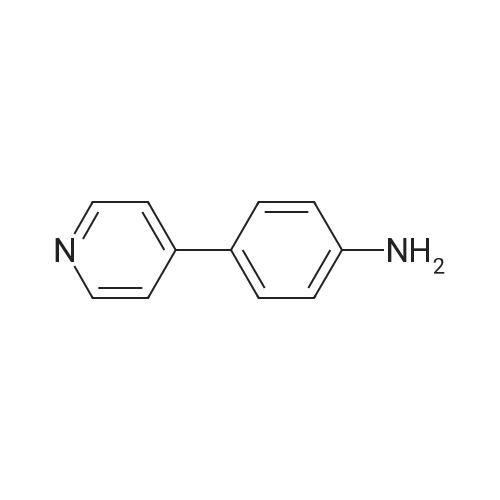
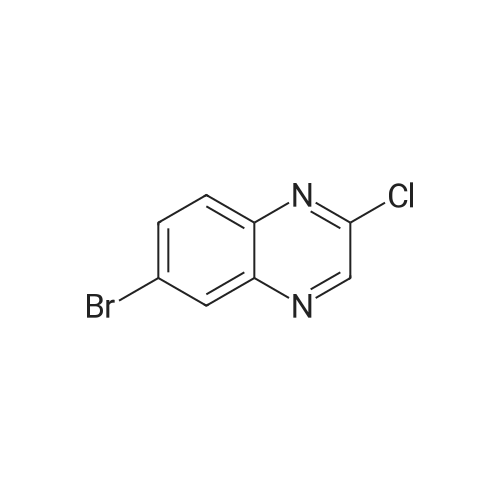
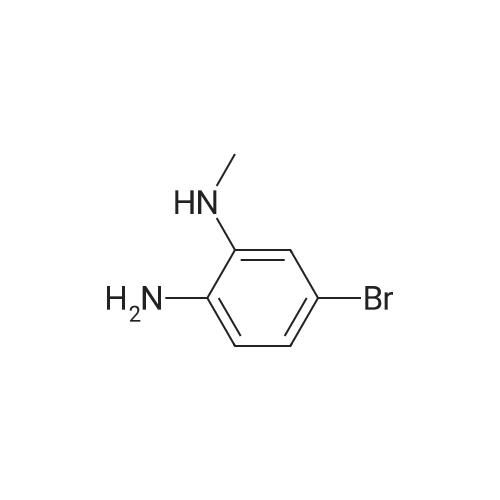
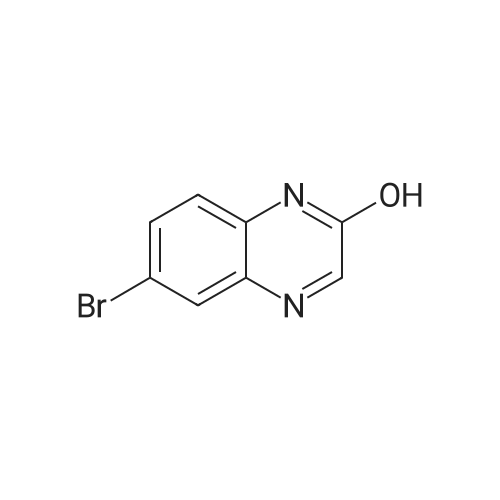
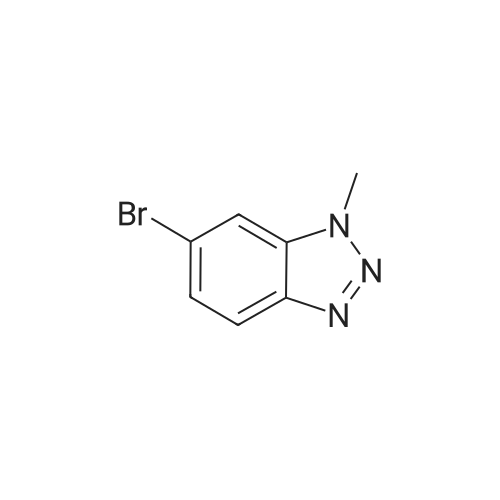
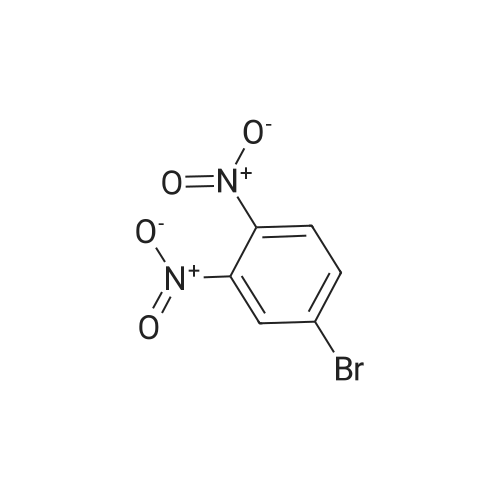
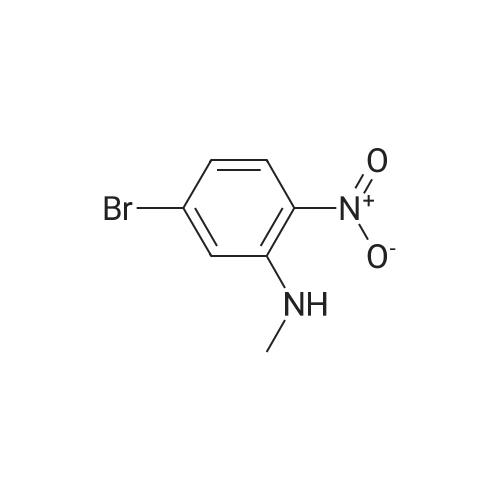
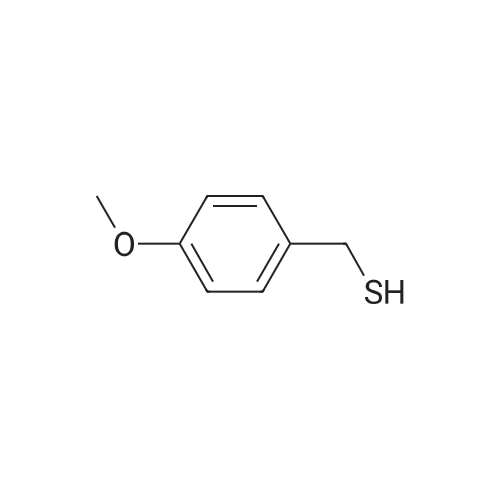
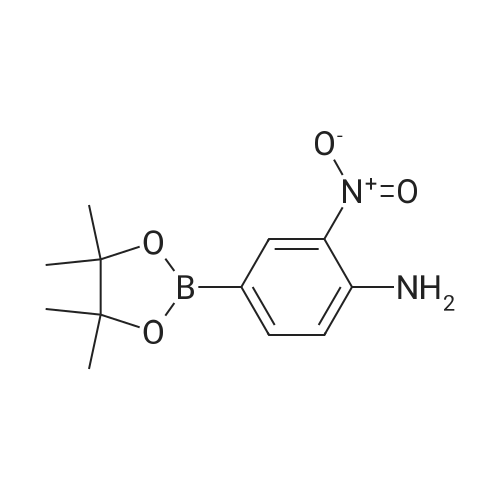
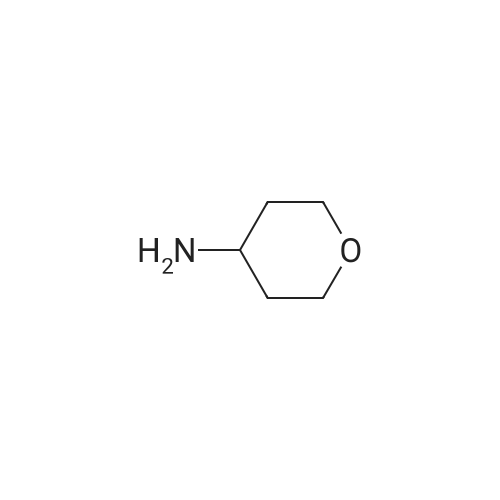
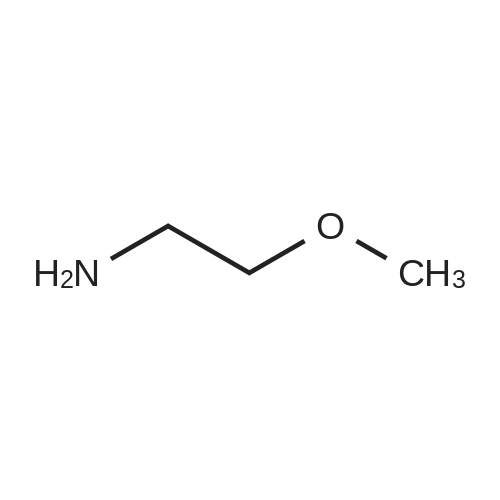
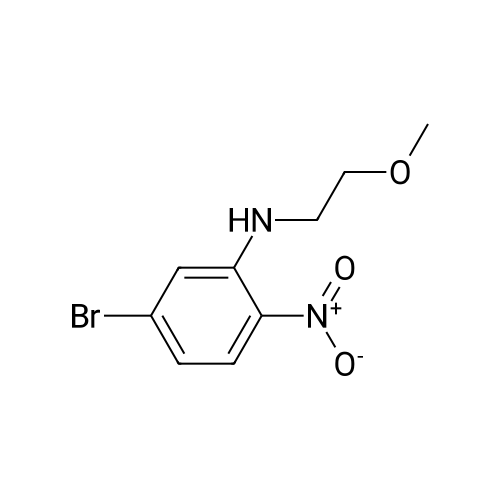
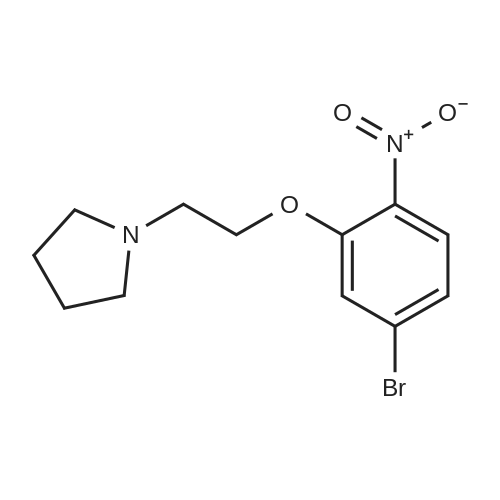
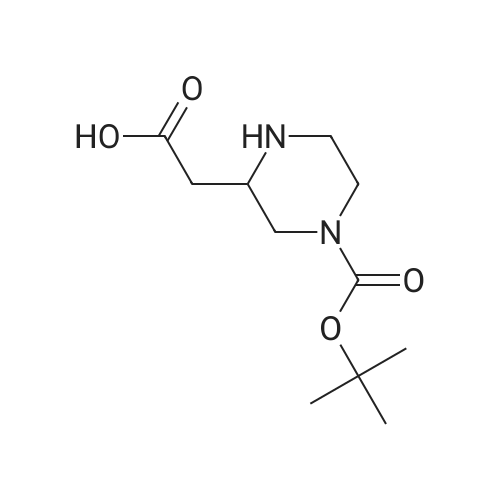
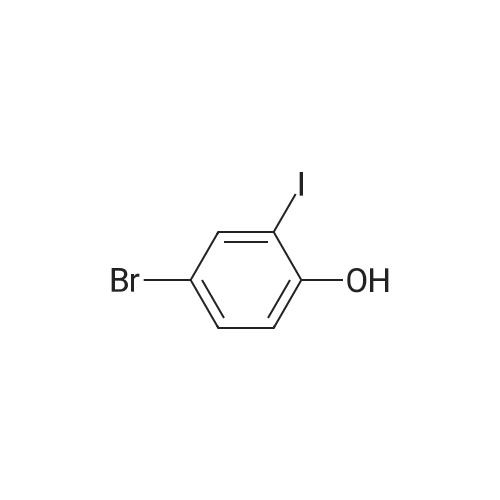
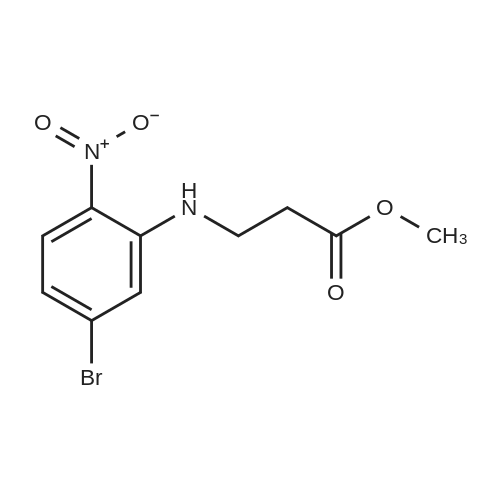
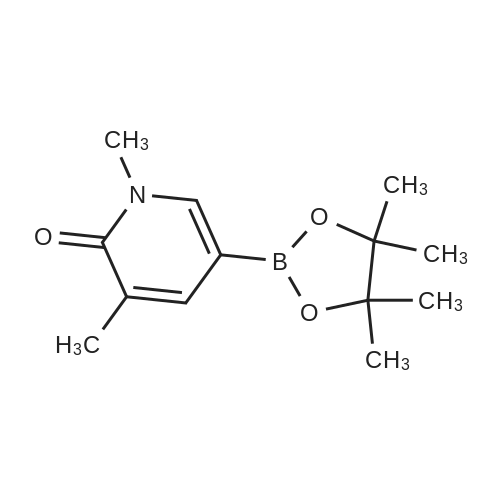
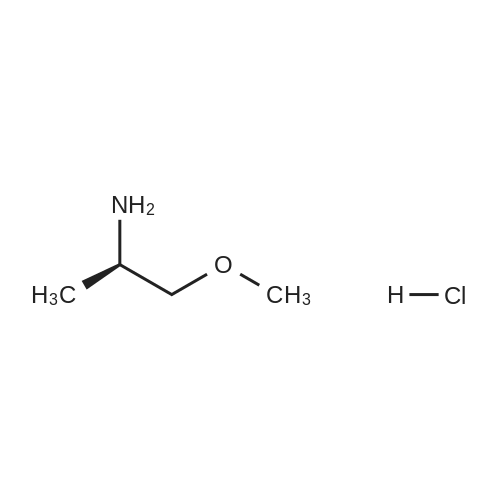
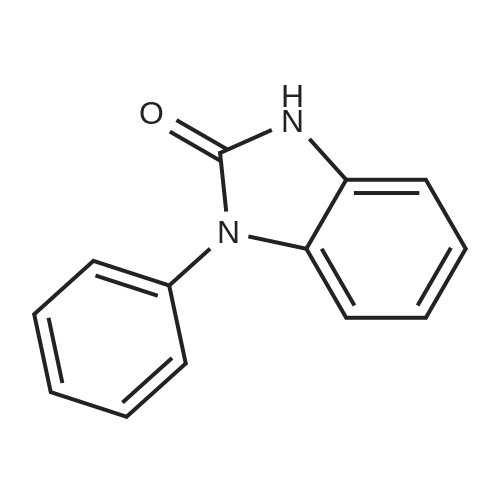
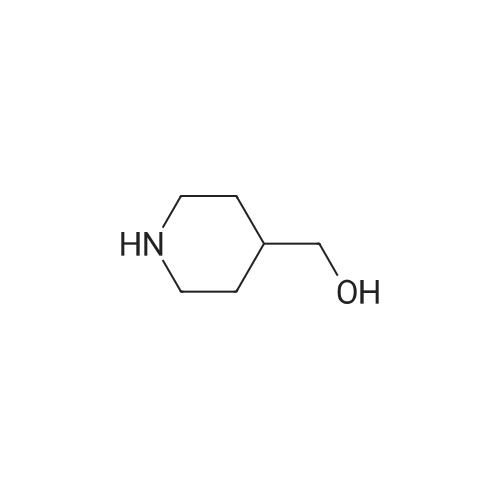
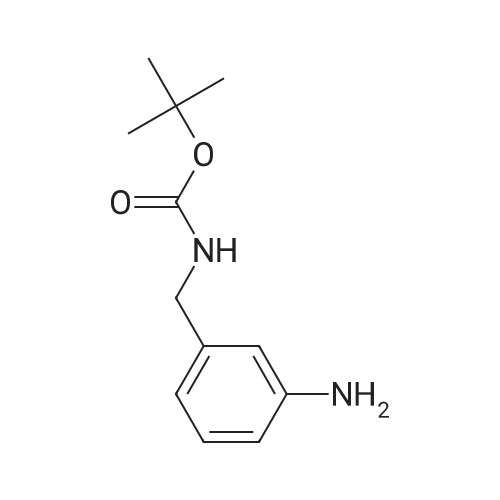
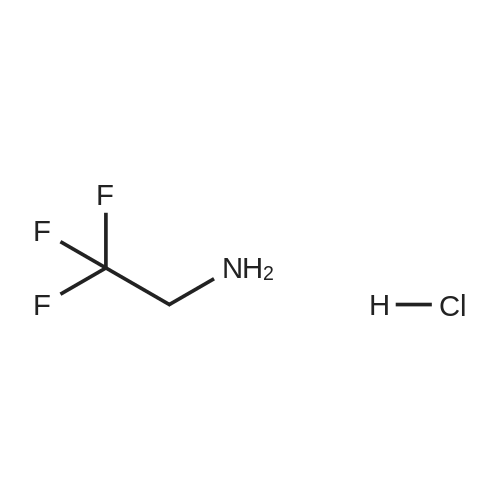
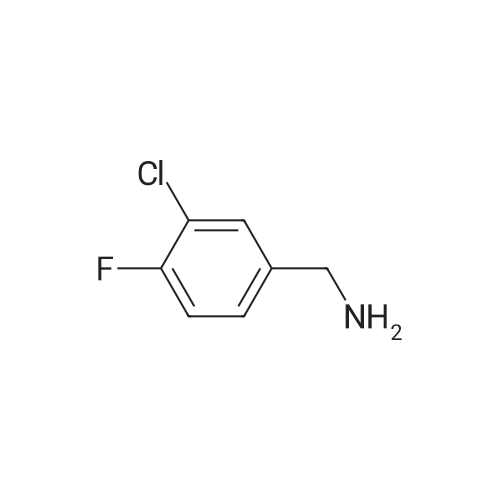
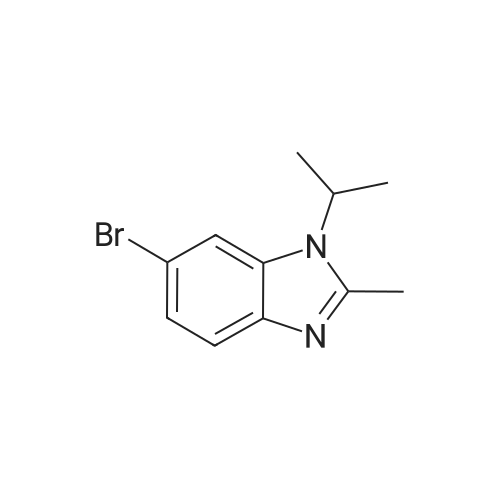
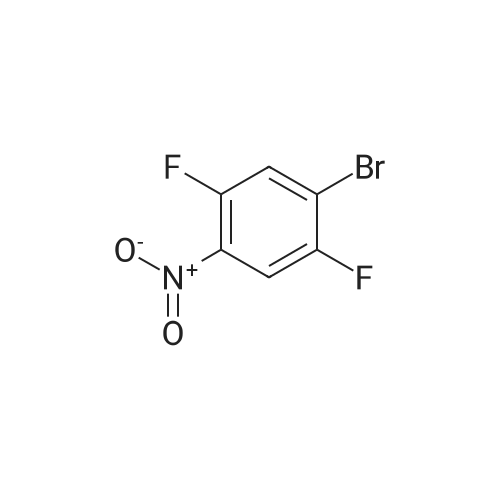
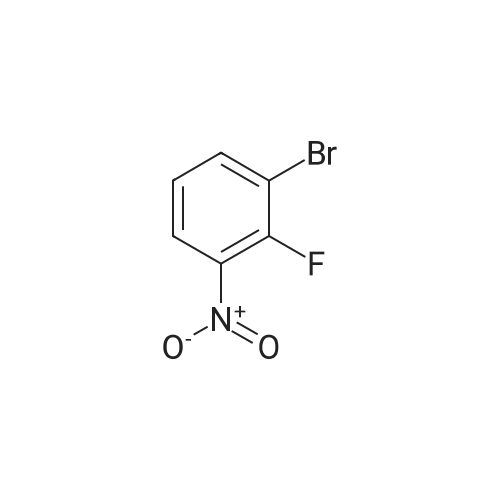
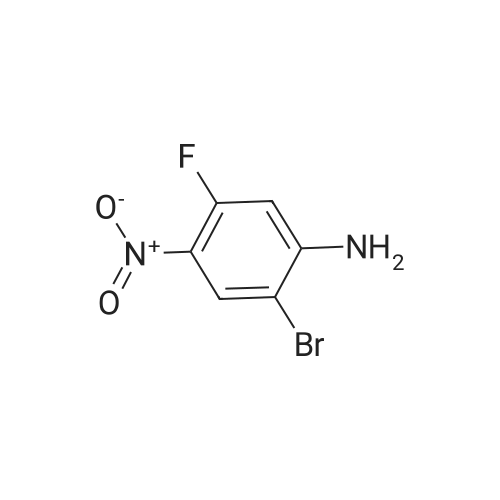
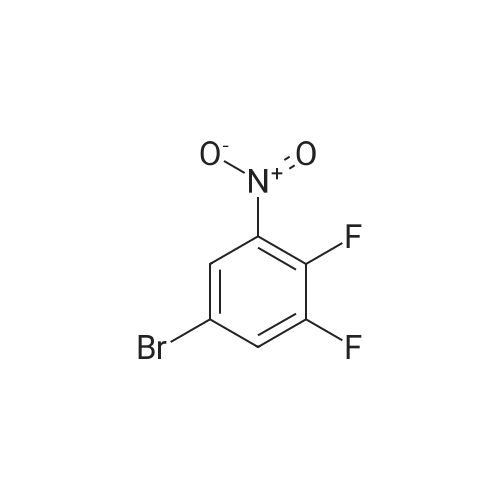
| 95% |
In ethanol; at 20℃; for 0.583333h; |
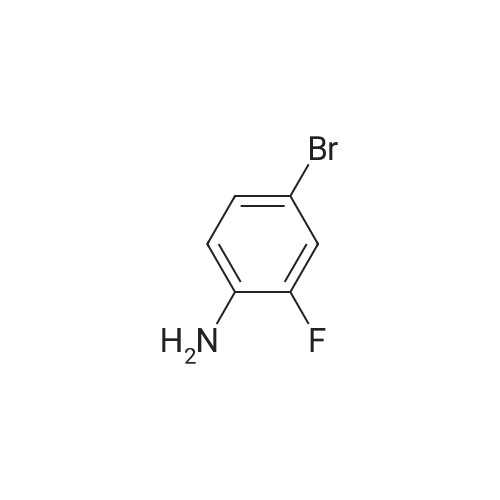
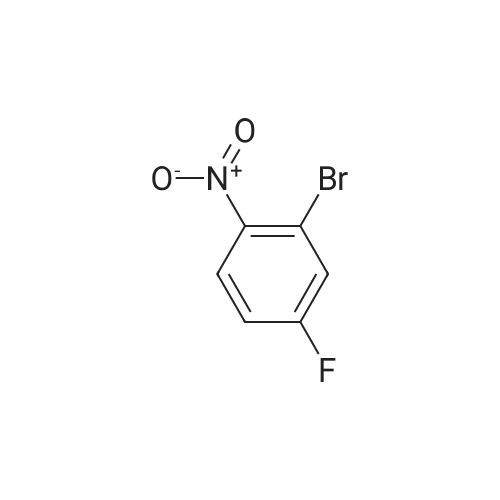
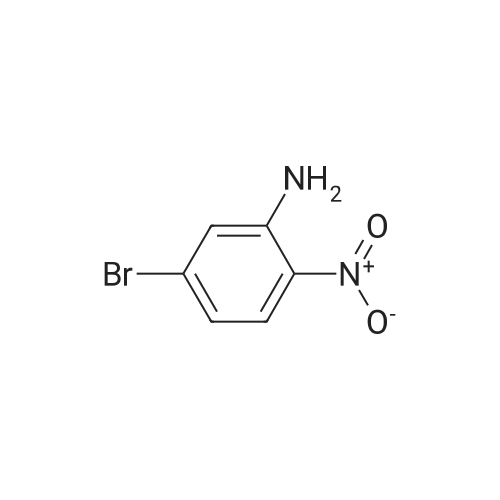
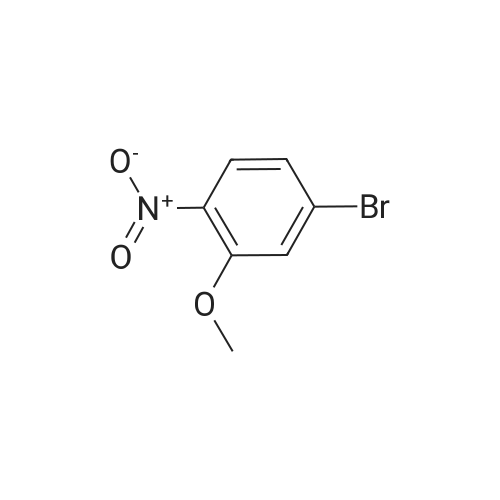
2M Methylamine in ethanol (4.9 ml; 9.73 mmol; 2.00 eqf.) is dropped in over 5 min at rt to the stirred solution of 4-bromo-2-fluoro-1 -nitrobenzene (1.07 g; 4.86 mmol; 1.00 eq.) in ethanol (10.00 ml). RM is stirred at rt for 30 min, then solvent is evaporated and residue is triturated with water to remove methylamine hydrofluoride. The remaining residue is collected by filtration, washed with water and dried at 60 C in an oven to give 5-bromo-N-methyl-2- nitroaniline (1.07 g; 4.62 mmol; yield 95.0%; 100% by UPLC) as orange thin needles. |
| 95% |
In ethanol; |
4-Bromo-2-fluoro-l -nitrobenzene (23 g, 105 mmol) was dissolved in EtOH(20 mL), Με Η2 (250 mL, 33%,in EtOH) was added, the mixture was stirred at R.T overnight, The mixture was detected by LC-MS, the starting material was consumed, evaporated the solvent, the crude was dissolved in EtOAc (300 mL) and washed with water (200 mL x 2) and brine (200 mL),dried with Na2S04, filtered and the organic phase was evaporated the solvent, 23 g of the target compound was obtained as yellow solid (95 % yield). NMR (400 MHz, CDCb) δ 8.03 (d, J= 9.2 Hz, 2H), 7.01(s, 1H), 6.76 (d, J= 9.2 Hz, 1H), 3.02(s, 3H); LC-MS (ESI+): m/z 232.1 (M+H)+ . |
| 95% |
In ethanol; at 20℃; |
To a solution of 4-bromo-2-fluoro-1-nitrobenzene (23 g, 105 mmol) in EtOH (50 mL) was added MeNH2 (250 ml, 33% in EtOH). After the addition, the mixture was stirred at rt overnight. The reaction mixture was concentrated under reduce pressure. The residue was dissolved in EtOAc (300 mL), washed with water (200 mL×2) and brine (200 mL), dried over anhydrous Na2SO4, filtered and concentrated to afford the title compound (23 g, 95% yield) as a yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.03 (d, J=9.2 Hz, 2H), 7.01 (s, 1H), 6.76 (d, J=9.2 Hz, 1H), 3.02 (s, 3H). LC/MS (ESI, m/z): [M+1]+=231.1 |
| 95% |
With potassium carbonate; In tetrahydrofuran; dichloromethane; at 20℃; for 16h; |
To a stirred solution of 4-bromo-2-fluoro-1-nitrobenzene (1, 300 g, 1.36 mol) in DCM (3 L) were added K2CO3 (0.94 Kg, 6.8 mol) and methylamine (2M in THF) (2.04 L, 4.09 mol) at RT and stirred for 16 h. Two batches of the reaction were combined. After completion of reaction, the reaction mixture was diluted with water (3.0 L) and extracted with DCM (2.5 L x 2). The combined organic layer was washed with saturated sodium bicarbonate solution (1.5 L x 2) and brine (1.5 L x 2). The organic layer was dried over sodium sulphate, filtered and solvent removed under reduced pressure to obtain 5-bromo-N-methyl-2-nitroaniline (2, 600 g, 95% yield) as a yellow solid. LCMS (ES+): m/z 231.1 [M+H]+ |
| 95% |
With potassium carbonate; In tetrahydrofuran; dichloromethane; at 20℃; for 16h; |
To a stirred solution of 4-bromo-2-fluoro-l -nitrobenzene (300 g, 1.36 mol) in DCM (3 L) were added K2CO3 (0.94 Kg, 6.8 mol) and methylamine (2M in THF) (2.04 L, 4.09 mol) at room temperature and stirred for 16 h. Two batches of the reaction were combined. After completion of reaction, the reaction mixture was diluted with water (3.0 L) and extracted with DCM (2 x 2.5 L). The combined organic layer was washed with saturated sodium bicarbonate solution (2 x 1.5 L) and brine (2 x 1.5 L). The organic layer was dried over sodium sulphate, filtered and solvent removed under reduced pressure to obtain 5-bromo-N-methyl-2-nitroaniline (600 g, 95% yield) as a yellow solid. LCMS (ES+): 231.1 [M+H]+ |
| 95% |
In tetrahydrofuran; dichloromethane; at 20℃; for 18h; |
To a 1 L round bottomed flask were added 4-bromo-2-fluoro-l -nitrobenzene (16 g, 73 mmol) and DCM (400 mL). To this mixture was added methylamine (2.0 M solution in THF) (88 mL, 180 mmol). Upon addition of amine, the solution rapidly changed from a pale yellow to a bright orange solution. The reaction mixture was stirred at room temperature under a closed system. After 18 h, the reaction mixture was filtered through a plug of SiCh (250 g), with 2: 1 Hex:EtOAc as eluent. The filtrate was concentrated and dried in vacuo to afford the title compound (16 g, 69 mmol, 95 % yield) as a yellow solid. ’H NMR (500 MHz, METHANOL-d4) 6 8.05 (d, J=9.1 Hz, 1H), 7.18 (d, J=1.9 Hz, 1H), 6.85-6.78 (m, 1H), 3.02 (s, 3H). Analytical LC/MS (Method 3): Observed Mass: 232.9; Retention Time: 0.97 min. |
| 95% |
In tetrahydrofuran; dichloromethane; at 20℃; for 18h; |
To a 1 L round bottomed flask were added 4-bromo-2-fluoro-l -nitrobenzene (16 g, 73 mmol) and DCM (400 mL). To this mixture was added methylamine (2.0 M solution in THF) (88 mL, 180 mmol). Upon addition of amine, the solution rapidly changed from a pale yellow to a bright orange solution. The reaction mixture was stirred at room temperature under a closed system. After 18 h, the reaction mixture was filtered through a plug of SiCh (250 g), with 2: 1 Hex:EtOAc as eluent. The filtrate was concentrated and dried in vacuo to afford the title compound (16 g, 69 mmol, 95 % yield) as a yellow solid. ’H NMR (500 MHz, METHANOL-d4) 6 8.05 (d, J=9.1 Hz, 1H), 7.18 (d, J=1.9 Hz, 1H), 6.85-6.78 (m, 1H), 3.02 (s, 3H). Analytical LC/MS (Method 3): Observed Mass: 232.9; Retention Time: 0.97 min. |
| 94% |
In ethanol; at 20℃; for 1h; |
To a solution of 11 (25.0 g, 114 mmol) in EtOH (100 mL) was added methylamine (40% in MeOH, 34.8 mL, 341 mmol) at rt. The mixture was stirred at rt for 1 h and then cooled to 0 C. The precipitate was collected by filtration, and washed with EtOH and IPE successively to give the title compound (24.8 g, 94%) as a yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 2.95 (3H, d, J = 4.9 Hz), 6.83 (1H, dd, J = 9.1, 1.9 Hz), 7.17 (1H, d, J = 1.9 Hz), 7.98 (1H, d, J = 9.1 Hz), 8.23 (1H, br s). |
| 93% |
In dichloromethane; at 13℃; for 10h; |
The methylamine aqueous solution (30 ml, 250 mmol, 33 wt %) is added to the 4 - bromo -2 - fluoro -1 - nitrobenzene (10.22 g, 46 . 45 mmol) in DCM (50 ml) solution, room temperature (13 C) reaction under 10 h. The reaction solution concentrated under reduced pressure, to obtain the product as a yellow solid (10 g, 93%). |
| 93% |
In ethanol; at 20℃; for 16h;Sealed tube; |
Into a sealed tube were added 4-bromo-2-fluoro-1-nitrobenzene (46 g, 209 mmol) and methylamine (30% in ethanol, 500 mL) at room temperature. After stirring for an additional 16 h, the resulting solution was concentrated under reduced pressure. The residue was triturated with water (1 L) and filtered. The filter cake was collected and dried under vacuum to afford 5-bromo- N-methyl-2-nitroaniline (45 g, 93%) as an orange solid. 1H NMR (400 MHz, DMSO-d6) d 8.25 (br s, 1H), 8.02-7.94 (m, 1H), 7.17 (t, J = 2.9 Hz, 1H), 6.83 (dd, J = 9.1, 2.2 Hz, 1H), 2.95 (d, J = 4.7 Hz, 3H). LC/MS (ESI, m/z): [(M + 1)]+ = 231.15, 233.15 |
| 92% |
In ethanol; at 23℃; for 0.5h;Inert atmosphere; |
Methylamine (56.6 mL, 455 mmol, 33% wt in EtOH) was added to a mixture of 4-bromo-2-fluoro-1-nitro-benzene (50.0 g, 227 mmol) in EtOH (455 mL) at 23 C under nitrogen. The mixture was stirred at 23 C for 30 min, filtered and washed with cold EtOH (200 mL) to provide the title compound as a solid (48.2 g, 92%). m/z (ES+) [M+H]+= 231.0, LCMS (A05); tR = 2.51 min; .1H NMR (400 MHz, DMSO-d6) δ 8.23 (d, J = 4.3 Hz, 1H), 7.98 (d, J = 9.1 Hz, 1H), 7.17 (d, J = 2.0 Hz, 1H), 6.82 (dd, J = 9.1, 2.1 Hz, 1H), 2.95 (d, J = 5.0 Hz, 3H) |
| 90.6% |
With potassium carbonate; potassium iodide; In dichloromethane; at 80℃; for 18h; |
4-bromo-2-fluoro-1-nitrobenzene (5.000 g, 22.727 mmol), methanamine (0.777 g, 25.000 mmol), potassium carbonate (6.282 g, 45.455 mmol) and potassium iodide (0.377 g, 2.273 mmol) were dissolved in dichloromethane (100 mL) at room temperature, after which the resulting solution was stirred at 80 for 18 hours, and then a reaction was finished bylowering a temperature to room temperature. Hexane was put into the reaction mixture and stirred, after which a precipitated solid was filtered and dried to obtain a title compound (4.760 g, 90.6%) in a yellow solid form. |
| 83% |
In tetrahydrofuran; at 15℃; for 0.166667h; |
4-bromo-2-fluoro-1-nitro-benzene (230 g, 1.05 mol, CAS321-23-3) was added to a solution of methylamine in tetrahydrofuran (2 M, 1.51 L). The mixture was stirred at 15 C. for 10 minutes. On completion, the mixture was diluted with H2O (250 mL) and extracted with EtOAc (3×300 mL). The combined organic layers were washed with brine (300 mL), dried over Na2SO4, filtered and concentrated in vacuo to give the title compound (200 g, 83% yield) as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.22 (s, 1H), 7.98 (d, J=9.2 Hz, 1H), 7.16 (d, J=1.6 Hz, 1H), 6.82 (dd, J=8.4, 1.6 Hz, 1H), 2.95 (d, J=4.8 Hz, 3H). |
| 83% |
at 15℃; for 0.166667h;Inert atmosphere; |
4-bromo-2-fluoro-1-nitro-benzene (230 g, 1.05 mol, CAS321-23-3) was added to a solution of mehylamine in tetrahydrofuran (2 M, 1.51 L). The mixture was stirred at 15 C for 10 minutes. On completion, the mixture was diluted with H2O (250 mL) and extracted with EtOAc (3 X 300 mL). The combined organic layers were washed with brine (300 mL), dried over Na2SO4, filtered and concentrated in vacuo to give the title compound (200 g, 83% yield) as a yellow solid.1H NMR (400MHz, DMSO-d6) d 8.22 (s, 1H), 7.98 (d, J = 9.2 Hz, 1H), 7.16 (d, J = 1.6 Hz, 1H), 6.82 (dd, J = 8.4, 1.6 Hz, 1H), 2.95 (d, J = 4.8 Hz, 3H). |
| 83% |
at 15℃; for 0.166667h;Inert atmosphere; |
4-bromo-2-fluoro-1-nitro-benzene (230 g, 1.05 mol, CAS321-23-3) was added to a solution of mehylamine in tetrahydrofuran (2 M, 1.51 L). The mixture was stirred at 15 C for 10 minutes. On completion, the mixture was diluted with H2O (250 mL) and extracted with EtOAc (3 X 300 mL). The combined organic layers were washed with brine (300 mL), dried over Na2SO4, filtered and concentrated in vacuo to give the title compound (200 g, 83% yield) as a yellow solid. 1H NMR (400MHz, DMSO-d6) d 8.22 (s, 1H), 7.98 (d, J = 9.2 Hz, 1H), 7.16 (d, J = 1.6 Hz, 1H), 6.82 (dd, J = 8.4, 1.6 Hz, 1H), 2.95 (d, J = 4.8 Hz, 3H). |
| 83% |
In tetrahydrofuran; at 15℃; for 0.166667h; |
4-bromo-2-fluoro-1-nitro-benzene (230 g, 1.05 mol, CAS321-23-3) was added to a solution of mehylamine in tetrahydrofuran (2 M, 1.51 L). The mixture was stirred at 15 C for 10 minutes. On completion, the mixture was diluted with H2O (250 mL) and extracted with EtOAc (3 X 300 mL). The combined organic layers were washed with brine (300 mL), dried over Na2SO4, filtered and concentrated in vacuo to give the title compound (200 g, 83% yield) as a yellow solid.1H NMR (400MHz, DMSO-d6) δ 8.22 (s, 1H), 7.98 (d, J = 9.2 Hz, 1H), 7.16 (d, J = 1.6 Hz, 1H), 6.82 (dd, J = 8.4, 1.6 Hz, 1H), 2.95 (d, J = 4.8 Hz, 3H). |
| 83% |
In tetrahydrofuran; at 15℃; for 0.166667h; |
4-bromo-2-fluoro-1-nitro-benzene (230 g, 1.05 mol, CASNo.321-23-3) was added to a solution of mehylamine in tetrahydrofuran (2 M, 1.51 L). The mixture was stirred at 15 C for 10 minutes. On completion, the mixture was diluted with H2O (250 mL) and extracted with EtOAc (3 X 300 mL). The combined organic layers were washed with brine (300 mL), dried over Na2SO4, filtered and concentrated in vacuo to give the title compound (200 g, 83% yield) as a yellow solid. 1H NMR (400MHz, DMSO-d6) δ 8.22 (s, 1H), 7.98 (d, J= 9.2 Hz, 1H), 7.16 (d, J= 1.6 Hz, 1H), 6.82 (dd, J= 8.4, 1.6 Hz, 1H), 2.95 (d, J = 4.8 Hz, 3H). |
| 83% |
In tetrahydrofuran; at 15℃; for 0.166667h; |
4-bromo-2-fluoro-1-nitro-benzene (230 g, 1.05 mol, CASNo.321-23-3) was added to a solution of mehylamine in tetrahydrofuran (2 M, 1.51 L). The mixture was stirred at 15 C for 10 minutes. On completion, the mixture was diluted with H2O (250 mL) and extracted with EtOAc (3 X 300 mL). The combined organic layers were washed with brine (300 mL), dried over Na2SO4, filtered and concentrated in vacuo to give the title compound (200 g, 83% yield) as a yellow solid. 1H NMR (400MHz, DMSO-d6) δ 8.22 (s, 1H), 7.98 (d, J= 9.2 Hz, 1H), 7.16 (d, J= 1.6 Hz, 1H), 6.82 (dd, J= 8.4, 1.6 Hz, 1H), 2.95 (d, J = 4.8 Hz, 3H). |
| 77% |
With potassium carbonate; In N,N-dimethyl-formamide; at 20℃; for 20h; |
j00417j To a solution of 4-bromo-2-fluoro-1-nitrobenzene (5.0 g, 22.7 mmol) in N,Ndimethylformamide (80 mL) was added methylamine (2 M in tetrahydrofuran, 22.7 mL) and potassium carbonate (3.93 g, 28.4 mmol). The mixture was stirred at room temperature for 20 hours, then poured into water (50 mL) and extracted with ethyl acetate (3 x 150 mL). The combined organic layers were dried over anhydrous sodium sulfate and concentrated in vacuo to give compound B-73 (4.0 g, 77% yield) as a yellow solid. LCMS (B): tR=0.777 mi, (ES) mlz (M+H) =231.0. ‘H-NMR (CD3C1, 400 MHz): 8.03 (d, J=9.2 Hz, 2H), 7.02 (d, J=1.6 Hz, 1H), 6.78 (dd, J11.2 Hz, J21.2 Hz, 1H), 3.02 (s, 3H). |
| 74.3% |
With potassium carbonate; In tetrahydrofuran; at 90℃; for 6h; |
Step A5-bromo-N-methyl-2-nitroaniline[00302] An orange mixture of 4-bromo-2-fluoro-1 -nitrobenzene (2 g, 9.09 mmol), a 2 M methylamine solution in THF (5.00 mL, 10.00 mmol) and K2C03 (2.51 g, 18.18 mmol) in DMF (20 mL) was heated at 90 C for 3 h. More methylamine solution in THF (10.00 mL, 20.00 mmol) was added and the reaction mixture was heated at 90 C for 3 h. The resulting mixture was allowed to cool to room temperature, diluted with EtOAc (100 mL) and washed with a 5% aq. LiCI solution (3x100 mL). The organic layer was concentrated onto Celite and purified by column chromatography (silica gel, 0-40% EtOAc/hexane) to obtain 5-bromo-N-methyl-2- nitroaniline (1.56 g, 6.75 mmol, 74.3 % yield) as an orange solid: 1H NMR (400 MHz, DMSO-d6) δ ppm 2.94 (d, 3 H) 6.82 (dd, J=9.07, 2.05 Hz, 1 H) 7.16 (d, =1.95 Hz, 1 H) 7.98 (d, J=9.07 Hz, 1 H) 8.24 (d, .7=4.19 Hz, 1 H). |
|
In ethanol; at 0 - 20℃; for 0.416667h; |
To a solution of 4-bromo-2-fluoro-1 -nitrobenzene (10 g, 45.45 mmol) in 20 ml of ethanol under 00C, 30 ml methylamine (33 wt% in ethanol) was added dropwise. After addition, reaction mixture was gradually warmed up to room temperature and stirred for 25 minutes. The mixture was concentrated under reduced pressure to yield N-methyl-(5-bromo- 2-nitrophenyl)amine (9.13 g). 400 MHz 1H NMR (CDCI3) δ 8.0 (d, 1H), 7.0 (d, 1H), 6.7 (dd, 1 H); MS+ 230, 232. |
|
In ethanol; acetonitrile; at 20℃; for 4h; |
To a stirred partial solution of 4-bromo-2-fluoronitrobenzene (9.2 g; Aldrich) in acetonitrile (140 ml) was added a solution of 8M methylamine in ethanol (86 ml). The reaction was stirred at room temperature for 4 hr and then evaporated to dryness. The residue was partitioned between EtOAc (500 ml) and water (500 ml). The aqueous phase was separated off and extracted with EtOAc (500 ml). The organic phases were combined, washed with water (500 ml) and brine, dried (magnesium sulphate) and evaporated to give the title compound (9.47 g) as an orange-brown solid. |
|
In 1,4-dioxane; methanol; at 90℃; for 3.5h;Inert atmosphere; |
To a round bottom flask was added added 4-bromo-2-fluoro- 1 -nitrobenzene (11.8 g, 53.7 mmol), anhydrous 1,4 Dioxane (100 mL), and a 2.0 M solution of methyl amine in methanol (56.4 mL, 113 mmol). The reaction mixture was then heated to 90C while stirring in a hot oil bath with a water cooled reflux condenser attached under an atmosphere of nitrogen for 3.5 hours. The crude reaction mixture was then allowed to cool to room temperature, and concentrated to give 5-bromo-N-methyl-2-nitroaniline (1-1). MS (M)+: observed = 230.9, calculated = 231.05. |
|
In methanol; ethanol; at 23℃; for 13h; |
S-bromo-N-methyl-2-nitroaniiine (1-2)A solution of 4-Bromo-2-ftuoronitrobenzene (1-1, 10.5 g, 47.7 mmol) in EtOH(100 ml) was treated with Methylamine (2M in MeOH, 28.6 ml, 573 mmol, 1.2 eq) and the resulting dark maroon solution was stirred at 23 deg C for 13 h. The reaction was then concentrated in vacuo, and the residual yellow-orange solid was partitioned between EtOAc (2x250 ml) and water (300 ml). The combined organic layers were dried over Na2S04 and concentrated, leaving the title compound, 5-bromo-N-methyl-2-nitroaniline (1-2), as a bright orange solid. 1HNM (300 MHz, CDC13) δ 8.03 (d, IH, J=9.15 Hz), 7.01 (sd, IH, J=l,83 Hz), 6.77 (dd, IH, J=7.02 Hz), 3.02 (d, 3H, J=4.88 Hz). LRMS m/z: Calc'd for C7H7BrN202 (M+H) 232.1, found 232.8. |
|
With potassium carbonate; In tetrahydrofuran; N,N-dimethyl-formamide; at 20℃; |
Intermediate IX(5-Bromo-2-nitro-phenyl)-methyl-amine Methylamine (2 M in tetrahydrofuran, 11.4 mL) was added to a mixture of 4-bromo-2-fluoro-1-nitro-benzene (2.50 g) and K2CO3 (1.90 g) in N,N-dimethylformamide (40 mL). The resulting mixture was stirred at ambient temperature overnight. Then, the mixture was concentrated under reduced pressure and dichloromethane was added. The resulting mixture was washed with 0.5 M aqueous HCl solution and brine and dried (MgSO4). The solvent was removed to give the product as a solid.Yield: 2.60 g (99% of theory); Mass spectrum (ESI+): m/z=231/233 (Br) [M+H]+. |
|
With N-ethyl-N,N-diisopropylamine; In N,N-dimethyl-formamide; at 85℃;Sealed vial; |
[000202] 4-Bromo-2-fluoronitrobenzene (Aldrich, 1 eqvt), methylamine (2 eqvt), and hunig's base (3 eqvt) in DMF (1 Molar in substrate) were placed in a sealed vial and heated to 85 C overnight. The crude mixture was diluted with water, and the product collected by filtration. Purification by chromatography (gradient: 92:8 hexanes:ethyl acetate to 68:32 hexanes:ethyl acetate) provided the title compound. 1HNMR (DMSO-d6, 400 MHz) δ 8.22 (bd, J = 4.4 Hz, 1H), 7.95 (d, J = 8.8 Hz, 1H), 7.14 (d, J = 2 Hz, 1H), 6.80 (dd, J = 9.2, 2.4 Hz, 1H), 2.92 (d, J = 4.8 Hz). |
| 24.8 g |
In methanol; ethanol; at 20℃; for 1h; |
Example 2 1- (2-Cyclopropyl-l-methyl-lH-benzimidazol-6-yl) -4- ( (4- fluorobenzyl ) oxy) pyridin-2 ( 1H) -one A) 5-Bromo-N-methyl-2-nitroaniline To a solution of 4-bromo-2-fluoro-l-nitrobenzene (25 g) in EtOH (100 ml) was added methylamine (40% in MeOH, 34.8 ml) at room temperature. The mixture was stirred at room temperature for 1 h. After being stirred, the reaction mixture was cooled to 0C, and the precipitate was collected by filtration and washed with EtOH (0C) and IPE. The solid was dried to give the title compound (24.8 g) as a yellow solid. XH NMR (300 MHz, DMSO-d6) : δ 2.95 (3H, d, . J = 4.9 Hz), 6.83 (1H, dd, J = 1.9, 9.1 Hz), 7.17 (1H, d, J = 1.9 Hz), 7.98 (1H, d, J = 9.1 Hz) , 8.23 (1H, brs) . |
| 24.8 g |
In methanol; ethanol; at 20℃; for 1h; |
A) 5-Bromo-N-methyl-2-nitroaniline [0294] To a solution of 4-bromo-2-fluoro-1-nitrobenzene (25 g) in ethanol (100 mL) was added methylamine (40% in methanol, 34.8 mL) at room temperature, and the mixture was stirred for 1 hr. The obtained mixture was cooled to 0C, and the resulting precipitate was collected by filtration and washed with ice-cooled ethanol and diisopropyl ether. The obtained solid was dried to give the title compound (24.8 g) as a yellow solid. 1H NMR (300 MHz, DMSO-d6): δ 2.95 (3H, d, J = 4.9 Hz), 6.83 (1H, dd, J = 9.1, 1.9 Hz), 7.17 (1H, d, J = 1.9 Hz), 7.98 (1H, d, J = 9.1 Hz), 8.23 (1H, brs). |
|
With N-ethyl-N,N-diisopropylamine; In tetrahydrofuran; N,N-dimethyl-formamide; at 20℃; for 2h; |
[0007991 To a stirred solution of the compound 1 (2 g, 1 eq)) in DMF (20 mL), methyl amine 2 M solution in THF (9.19 mL, 2 eq) and DIPEA (3.53 g, 3 eq) were added and stirred at room temperature for 2 h. The progress of the reaction was monitored by TLC. After completion of the reaction, the reaction mixture was diluted with water and extracted with ethyl acetate (3 X 25 mL). Combined organic extracts were washed with brine and dried over anhydrous sodium sulfate and evaporated under reduced pressure to afford the crude product 2 (2 g) and used as such for the next step without further purification. |
|
In methanol; at 80℃; for 15h; |
A mixture of 4-bromo-2-fluoro-1-nitrobenzene (4.0 g, 18.26 mmol) in 30 mLMeNH2/MeOH was stirred at 80 C for 15 h, and poured into H20 (100 mL). The mixture wasextracted with EA (30 mL x 3), dried over anhydrous Na2SO4, and filtered. The filtrate was concentrated in vacuo to afford 5-bromo-N-methyl-2-nitroaniline (3.8 g, 90 %) as a yellow solid. LC-MS m/z: 232.9 [M+H]+. Purity (214 nm): 88.0%; tR = 1.89 min |
|
In ethanol; at 0 - 20℃; for 0.416667h; |
To a solution of bromo-2-fluoro-1- nitrobenzene (10 g, 45.45 mmol) in 20 ml of ethanol under O0C, 30 ml methylamine (33 wt% <n="67"/>in ethanol) was added dropwise. After addition, reaction mixture was gradually warmed up to room temperature and stirred for 25 minutes. The mixture was concentrated under reduced pressure to yield N-methyl-(5-bromo-2-nitrophenyl)amine (9.13 g). 400 MHz 1H NMR (CDCI3) δ 8.0 (d, 1 H), 7.0 (d, 1 H), 6.7 (dd, 1 H); MS+ 230, 232. |
|
In ethanol; |
4-Bromo-2-fluoro-1-nitrobenzene (23 g, 105 mmol) was dissolved in EtOH (20 mL), Me Eh (250 mL, 33% in EtOH) was added, the mixture was stirred at R.T overnight. When LC- MS showed the starting material was consumed, the solvent was evaporated and the resulting crude was dissolved in EtOAc (300 mL) and washed with water (200 mL x 2) and brine (200 mL), dried over anhydrous Na2S04. The solid was filtered and the filtrate was concentrated to give the crude product 5-bromo-N-methyl-2-nitroaniline as yellow solid (23 g, 95 % yield). 1H NMR (400 MHz, CDCb) δ 8.03 (d, J= 9.2 Hz, 2H), 7.01 (s, 1H), 6.76 (d, J= 9.2 Hz, 1H), 3.02 (s, 3H). LC-MS (ESI+): m/z 232.1 (M+H)+. |
|
In methanol; ethanol; at 20℃; for 1h; |
To a solution of 4-bromo-2-fluoro-1 -nitrobenzene (0.50 g, 2.28 mmol), in EtOH (10 ml_), methylamine (40% wt/v in methanol, 1 .0 ml.) was added and stirred at room temperature for 1 hour. The precipitated material was filtered under vacuum, washed with hexanes and dried to obtain 5-bromo-N-methyl-2-nitroaniline. 1 H NMR (400 MHz, DMSO-d6) d: 8.24 (brs,1 H), 7.98 (d, J = 8.8 Hz, 1 H), 7.16 (d, J = 2 Hz 1 H), 6.82 (dd, J = 2 Hz and 9.2 Hz, 1 H), 2.94 (d, J = 4.8 Hz, 3H). |
|
With potassium carbonate; In tetrahydrofuran; N,N-dimethyl-formamide; at 25℃; for 18h;Inert atmosphere; |
Step A: 5-Bromo-N-methyl-2-nitroaniline Under a protection of nitrogen, to a solution of 4-bromo-2-fluoro-1-nitroaniline (15.0g, 68.18mmol) and potassium carbonate (11.31g, 81.82mmol) in DMF (250mL) was added dropwise methylamine in tetrahydrofuran (2M, 68.18mL) at 25C, and it was stirred at 25C for 18 hours. The reaction solution was poured into 500mL water, and it was stirred for 10 minutes. The precipitated solid was filtered and dried to give the title compound. 1H NMR (400MHz, CHLOROFORM-d) δ=8.03 (d, J=9.4 Hz, 2H), 7.01 (s, 1H), 6.77 (dd, J=1.8, 9.2 Hz, 1H), 3.02 (d, J=5.1 Hz, 3H). |
|
In tetrahydrofuran; at 20℃; |
To a solution of 4-bromo-2-fluoro-1-nitro-benzene (1 g, 4.55 mmol) in THF (15 mL) was added methanamine (6.82 mL, 13.64 mmol) (2 M solution in THF), and the mixture was stirred at 20 C. for 2 hours. (0305) The mixture was concentrated to give the crude product (1200 mg, 4.97 mmol) as a solid. LCMS Rt=0.815 min in 1.5 min chromatography, MS ESI calcd. for C7H8BrN2O2 [M+H]+ 231.0, found 230.7. |
|
In tetrahydrofuran; at 20℃; |
To a solution of 4-bromo-2-fluoro-1-nitro-benzene (1 g, 4.55 mmol) in THF (15 mL) was added methanamine (6.82 mL, 13.64 mmol) (2 M solution in THF), and the mixture was stirred at 20 C. for 2 hours. (0305) The mixture was concentrated to give the crude product (1200 mg, 4.97 mmol) as a solid. LCMS Rt=0.815 min in 1.5 min chromatography, MS ESI calcd. for C7H8BrN2O2 [M+H]+ 231.0, found 230.7. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping