 Hz), 7.46 (2H, d, J = 8.2 Hz), 8.01 (2H, d, J = 1.8 Hz), 8.46 (1H, t, J = 1.8 Hz), 8.75 (1H, d, J = 6.9 Hz), 9.10-9.28 (1H, br), 10.18 (1H, br), 10.29 (1H, s). MS (ESI) m/z: 548 (M+H)+. Anal.: C24H30ClN7O4S·C7H8O3S·H2O Theoretical: C; 50.43, H; 5.46, N; 13.28, Cl; 4.80, S; 8.69. Found: C; 50.25, H; 5.36, N; 13.32, Cl; 4.93, S; 8.79. mp (dec.): 245-248°C. |
| 7.4 g |
In ethanol; dichloromethane |
8 N1-(5-chloropyridin-2-yl)-N2-((1S,2R,4S)-4-[(dimethylamino)carbonyl]-2-[(5-methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-yl)carbonyl]amino}cyclohexyl)ethanediamide mono-p-toluenesulfonate monohydrate (E-a)
Reference Example 8 N1-(5-chloropyridin-2-yl)-N2-((1S,2R,4S)-4-[(dimethylamino)carbonyl]-2-[(5-methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-yl)carbonyl]amino}cyclohexyl)ethanediamide mono-p-toluenesulfonate monohydrate (E-a) N1-(5-Chloropyridin-2-yl)-N2-((1S,2R,4S)-4-[(dimethylamino)carbonyl]-2-[(5-methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-yl)carbonyl]amino}cyclohexyl)ethanediamide (E) (6.2 g) was dissolved in methylene chloride (120 ml). To the solution, a 1 mol/L solution of p-toluenesulfonic acid in ethanol (11.28 ml) was added, and the solvent was distilled off. To the residue, 15% aqueous ethanol (95 ml) was added, and the mixture was dissolved by stirring at 60° C. Then, the mixture was cooled to room temperature and stirred for 1 day. The precipitated crystals were collected by filtration, washed with ethanol, and then dried under reduced pressure at room temperature for 2 hours to obtain the title compound (E-a) (7.4 g). 1H-NMR (DMSO-d6) δ: 1.45-1.54 (1H, m), 1.66-1.78 (3H, m), 2.03-2.10 (2H, m), 2.28 (3H, s), 2.79 (3H, s), 2.91-3.02 (1H, m), 2.93 (3H, s), 2.99 (3H, s), 3.13-3.24 (2H, m), 3.46-3.82 (2H, m), 3.98-4.04 (1H, m), 4.43-4.80 (3H, m), 7.11 (2H, d, J=7.8 Hz), 7.46 (2H, d, J=8.2 Hz), 8.01 (2H, d, J=1.8 Hz), 8.46 (1H, t, J=1.8 Hz), 8.75 (1H, d, J=6.9 Hz), 9.10-9.28 (1H, br), 10.18 (1H, br), 10.29 (1H, s). MS (ESI) m/z: 548 (M+H)+. |
| 102.9 g |
In ethanol; water at 70℃; for 1h; |
6 Synthesis of N’-(5-chloropyridin-2-yl)-N2-((1S,2R, 45)-4-[(dimethylamino)carbonyl]-2-{ [(5-methyl-4,5, 6,7-tetrahydrothiazolo[5,4-c]pyridin-2-yl)carbonyl]amino }cyclohexyl)ethanediamide p-toluenesulfonic acid monohydrate (X-a) (the Method Described in International Publication No. WO 07/032498)
10100] N’ -(5-Chloropyridin-2-yl)-N2-((1 S,2R,45)-4-[(dimethylamino)carbonyl] -2-{ [(5-methyl-4,5,6,7-tetrahy-drothiazolo[5,4-c]pyridin-2-yl)carbonyl]amino}cyclohexyl)ethanediamide (86.8 g) was dissolved in 30% aqueous ethanol (418 ml) at 60° C., and a solution ofp-toluenesulfonic acid monohydrate (29.0 g) in 30% aqueous ethanol (167 ml) was then added to the above obtained solution. The reaction mixture was stirred at 70° C. for 1 hour, and thereafier, it was gradually cooled to room temperature. Afier that, ethanol was added to the mixture, and the obtained mixture was stirred for 16 hours. The reaction solution was stirred under cooling on ice for 1 hour, and a crystal was collected by filtration to obtain 102.9 g of the title compound. |
| 7.4 g |
In ethanol; water at 20 - 60℃; for 24h; |
11 N1-(5-chloropyridin-2-yl)-N2-((1S,2R,4S)-4-[(dimethylamino)carbonyl]-2-[(5-methyl-4,5,6,7-tetrahydrothiazolone[5,4-c]pyridin-2-yl)carbonyl]amino}cyclohexyl)ethanediamide mono p-toluenesulfonic acid salt monohydrate
N1-(5-chloropyridin-2-yl)-N2-((1S,2R,4S)-4-[(dimethylamino)carbonyl]-2-[(5-methyl-4,5,6,7-tetrahydrothiazolone[5,4-c]pyridin-2-yl)carbonyl]amino}cyclohexyl)ethanediamide (X) (6.2g), It dissolved in the methylene chloride (120 ml), the p-toluenesulfonic acid/ethanol solution (1 mol/L solution: 11.28 ml) was added, and the solvent was distilled off. Hydrated ethanol (95 ml) was added to residue 15%, and it agitated and dissolved at 60 degrees C. Then, to the room temperature, it cooled and agitated on the 1st. Precipitated crystal was separated, and it dried in vacuum at the room temperature after washing by ethanol for 2 hours, and obtained the title compound (X-a) (7.4g). |
| 23 g |
In ethanol; dichloromethane at 38℃; for 1h; |
2 ((1S, 2R, 4S) -4 - [(dimethylamino) carbonyl] -2 - [(5-methyl-Tetrahydrothiazolo [5,4-c] pyridin-2-yl) carbonyl] amino} cyclohexyl) ethanediamide p-toluenesulfonic acid monohydrate,Preparation of Compound (1-a)
A 500 mL autoclave was charged with 220 mL of methylene chloride, 20.9 g of N- (5-chloropyridin-2-yl) -N '- ((1S, 2R,4S) -4 - [(Dimethylamino) carbonyl] -2 - [(5-methyl- 4,5,6,7-tetrahydrothiazolo [5,4-c] Carbonyl]Amino} cyclohexyl) ethanediamide, stirred to dissolve all solids,25 ± 2 was added 38.2ml1mol / L of p-toluenesulfonic acid ethanol solution, the reaction 1h, the reaction was completed, the net solvent was removed under reduced pressure to the residue was added 313.5mL of 85% aqueous ethanol, 60 ± 2 incubated 1h, Cooling to 10 ° C suction filtration, 25 ° C under vacuum to constant weight to give 23.0g white solid. |
|
In water; acetonitrile at 2 - 70℃; for 5.5h; |
1a Example 1 a: Preparation of edoxaban tosylate monohydrate in ACN/H?Q
N1-(5-Chloropyridin-2-yl-)-N2-((1S,23,4S)-4-[(dimethylamino)carbonyl]-2-[(5-methyl-9,5,6,7-tetrahydrothiazolo[5,4-c]pyridine-2-yl)carbonyl]amino}cyclohexyl)ethanediamide (1 .6 g) (purity by HPLC: 98.1 %a/a, Imp. A: 0.69%a/a) and p-toluenesulfonic acid monohydrate (0.58 g) were mixed with a mixture of ACN and water 1 : 1 (6.4 ml). The mixture was heated to 70 °C (0.47 °C/min) and after 30 minutes at this temperature a mixture of ACN and water 1 : 1 (1 .6 ml) was added. The temperature was reduced to 50 °C (0.2 °C/min) and after 120 minutes at this temperature, to 2 °C (0.1 °C/min). Water (12 ml) was added at 2 °C in 120 minutes to the suspension. Then the reaction was stirred 60 minutes at 2 °C and filtered by vacuum. The crystals were washed with a solution of H20: ACN 8:2 and dried at 50 °C under reduced pressure. The outcome yield of the compound N 1-(5-Ch loropyrid in-2-yl-)-N2-(( 1 S,23 ,4S)-4-[(d imethylamino)carbonyl]-2-[(5- methyl-9 ,5,6 ,7-tetrahyd roth iazolo[5,4-c]pyrid ine-2-yl)carbonyl]amino}cyclohexyl ) ethanediam ide p-toluenesulfonate monohydrate was 1 .835 g (84.9%). Analysis by XRPD gave a diffractogram essentially the same as that represented by Figure 1 . Purity by HPLC: 99.6%a/a, imp. A: 0.05%a/a. |
| 102.9 g |
In ethanol at 70℃; for 18h; |
1 Reference Example 1 (0050) Synthesis of N1-(5-chloropyridin-2-yl)-N2-((1S,2R,4S)-4-[(dimethylamino)carbonyl]-2-[(5-methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-yl)carbonyl]aminolcyclohexyl)ethanediamide p-toluenesulfonic acid monohydrate (1a)
Reference Example 1 (0050) Synthesis of N1-(5-chloropyridin-2-yl)-N2-((1S,2R,4S)-4-[(dimethylamino)carbonyl]-2-[(5-methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-yl)carbonyl]aminolcyclohexyl)ethanediamide p-toluenesulfonic acid monohydrate (1a) (the method described in International Publication No. WO 07/032498) (0051) N1-(5-Chloropyridin-2-yl)-N2-((1S,2R,4S)-4-[(dimethylamino)carbonyl]-2-[(5-methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-yl)carbonyl]aminolcyclohexyl)ethanediamide (86.8 g), which had been produced by the method described in International Publication No. WO 07/032498, was dissolved in 70% aqueous ethanol (418 ml) at 60° C., and thereafter, a solution of p-toluenesulfonic acid monohydrate (29.0 g) in 70% aqueous ethanol (167 ml) was added to the above obtained solution. The reaction mixture was stirred at 70° C. for 1 hour, and thereafter, the reaction solution was gradually cooled to room temperature. Ethanol was added to the reaction solution, and the obtained mixture was then stirred for 16 hours. Thereafter, the reaction solution was stirred for 1 hour under cooling on ice, and crystals were then collected by filtration to obtain 102.9 g of the title compound. (0052) It was found that the obtained compound had an absorption peak with the same strength at the same wave number as that of a standard preparation (known compound) in the IR. (0053) Moreover, as a result of the analysis of the obtained compound using HPLC, a plurality of impurity peaks (all of which were 0.03% by weight or less) were observed as impurities, and the total content of all impurities was 0.16% by weight. Thus, the purity thereof was found to be 99.84% (wherein % by content indicates % with respect to the HPLC area value of a free form of the compound represented by formula (1a)). (0054) H-NMR (DMSO-d6) δ: 1.45-1.54 (1H, m), 1.66-1.78 (3H, m), 2.03-2.10 (2H, m), 2.28 (3H, s), 2.79 (3H, s), 2.91-3.02 (1H, m), 2.93 (3H, s), 2.99 (3H, s), 3.13-3.24 (2H, m), 3.46-3.82 (2H, m), 3.98-4.04 (1H, m), 4.43-4.80 (3H, m), 7.11 (2H, d, J =7.8 Hz), 7.46 (2H, d, J =8.2 Hz), 8.01 (2H, d, J=1.8 Hz), 8.46 (1H, t, J=1.8 Hz), 8.75 (1H, d, J=6.9 Hz), 9.10-9.28 (1H, br. s), 10.18 (1H, br. s), 10.29 (1H, s). (0055) Elemental analysis: Anal. Calcd. For: C; 50.43%, H; 5.46%, N; 13.28%. (0056) Found: C; 50.25%, H; 5.36%, N; 13.32% |
|
Stage #1: edoxaban With triethylamine In dichloromethane at 55℃; for 1h;
Stage #2: toluene-4-sulfonic acid |
|
| 95.5 % |
In ethanol; dichloromethane at 20℃; |
1.3; 2.5 Example 1.3 Preparation of edoxaban tosylate monohydrate
To 11.0 g (20.1 mmol, 1.0 eq) of edoxaban obtained in Example 1.2, methylene chloride (MC) 55 mL (5 v / v), 95% ethanol (EtOH) 55 mL (5 v / v) was added. ρ-toluenesulfonic acid monohydrate 4.2g (22mmol, 1.1eq) was added and dissolved at room temperature. The reaction solution was filtered and washed with 10 mL of MC. The filtrate was concentrated at 40° C. or lower. EtOH 72.6mL (6.6v/v) and purified water 26.4mL (2.4v/v) were added and dissolved by heating at 70°C. After cooling to room temperature, crystals were precipitated, and 165 mL of EtOH (15 v/v) was added. After stirring at room temperature for 12 hours or more (overnight), the mixture was stirred at 5° C. or less for 1 hour. The product was filtered and washed with 40.0 mL EtOH. The filtrate was dried at 45° C. to obtain 14.19 g (19.2 mmol, 95.5% yield, purity 98.883%) of a white solid compound. |
| 92 % |
In ethanol; water at 0 - 75℃; |
8-9 Preparation of edoxaban p-toluenesulfonate monohydrate (compound of formula 7)
In the reaction flask, add edoxaban (compound of formula 6) (5.0g, 9.1mmol), 25mL of absolute ethanol and 10mL of purified water in sequence, and add p-toluenesulfonic acid monohydrate (1.6g, 8.4mmol ), heated up to 70-75°C and stirred to dissolve, then lowered the temperature, and when the system dropped to 55-60°C, added p-toluenesulfonic acid monohydrate (0.3g, 1.6mmol) to the system. Add 50mL of absolute ethanol dropwise to the system, then lower it to 20-25°C and stir for 1 hour, then lower it to 0-10°C and stir for 1 hour, filter with suction, wash with 2×10mL of absolute ethanol and dry under reduced pressure at 45°C to obtain the compound (compound of formula 7), 6.2 g, yield 92%. |
| 95.5 % |
In ethanol; dichloromethane at 20℃; |
1.3; 2.5 Example 1.3 Preparation of edoxaban tosylate monohydrate
To 11.0 g (20.1 mmol, 1.0 eq) of edoxaban obtained in Example 1.2, methylene chloride (MC) 55 mL (5 v / v), 95% ethanol (EtOH) 55 mL (5 v / v) was added. ρ-toluenesulfonic acid monohydrate 4.2g (22mmol, 1.1eq) was added and dissolved at room temperature. The reaction solution was filtered and washed with 10 mL of MC. The filtrate was concentrated at 40° C. or lower. EtOH 72.6mL (6.6v/v) and purified water 26.4mL (2.4v/v) were added and dissolved by heating at 70°C. After cooling to room temperature, crystals were precipitated, and 165 mL of EtOH (15 v/v) was added. After stirring at room temperature for 12 hours or more (overnight), the mixture was stirred at 5° C. or less for 1 hour. The product was filtered and washed with 40.0 mL EtOH. The filtrate was dried at 45° C. to obtain 14.19 g (19.2 mmol, 95.5% yield, purity 98.883%) of a white solid compound. |
| 96 % |
In ethanol; water at 70 - 80℃; Inert atmosphere; |
5 Example 5Synthesis of Edoxaban p-Toluenesulfonate Monohydrate (Compound 9)
Under nitrogen protection, in a 500ml reaction flask equipped with a condensing device, add 61.2g of purified water, 171.5g of 95% ethanol, 30g of compound 8 solid (0.064mol, 1.0Eq), start stirring, and add p-toluenesulfonic acid monohydrate thing12.8g (0.067mol, 1.05Eq),Start heating to 70-80°C to dissolve, start hot filtration, add 203.0g of 95% ethanol to the filtrate, continue heating to 75-80°C to dissolve, keep stirring for 5-10 minutes, stop heating, and slowly cool down to - Suction filtration at 5-0°C, soak the filter cake in 95% ethanol, filter, and drain to obtain a wet product of the filter cake, put it in a vacuum oven at 50-55°C to dry, and obtain solid Edoxaban p-toluenesulfonate monohydrate , 96% molar yield. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +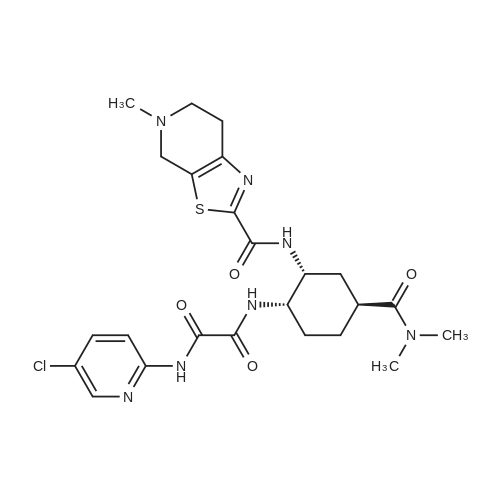

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping