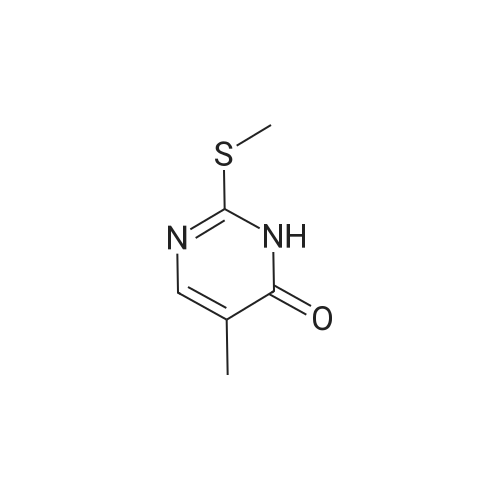
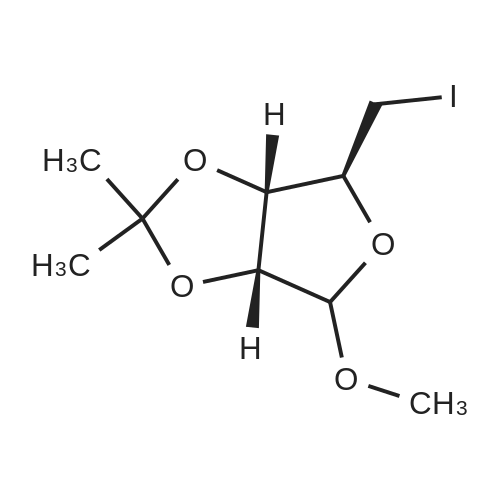
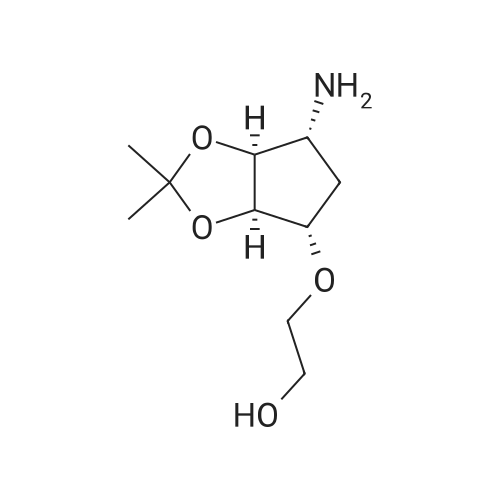
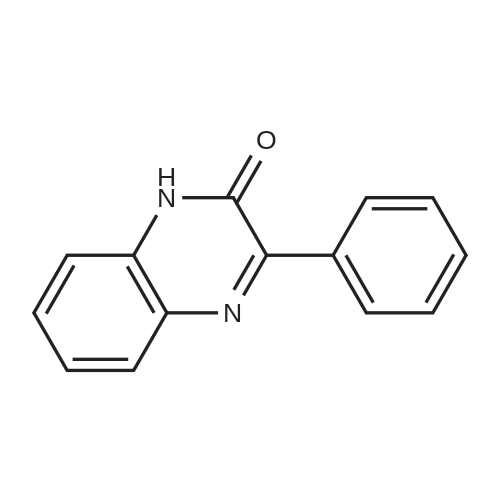
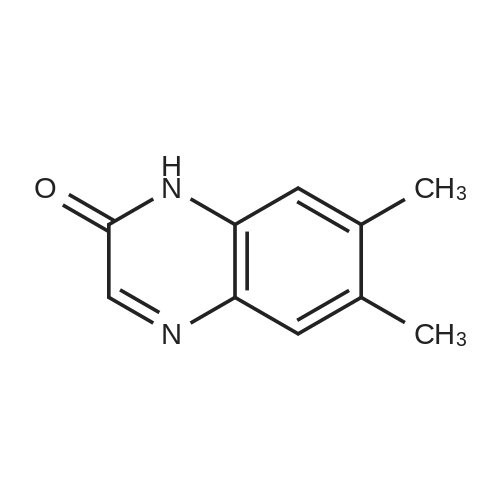
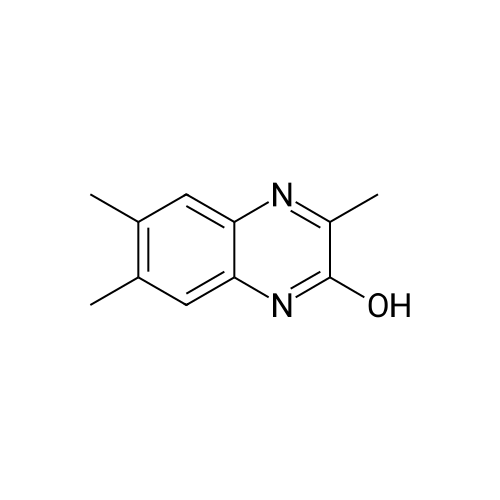
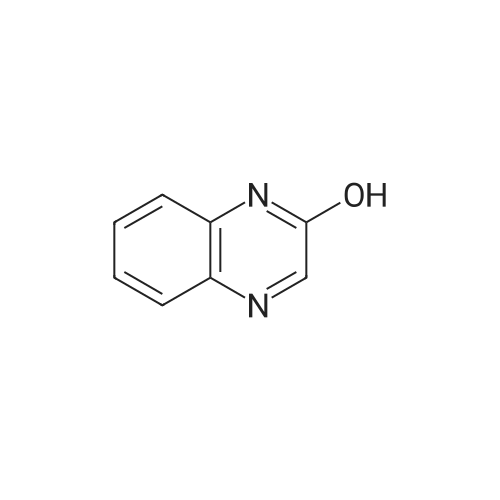
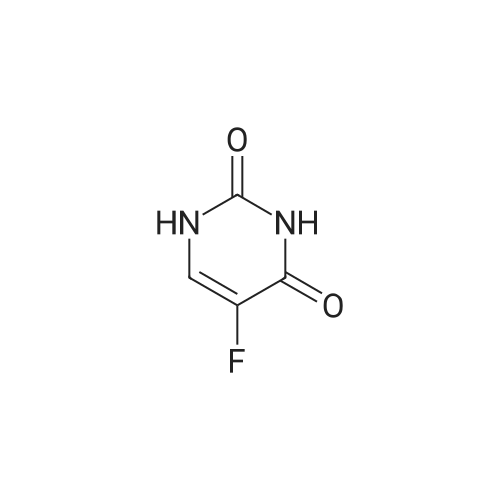
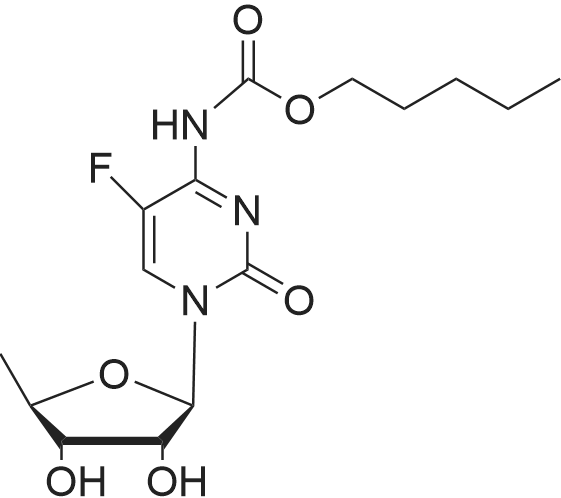
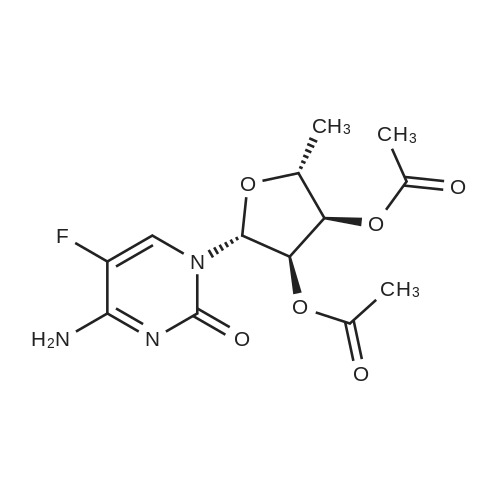
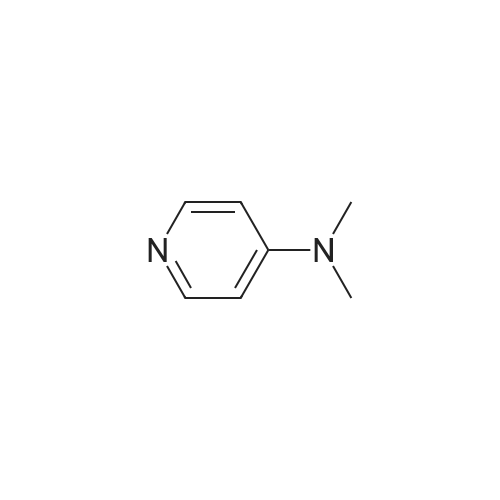
| 90% |
With pyridine; dmap at 20℃; Inert atmosphere; Cooling with ice; |
|
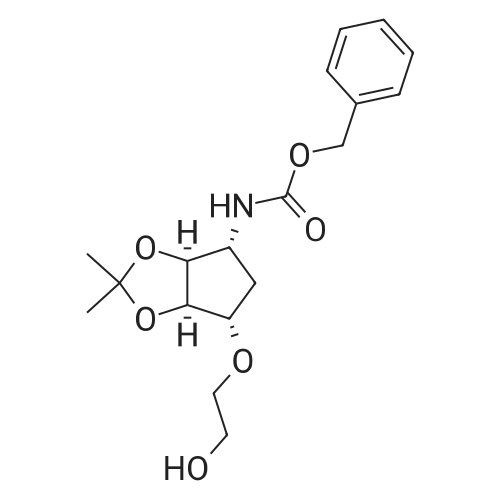
| 90% |
With pyridine at 20℃; for 2h; Inert atmosphere; |
|
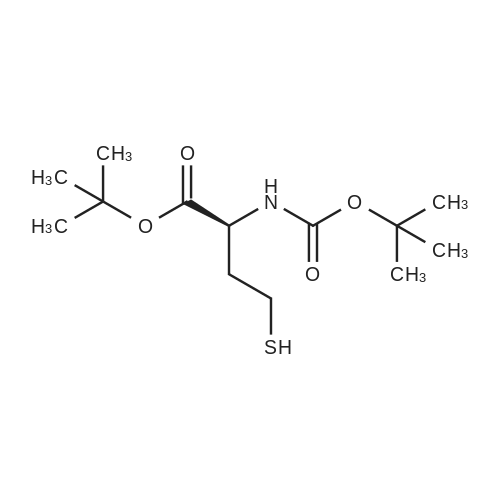
| 90% |
With dmap In pyridine at 0 - 25℃; for 5h; Inert atmosphere; |
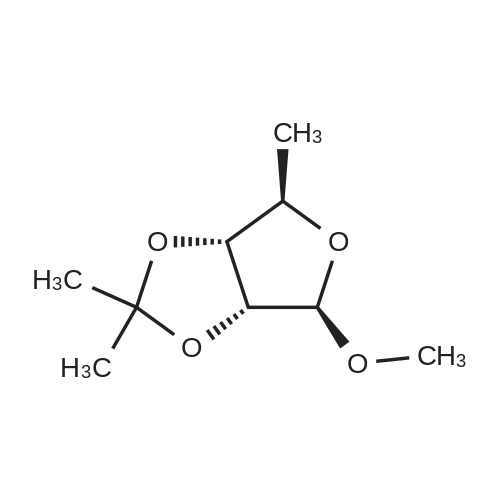
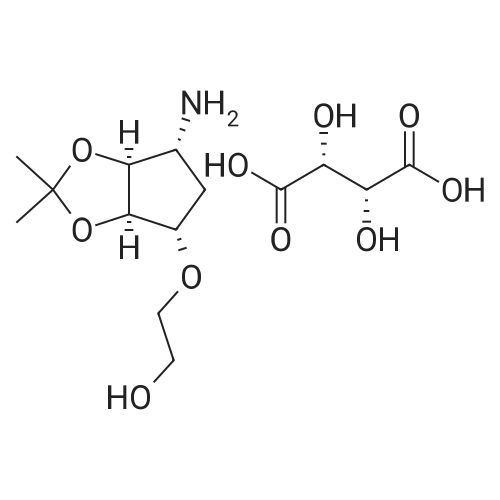
3.3. 1-O-methyl-2,3-O-isopropylidene-5-O-p-toluenesulfonyl-b-Dribofuranose(3)
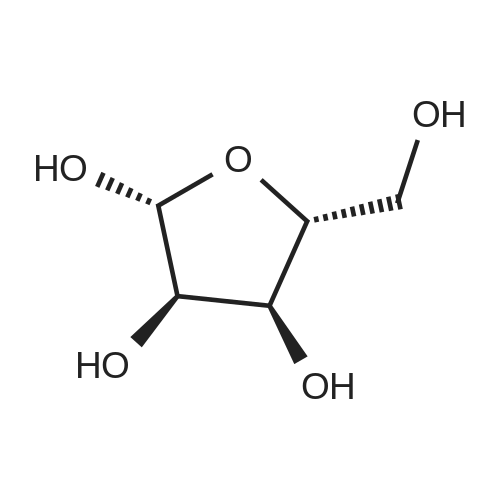
To a solution of 1-O-methyl-2,3-O-isopropylidene-b-D-ribofuranose(4.5 g, 0.022 mol) in dry pyridine (8.5 mL), externally cooledwith ice, was added p-toluenesulfonyl chloride (5.3 g, 0.027 mol)and catalytic amount of DMAP. The reaction mixture was stirred atroom temperature under nitrogen atmosphere for 5 h. After thistime, 2mL of distilledwaterwas added and the stirringwas kept formore 30 min. This mixture was extracted with chloroform (3 x 25mL) and the combined organic phases was washed with aqueoussolution of CuSO4 (10% w/v), saturated solution of NaHCO3, waterand dried over anhydrous sodium sulfate. Removal of the solventunder reduced pressure furnished product (3) as a white powder(90%, 7.09 g) with m.p. 85-86 °C. FT-IR n(cm1): 1358 and 1180; 1HNMR (CDCl3, 600 MHz) δ 1.29 (s, 3H, CH3), 1.45 (s, 3H, CH3), 2.46 (s,3H, CH3-aryl), 3.24 (3H, s, OCH3), 4.02 (dd, J 1.5 and 7.5 Hz, 2H, H-5 and H-50), 4.31 (ddd, J 0.6, 6.9 and 7.2 Hz, 1H, H-4), 4.53 (d,J 6.0 Hz, 1H, H-2), 4.60 (dd, J 0.3 and 5.7 Hz, 1H, H-3), 4.93 (s, 1H,H-1), 7.36 (dd, J 0.3 and 8.7 Hz, 2H, H-30), 7.81 (dd, J 0.6 and8.7 Hz, 2H, H-2’); 13C NMR (CDCl3, 150 MHz) δ 21.5, 24.7, 26.2, 54.9,69.1, 81.2, 83.4, 84.7, 109.3, 112.6, 127.8, 129.8, 132.6, 144.9. GC/MS(EI, 70 eV): 343 (M 15, 100%). |
| 89% |
With pyridine In dichloromethane |
|
| 86% |
With pyridine; dmap at 20℃; for 2h; Inert atmosphere; Cooling with ice; |
Methyl 2,3-O-isopropylidene-5-O-p-toluenesulfonyl-b-D-ribofuranoside (10)
To a solution of 8 (1.00 g, 4.90mmol) in dry pyridine (5mL) externally cooledwith an ice bath were added p-toluenesulfonyl chloride (3.00 g, 15.75mmol)and a catalytic amount of DMAP. The reaction mixture was stirred at rt undera nitrogen atmosphere for two h. After this time, 2mL of distilled water wasadded, and the stirring was kept for an additional 30 min. This mixture wasextracted with chloroform (325mL) and the combined organic phases werewashed with aqueous HCl solution (10% v/v), aqueous CuSO4 solution (10%w/v), cold water, saturated NaHCO3 solution, cold water, and dried over anhydroussodium sulfate. Removal of the solvent under reduced pressure furnisheda white powder (86%, 1.51 g) with m.p. 85-86 C (lit. 84-85 C). Rf 0.40(hexane/ethyl acetate: 7/3); IR (KBr pellet) m(cm1): 1596 (Ph), 1359 (SO2) and1180 (SO2). 1H NMR (CDCl3, 400.13MHz): d 1.29 (s, 3H, CH3), 1.45 (s, 3H,CH3), 2.46 (s, 3H, CH3-aryl), 3.23 (3H, s, OCH3), 4.01 (d, 1H, J5a,5b 2.0Hz,H-5), 4.02 (d, 1H, J5a,5b 2.0Hz, H-5) , 4.31 (t, 1H, J4,5 7.2Hz, H-4), 4.53 (d,1H, J2,3 5.9Hz, H-2), 4.60 (d, 1H, J3,2 5.9Hz, H-3), 4.93 (s, 1H, H-1), 7.36(d, 2H, J30,20 8.2Hz, H-30), 7.81 (dd, 2H, J20,30 8.2Hz, H-20). 13C NMR(CDCl3, 100.61MHz): d 21.8 (CH3-aryl), 25.1 and 26.5 (2 x CH3), 55.2(OCH3), 69.4 (C-5), 81.6 (C-3), 83.8 (C- 4), 85.1 (C-2), 109.7 (C-1), 112.9(C-6), 128.2 (C-20), 130.1 (C-30), 133.0 (C-40), 145.3 (C-10). These data are inagreement with that in the literature.[5a] |
| 85% |
In pyridine; dichloromethane Ambient temperature; |
|
| 83% |
With pyridine at 0℃; for 2h; |
|
| 82% |
With pyridine at 0 - 20℃; for 12h; Inert atmosphere; |
General procedure for synthesis of tosyl-sugars (2a-e)
General procedure: The stirring solutions of compounds 1a-e in pyridine at 0 °C were added with p-toluene sulphonyl chloride under anhydrous condition. The reactions was allowed to come at room temperature and further stirred for 12 h. After completion (monitered by TLC), the reaction mixtures were in vacuo concentrated and the crude obtained were purified by flash column chromatography to afford tosyl-sugars 2a-e in good yields. |
| 81% |
With pyridine; dmap at 20℃; for 5h; Cooling with ice; Inert atmosphere; |
|
| 80% |
With pyridine at 20℃; |
|
| 80% |
With pyridine at 20℃; for 20h; |
|
| 76% |
With pyridine Further stages; |
|
| 72.86% |
With pyridine In dichloromethane at 0 - 5℃; for 20h; |
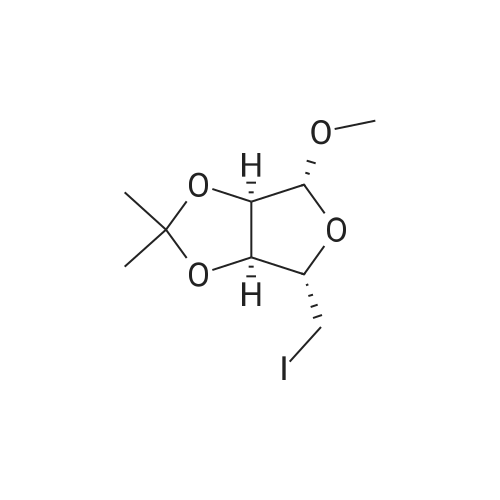
1 Methyl-2,3-O-isopropylidene-5-O-tosyl-β-D-ribofuranoside (2)
Methyl-2,3-O-isopropylidene-5-O-tosyl-β-D-ribofuranoside (2) To a cold solution containing Methyl-2,3-O-isopropylidene-β-D-ribofuranoside (1) (108.4g, 531mmol) in CH2Cl2 (1000ml) dropwise adding 300ml of toluene-4-sulfonyl chloride (140g, 734mmol) in anhydrous pyridine solution. With vigorous stirring, the reaction was carried out at 0-5°C for 20 h. The resulting solution was washed with NaHCO3 aqueous solution, brine and evaporated to yield a syrupy mass (185g, 97.2%), which can be further crystallized from hexane and dried under high vacuum to give pure methyl-2,3-O-isopropylidene-5-O-tosyl-β-D-ribofuranoside (138.8g, 72.86% yield) as a white solid crystals. 1H NMR (CDCl3): δ1.28 and 1.45 (2s, each 3H, CMe2), 2.46 (s, 3H, aromatic Me), 3.24 (s, 3H, -OMe), 4.01 (d, 2H, J=7.2Hz, H-5), 4.31 (dt, 1H, J=7.3Hz, H-4), 4.53 (d, 1H, J=6.0Hz, H-2), 4.60(dd, 1H, J=5.6Hz, H-3), 4.93 (s, 1H, H-1), 7.36 (d, 1H, J=8.0Hz, aromatic H), 7.81 (d, 2H, J=8.2Hz, aromatic H). |
| 70% |
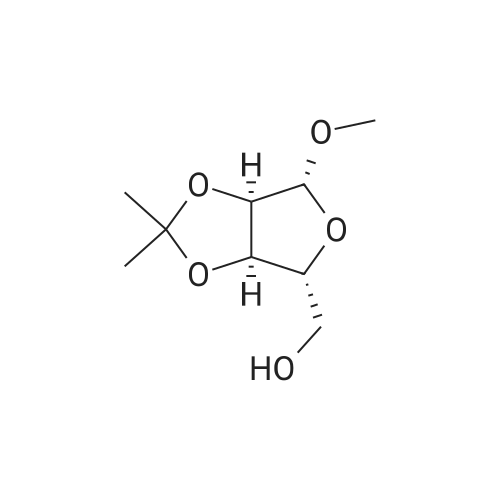
Stage #1: ((3aR,4R,6R,6aR)-6-methoxy-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methanol With pyridine In dichloromethane at 0℃; for 0.166667h;
Stage #2: p-toluenesulfonyl chloride In dichloromethane at 20℃; for 24h; |
|
|
With pyridine at 0℃; |
Methyl 2,3-O-isopropyliden-5-O-tosyl-β-D-ribofuranoside (6); A solution of methyl 2,3-O-isopropyliden-β-D-ribofuranoside (5; 22,6 g, 0,11 mol) in pyridine (25 mL) cooled at 0°C was treated with tosyl chloride (31,6 g, 0,17 mol) added in 4 similar parts. The reaction mixture was stirred for 3,5 hours at 0°C. Then it was poured into a mixture of 250 g of ice and 250 g of water. The ice was let to melt by stirring and the mixture was filtered. The filtrate was washed with cold water (2 x 100 mL) and it was dried with the desiccator. The compound 6 was obtained as a white crystalline solid (35,6 g, 89% yield starting from d-ribose) 1H RMN (CDCl3, 500 MHz) δ 1.29 (s, 3H, CCH3), 1.45 (s, 3H, CCH3), 2.46 (s, 3H, PhCH3), 3.23 (OCH3), 4.00 (dd, J = 10.0, 6.8 Hz, 1H, H-5'a), 4.03 (dd, J = 9.9, 7.6 Hz, 1H, H-5'b), 4.31 (t, J = 7.2 Hz, 1H), 4.53 (d, J = 5.9 Hz, 1H), 4.60 (d, J = 5.9 Hz, 1H), 7.36 (d, J = 8.1 Hz, 2H, H-3"), 7.81 (d, J = 8.3 Hz, 2H, H-2"). |
| 0.984 g |
With pyridine at 0 - 20℃; for 26h; Inert atmosphere; |
1-O-methyl-2,3-O-isopropylidene-5-O-(p-toluenesulfonyl)-β-D-ribofuranoside (8d)
Compound8d was prepared from protectedribose derivative 5 according toliterature procedures.3 Toluenesulfonyl chloride (1.413 g, 7.414mmol) was added to a stirred solution of the protected ribose derivative 5 (0.757 g, 3.707 mmol) in pyridine (8mL) at 0 °C. The reactiongradually came to RT while stirring for 26 hours. Monitoring by TLC (1:1 EtOAc/hexanes)revealed no visible starting material remaining, and the pyridine was co-evaporatedwith three portions of toluene (8 mL) by rotary evaporation. The crude product was taken up in EtOAc (60mL), washed with two portions of saturated aqueous NaHCO3 (25 mL),and one portion of brine (25 mL). Theorganic layer was dried over MgSO4, filtered, and solvent wasremoved by rotary evaporation. Productisolated as fibrous white crystals after recrystallization from hot ethanol (0.984g, 2.745 mmol, 74%). |
|
With pyridine |
|
| 666g |
With pyridine In dichloromethane at 0 - 20℃; for 5.5h; |
2 Example 2 Preparation of Compound II
To a 2000 ml three-necked flask was added 426 g of Compound I,342 ml of pyridine,Salt ice water bath cooling to below 0 ,A solution of p-toluenesulfonyl chloride in dichloromethane (422 g: 933 ml) was added dropwise,About 30min drop finished.The three-necked flask was then moved to room temperature,After stirring for 5 h, 311 ml of water was added dropwise,After stirring for an additional 30 min, the reaction system was transferred to a 2000 ml separatory funnel,Taking organic layer,Vacuum distillation to dryness,To obtain 732 g of the crude product of the beige solid compound II,Yield 98%. The crude product of compound II was placed in 2928ml (4V) of ethanol, stirred and heated to reflux, completely dissolved, and decolorized with 60g activated charcoal for 15min, filtered, cooled, crystallized, filtered and dried to obtain 666g of Compound II %). |
|
With pyridine at 25℃; |
|
| 952 g |
With dmap; sodium hydroxide In toluene at 5 - 15℃; |
2 Example 2: Preparation of a compound of formula 2
Toluene solution of the compound of the formula 1 obtained in Example 1 and 4-dimethylaminopyridine (0.3 g) were sequentially added to a four-necked flask.Sodium hydroxide (195.0g), control temperature 5~15 °C,A solution of p-toluenesulfonyl chloride (560.0 g) in toluene (600 g) was added dropwise.After the addition is completed, the temperature control reaction is 1 to 5 hours.After TLC monitors the reaction, add water (2kg), dispense,The aqueous phase was extracted once with toluene (1.0 kg).The organic phase was combined and washed once with saturated brine (2 kg).The organic layer was concentrated, and then ethanol (2.5 kg) was added to raise the temperature to 60-75 ° C.After dissolution, the temperature was lowered to 0 to 10 °C. filter,The filter cake is rinsed once with ethanol.The blast was dried to give a white solid compound 2 952.0 g,The total yield of the two steps is 79.8%.The HPLC purity was 99.8%. |
|
With pyridine at 0 - 5℃; for 16h; |
|






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping