| 67% |
|
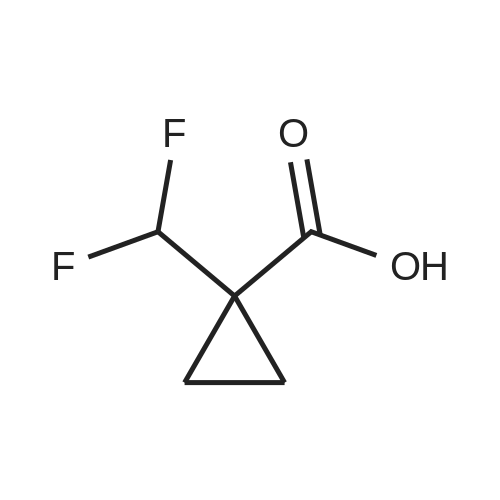
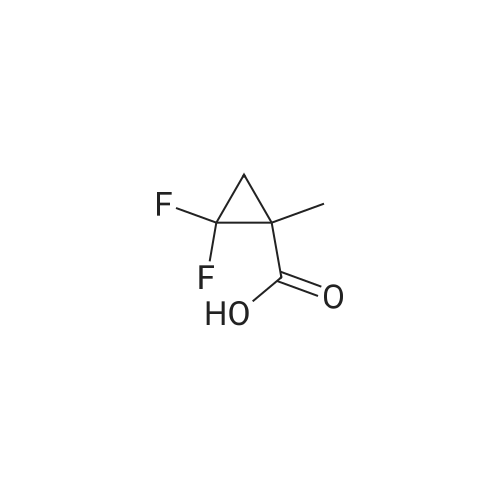
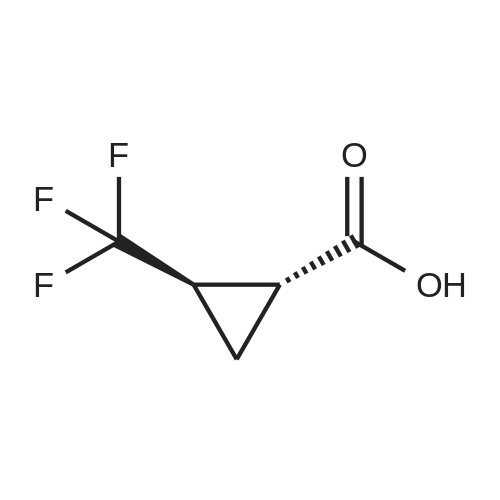
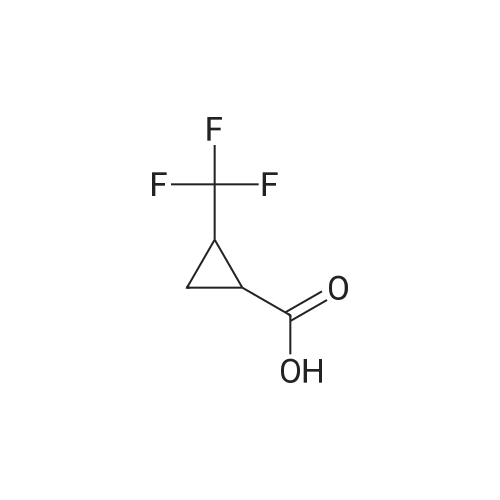
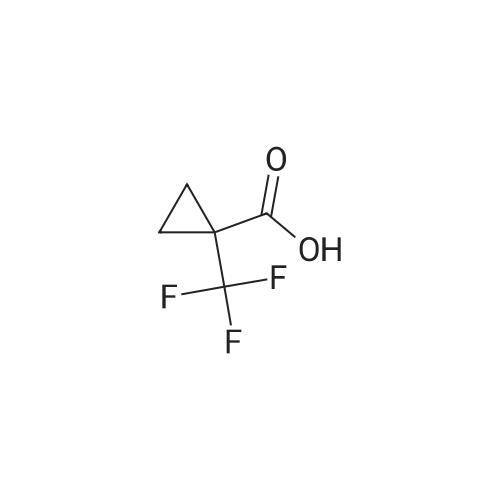
A solution of l-(trifluoromethyl)cyclopropanecarboxylic acid (37.0 mg, 240 muiotaetaomicron) in N,N- dimethylformamide (800 mu) was treated with HATU (91.4 mg, 240 muiotaetaomicron) and stirred 15 min at room temperature. 4-[2-Amino-3,3,3-trifluoropropyl]-5-(4-chlorophenyl)-2-{ [l-(3-chloropyridin-2- yl)-lH-l,2,4-triazol-3-yl]methyl}-2,4-dihydro-3H-l,2,4-triazol-3-one (Example 11 A, 80.0 mg, 160 muiotaetaomicron) was then added followed by N,N-diisopropylethylamine (84 mu, 480 muiotaetaomicron). The resulting mixture was stirred 1 h at room temperature and evaporated. The residue was purified by preparative HPLC (Method 4) affording 68.1 mg (67% of th.) of the title compound. (0656) LC-MS (Method 1): Rt = 1.04 min; MS (ESIpos): m/z = 635.2 [M+H]+ (0657) -NMR (400 MHz, DMSO-d6) delta [ppm] : 9.05 (s, 1H), 8.58 (dd, 1H), 8.39 (d, 1H), 8.29 (dd, 1H), 7.80-7.53 (m, 5H), 5.12 (d, 2H), 4.87-4.66 (m, 1H), 4.38-4.00 (m, 2H), 1.43-1.06 (m, 4H). (0658) The two enantiomers were separated by preparative chiral HPLC [sample preparation: 50 mg dissolved in 1 ml ethanol and 1 ml 2-propanol; injection volume: 250 mu; column: Daicel Chiralpak ID 5 muiotaeta, 250 x 20 mm; eluent: n-heptan/2-propanol 50:50; flow rate: 20 ml/min; temperature: 23C; UV detection: 220 nm] . After separation, 19 mg of enantiomer 1 (Example 42), which eluted first, and 18 mg of enantiomer 2 (Example 43), which eluted later, were isolated. (0659) Example 42 (0660) N- { 3- [3-(4-Chlorophenyl)- 1 - { [ 1 -(3-chloropyridin-2-yl)- 1 H- 1 ,2,4-triazol-3-yl] methyl } -5-oxo- 1 ,5- dihydro-4H- 1 ,2,4-triazol-4-yl] -1,1,1 -trifluoropropan-2-yl } - 1 -(trifluoromethyl)cyclopropane- carboxamide (enantiomer 1) (0661) For separation conditions see Example 41. (0662) Analytical chiral HPLC: Rt = 1.48 min, e.e. = 100% [column: Daicel Chiralpak ID-3 3 muiotaeta, 50 x 4.6 mm; eluent: iso-hexane/ethanol 50:50; flow rate: 1.0 ml/min; temperature: 23C; UV detection: 220 nm]. (0663) LC-MS (Method 1): Rt = 1.07 min; MS (ESIpos): m/z = 635.2 [M+H]+ -NMR (400 MHz, DMSO-d6) delta [ppm] : 9.05 (s, IH), 8.58 (dd, IH), 8.39 (d, IH), 8.29 (dd, IH), 7.74-7.55 (m, 5H), 5.12 (d, 2H), 4.83-4.71 (m, IH), 4.27 (dd, IH), 4.08 (dd, IH), 1.42-1.10 (m, 4H). (0664) Example 43 (0665) N- { 3- [3-(4-Chlorophenyl)- 1 - { [ 1 -(3-chloropyridin-2-yl)- 1 H- 1 ,2,4-triazol-3-yl] methyl } -5-oxo- 1 ,5- dihydro-4H- 1 ,2,4-triazol-4-yl] -1,1,1 -trifluoropropan-2-yl } - 1 -(trifluoromefhyl)cyclopropane- carboxamide (enantiomer 2) (0666) For separation conditions see Example 41. (0667) Analytical chiral HPLC: Rt = 2.00 min, e.e. = 100% [column: Daicel Chiralpak ID-3 3 muiotaeta, 50 x 4.6 mm; eluent: iso-hexane/ethanol 50:50; flow rate: 1.0 ml/min; temperature: 23C; UV detection: 220 nm] . (0668) LC-MS (Method 1): Rt = 1.08 min; MS (ESIpos): m/z = 635.1 [M+H]+ (0669) -NMR (400 MHz, DMSO-d6) delta [ppm] : 9.05 (s, IH), 8.58 (dd, IH), 8.39 (d, IH), 8.29 (dd, IH), 7.80-7.47 (m, 5H), 5.12 (d, 2H), 4.91-4.61 (m, IH), 4.38-3.96 (m, 2H), 1.37-1.11 (m, 4H). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping