| 92.7% |
|
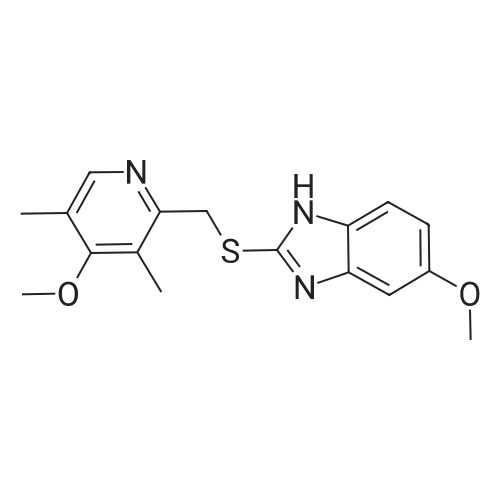
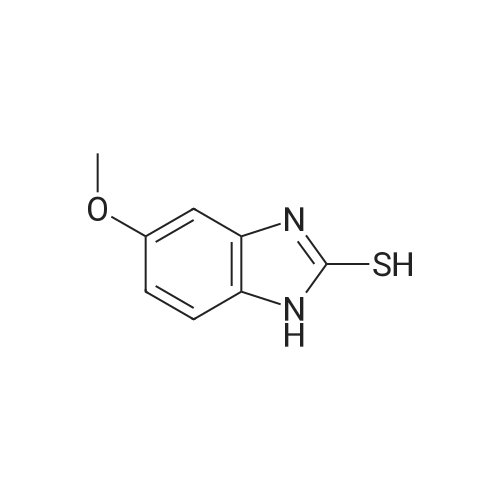
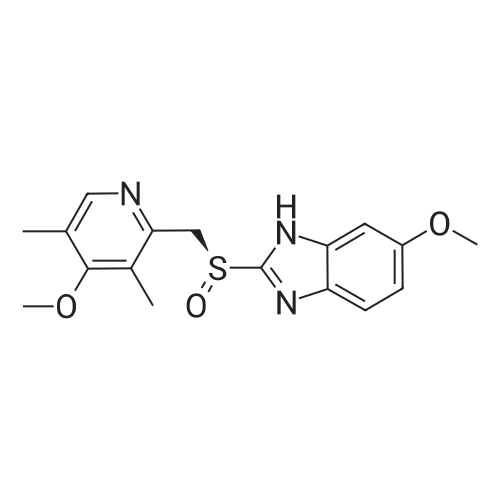
5-Methoxy-2-[[(4-methoxy-3,5-dimethyl-2-pyridyl)methyl]thio]-lH- benzimidazole (30 g, 90.19 mmol), toluene (242 mL) and D-tartaric acid di-w-propionamide (12.96 g, 54.71 mmol) were added into a reaction flask. The reaction mixture was warmed to 70 <n="40"/>0C and stirred for 10 minutes and then cooled to 55-60 0C. To the resulting solution was added titanium (IV) tetraisopropoxide (7.78 g, 27.36 mmol). The resulting mixture was stirred for 60 minutes and then water (0.16 mL, 9.019 mmol) was added thereinto. The resulting solution was further stirred for 30 minutes and cooled 30 0C. To the solution was added triethylamine (3.8 mL, 27.36 mmol). The resulting solution was stirred for 30 minutes and then cumyl hydroperoxide (19.4 mL, 107.8 mmol) was added thereinto. The reaction was kept at 30 0C over 43 hours. To the resulting solution was added 10% aqueous sodium hydroxide. The aqueous phase was separated and extracted twice with toluene. The pH of the aqueous layer was adjusted to about 9 with glacial acetic acid. The resulting aqueous phase was extracted with dichlormethane. The resulting organic phase was washed twice with saturated aqueous brine, dried over anhydrous sodium sulfate and filtered. The combined organic phase was evaporated to dryness under reduced pressure to give S-(-)-levo-omeprazole (29.2 g, 92.7%, ee: 95.2%). |
| 92% |
|
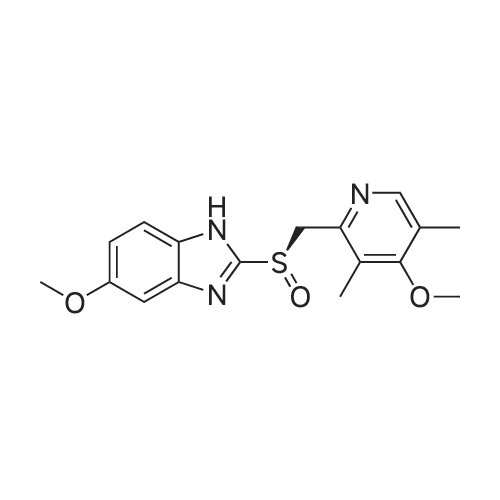
General procedure: In a typical experiment, Ti(Oi-Pr)4 (0.9 mL, 3 mmol) was added to a solution of (S,S)-DBzTA 5i (1.969 g, 6 mmol) and pyrmetazol (3.29 g, 10 mmol) in toluene (20 mL) at 80 C. The solution was stirred for 60 min., after which water (54 mg, 3 mmol) was added to the mixture, and the solution was stirred for another 60 min. Next, the temperature was adjusted to 30 C, after which cumene hydroperoxide (80 %, 3.6 mL, 20 mmol) was slowly added. After 1 h at 30 C, the solution was added to aqueous sodium hydroxide (1.2 g NaOH in 20 mL water), stirred for 1 h, and extracted with water (20 mL × 3). The aqueous phase was adjusted to pH 8 with acetic acid, separated, and extracted with dichloromethane (20 mL × 3). The combined organic solutions were dried over anhydrous Na2SO4, filtered, and the solvent was removed in vacuo. The residue was purified by chromatography on silica gel with ethyl acetate/petroleum ether (2:1) as the eluent to afford the sulfoxide. |
| 92% |
With AD-mix beta; dichloro[(-)-sparteine-N,N']copper(II); Cumene hydroperoxide; C14H17F6N3S; In ethyl acetate; at 20℃; for 1.5h;Catalytic behavior; |
General procedure: (1), in cleanliness,Add 5 mol of toluene to the dried glass reaction flask.Turn on the stirring and add the intermediate product Ome Thiol.0.5 mol chiral thiourea catalyst (R1 = R2 = CF3, R3 = ethyl),Add 0.5 mol of chiral hawkskin-copper complex,Add 0.4mol of CAA6,0.9 mol of cumene hydroperoxide was added dropwise at room temperature.Stir for 1.5 h. (2), after treatment: ammonia extraction twice,The aqueous layer was collected and 25 mol of dichloromethane was added.The glacial acetic acid is adjusted to pH=8-9, and the organic layer is collected.Dry over anhydrous sodium sulfate,Filtration and concentration of the filtrate under reduced pressure until solventless distillation.Add 20 mol of ethyl acetate,Stir to a homogeneous phase,Then cool down to 0 ~ 10 C, slowly add 10mol of drinking water,There is a lot of solid precipitation,filter,An esomeprazole solid is obtained. |
| 91.5% |
With titanium(IV) isopropylate; tert.-butylhydroperoxide; (R,R)-hydroxybenzoin; isopropyl alcohol; In dichloromethane; water; at 5 - 20℃; |
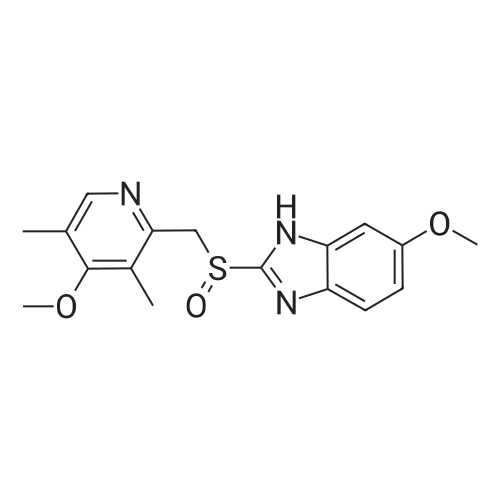
(R, R) -1,2-diphenyl-1,2-ethanediol, 2.14 g (10mmol) was added to 100 ml of dichloromethane, 1.42 g (5 mmol) of titanium tetraisopropoxide and 1.2 g of isopropyl alcohol (20 mmOl) were added dropwise, and 33.0 g of omeprazole sulfur Ether (0.1 m omicron 1), dissolved at room temperature after stirring, cooled to 5 -10 C, dropping 7 0% t-butyl hydroperoxide aqueous solution (0.2 mol), the mixture was stirred at 5-10 C for 2.0-2.5 hours and 10 ml of aqueous ammonia (10%) was added to terminate the reaction. 10% acetic acid aqueous solution to adjust rhoH- = 8-9, stratified, the water phase extracted with dichloromethane 3 times, combined with organic phase, Na2S04 dry Filtered and concentrated to give 31.6 g of a white solid, 91.5% yield; 98.5% purity and 0.56% impurity sulfone. value of 99.65%. |
| 90% |
With tert.-butylhydroperoxide; In water; toluene; at -20℃; for 12h; |
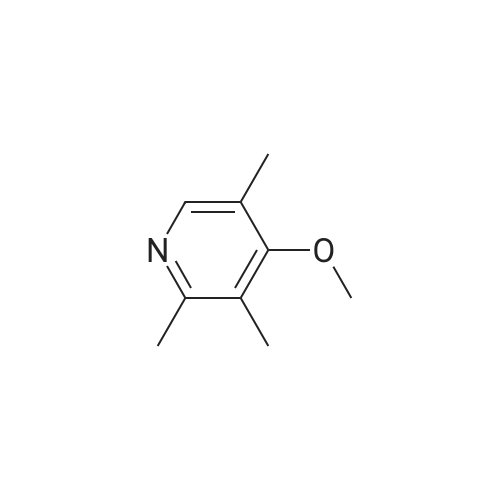
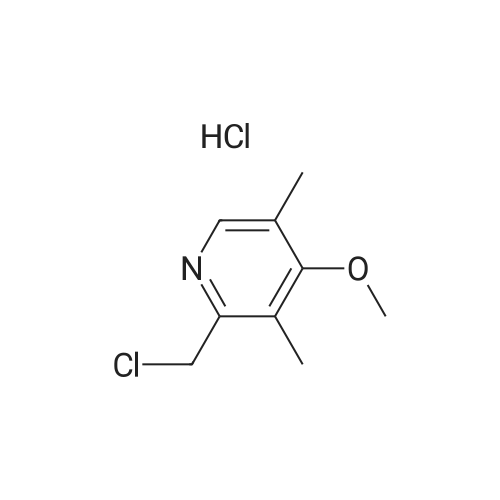
Titanium tetraisopropoxide (4.5 mg, 0.016 mmol) was added to a solution of (R,R)-1,2-bis-(2-bromophenyl)ethane-1,2-diol (12 mg, 0.032 mmol) in toluene (2 ml) at 25 C. The solution was stirred for 10 minutes, water (5.7 mg, 0.32 mmol) was added, and the solution was then stirred for another 10 minutes. 5-Methoxy-2-[[(4-methoxy-3,5-dimethyl-2-pyridinyl)-methyl]thio]-1H-benzimidazole (105 mg, 0.32 mol) was subsequently added to the solution, and the temperature was adjusted to -20 C. Thereafter, t-butyl hydroperoxide (70%, 96 mul, 0.064 mmol) was slowly added. After 12 hours at -20 C., the solution was extracted three times with aqueous ammonium hydroxide (12.5% NH3, 3×5 ml). Thereafter, methyl isobutyl ketone (5 ml) was added to the combined aqueous extracts. Then, the pH of the aqueous phase was adjusted using acetic acid, the aqueous phase was separated and extracted with an additional amount of methyl isobutyl ketone (5 ml). The organic solution was cooled to -10 C. over night, and the neutral form of S-omeprazole was precipitated as a solid to obtain the title compound (99 mg, 90% yield). The enantiomeric excess of S-omeprazole was 94%. Purification using methyl isobutyl ketone yielded S-omeprazole, and the enantiomeric excess was >99%. |
| 87% |
With Aspergillus niger catalase; oxygen; NADP; In aq. phosphate buffer; isopropyl alcohol; at 25℃; for 48h;pH 9.0;Enzymatic reaction; |
Example 6 Process for Production of Esomeprazole at 30 g Scale Using an Engineered CHMO Polypeptide (0318) This example illustrates a process for preparing esomeprazole in enantiomeric excess at a 30 g scale via a biocatalytic conversion of the substrate pyrmetazole using an engineered CHMO polypeptide of the disclosure (e.g., a polypeptide of SEQ ID NO: 8, 10, 12, 14, 16, 18, 20, 22, 24, 26, 28, 30, 32, 34, 36, 38, 40, 42, 44, 46, 48, 50, 52, 54, 56, 58, 60, 62, 64, 66, 68, 70, 72, 74, 76, 78. 80. 82, 84, 86, 88, 90, 92, 94, 96, 98, 100, 102, 104, 106, 108, 110, 112, 114, 116, 118, 120, 122, 124, 126, 128, 130, 132, 134, 136, 138, 140, 142, 144, 146, 148, 150, 152, 154, 156, or 158). (0319) A. Biocatalytic Reaction Protocol: (0320) A 1 L multi-neck jacketed reactor vessel equipped with baffle and overhead stirrer, fitted with an anchor shaped agitator was charged with a pre-mixed powder of 30 g pyrmetazole (from Sinojie (HK) Ltd.) and 1.5 g of an esomeprazole ?seed? (prepared in a previous enzymatic reaction), and then 517 mL of 0.05 M potassium phosphate at pH 9.0 (?buffer solution?). This reaction mixture was stirred at 150 rpm at 25 C. for 10 min in order to obtain a well-suspended slurry. A three way tap was attached to the necks of the vessel. The tap was fitted with 2 rubber balloons filled with oxygen and a vacuum line for conducting degassing steps prior to the start of the reaction. Vacuum was applied to the vessel (30 mbar, obtained within 5 min) and the evacuated flask was filled with oxygen via the three way tap. This evacuation-gas-filling cycle was repeated two more times. The reaction vessel under positive oxygen pressure then was charged sequentially with the following: 24 mL IPA (HPLC grade); 60 mg NADP in 4 mL buffer solution (pH 9.0); 300 mg ketoreductase of SEQ ID NO: 268 in 15 mL buffer solution (pH 9.0); 600 mg CHMO of SEQ ID NO: 158 in 40 mL buffer solution (pH 9.0); and 1.2 mL of catalase (Aspergillus niger catalase solution in buffer stabilized with sodium chloride and sorbitol with stated activity of 25000 ClU/g; available from Sigma-Aldrich). Catalase is added to neutralize peroxide that may form during the reaction and negatively affect the performance of the CHMO. The above reaction mixture was stirred at 25 C. for 48 hours. The stir rate was 300 rpm in the beginning and increased stepwise as shown in Table 8 below. The course of the reaction was followed by taking periodic 0.3 mL samples from the reaction mixture which were quenched in 10 mL MeOH and analyzed using HPLC as described below. For the purpose of tracking the process, t=0 was set at the time at which the CHMO was added. Samples were also taken and tested for peroxide during course of reaction but no peroxide was detected. The in-process reaction profile based on the sample analyzes is summarized in Table 8 below. A % conversion of >99% within 36 hours can be estimated from the kinetic profile of the reaction. In comparable runs a conversion of 98% was determined after 32 h (with a rate of conversion of 1%/h constantly at the latter stage of the reaction) and similar kinetic profile could be obtained in repetitive runs with the described experimental set-up. The reaction mixture 48 hours after start was taken for product work-up and isolation as described below. [table-us-00009-en] TABLE 8 Reaction Profile Time Stirring speed (h) % Conversion (rpm) 0 5 300 2 17.6 350 4 30.7 350 6 43.3 350 7.5 50.1 400 22.5 87.8 450 26 92.3 450 28 94.3 450 30 96.1 450 44 99.6 450 (0322) B. Reaction Work-Up Protocol: (0323) To the reaction mixture was added 165 mL (5.5 volumes) of methyl isobutyl ketone (?MIBK?), the jacket temperature was adjusted to 48 C. and the mixture was stirred at 300 rpm. After 25 min the internal temperature showed 45 C. and the slurry was completely dissolved. Mixing was stopped and after 20 min the phases were separated. The lower aqueous layer was slightly turbid with a yellowish color. The upper organic layer appeared to be an emulsion and was brown in color. The aqueous layer was drained and collected. The organic layer was subsequently drained and submitted to a warm filtration over Celite applying vacuum (the temperature of the jacket filter was adjusted to 45 C.). The aqueous phase was transferred back to the heated vessel and 45 mL of MIBK was added. Stirring at 300 rpm for 30 min and phase separation within 20 min afforded a lower slightly turbid, yellowish aqueous layer and a brownish upper organic phase. The aqueous layer was drained and discarded. The organic layer was drained and collected. The solution was submitted to warm Celite filtration after the first filtration was completed (same filter and Celite layer). The organic phases were combined and separated from the aqueous layer that was formed during filtration. The aqueous layer was discarded and the organic phase was transferred back to the vessel. The temperature was adjusted to 15 C. and the solution stirred at 150 rpm for 1 hour. The p... |
|
|
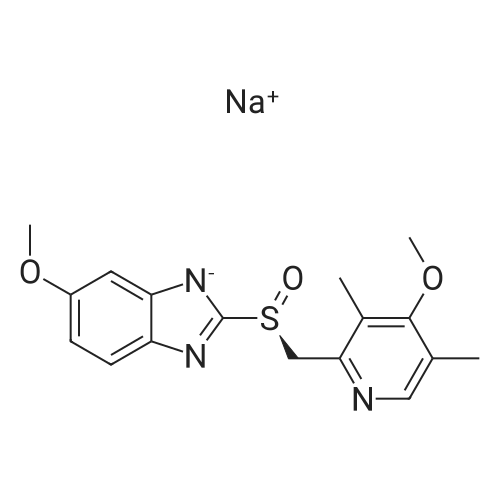
Mix, S- (+)-Methyl mandelate 60.6 g, Toluene 250 ml and Titanium isopropoxide 15 ml, into a 500 ml R. B. Flask assembly and stir to make a clear solution under Nitrogen atmosphere. Heat the reaction mixture to 40C and maintain for 17 hr. Cool to 25-30C and charge Omeprazole sulfide 50 g and Diisopropylethylamine 1.35 ml to it and stir for 10-15 minutes. Add Cumene hydroperoxide (-80 % solution in cumene) 28 ml slowly through addition funnel to the reaction mixture at 25-30C. Stir the reaction mixture at 25-30C for 2.0 hr. Filter the solid and wash with Toluene 50 ml. Collect the filtrate and charge 12.5 % ammonia solution 300 ml, to the filtrate and stir the mixture for 15 minutes. Back extract the Toluene layer with 12.5% Ammonia solution; 200 ml and add Methyl isobutyl ketone 250 ml, to the combined aqueous layer. Adjust the pH of the solution to 7.3-7. 6 by adding Glacial Acetic acid at 25-30 C. Stir the content for 30 min. Separate the organic layer and extract the aq. layer with Methyl isobutyl ketone ; 50 ml. Charge sodium hydroxide solution (50% w/v) 11 ml to combined organic layer and stir the mixture for 15 minutes. Distil off solvent completely under vacuum at 55-60 C. Add Acetonitrile; 300 ml, to the residue and stir the content at 25-30C. Filter the product under nitrogen atmosphere and wash the cake with Acetonitrile 50 ml. Dry the product in vacuum at 50-55C. Yield of Esomeprazole sodium (22.4 g) with enantiomeric excess > 98%. |
|
|
Mix, Toluene 120 L, Omeprazole sulfide 30 Kg, followed by 204 ml Water, D- (-)- Diethyl tartrate 9.4 L and Titanium isopropoxide 7.98 Kg, into a 500 L reaction assembly and stir to make a homogenous suspension under Nitrogen atmosphere. Heat the reaction mixture to 50-52C and maintain for 1 hour. Cool to 15-20C and charge Diisopropylethylamine 3.56 Kg to it and stir for 10-15 minutes. Add Cumene hydroperoxide (-80 % solution in cumene) 16.37 Kg slowly through addition funnel to the reaction mixture at 0 to 5C. Stir the reaction mixture at 25-30C for 2.0 hr. Charge 12.5 % ammonia solution 240 L, to it and stir for 10-15 minutes. Back extract the Toluene layer with 12.5% Ammonia solution; 60 L and add Methyl isobutyl ketone 120 L, to the combined aqueous layer. Adjust the pH of the solution to 7.3-7. 6 by adding Glacial Acetic acid at 25-30 C. Stir the content for 30 min. Separate the organic layer and extract the aq. layer with Methyl isobutyl ketone ; 30 L. Charge sodium methoxide solution (30% w/v) about 15 Kg to combined organic layer and stir the mixture for 15 minutes. Distil off solvent completely under vacuum at 55-60 C. Add Acetonitrile; 90 L to the residue and stir the content at 25-30C. Filter the product under nitrogen atmosphere and wash the cake with Acetonitrile 60 L. Dry the product in vacuum at 50-55C. Yield of Esomeprazole sodium = about 18 Kg. Sulfone content: 4.0 % |
|
|
EXAMPLE 1; Preparation of (S)-5/6-methoxy-1-benzyloxycarbonyl-2-[[(4-methoxy-3,5-dimethyl-2-pyridinyl)methyl]sulfinyl]-1H-benzimidazoleTo a solution of 2-[2-(3,5-dimethyl-4-methoxypyridyl)methylthio]-5-methoxy-benzimidazole 2 (10 g) in 50.0 mL toluene under an inert atmosphere, was added (D)-diethyl tartrate (2.75 g). The mixture was heated to 50-55 C. and stirred for 30 minutes. Titanium (IV) isopropoxide (1.73 g) was added and the temperature was maintained at 50-55 C. for an additional 60 minutes. The reaction mixture was cooled to 0-5 C. whereupon diisopropylethylamine (1.33 g) and 80% cumene hydroperoxide (6.93 g) were added while keeping the temperature below 10 C. The reaction mixture was stirred at 0-10 C. for 2-4 hours until the reaction was complete. The reaction mixture was warmed to room temperature, filtered through Celite and extracted with 12-14% ammonium hydroxide. The aqueous and methyl isobutyl ketone (MIBK, 30 mL) phases were cooled to 0-5 C. The pH was adjusted to 7.3 to 7.8 with acetic acid and phases were separated. The aqueous phase was extracted with MIBK. The combined organic phases were washed with brine and vacuum distilled to 40 mL to give a solution of (S)-(-)-5-methoxy-2-[[4-methoxy-3,5-dimethyl-2-pyridinyl)methyl] sulfinyl]-1H-benzimidazole in MIBK. The sulfoxide solution was diluted with dichloromethane (30 mL) and triethylamine (4.61 g). The mixture was cooled to 0-10 C. and 95% benzyl chloroformate (6.0 g) was added while keeping the temperature below 10 C. After stirring for 1-4 hours, water (30 mL) and ethyl acetate (30 mL) were added. The phases were separated and the aqueous phase was extracted with ethyl acetate. The combined organic phases was washed with brine and saturated sodium bicarbonate, vacuum distilled to 30 mL and filtered through Celite. The filtrate was stirred while 80 mL of heptanes was added dropwise whereupon the suspension was cooled to 0-5 C. and maintained at this temperature for 1-2 hours. The suspension was filtered, washed with heptanes/ethyl acetate (4/1) and dried under vacuum at room temperature to afford (S)-5/6-methoxy-3-benzyloxycarbonyl-2-[[4-methoxy-3,5-dimethyl-2-pyridinyl)-methyl]sulfinyl]-1H-benzimidazole. Weight: 11.5 g. Purity: 99% by HPLC. Chiral purity: 99.5% (S-form) by HPLC. Ratio of 5- and 6-methoxy products: 1:1. The analytical data were consistent with the assigned structure.H-NMR (400 MHz, CDCl3):5-Methoxy isomer: delta/ppm=2.18 (3H, s), 2.32 (3H, s), 3.73 (3H, s), 3.76 (3H, s), 4.67 (2H, dd, J=13, 38 Hz), 5.54 (2H, s), 6.95-7.01 (1H, m), 7.38 (1H, d, J=2 Hz), 7.40-7.43 (2H, m), 7.47-7.59 (2H, m), 7.68 (1H, d, J=9 Hz), 8.05 (1H, s);6-Methoxy isomer: 6/ppm=2.18 (3H, s), 2.32 (3H, s), 3.73 (3H, s), 3.83 (3H, s), 4.67 (2H, dd, J=13, 38 Hz), 5.53 (2H, s), 6.95-7.01 (1H, m), 7.29 (1H, d, J=2 Hz), 7.40-7.43 (2H, m), 7.47-7.59 (2H, m), 7.75 (1H, d, J=9 Hz), 8.05 (1H, s). |
|
|
In a 500 ml flask, 18.8 gm diethyl (-)-D-tartrate, 12.9 gm titanium(IV) isopropoxide and 0.33 ml water were taken at room temperature, and 50 gm 5-methoxy[(2-(4-methoxy)- 3,5-dimethyl-2-pyridinyl] methylsulfenyl]-lH-benzimidazole (also termed as pyrmetazole or PMT) was added and the mixture was heated to 70-75 degrees for 1 hour. After cooling to 40 degrees, 3 gm diisopropyl ethyl amine and 34.6 gm (80%) cumene <n="24"/>hydroxide were added and maintained mixture under stirring. The reaction was monitored, after completion of reaction 100 ml toluene and 31.6 gm NaOH solution were added to the mixture. The mixture then stirred for 0.5 hours at heating and then cooled to room temperature and further to 0-5 degree Celsius. Layers were separated and toluene layer washed with water. The aqueous layer neutralized with cone. Hydrochloric acid and esomeprazole free base was extracted using dichloromethane. The dichloromethane layers, dried, and evaporated to obtain esomeprazole free base. |
| 5.6 g |
|
The 5 - methoxy -2 - (4 - methoxy - 3, 5 - dimethyl -2 - pyridyl) methyl thio - 1H - benzimidazole 5.67g added to the 100 ml three-opening in the bottle, the toluene is added to 25 ml, stirring, heated to 65 C, then adding purified water 90uL, D - tartaric acid diethyl ester 2.3g and tetraisopropyl titanate 28g, stirring 1h, cooling. T=30 C when, adding N, N - diisopropyl ethylamine 1.6g, after 15min, for 30min slowly dropping hydrogen peroxide in 2.5 ml. TLC detection after the reaction, a small amount of raw material remaining. For 12% of ammonia water extraction 3 times, the combined aqueous phase, by adding dichloromethane, then adjusting the pH to acetic acid for 8 the left and the right, liquid, organic phase with pure water and saturated salt water all 20 ml wash once, dried with anhydrous sodium sulfate, filtered, concentrated to obtain brown oil of 5.6g. |
| 36.7 g |
|
Omeprazole thioether 50 g (0.152 mol)Toluene 220g mixed with stirring,The solution layer was a white suspension.And stirred at 20 to 25 CDiethyl D-tartrate (63 g, 0.304 mol)And 43 g (0.152 mol) of (IV) isopropyl ester,The reaction solution was yellow turbid liquid.after that0.68 g (0.038 mol) of purified water was added,20 ~ 25 and then stirring 10min.After heating to 45 ~ 55 ,Insulation reaction 1 hour,The color gradually darkens to brown.Cooling to 25 ~ 30 ,20 g (0.155 mol) of N, N-diisopropylethylamine was added,Stir for 0.5 hours.Continue to cool,And stirred at -2 to 4 C for 1 hour.Then, 25 g (0.135 mol) of cumene hydroperoxide (85%) was added with stirring,The stirring was continued at -2 to 4 C for 7 hours,Simultaneously, the reaction solution was detected by HPLC,Sulfone content of 3.8%,Omeprazole thioether content of 11.2%Stop the reaction. To the above reaction solution was added 100 g of triethylamine,Stirring 10min after adding purified water 150g,Stir quickly for 30 minutes,Static stratification,The organic phase was extracted twice with the same amount of triethylamine and water as described above.Combine the aqueous phase and filter.12 g of glacial acetic acid was added dropwise at -3 to 2 C,Adjust the material liquid pH = 8 ~ 9,And maintain the pH value of half hour re-measured the same,The same temperature to continue stirring 6h,Precipitate a large amount of white solid.Filtered and washed three times with 100 g x 3 purified water,Filter dry.Dried to give esomeprazole 36.7 g. |
|
|
The resulting organic phase was heated to 55 C with stirring,Adding 6.2 grams of diethyl D-tartrate, 4.3 g of titanium isopropoxide, Keep heating for 1 hour, remove the heating, down to room temperature, plus N,N-diisopropyl Diamine 1.9 g,11.4 g of cumene hydroperoxide was slowly added dropwise, and the reaction was stirred for 4 hours(Sampling detection,EsomeprazoleThe thioether content is in1.0% or less),To the reaction solution was added 15% aqueous ammonia 100 ml twice in two mixing, combined ammonia water, ammonia phase with 100 ml of methylene chloride backwash once. |
| 148.2 g |
|
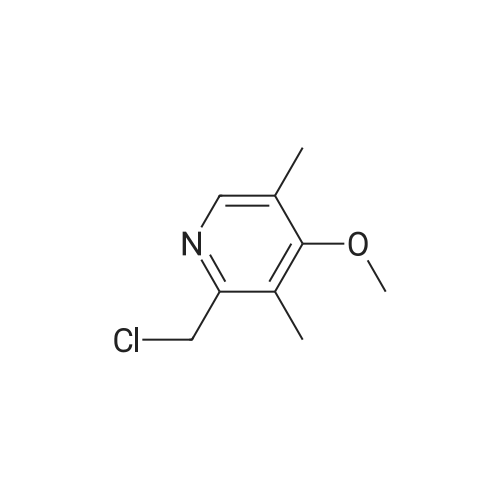
In a 3000 mL three-necked round bottom flask,Add 270 ml of methanol,2-Mercapto-5-methoxybenzimidazole 90. 0 g (0.50 mol),Stirring,Add an aqueous sodium hydroxide solution (40.0 g sodium hydroxide/100 ml purified water).Stir the reaction at room temperature for 1 hour.2-chloromethyl-3,5-dimethyl-4-methoxypyridine hydrochloride 110 g (0.50 mol) was added,Methyl tert-butyl ether (900 ml) was heated to 35-45 C for 3 hours.After the reaction,Cool the feed to room temperature,Purified water 250 ml*2 was added twice to the remaining stock solution.Obtained methyl sulfide tert-butyl ether solution,The water bath is heated to 50-60C,Addition of 62.5 g (0.30 mol) of D-(-)-diethyl tartrate,Tetraisopropyl titanate 43.0 g (0.15 mol),Insulation for 1 hourAfter cooling to 25-35C,Triethylamine 15.0 g (0.15 mol) was added,72.0 g (0.475 mol) of cumene hydroperoxide was added dropwise.5h, heat preservation reactionAfter the reaction,Add 15% ammonia water 400ml to the reaction solution.Stir for 20min,Extraction liquid, The organic layer was extracted twice with 15% aqueous ammonia 400ml*2.Combined ammonia layer,Add activated carbon 12 g to decolorize at room temperature for 4 hours.filter,Ammonia layer is added with ethyl acetate 900 ml,Ice bath cooling to 5-15 C,The glacial acetic acid was added dropwise to adjust the pH to 7. 5-8. 5,Dividing fluid,The aqueous layer was extracted with ethyl acetate 450ml for the second extraction.Combine the ethyl acetate layers,Wash 400 ml*2 twice with saturated saline solution.Ethyl acetate extraction phase,Temperature control 35 ~ 45 C vacuum concentration,Light brown oil 148.2 g,Add acetone 900 ml,Stir so that the oil is completely dissolved,Transfer the feed solution to a 2000 ml three-neck round bottom flask.Sodium hydroxide solution (18.1 g of sodium hydroxide/1. 8 g of purified water) was added,Heating reflux 60min,The crystal was dropped to 0-10C and stirred for 6h.Suction filtrationAcetone 50ml wash cake,Filter cake 35-45 C / -0. 095MPa vacuum drying 12h,120.7 g of esomeprazole sodium was obtained.Yield 65.7%.HPLC: 99.6%,Ee%: 99.7%. |
| 163 g |
|
2) adding 38.5 g of titanium tetraisopropoxide to the dichloromethane phase,6-g D-tartrate, heated to refluxAfter reacting for 1 h, the temperature was lowered to 20 C and 15.2 g of triethylamine was added.Temperature control -20 ~ -15 C,Adding 95g of 80wt% cumene hydroperoxide anda solution of 95 ml of dichloromethane,2h drop,Reaction 2h. Wash the dichloromethane phase by adding 300 ml of water.Set the liquid to obtain the methylene chloride phase,The methylene chloride phase was concentrated under reduced pressure at a temperature of 20 to 25 C until no droplets were distilled off.An oil of 163 g was obtained. |
|
With titanium(IV) isopropylate; diethyl (2S,3S)-tartrate; In water; toluene; at 50℃; for 1.33333h;Inert atmosphere; |
(1) 21.62 kg of the intermediate I obtained in Example 1 (containing 64.54 mol of omeprazole thioether) was added to 87.7 kg.Toluene (the amount of toluene is 4 times the mass of omeprazole thioether), the temperature is raised to 50 C, stirred and dissolved, and then transferred to a 500 L reactor.Turn on nitrogen protection.(2) The temperature was raised to 50±3C, and 169g of purified water and 5.47kg of D-tartrate dibasic acid were added, and the reaction was kept for 20 minutes.Slowly add dropwise a solution of isopropyl titanate toluene (5.79 kg of isopropyl titanate dissolved in 7.4 kg of toluene) to maintain the temperature of the reaction solution.The reaction was stirred at 50 ± 3 C for 50 min to 60 min. Among them, water added, D-diethyl tartrate, isopropyl titanate and omeprazole sulfurThe molar ratio of ether was 0.14:0.4:0.3:1.(3) Cool down to 15 to 20 C and add 2.66 kg of triethylamine. Continue to cool down, slowly add peroxide at 10~15 CHydrogen cumene toluene solution (11.28 kg of cumene hydroperoxide dissolved in 29.3 kg of toluene), after the addition is completed, the temperature is raised to 25 ~The reaction was carried out at 30 C to obtain esmeprazole. The molar ratio of triethylamine to omeprazole thioether added was 0.3:1.After 2 hours, the sample was monitored by TLC method (extension agent ethyl acetate), and the concentration of intermediate I in the reaction solution was low.In the control solution, the concentration of the intermediate I was spotted, and the chiral oxidation reaction was terminated to obtain Esomeprazole, which was used to enter the potassium salt.should. If the spot concentration of the intermediate I is higher than the concentration of the intermediate I spot in the control solution, the reaction is continued. Sampling every 30 minutes,Until the concentration of the control solution is lower than the spot concentration. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping