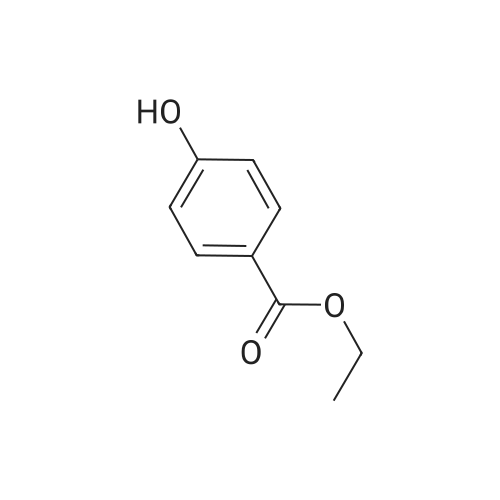
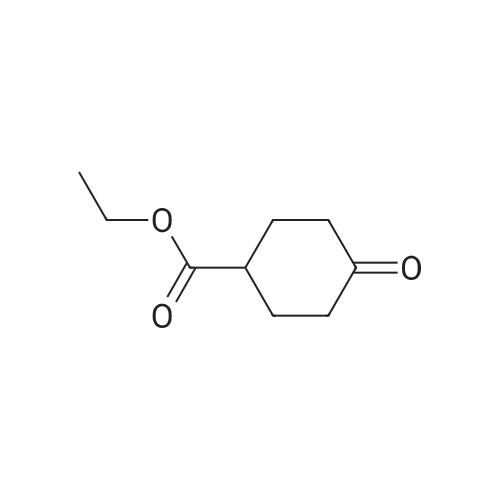
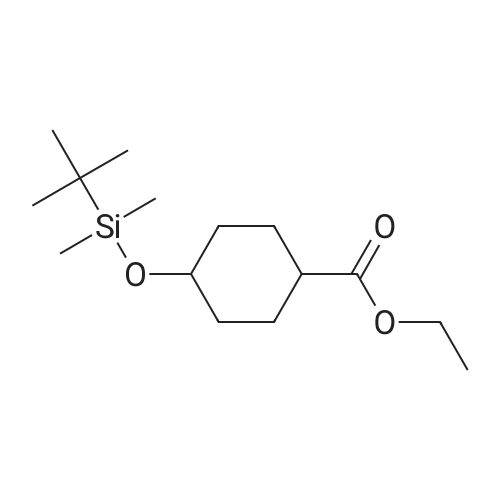
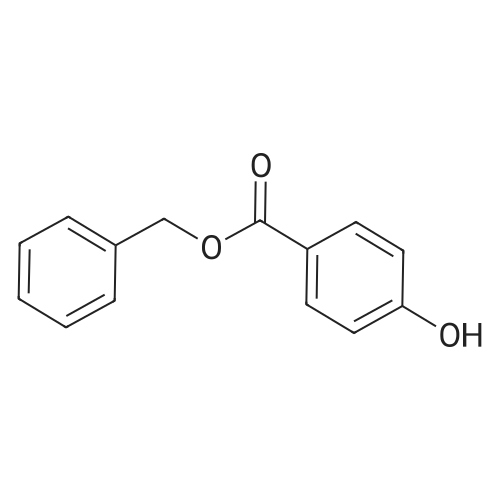
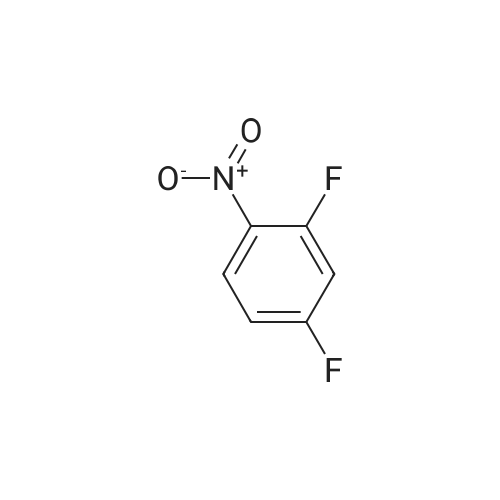
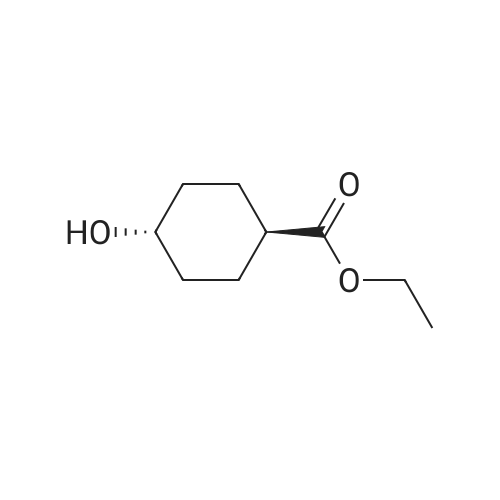
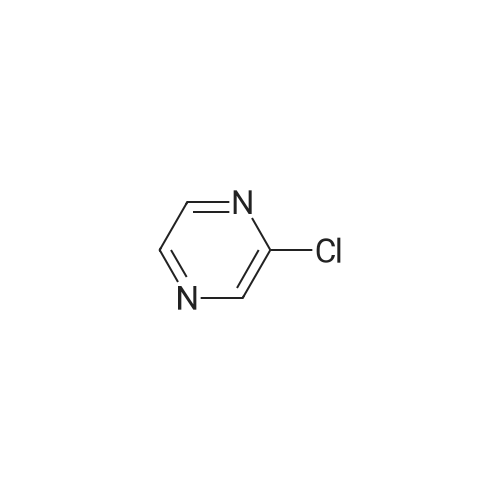
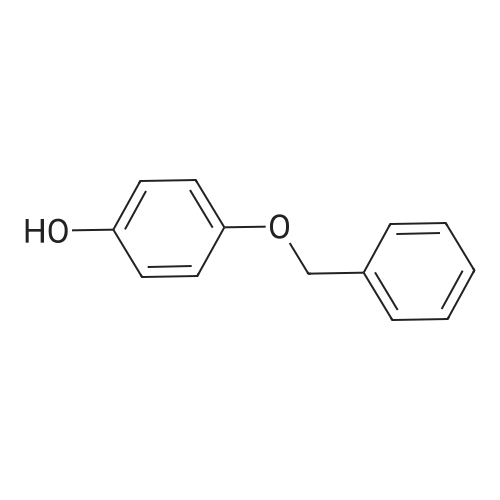
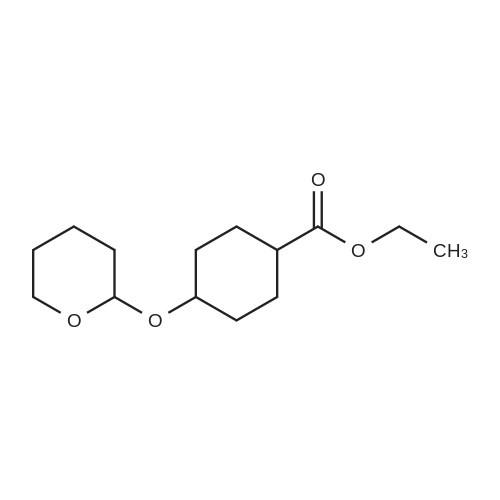
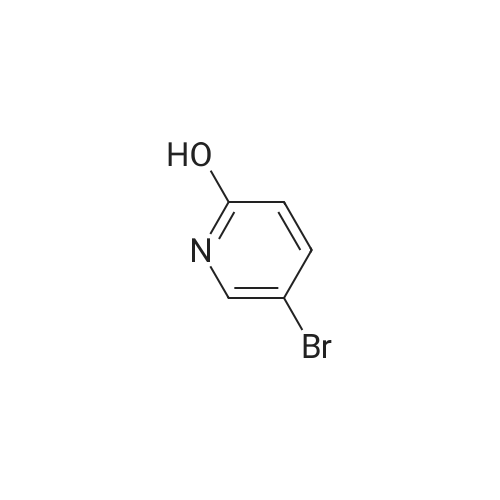
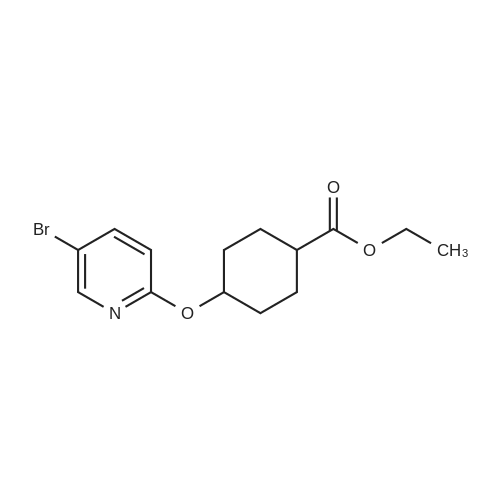
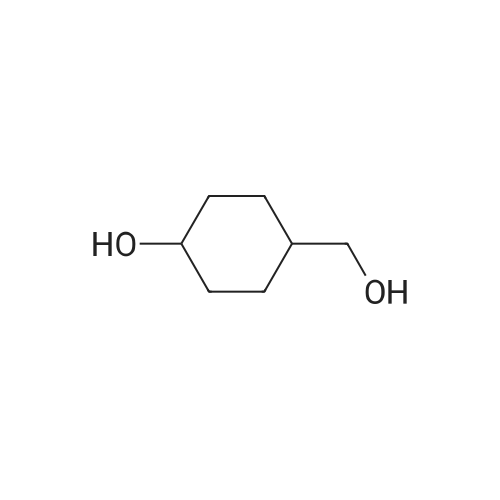
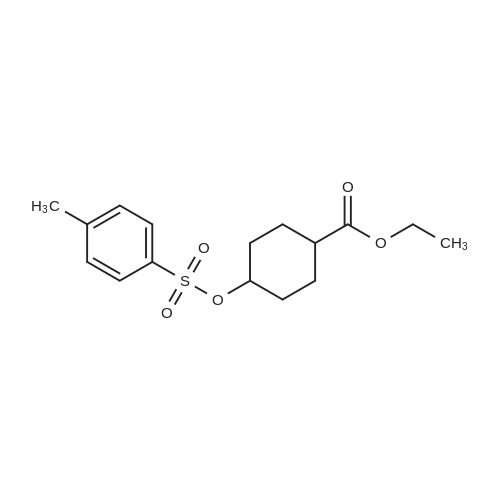
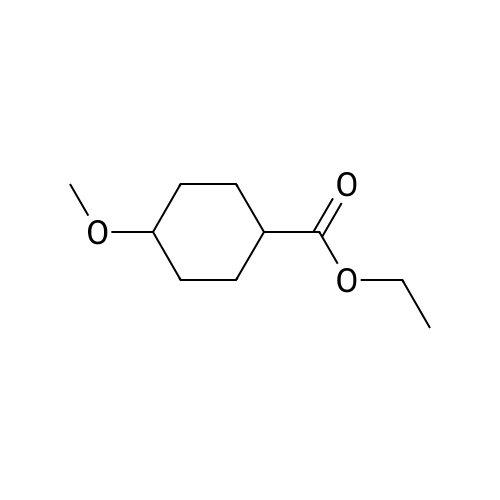
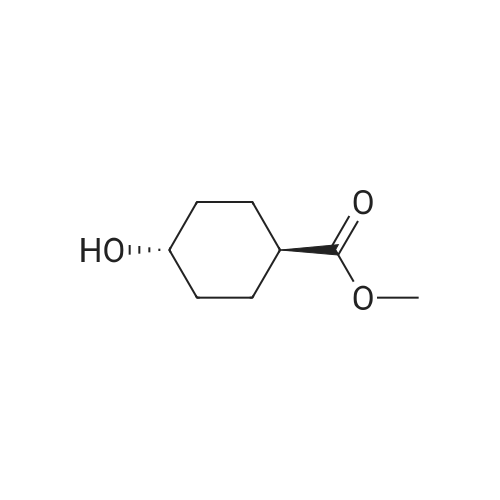
| 74% |
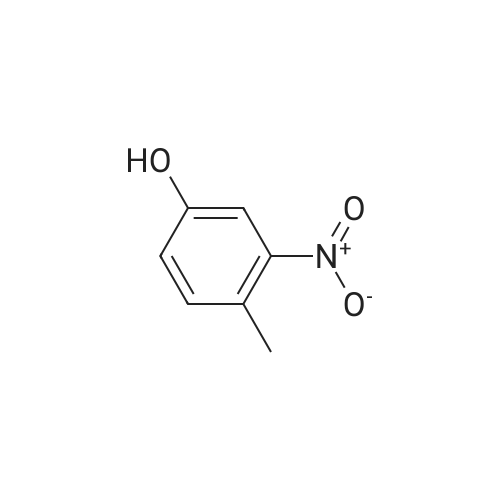
With 1H-imidazole In N,N-dimethyl-formamide at 23℃; for 4h; |
|
| 69.8% |
With 1H-imidazole In N,N-dimethyl-formamide at 20℃; for 48h; |
Intermediate 3-h: 4- ((4- ((tert-butyldimethylsilyl) oxy) cyclohexyl) methoxy) -3-nitrobenzenesulfonamide Step 1: ethyl 4- ((tert-butyldimethylsilyl) oxy) cyclohexane-1-carboxylate
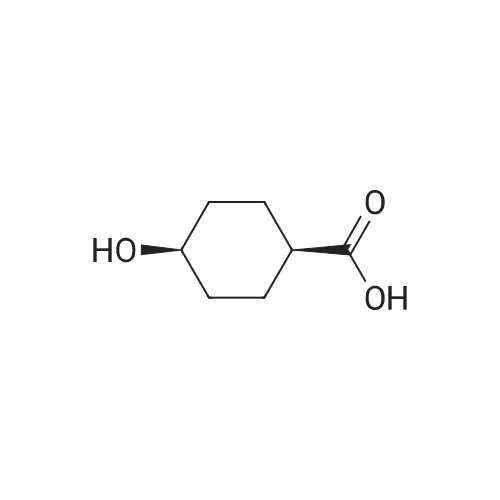
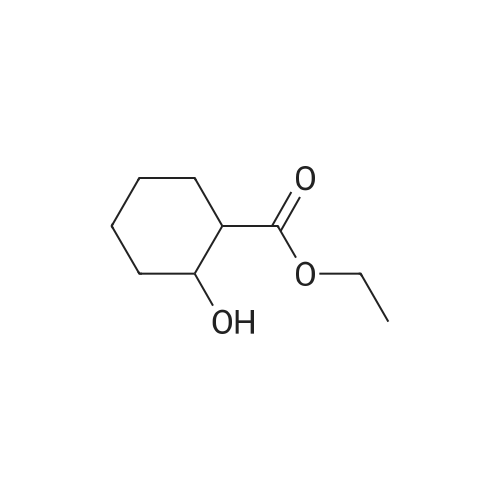
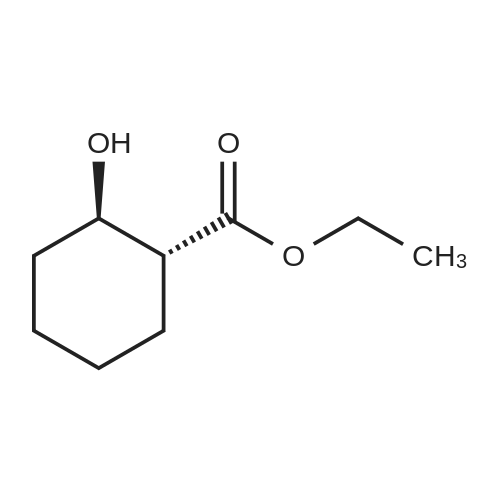
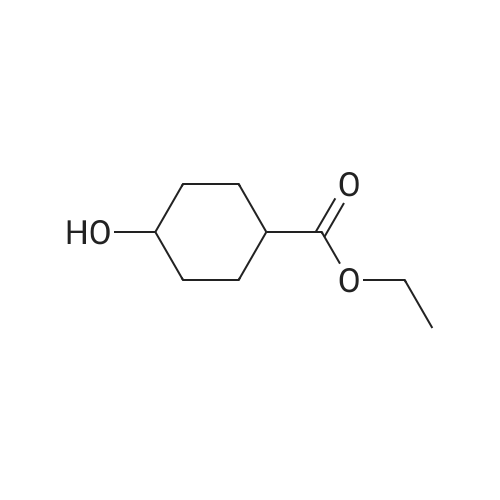
To a solution of ethyl 4-hydroxycyclohexane-1-carboxylate (2 g, 11.61mmol) in DMF (50 ml) were added tert-butylchlorodimethylsilane (1.575 g, 10.4 mmol) and imidazole (1.58 g, 23.22 mmol). The mixture was stirred at r.t. for 2 days. The mixture was concentrated. The residue was dissolved with DCM (200 ml), washed with brine, dried over Na 2SO 4 and concentrated. The residue was purified by chromatography column on silica (eluent: EA/PE = 1/40) to give the product (2.32 g, 69.8%). |
| 45% |
Stage #1: 4-hydroxycyclohexyl carboxylic acid ethyl ester With triethylamine In dichloromethane at 20℃; for 0.333333h;
Stage #2: tert-butyldimethylsilyl chloride In dichloromethane at 20℃; for 40h; |
Ethyl 4-[(tert-butyldimethylsilyl)oxy]cyclohexane-1-carboxylate
To a solution of ethyl 4-hydroxycyclohexane-1-carboxylate (10 g, 58.06 mmol) in dichloromethane (25 mL) was added triethylamine (13 g, 128.47 mmol) slowly at room temperature. After stirring for additional 20 min, TBDMSC1 (24.9 g, 87.09 mmol) was slowly added. The resulting reaction mixture was then stirred at room temperature for 40 h. The reaction mixture was quenched by the addition of water (100 mL) and extracted with dichloromethane (100 mL×2). The organic phases were combined, washed with brine and dried over Na2SO4. The solvent was removed under reduced pressure and the residue was purified by flash chromatography eluting with EtOAc in hexane (1% to 10% gradient) to yield ethyl 4-[(tert-butyldimethylsilyl)oxy]cyclohexane-1-carboxylate as yellow oil (7.5 g, 45%). |
| 2.25 g (79%) |
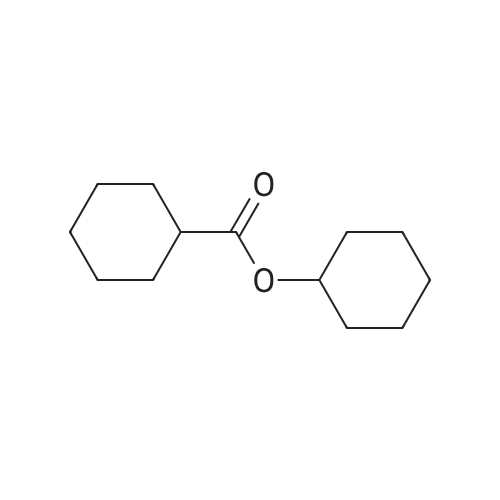
With dmap; triethylamine In N,N-dimethyl-formamide at 20℃; for 18h; |
5 5-Butyl-9-[1-(4,6-dimethyl-pyrimidine-5-carbonyl)-4-methyl-piperidin-4-yl]-3-(trans-4-hydroxy-cyclohexylmethyl)-1-oxa-3,9-diaza-spiro[5.5]undecan-2-one (I-4)
EXAMPLE 5 5-Butyl-9-[1-(4,6-dimethyl-pyrimidine-5-carbonyl)-4-methyl-piperidin-4-yl]-3-(trans-4-hydroxy-cyclohexylmethyl)-1-oxa-3,9-diaza-spiro[5.5]undecan-2-one (I-4) 4-(tert-Butyl-dimethyl-silanyloxy)-cyclohexanecarboxylic acid ethyl ester (38)-To a solution of 4-oxy-cyclohexanecarboxylic acid ethyl ester (CASRN 17159-80-7, 1.7 g, 0.01 mol) in DMF (14 mL) was added DMAP (58 mg, 0.47 mmol), TEA (1.54 mL) and tert-butyl-dimethylsilyl chloride (1.65 g, 0.011 mol). The reaction mixture was stirred at RT for 18 h, poured into ice (10 g), extracted with EtOAc (3*50 mL). The organic extracts were dried (MgSO4) filtered and concentrated. The residue was purified by SiO2 chromatography eluding with EtOAc/hexane (1:20) to afford 2.25 g (79%) of 4-(tert-butyl-dimethyl-silanyloxy)-cyclohexanecarboxylic acid ethyl ester (38) as colorless oil. |
|
With 1H-imidazole In N,N-dimethyl-formamide at 20℃; for 18h; |
15
tert-Butyldimethylchlorosilane (9.71 g, 64.45 mmol) was added portion wise to ethyl 4- hydroxycyclohexanecarboxylate (CAS no. 17159-80-7) (10 g, 58 mmol) and imidazole (7.9 g, 116 mmol) in DMF (58 mL) under nitrogen. The resulting solution was stirred at room temperature for 18 hours. The reaction mixture was diluted with diethyl ether (250 mL), and washed with saturated brine (500 mL). The organic layer was dried (MgSO4), filtered and evaporated to afford the product (16.3 g). This was used without any further purification.IH NMR (300 MHz, CDCl3) δ 0.02 - 0.07 (5H, m), 0.87 - 0.90 (9H, m), 0.92 (IH, s), 1.22 - 1.29 (3H, m), 1.30 - 1.54 (3H, m), 1.59 - 1.70 (2H, m), 1.87 - 2.04 (3H, m), 2.21 - 2.32 (IH, m), 3.70 - 3.89 (IH, m), 4.07 - 4.16 (2H, m). |
|
With 1H-imidazole In N,N-dimethyl-formamide at 20 - 40℃; |
1
Chloro(1 ,1-dimethylethyl)dimethylsilane (1 15 g; 0.76 mol) was added in portions over 1 hour to a solution of commercially available ethyl 4-hydroxycyclohexanecarboxylate (118 g; 0.68 mol), imidazole (103 g; 1.52 mol) and dimethylformamide (400 ml.) stirred under an atmosphere of argon. A small exotherm was observed resulting in the reaction mixture temperature increasing to -40 0C. The mixture was stirred at room temperature overnight then poured into 10% citric acid solution (2 L) and extracted with diethyl ether (2 x 800 ml_). The ether extracts were washed with water, brine and then dried (Na2SC>4) and the solvent was removed to give the title compound as an oil (198.4 g) 1H NMR δ (CDCI3, 400 MHz): 0.01 (6H, m), 0.85 (9H, s), 1.2 (3H, m), 1.3-1.5 (2H, m), 1.6 (2H, m), 1.85-2 (3H, m), 2.15-2.3 (1 H, m) 3.5 (0.4H, m) 3.86 (1 H, m) 4.1 (1 H, m). |
|
With 1H-imidazole In N,N-dimethyl-formamide at 20℃; for 20h; |
2.a
PREPARATION 2: l-(2-(Benzyloxy)ethyl)-4-(/- butyldimethylsilyloxy)cyclohexanecarbaldehyde; Step a: Ethyl 4-(tert-butyldimethylsilyloxy)cyclohexanecarboxylate; [00164] A mixture of ethyl 4-hydroxycyclohexane-carboxylate (50 mL, 0.31mol), imidazole (50.1 g, 0.74 mol), and t-butyldimethylsilyl chloride (56 g, 0.37mol) in DMF (580 mL) was stirred at room temperature for 20 hrs under atmosphere of nitrogen. Water (100 mL) was added to the mixture, and the mixture was extracted with ether (600 mL). The extract was washed with water (400 mL) and brine (500 mL), dried over anhydrous Na2SO4, and filtered. The filtrate was concentrated to give compound 2 as a colorless oil (102 g), which was used for the next step without further purification. |
|
With 1H-imidazole In DMF (N,N-dimethyl-formamide) at 20℃; for 16h; |
45A Example 45 (2S, [5R)-5-ETHYNYL-L- (N- (4-HYDROXY-L-METHYLCYCLOHEXYL)] glycyl) [PYRROLIDINE-2-CARBONITRILE] Example 45A [4- (TERT-BUTYL-DIMETHYL-SILANYLOXY)-CYCLOHEXANECARBOXYLIC] acid ethyl ester
To a solution of 4-hydroxy-cyclohexanecarboxylic acid ethyl ester (10.32 g, 59.92 mmol) in dimethylformamide (50 mL) was added imidazole (8.16 g, 119.8 mmol), followed by tert-butyldimethylsilyl chloride (9.94 g, 65.9 mmol). The resulting mixture was stirred at room temperature for 16 hours. Diethyl ether was added (150 mL), and the mixture was washed with 1M [HC1] (150 mL). The aqueous layer was extracted with diethyl ether (150 mL). The combined organic layers were washed with 1M [HC1] (100 mL) and saturated sodium chloride solution (100 mL), dried over magnesium sulfate, filtered and concentrated to afford a clear oil. MS (CI) m/z 287 (M+1) [+] |
| 34.94 g |
With 1H-imidazole In N,N-dimethyl-formamide at 20℃; for 15h; Cooling with ice; |
53.3
Compound 5 (20.04 g) and imidazole (15.84 g) were dissolved in N,N-dimethylformamide (116 mL), tert-butyldimethylsilylchloride (19.47 g) was added under ice cooling, and the mixture was stirred at room temperature for 15hours. The reaction solution was added to ice water, and the mixture was extracted with diethyl ether. The extract waswashed with water and saturated brine. The solution was dried over anhydrous magnesium sulfate, and concentratedunder reduced pressure to obtain Compound 6 (34.94 g) as a colorless oil.MS (m/z): 287 [M+H]+ |
|
With 1H-imidazole In N,N-dimethyl-formamide at 20℃; Inert atmosphere; |
2 4.1.2. 4-(tert-Butyldimethylsilanoxy)cyclohexane carboxylic acid (3)
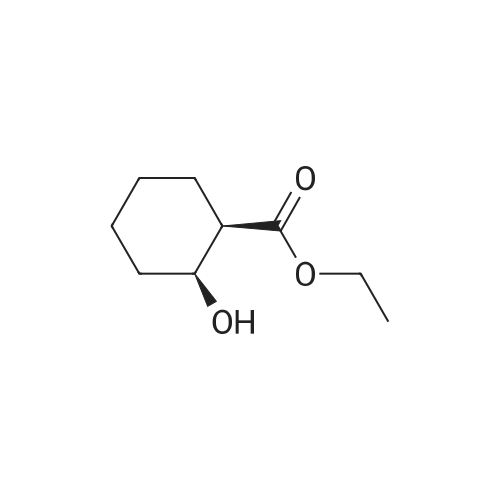
To a solution of ethyl 4-hydroxycyclohexanecarboxylate 2 (mixture of cis and trans) (2.5 g, 14.5 mmol) and imidazole (1.97 g, 29 mmol) in anhydrous DMF (20 mL), tert-butyldimethylsilyl chloride (2.63 g, 17.5 mmol) was added. After stirring overnight, Et2O (60 mL) and 1M aqueous HCl (60 mL) were added. After extraction with Et2O, the combined organic phases were washed with 1M aqueous HCl and brine, dried over Na2SO4, filtered and concentrated under reduced pressure to give a TBS-protected alcohol as a colorless oil (4.14 g), which was used in the next step without further purification. To a solution of the former alcohol (4.14 g, 14.5 mmol) in THF (20 mL) and methanol (12 mL), lithium hydroxide (0.7 g, 29 mmol) was added. The mixture was then stirred at 60 °C for 3 h. After removal of the solvents, water and Et2O were added and the aqueous layer was extracted with diethyl ether, cooled in an ice bath then acidified to pH=1 with 1M aqueous HCl. After extraction with ethyl acetate, the organic phases were dried over Na2SO4, filtered and concentrated. The residue was purified by flash chromatography on silica gel (cyclohexane/ethyl acetate, 50:50) to give the title compound 3 as a mixture of cis/trans isomers (3.48 g, 100% yield) as a colorless oil; δH (400 MHz, CDCl3) 10.84 (2H, m, 2*COOH), 3.89 and 3.56 (2*1H, 2*bs, 2*H-4), 2.41-2.18 (2H, m, 2*H-1), 2.10-1.83 (6H, m), 1.74-1.59 (4H, m), 1.58-1.23 (6H, m), 0.88 and 0.87 (2*9H, 2*s, 2*(CH3)3CSi), 0.05 and 0.03 (2*6H, 2*s, 2*(CH3)2Si); δC (100 MHz; CDCl3) 181.8 (2*COOH), 70.5 and 66.7 (2*C-4), 42.0 and 41.9 (2*C-1), 34.8 (2*CH2), 32.9 (2*CH2), 27.1 (2*CH2), 23.4 (2*CH2), 26.1 and 26.0 (2*(CH3)3CSi), 18.3 and 18.2 (2*(CH3)3CSi), -4.5 and -4.7 (2*(CH3)2Si). |
| 43.1 g |
With 1H-imidazole In N,N-dimethyl-formamide at 20℃; |
I56 ethyl 4-[tert-butyl(dimethyl)silyl]oxy}cyclohexanecarboxylate(mixture of cis-/trans isomers)
tert-Butyldimethylsilyl chloride (26.3 g, hydroxycyclohexanecarboxylate (25.0 g, 171 59-80-7) and imidazole (24.7 g, 363174 mmol) was added to a solution of ethyl 4-145 mmol, mixture of cis-/trans-isomers, Gas Nommol) in N,N-dimethylformamide (36 ml) and the mixture was stirred over night at room temperature. For work-up, water was added and the mixture was extracted with tert-butyl methyl ether (3x). The combined organic phases were washed with brine, filtrated through a silicone filter and concentrated under reduced pressure to yield ethyl 4-[tert-butyl(dimethyl)silyl]oxy}cyclohexanecarboxylate (43.1 g, 104% yield) which was used in the next step without further purification. 1HNMR (400 MHz, DMSO-d6, mixture of isomers): 6 [ppm] = 4.10-3.99 (m, 2H), 3.93-3.86 (m, 0.7H), 3.63-3.51 (m, 0.3H), 2.39-2.28 (m, 0.8H), 2.27-2.14 (m, 0.3H), 1.91-1.21 (m, 8H), 1.20-1.13 (m, 3H), 0.89-0.79 (m, 9H), 0.08-0.00 (m, 6H). |
|
With 1H-imidazole In N,N-dimethyl-formamide at 20℃; |
3 ethyl 4-[tert-butyl(dimethyl)silyl]oxy}cyclohexanecarboxylate (mixture of cis-/trans-isomers)
tert-Butyldimethylsilyl chloride (26.3 g, 174 mmol) was added to a solution of ethyl 4-hydroxycyclohexanecarboxylate (25.0 g, 145 mmol, mixture of cis-/trans-isomers, Cas No 17159-80-7) and imidazole (24.7 g, 363 mmol) in N,N-dimethylformamide (36 ml) and the mixture was stirred over night at room temperature. For work-up, water was added and the mixture was extracted with tert-butyl methyl ether (3x). The combined organic phases were washed with brine, filtrated through a silicone filter and concentrated under reduced pressure to yield ethyl 4-[ferf-butyl(dimethyl)silyl]oxy}cyclohexanecarboxylate (43.1 g, 104% yield) which was used in the next step without further purification.1H-NMR (400 MHz, DMSO-cfe, mixture of isomers): δ [ppm] = 4.10-3.99 (m, 2H), 3.93-3.86 (m, 0.7H), 3.63-3.51 (m, 0.3H), 2.39-2.28 (m, 0.8H), 2.27-2.14 (m, 0.3H), 1.91-1 .21 (m, 8H), 1.20-1 .13 (m, 3H), 0.89-0.79 (m, 9H), 0.08-0.00 (m, 6H). |
|
With 1H-imidazole In N,N-dimethyl-formamide at 20℃; |
Intermediate 1 ethyl 4-[tert-butyl(dimethyl)silyl]oxy}cyclohexanecarboxylate (mixture of cis-/trans-isomers)
tert-Butyldimethylsilyl chloride (26.3 g, 174 mmol) was added to a solution of ethyl 4- hydroxycyclohexanecarboxylate (25.0 g, 145 mmol, mixture of cis-/trans-isomers, Gas No 17159-80-7) and imidazole (24.7 g, 363 mmol) in DMF (36 ml) and the mixture was stirred overnight at room temperature. For work-up, water was added and the mixture was extracted with tert-butyl methyl ether (3x). The combined organic phases were washed with brine, filtered through a silicone filter and concentrated under reduced pressure to yield ethyl 4-[tert- butyl(dimethyl)silyl]oxy}cyclohexanecarboxylate (43.1 g, 104% yield) which was used in the next step without further purification.1HNMR (400 MHz, DMSO-d6, mixture of isomers): 6 [ppm] = 4.10-3.99 (m, 2H), 3.93-3.86 (m,0.7H), 3.63-3.51 (m, 0.3H), 2.39-2.28 (m, 0.8H), 2.27-2.14 (m, 0.3H), 1.91-1.21 (m, 8H), 1.20-1.13 (m, 3H), 0.89-0.79 (m, 9H), 0.08-0.00 (m, 6H). |
|
With 1H-imidazole In N,N-dimethyl-formamide at 20 - 40℃; |
50
Chloro(1 ,1-dimethylethyl)dimethylsilane (115g; 0.76mol) was added in portions over 1 hour to a solution of ethyl 4-hydroxycyclohexanecarboxylate (118g; 0.68mol), imidazole(103g; 1.52mol) and dimethylformamide (400ml) stirred under an atmosphere of argon. A small exotherm was observed resulting in the reaction mixture temperature increasing to~40°C. The mixture was stirred at room temperature overnight then poured into 10% citric acid solution (2L) and extracted with diethyl ether (2 x 800ml). The ether extracts were washed with water, brine and then dried (Na2SO4) and the solvent was removed to give the title compound as a oil (198.4g) 1H NMR δ (CDCI3, 400 MHz): 0.01 (6H, m), 0.85 (9H, s), 1.2 (3H, m), 1.3-1.5 (2H, m), 1.6 (2H, m), 1.85-2 (3H, m), 2.15-2.3 (1 H, m) 3.5 (0.4H, m) 3.86 (1 H, m) 4.1 (1 H, m). |
|
With 1H-imidazole In N,N-dimethyl-formamide at 20 - 40℃; |
18
Description 18.; cis/trans-Ethy 4-[(1 ,1- dimethylethyl)(dimethyl)silyl]oxy}cyclohexanecarboxylate (D18); Chloro(1 ,1-dimethylethyl)dimethylsilane (1 15g) was added in portions over 1 hour to a solution of ethyl 4-hydroxycyclohexanecarboxylate (118g), imidazole (103g) and dimethylformamide (400ml) stirred under an atmosphere of argon. A small exotherm was observed resulting in the reaction mixture temperature increasing to -4O0C. The mixture was stirred at room temperature overnight then poured into 10% citric acid solution (2000ml) and extracted with diethyl ether (2 x 800ml). The ether extracts were washed with water, brine and then dried (Na2SO4) and the solvent was removed to give the title compound as a oil (198.4g)1H NMR δ(CDCI3, 400 MHz): 0.01 (6H, m), 0.85 (9H, s), 1.2 (3H, m), 1.3-1.5 (2H, m), 1.6 (2H, m), 1.85-2 (3H, m), 2.15-2.3 (1 H, m) 3.5 (0.4H, m) 3.86 (1 H, m) 4.1 (1 H, m). |
|
With 1H-imidazole In dichloromethane at 20℃; |
2
tert-Butyldimethylsilyl chloride (8.8 g, 58 mmol) was added to a solution of ethyl 4- hydroxycyclohexanecarboxylate (10 g, 58 mmol) and imidazole (4.3 g, 64 mmol) in DCM (300 ml), and the mixture was stirred overnight at 20 0C. After washing with water (2 X 100 ml), the organic phase was dried (MgSO4) and evaporated to dryness. The residue was dissolved in dry THF (300 ml) and cooled to -10 0C. Dibal-H (1.0 M in toluene, 174 ml, 174 mmol) was added dropwise over a period of 60 min, while the temperature was kept at - 100C. After stirring for 2 hrs the reaction was quenched by slow addition of a saturated ammonium chloride solution (30 ml). The resulting suspension was filtered, and the filtrate was concentrated in vacuo to give 13 g of the title compound as a mixture of two isomers. LC-MS {m/z) : 245 (M + l). |
|
With 1H-imidazole In N,N-dimethyl-formamide at 20 - 40℃; |
60
Description 60. c/s/frans-Ethyl 4-[(1,1 - dimethylethyl)(dimethyl)silyl]oxy}cyclohexanecarboxylate (D60); Chloro(1 ,1-dimethylethyl)dimethylsilane ( 115g ) was added in portions over 1 hour to a solution of ethyl 4-hydroxycyclohexanecarboxylate (118g), imidazole (103g) and dimethylformamide (400ml) stirred under an atmosphere of argon. A small exotherm was observed resulting in the reaction mixture temperature increasing to -4O0C. The mixture was stirred at room temperature overnight then poured into 10% citric acid solution (2000ml) and extracted with diethyl ether (2 x 800ml). The ether extracts were washed with water, brine and then dried (Na2SU4) and the solvent was removed to give the title compound as a oil (198.4g)1H NMR D(CDCI3, 400 MHz): 0.01 (6H, m), 0.85 (9H, s), 1.2 (3H, m), 1.3-1.5 (2H, m), 1.6 (2H, m), 1.85-2 (3H, m), 2.15-2.3 (1 H, m) 3.5 (0.4H, m) 3.86 (1 H, m) 4.1 (1 H1 m). |
|
With 1H-imidazole In N,N-dimethyl-formamide at 20 - 40℃; |
27; 38
Description 27. Ethyl 4-[(1 ,1- dimethylethyl)(dimethyl)silyl]oxy}cyclohexanecarboxylate (D27); Chloro(1 ,1-dimethylethyl)dimethylsilane ( 115g ) was added in portions over 1 hour to a solution of ethyl 4-hydroxycyclohexanecarboxylate (118g), imidazole (103g) and dimethylformamide (400ml) stirred under an atmosphere of argon. A small exotherm was observed resulting in the reaction mixture temperature increasing to -4O0C. The mixture was stirred at room temperature overnight then poured into 10% citric acid solution(2000ml) and extracted with diethyl ether (2 x 800ml). The ether extracts were washed with water, brine and then dried (Na2SO4) and the solvent was removed to give the title compound as a oil (198.4g)1H NMR D(CDCI3, 400 MHz): 0.01 (6H, m), 0.85 (9H, s), 1.2 (3H, m), 1.3-1.5 (2H, m), 1.6 (2H, m), 1.85-2 (3H, m), 2.15-2.3 (1 H, m) 3.5 (0.4H, m) 3.86 (1 H, m) 4.1 (1 H, m). Description 38. c/s/frans-Ethyl 4-[(1,1 - dimethylethyl)(dimethyl)silyl]oxy}cyclohexanecarboxylate (D38)Chloro(1 ,1-dimethylethyl)dimethylsilane ( 115g ) was added in portions over 1 hour to a solution of ethyl 4-hydroxycyclohexanecarboxylate (118g), imidazole (103g) and dimethylformamide (400ml) stirred under an atmosphere of argon. A small exotherm was observed resulting in the reaction mixture temperature increasing to ~40°C. The mixture was stirred at room temperature overnight then poured into 10% citric acid solution(2000ml) and extracted with diethyl ether (2 x 800ml). The ether extracts were washed with water, brine and then dried (Na2SO4) and the solvent was removed to give the title compound as a oil (198.4g).1H NMR δ (CDCI3, 400 MHz): 0.01 (6H, m), 0.85 (9H, s), 1.2 (3H, m), 1.3-1.5 (2H, m), 1.6 (2H, m), 1.85-2 (3H, m), 2.15-2.3 (1 H, m) 3.5 (0.4H, m) 3.86 (1 H, m) 4.1 (1 H, m). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping