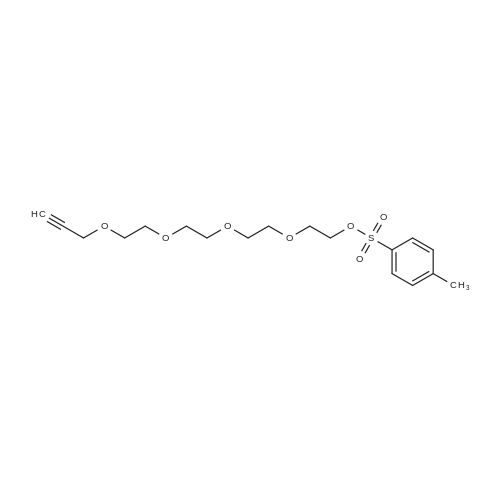
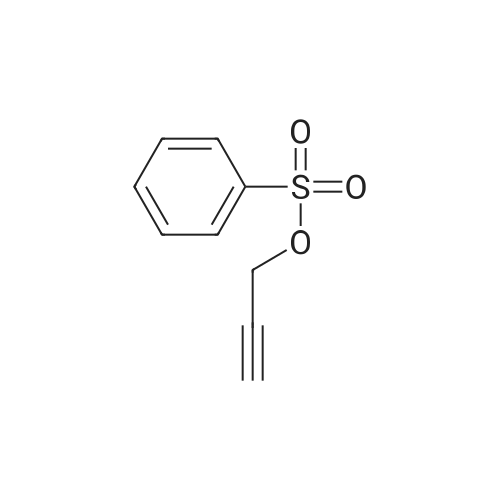
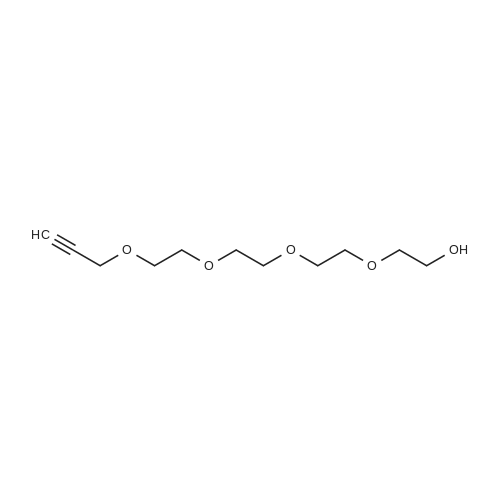
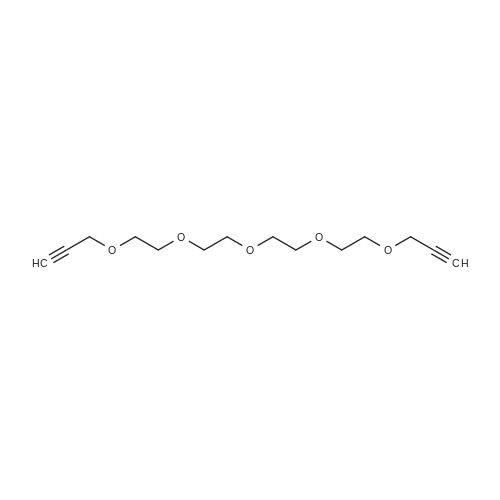
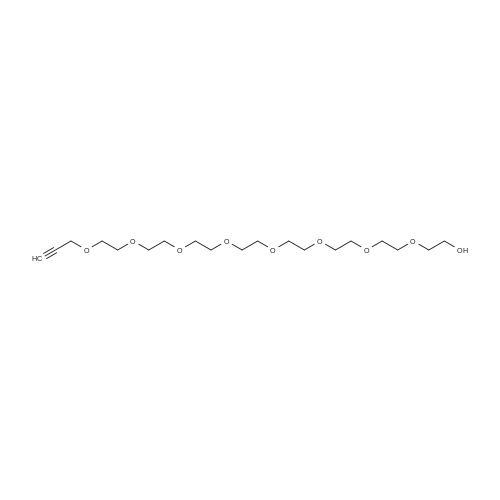
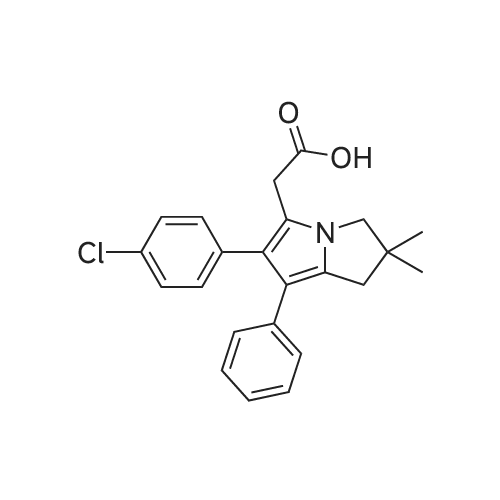
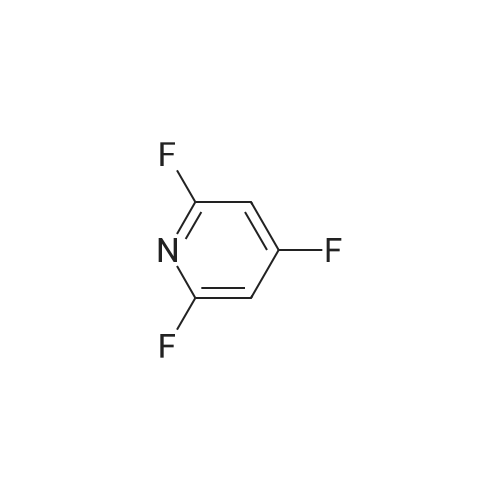
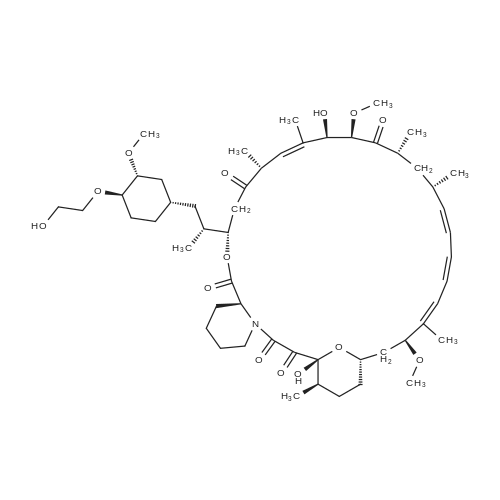
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Reference:
[1] Phosphorus, Sulfur and Silicon and the Related Elements, 2006, vol. 181, # 1, p. 219 - 225
[2] Russian Journal of Organic Chemistry, 2012, vol. 48, # 10, p. 1345 - 1352[3] Zh. Org. Khim., 2012, vol. 48, # 10, p. 1350 - 1357,8
[4] Polyhedron, 2018, vol. 141, p. 385 - 392
2
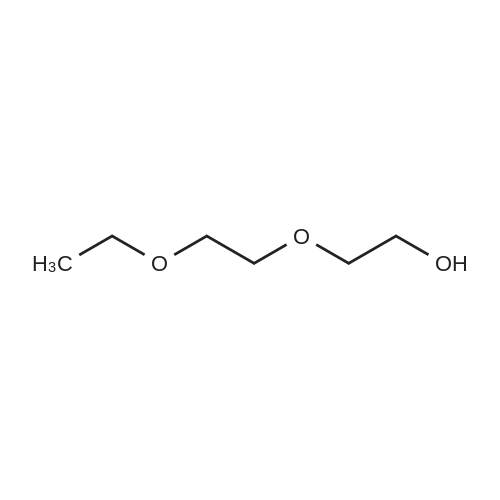
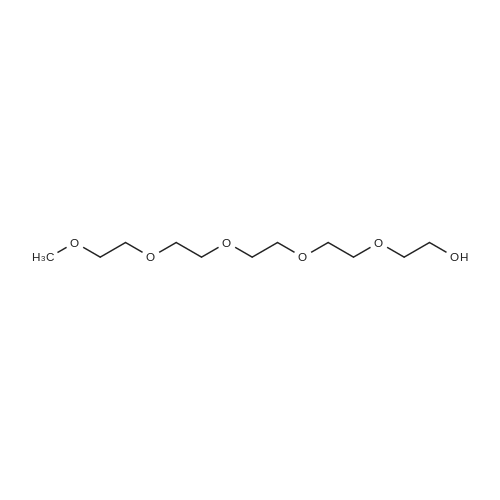
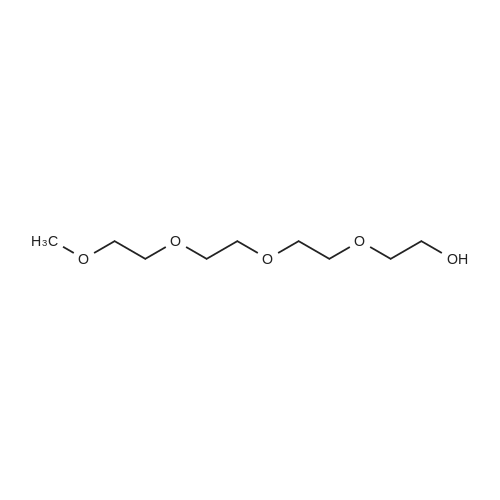
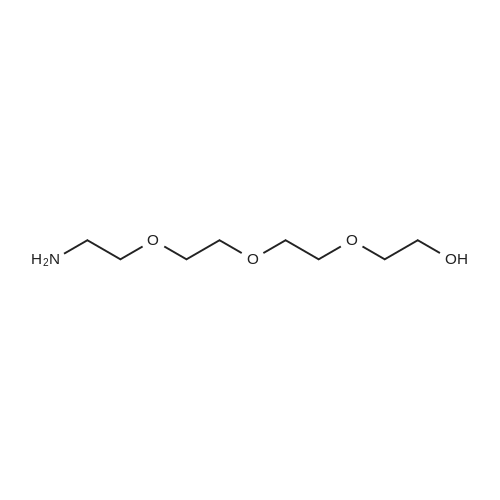
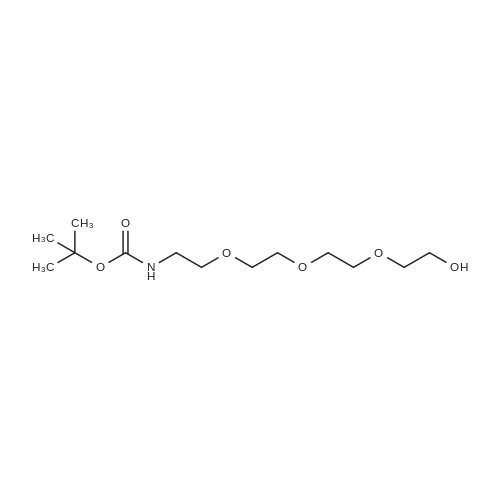
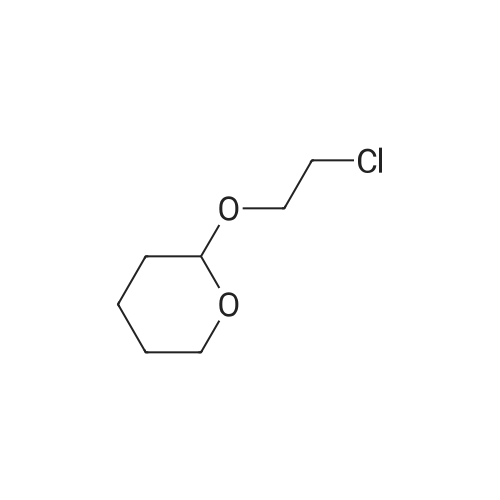
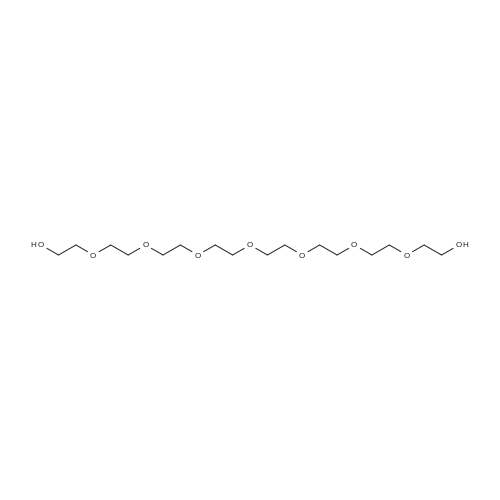
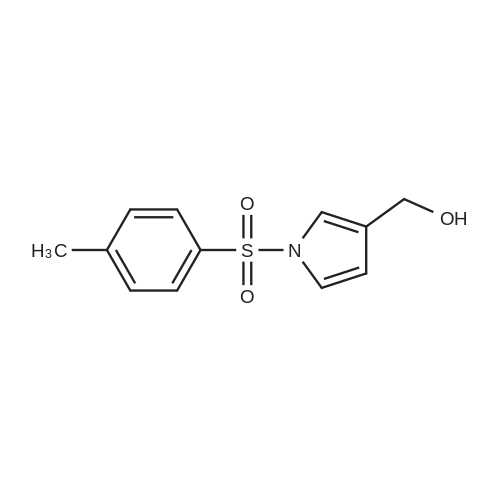
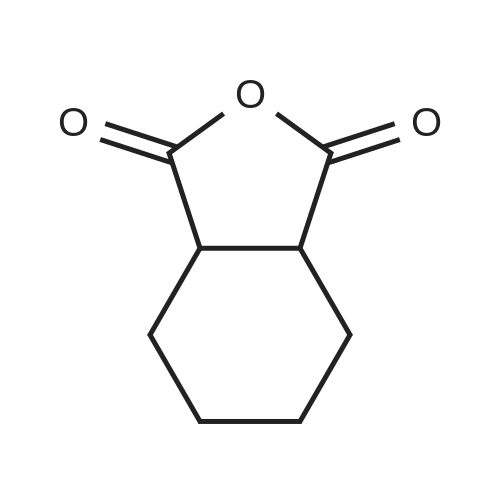
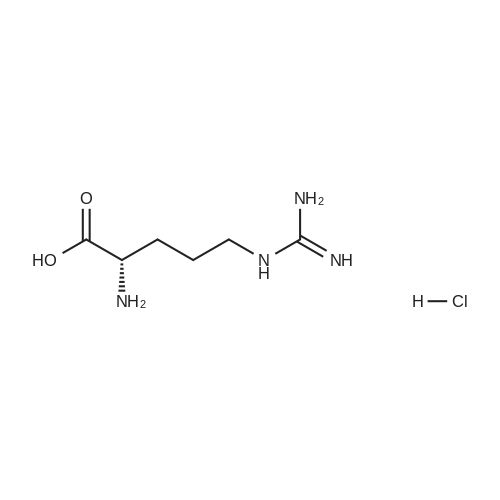
[ 112-60-7 ]
[ 98-59-9 ]
[ 37860-51-8 ]
[ 77544-60-6 ]
Yield
Reaction Conditions
Operation in experiment
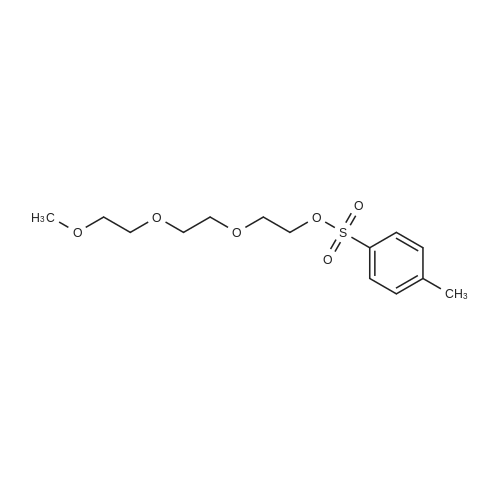
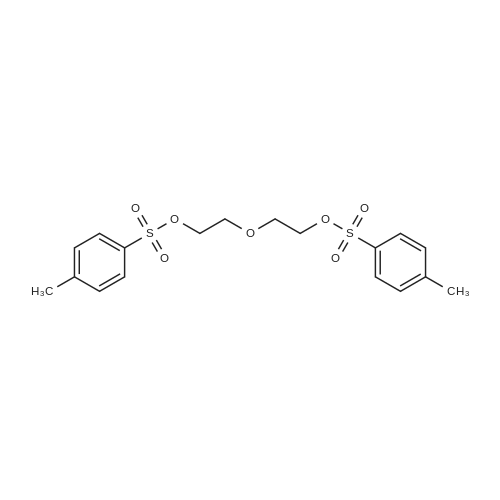
35.9%
With triethylamine In acetonitrile at 0 - 25℃; for 1 h; Cooling with ice
20.0 g of triethylene glycol and 10.2 g of triethylamine were charged into a reaction flask, 300 ml of acetonitrile was added thereto, and the mixture was cooled in an ice bathTo 0-5 ° C, 19.0gTsCl dissolved in 100ml acetonitrile slowly dripping into the solution, 1h drops finished, finished after the drop to 20-25Deg.] C for 12-14 h, the solvent was removed by rotary column chromatography, and eluted with n-hexane: ethyl acetate volume ratio= 4: 1 and 1.5: 1 gradient elution, in n-hexane: ethyl acetate volume ratio = 4: 1 eluentTo give 12.9 g of an oily product (compound thirteen) in a yield of 35.9percent in n-hexane: ethyl acetate volume ratio = 1.5: 1To give 5.4 g of an oil (Compound 14) in a yield of 10.4percent.
Reference:
[1] Organic Letters, 2002, vol. 4, # 14, p. 2329 - 2332
[2] Journal of Organic Chemistry USSR (English Translation), 1990, vol. 26, # 11, p. 2094 - 2100[3] Zhurnal Organicheskoi Khimii, 1990, vol. 26, # 11, p. 2425 - 2433
[4] Patent: CN105541736, 2016, A, . Location in patent: Paragraph 0044; 0052
[5] Journal of the Chemical Society, Perkin Transactions 2, 2001, # 9, p. 1573 - 1584
[6] Patent: WO2009/108484, 2009, A1, . Location in patent: Page/Page column 22; 23
[7] Patent: US2012/4423, 2012, A1, . Location in patent: Page/Page column 10
3
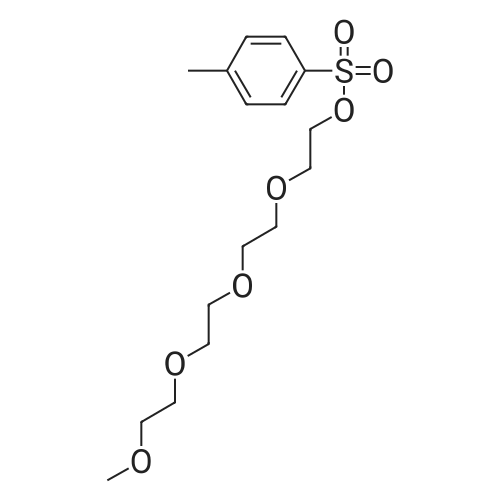
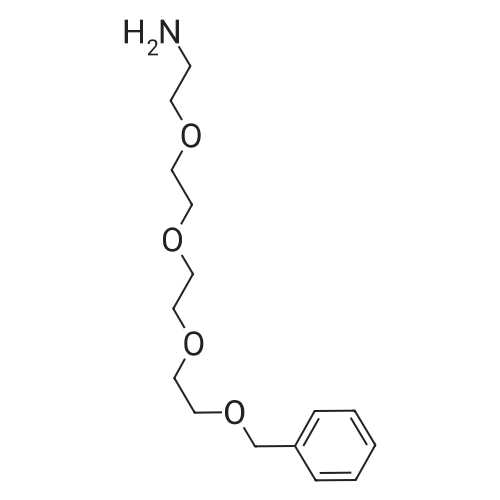
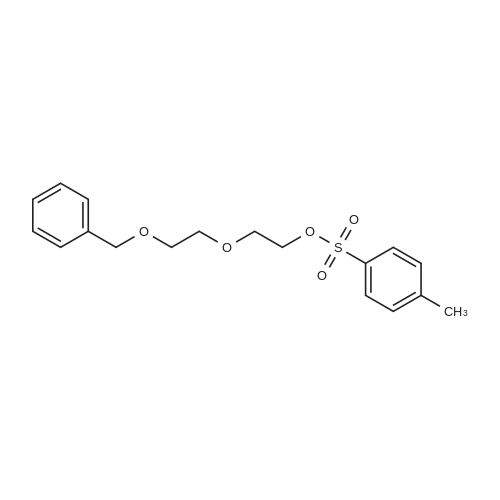
[ 112-60-7 ]
[ 98-59-9 ]
[ 37860-51-8 ]
Yield
Reaction Conditions
Operation in experiment
98%
With potassium hydroxide In dichloromethane at 0 - 10℃; for 3 h;
General procedure: Compounds 2a–c were prepared by a modified procedure known from the literature1. Oligo ethylene glycol(37.7 mmol) and p-toluenesulfonyl chloride (14.4 g, 75.4 mmol, 2 equiv) were dissolved in dichloromethane (36 mL)and this solution was cooled to 0 °C. During permanent stirring, milled potassium hydroxide (17 g, 302 mmol, 8equiv) was added and the solution was then stirred for 3 hours while the temperature was kept from 0 to 10 °C. This reaction was monitored by TLC (UV detection). After warming up to room temperature the reaction mixture was diluted by chloroform and extracted with water (3 × 1:1). Chloroform extracts were collected, dried over magnesium sulfate overnight, filtered and solvents were evaporated under reduced pressure.
98%
With potassium hydroxide In dichloromethane for 1.5 h; Cooling with ice
The 10mL (0.068mol) of tetraethylene glycol,300ml dichloromethane,25.8g (0.136mol)MethylBenzenesulfonyl chloride,Added to a 500ml round-bottomed flask,Under ice-cooling,Was slowly added 30.4g (0.544mol) of potassium hydroxide,1.5h the reaction is substantially complete.The reaction solution was washed three times with water (3 × 100ml),The organic layer was collected,Dried over anhydrous sodium sulfate.filter,Spin dry solvent,To give the crude product as an oil.Column chromatography (petroleum ether: ethyl acetate = 1:4) to give a pale yellow oily liquid 34g.The yield was 98percent.
95%
With 1H-imidazole; triethylamine In dichloromethane at 0 - 20℃;
0-Ditosylate Tetraethylne Glycol (32). Compound 2 (5.00 mL, 28.95 mmol) was added dropwise to a stirred solution of /Moluenesulfonyl chloride (12.15 g, 63.73 mmol), imidazole (0.04 g, 0.65 mmol) and Et3N (16.15 mL, 108.30 mmol) in CH2Cl2 (30 mL) at 0 0C. The reaction mixture was allowed to stir at room temperature (12 h), and then Et2O (80 mL) was added and the reaction washed with H2O (5 x 60 mL). The organic layer was dried (Na2SO4) and evaporated in vacuo, and then the crude product was purified by column chromatography (SiO2; 3/1 EtOAc/hexanes) to yield 13.81 g (95percent) of 32 as a clear oil (Busch et al., 2002): Rf = 0.65 (3/1 EtOAc/hexanes); 1H NMR (CDCl3) δ 2.44 (s, 2 CH3), 3.53-3.59 (m, 2 OCH2CH2O), 3.68 (t, J = 4.8 Hz, OCH2CH2OTs), 4.15 (t, J = 4.8 Hz, OCH2CH2OTs), 7.34 (d, J= 8.4 Hz, 4 ArH), 7.79 (d, J= 8.4 Hz, 4 ArH); 13C NMR (CDCl3) δ 21.7 (CH3), 68.8 , 69.4, 70.6, 70.8 (OCH2CH2O, OCH2CH2OTs), 128.1, 130.1, 133.1, 145.0 (C6H4).
89%
Stage #1: With potassium hydroxide In dichloromethane at 0 - 20℃; Stage #2: at 20℃; for 10 h;
General procedure: KOH (20 g, 356.5 mmol for 9; and 10 g, 181.5 mmol for 10–11) was added to 300 mL of a CH2Cl2 solution containing the oligoethylene glycol (10 g,94 mmol for 9, 66.6 mmol for 10, and 51.5 mmol for 11) at 0 °C. After stirring for 30 min at room temperature, TsCl (45 g, 236 mmol for 9–11) was added and the resulting solution was stirred for 10 h at room temperature. The resulting solution was filtered and then washed with aq. K2CO3. The organic layer was dried with MgSO4, filtered, and concentrated in vacuo. The resulting residue was then subjected to silicagel column chromatography (EtOAc:Hex = 1:1) to yield 35 g of 9 (90percent), 27 g of 10 (88percent), and 23 g of 11 (89percent).
74.4%
With triethylamine In dichloromethane at 0 - 20℃;
To a solution of 2-{2-[2-(2-hydroxyethoxy)ethoxy]ethoxy}ethan-l-ol (13 g, 67.1 mmol), Et3N (13 mL) in DCM (100 mL) was added TsCl (25.5 g, 134.2 mmol) in portions at 0°C. The resulting mixture was allowed to stir at room temperature overnight. TLC showed the reaction completed. The mixture was partitioned between DCM and H20. The organic phase was washed with brine, dried over magnesium sulfate and evaporated to dryness. The crude product was purified by silica gel chromatography using with 10-20percent EtOAc in hexane as eluent to afford the desired compound (25.0 g, 74.4percent).
73%
With triethylamine In dichloromethane at 25℃; for 12 h;
Tetraethylene glycol (3.88 g, 20 mmol) and triethylamine (TEA) (8.0 mL) were dissolved in dichloromethane (60 mL). Then, tosyl-chloride (9.50 g, 50 mmol) was added in one portion. The resulting mixture was stirred at 25 oC for 12 h. After washing with KHSO4 (1 M, 40 mL) and NaHCO3 (5percent, 40 mL), respectively and drying over Na2SO4, the crude product was obtained by evaporation and subsequently purification by column chromatography over silica gel (dichloromethane) to obtain the target product 7 as a colorless oil (7.5 g, 14.6 mmol, 73percent).
73%
With potassium hydroxide In tetrahydrofuran; water at 0 - 20℃; for 18 h; Inert atmosphere; Glovebox
A 1 L round bottom flask, charged with a magnetic stir bar, THF (400 mL), ptoluenesulfonylchloride(165.6 g, 0.868 mol) and tetraethylene glycol (56.4 g, 0.290 mol). Theflask was cooled to 0°C and a solution of KOH (104.8 g in 100 mL water, 1.87 mol) was addeddropwise through a dropping funnel over the course of 3 h. After addition, the mixture was allowedto stand at ambient temperature for 18 h. At this stage, the reaction mixture had generated a whiteprecipitate. The reaction was poured into 600 mL of water/CH2Cl2 mixture (70/30). The two-phasesolvent system was separated, and the aqueous phase was extracted four times with 100 mLCH2Cl2. Note: Before extraction the aqueous phase is denser than CH2Cl2. The organic phaseswere combined, dried over MgSO4 and solvent was evaporated under reduced pressure to yield acolorless oil. (53.3 g, 73percent). The NMR spectra are consistent with published data.
58%
With triethylamine In dichloromethane at 0 - 20℃; for 6 h;
Under room temperature, the methyl sulfonyl chloride (29.5g, 155mmol) by adding 2,2 ' - ((oxygen dihydrogenmethylenebisphosphonate (ethane -2,1-diyl)) double (oxy)) b ethanol 20a (10.0g, 51 . 5mmol) in dichloromethane (150 ml) solution, in 0 °C next, add triethylamine (32.6 ml, 232mmol), stirring 5 minutes, to the reaction room temperature for 6 hours. Added to the reaction solution 100 ml water quenching reaction, separation, of sequentially separated organic phase is washed with water (100 ml), saturated salt water washing (100 ml), then dried with anhydrous sodium sulfate, filtered, filtrate concentrated. The resulting residue is purified by silica gel column chromatography [petroleum ether/ethyl acetate (v/v)=10/1], to obtain title compound 20b (15g, pale yellow liquid), yield: 58.0percent.
43%
With dmap In dichloromethane at 5 - 20℃;
Into a 250-mL 3-necked round-bottom flask, was placed a solution of tetraethylene glycol (10 g, 51 55 mmol, 1 00 equiv) in DCM (100 mL) This was followed by the addition of a solution of 4-methylbenzene-l-sulfonyl chloride (21 4 g, 112 63 mmol, 2 20 equiv) in DCM (50 mL) dropwise with stirring at 5°C To this was added N,N-dimethylpyridin-4-amine (15 7 g, 128 69 mmol, 2 50 equiv) The resulting solution was stirred for 2 h at room temperature at which time it was diluted with 100 mL of water The resulting solution was extracted with 3x100 mL of DCM and the organic layers combined The resulting mixture was washed with 1x100 mL of brine and then concentrated under vacuum The residue was applied onto a silica gel column and eluted with ethyl acetate/petroleum ether (1 2) to afford H g (43percent) of the title compound as white oil.
21.4 g
at 0 - 10℃; for 2 h;
Example 4 19.1 g paratoluensulfonyl chloride and 40 mL pyridine are added in 250 mL three-neck flask, and cooled to 0° C. 9.7 g HO-PEG(n=4)-OH is mixed uniformly with 20 mL pyridine, and then dripped in the three-neck flash, and the temperature is controlled between 0 and 10° C. Stirring and reaction is continued at this temperature for two hours. TLC monitors that the reaction is complete. 300 mL cold water and 60 mL concentrated hydrochloric acid are added in the reaction liquid and slowly stirred for half an hour, then the reaction liquid is transferred into a 500 mL separating funnel, acetic ether is added to extract twice (300 mL+200 mL). The organic layers are merged and washed by water to neutrality, dried by anhydrous sodium sulfate for two hours. A rotary evaporator evaporates out solvent to obtain 21.4 g viscous liquid, which will be directly used for next reaction.
Reference:
[1] Bioorganic and Medicinal Chemistry, 2007, vol. 15, # 14, p. 4841 - 4856
[2] Inorganica Chimica Acta, 2011, vol. 365, # 1, p. 38 - 48
[3] Journal of Medicinal Chemistry, 2016, vol. 59, # 17, p. 7840 - 7855
[4] Journal of Organic Chemistry, 1999, vol. 64, # 18, p. 6870 - 6873
[5] Journal of the American Chemical Society, 2008, vol. 130, # 33, p. 10882 - 10883
[6] Journal of Organic Chemistry, 1992, vol. 57, # 24, p. 6678 - 6680
[7] Beilstein Journal of Organic Chemistry, 2016, vol. 12, p. 349 - 352
[8] Patent: CN105384745, 2016, A, . Location in patent: Paragraph 0084; 0085; 0086
[9] Journal of Organic Chemistry, 1999, vol. 64, # 3, p. 721 - 725
[10] Phosphorus, Sulfur and Silicon and the Related Elements, 2006, vol. 181, # 1, p. 219 - 225
[11] Russian Journal of Organic Chemistry, 2012, vol. 48, # 10, p. 1345 - 1352[12] Zh. Org. Khim., 2012, vol. 48, # 10, p. 1350 - 1357,8
[13] Chemical Communications, 2013, vol. 49, # 81, p. 9311 - 9313
[14] Organic and Biomolecular Chemistry, 2017, vol. 15, # 17, p. 3681 - 3705
[15] Collection of Czechoslovak Chemical Communications, 1987, vol. 52, # 8, p. 2057 - 2060
[16] Patent: WO2010/14236, 2010, A2, . Location in patent: Page/Page column 34
[17] Chemistry - A European Journal, 2014, vol. 20, # 40, p. 12894 - 12900
[18] Langmuir, 2015, vol. 31, # 49, p. 13410 - 13419
[19] Chemical Communications, 2018, vol. 54, # 10, p. 1249 - 1252
[20] Organic Letters, 2012, vol. 14, # 18, p. 4866 - 4869
[21] Bulletin of the Chemical Society of Japan, 1990, vol. 63, # 4, p. 1260 - 1262
[22] Journal of Organic Chemistry, 1983, vol. 48, # 25, p. 4864 - 4869
[23] Bulletin of the Chemical Society of Japan, 1990, vol. 63, # 4, p. 1260 - 1262
[24] Chemical Communications, 2016, vol. 52, # 45, p. 7310 - 7313
[25] Journal of the American Chemical Society, 1994, vol. 116, # 8, p. 3192 - 3196
[26] Tetrahedron, 2005, vol. 61, # 33, p. 7924 - 7930
[27] Journal of Physical Organic Chemistry, 2009, vol. 22, # 1, p. 1 - 8
[28] Tetrahedron, 1999, vol. 55, # 5, p. 1491 - 1504
[29] Tetrahedron, 2007, vol. 63, # 23, p. 5083 - 5087
[30] Organic Letters, 2011, vol. 13, # 22, p. 6006 - 6009
[31] Journal of Porphyrins and Phthalocyanines, 2013, vol. 17, # 1-2, p. 104 - 117
[32] Chinese Journal of Chemistry, 2013, vol. 31, # 5, p. 607 - 611
[33] Bulletin of the Korean Chemical Society, 2015, vol. 36, # 6, p. 1654 - 1660
[34] Journal of Organic Chemistry, 1995, vol. 60, # 24, p. 7984 - 7996
[35] ChemMedChem, 2010, vol. 5, # 5, p. 777 - 789
[36] Tetrahedron Asymmetry, 2005, vol. 16, # 12, p. 2119 - 2124
[37] Journal of the American Chemical Society, 2011, vol. 133, # 8, p. 2749 - 2759
[38] Journal of Organic Chemistry, 1984, vol. 49, p. 1408 - 1412
[39] Patent: WO2014/9429, 2014, A1, . Location in patent: Sheet 13/23
[40] Tetrahedron Letters, 1995, vol. 36, # 25, p. 4377 - 4380
[41] Canadian Journal of Chemistry, 1997, vol. 75, # 11, p. 1472 - 1482
[42] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1994, # 4, p. 447 - 460
[43] Organic and Biomolecular Chemistry, 2006, vol. 4, # 11, p. 2082 - 2087
[44] Journal of Medicinal Chemistry, 2012, vol. 55, # 7, p. 2981 - 2993
[45] Organometallics, 2014, vol. 33, # 16, p. 4323 - 4335
[46] Chemical Communications, 2015, vol. 51, # 8, p. 1524 - 1527
[47] Journal of Heterocyclic Chemistry, 1998, vol. 35, # 1, p. 209 - 215
[48] MedChemComm, 2013, vol. 4, # 10, p. 1400 - 1404
[49] Chemical Communications, 2013, vol. 49, # 86, p. 10097 - 10099
[50] Chemistry - A European Journal, 2012, vol. 18, # 52, p. 16689 - 16697
[51] Chemical Communications, 2013, vol. 49, # 22, p. 2195 - 2197
[52] Synlett, 2013, vol. 24, # 12, p. 1523 - 1528
[53] Patent: WO2017/30814, 2017, A1, . Location in patent: Paragraph 00240
[54] Molecular Crystals and Liquid Crystals Science and Technology Section A: Molecular Crystals and Liquid Crystals, 2001, vol. 365, p. 427 - 437
[55] Chinese Chemical Letters, 2016, vol. 27, # 11, p. 1655 - 1660
[56] Polyhedron, 2018, vol. 141, p. 385 - 392
[57] Organic Letters, 2010, vol. 12, # 13, p. 3050 - 3053
[58] Bioorganic and Medicinal Chemistry, 1999, vol. 7, # 9, p. 1881 - 1890
[59] Journal of Organic Chemistry, 1999, vol. 64, # 14, p. 5156 - 5161
[60] Patent: CN105461762, 2016, A, . Location in patent: Paragraph 0661; 0663; 0664; 0665; 0666
[61] Journal of the Chemical Society, Perkin Transactions 2: Physical Organic Chemistry (1972-1999), 1985, p. 607 - 624
[62] Macromolecules, 2004, vol. 37, # 13, p. 4761 - 4769
[63] Journal of the American Chemical Society, 2012, vol. 134, # 1, p. 83 - 86
[64] Patent: WO2010/78449, 2010, A2, . Location in patent: Page/Page column 279
[65] Liebigs Annalen der Chemie, 1983, # 5, p. 770 - 801
[66] Journal of Organic Chemistry, 1994, vol. 59, # 8, p. 2186 - 2196
[67] Journal of Heterocyclic Chemistry, 1994, vol. 31, # 4, p. 1047 - 1052
[68] Zeitschrift fuer Naturforschung, B: Chemical Sciences, 2002, vol. 57, # 1, p. 107 - 112
[69] Tetrahedron, 2003, vol. 59, # 50, p. 9939 - 9950
[70] Organic and Biomolecular Chemistry, 2005, vol. 3, # 12, p. 2255 - 2261
[71] Tetrahedron Letters, 2006, vol. 47, # 48, p. 8563 - 8566
[72] Journal of Organic Chemistry, 2006, vol. 71, # 26, p. 9884 - 9886
[73] Journal of the American Chemical Society, 2010, vol. 132, # 2, p. 656 - 666
[74] Chemistry Letters, 2010, vol. 39, # 2, p. 100 - 101
[75] Journal of Molecular Structure, 2010, vol. 982, # 1-3, p. 162 - 168
[76] Synthesis, 2012, vol. 44, # 5, p. 717 - 722
[77] Chemical Communications, 2012, vol. 48, # 45, p. 5650 - 5652
[78] Dalton Transactions, 2012, vol. 41, # 29, p. 8767 - 8769
[79] Langmuir, 2012, vol. 28, # 33, p. 12357 - 12363
[80] Journal of Materials Chemistry, 2012, vol. 22, # 33, p. 16927 - 16932
[81] Chemical Communications, 2013, vol. 49, # 38, p. 3982 - 3984
[82] Chinese Chemical Letters, 2014, vol. 25, # 12, p. 1643 - 1647
[83] Letters in Organic Chemistry, 2015, vol. 12, # 2, p. 85 - 90
[84] Bioorganic and Medicinal Chemistry, 2016, vol. 24, # 7, p. 1488 - 1494
[85] Patent: US2016/82117, 2016, A1, . Location in patent: Paragraph 0052
[86] Tetrahedron, 2016, vol. 72, # 38, p. 5744 - 5748
[87] Tetrahedron, 2014, vol. 70, # 50, p. 9545 - 9553
[88] Patent: KR101508710, 2015, B1, . Location in patent: Paragraph 0023-0032
[89] Journal of Medicinal Chemistry, 2017, vol. 60, # 7, p. 2890 - 2907
[90] New Journal of Chemistry, 2018, vol. 42, # 14, p. 11324 - 11333
[91] Tetrahedron, 2018, vol. 74, # 37, p. 4777 - 4789
[92] Journal of Heterocyclic Chemistry, 2018, vol. 55, # 9, p. 2172 - 2177
4
[ 107-21-1 ]
[ 111-44-4 ]
[ 112-60-7 ]
Reference:
[1] Journal of Organic Chemistry USSR (English Translation), 1980, vol. 16, p. 1124 - 1129[2] Zhurnal Organicheskoi Khimii, 1980, vol. 16, # 6, p. 1301 - 1307
[3] J. Appl. Chem. USSR (Engl. Transl.), 1972, vol. 45, p. 2708 - 2710[4] Zhurnal Prikladnoi Khimii (Sankt-Peterburg, Russian Federation), 1972, vol. 45, # 11, p. 2581 - 2583
Reference:
[1] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1981, p. 449 - 454
21
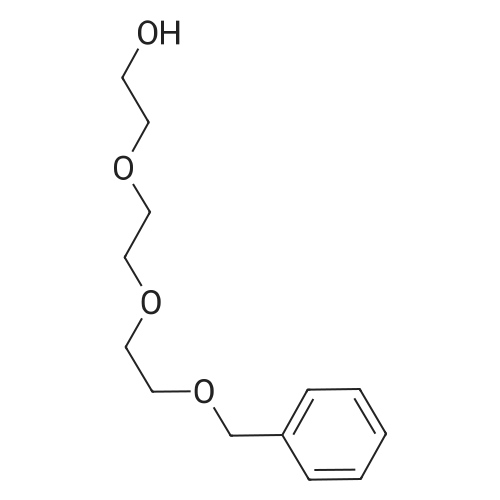
[ 7553-56-2 ]
[ 111-46-6 ]
[ 112-60-7 ]
[ 4792-15-8 ]
[ 112-27-6 ]
Reference:
[1] Canadian Journal of Research, Section B: Chemical Sciences, 1936, vol. 14, p. 80
22
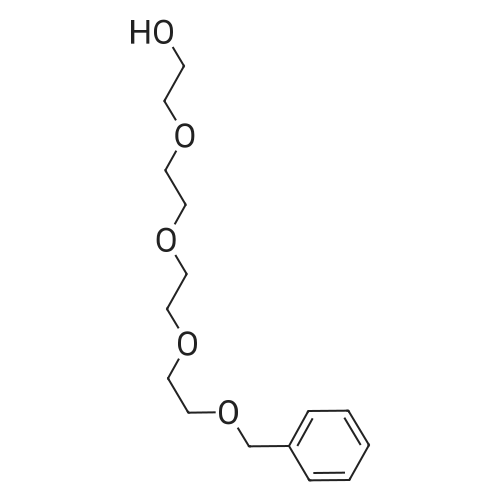
[ 107-21-1 ]
[ 106-93-4 ]
[ 112-60-7 ]
[ 4792-15-8 ]
[ 111-46-6 ]
[ 112-27-6 ]
Reference:
[1] Annales de Chimie (Cachan, France), 1863, vol. <3>67, p. 272
23
[ 75-21-8 ]
[ 111-46-6 ]
[ 112-60-7 ]
[ 4792-15-8 ]
[ 112-27-6 ]
Reference:
[1] Yakugaku Zasshi, 1944, vol. 64, p. 295 - 298[2] Chem.Abstr., 1951, p. 5104
24
[ 112-60-7 ]
[ 74654-05-0 ]
[ 4437-01-8 ]
Yield
Reaction Conditions
Operation in experiment
82%
Stage #1: With sodium hydride In DMF (N,N-dimethyl-formamide) at 20℃; for 1 h; Stage #2: at 20℃; for 3.5 h;
To a stirred solution of non-polydispersed compound 11 (35.7 mmol) in dry DMF (25.7 ML), under N2 was added in portion a 60percent dispersion of NaH in mineral oil, and the mixture was stirred at room temperature for 1 hour.To this salt 12 was added a solution of non-polydispersed mesylate 9 (23.36) in dry DMF (4 ml) in a single portion, and the mixture was stirred at room temperature for 3.5 hours.Progress of the reaction was monitored by TLC (12percent CH3OH-CHCl3).The reaction mixture was diluted with an equal amount of 1N HCl, and extracted with ethyl acetate (2*20 ml) and discarded.Extraction of aqueous solution and work-up gave non-polydispersed polymer 10 (82-84percent yield).
41%
Stage #1: With potassium <i>tert</i>-butylate In tetrahydrofuran for 1.5 h;
To a solution of non-polydispersed tetraethylene glycol (51.5 g, 0.27 mol) in THF (1L) was added potassium t-butoxide (14.8 g, 0.13 mol, small portions over 30 min). The reaction mixture was then stirred for 1 h and then 24 (29.15 g, 0.12 mol) dissolved in THF (90 mL) was added dropwise and the reaction mixture was stirred overnight. The crude reaction mixture was filtered through Celite (washed CH2Cl2, 200 mL) and evaporated to dryness. The oil was then dissolved in HCl (250 mL, 1 N) and washed with ethyl acetate (250 mL) to remove excess 24. Additional washings of ethyl acetate (125 mL) may be required to remove remaining 24. The aqueous phase was washed repetitively with CH2Cl2 (125 mL volumes) until most of the 25 has been removed from the aqueous phase. The first extraction will contain 24, 25, and dicoupled side product and should be back extracted with HCl (125 mL, 1N). The organic layers were combined and evaporated to dryness. The resultant oil was then dissolved in CH2Cl2 (100 mL) and washed repetitively with H20 (50 mL volumes) until 25 was removed. The aqueous fractions were combined, total volume 500 mL, and NaCl was added until the solution became cloudy and then was washed with CH2Cl2 (2.x.500 mL). The organic layers were combined, dried MgSO4, and evaporated to dryness to afford a the non-polydispersed title compound as an oil (16.9 g, 41percent yield). It may be desirable to repeat one or more steps of the purification procedure to ensure high purity.
41%
Stage #1: With potassium <i>tert</i>-butylate In tetrahydrofuran for 1.5 h; Stage #3: With hydrogenchloride In water
To a solution of monodispersed tetraethylene glycol (51.5 g, 0.27 mol) in THF (1L) was added potassium t-butoxide (14.8 g, 0.13 mol, small portions over 30 min). The reaction mixture was then stirred for 1 h and then 9 (29.15 g, 0.12 mol) dissolved in THF (90 mL) was added dropwise and the reaction mixture was stirred overnight. The crude reaction mixture was filtered through Celite (washed CH2Cl2, 200 mL) and evaporated to dryness. The oil was then dissolved in HCl (250 mL, 1 N) and washed with ethyl acetate (250 mL) to remove excess 9. Additional washings of ethyl acetate (125 mL) may be required to remove remaining 9. The aqueous phase was washed repetitively with CH2Cl2 (125 mL volumes) until most of the compound 18 has been removed from the aqueous phase. The first extraction will contain 9, 10, and dicoupled side product and should be back extracted with HCl (125 mL, 1N). The organic layers were combined and evaporated to dryness. The resultant oil was then dissolved in CH2Cl2 (100 mL) and washed repetitively with H2O (50 mL volumes) until 10 was removed. The aqueous fractions were combined, total volume 500 mL, and NaCl was added until the solution became cloudy and then was washed with CH2Cl2 (2.x.500 mL). The organic layers were combined, dried MgSO4, and evaporated to dryness to afford the monodispersed compound 10 as an oil (16.9 g, 41percent yield). It may be desirable to repeat one or more steps of the purification procedure to ensure high purity.
With sodium chloride In tetrahydrofuran; hydrogenchloride; dichloromethane; ethyl acetate
Example 20 Heptaethylene glycol monomethyl ether (25) To a solution of non-polydispersed tetraethylene glycol (51.5 g, 0.27 mol) in THF (1L) was added potassium t-butoxide (14.8 g, 0.13 mol, small portions over -30 min). The reaction mixture was then stirred for lh and then 24 (29.15 g, 0.12 mol) dissolved in THF (90 mL) was added dropwise and the reaction mixture was stirred overnight. The crude reaction mixture was filtered through Celite (washed CH2Cl2, ~200 mL) and evaporated to dryness. The oil was then dissolved in HCl (250 mL, 1 N) and washed with ethyl acetate (250 mL) to remove excess 24. Additional washings of ethyl acetate (125 mL) may be required to remove remaining 24. The aqueous phase was washed repetitively with CH2Cl2 (125 mL volumes) until most of the 25 has been removed from the aqueous phase. The first extraction will contain 24, 25, and dicoupled side product and should be back extracted with HCl (125 mL, 1N). The organic layers were combined and evaporated to dryness. The resultant oil was then dissolved in CH2Cl2 (100 mL) and washed repetitively with H2O (50 mL volumes) until 25 was removed. The aqueous fractions were combined, total volume 500 mL, and NaCl was added until the solution became cloudy and then was washed with CH2Cl2 (2*500 mL). The organic layers were combined, dried MgSO4, and evaporated to dryness to afford a the non-polydispersed title compound as an oil (16.9 g, 41percent yield). It may be desirable to repeat one or more steps of the purification procedure to ensure high purity.
41%
With sodium chloride In tetrahydrofuran; hydrogenchloride; dichloromethane; ethyl acetate
Example 20 Heptaethylene Glycol Monomethyl Ether (25) To a solution of non-polydispersed tetraethylene glycol (51.5 g, 0.27 mol) in THF (1L) was added potassium t-butoxide (14.8 g, 0.13 mol, small portions over ~30 min). The reaction mixture was then stirred for lh and then 24 (29.15 g, 0.12 mol) dissolved in THF (90 mL) was added dropwise and the reaction mixture was stirred overnight. The crude reaction mixture was filtered through Celite (washed CH2Cl2,~200 mL) and evaporated to dryness. The oil was then dissolved in HCl (250 mL, 1 N) and washed with ethyl acetate (250 mL) to remove excess 24. Additional washings of ethyl acetate (125 mL) may be required to remove remaining 24. The aqueous phase was washed repetitively with CH2Cl2 (125 mL volumes) until most of the 25 has been removed from the aqueous phase. The first extraction will contain 24, 25, and dicoupled side product and should be back extracted with HCl (125 mL, 1N). The organic layers were combined and evaporated to dryness. The resultant oil was then dissolved in CH2Cl2 (100 mL) and washed repetitively with H2O (50 mL volumes) until 25 was removed. The aqueous fractions were combined, total volume 500 mL, and NaCl was added until the solution became cloudy and then was washed with CH2Cl2 (2*500 mL). The organic layers were combined, dried MgSO4, and evaporated to dryness to afford a the non-polydispersed title compound as an oil (16.9 g, 41percent yield). It may be desirable to repeat one or more steps of the purification procedure to ensure high purity.
Reference:
[1] Organic and Biomolecular Chemistry, 2015, vol. 13, # 6, p. 1700 - 1707
28
[ 112-60-7 ]
[ 52995-76-3 ]
[ 4437-01-8 ]
Reference:
[1] Liebigs Annalen der Chemie, 1980, # 6, p. 858 - 862
[2] Journal of Mass Spectrometry, 2002, vol. 37, # 7, p. 699 - 708
29
[ 112-60-7 ]
[ 57602-02-5 ]
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2014, vol. 24, # 2, p. 636 - 643
30
[ 112-60-7 ]
[ 6482-24-2 ]
[ 23778-52-1 ]
Yield
Reaction Conditions
Operation in experiment
35%
Stage #1: With potassium <i>tert</i>-butylate In tetrahydrofuran at 20℃; for 0.5 h; Stage #2: at 20℃;
Tetra(ethylene glycol) (55 mmol, 10.7 g) was dissolved in 100 mL of tetrahydrofuran ("THF") and to this solution was added KOtBu (55 mL, 1.0 M in THF) at room temperature. The resulting solution was stirred at room temperature for 30 minutes, followed by dropwise addition of CH3OCH2CH2Br (55 mmol, 5.17 mL in 50 mL THF). The reaction was stirred at room temperature overnight, followed by extraction with H2O (300 mL)/CH2Cl2 (3.x.300 mL). The organic extracts were combined and then dried over anhydrous Na2SO4. After filtering off the solid drying agent and removing the solvent by evaporation, the recovered crude residue was purified by column chromatography using a silica gel column (CH2Cl2:CH3OH=60:140:1) to give pure penta(ethylene glycol) monomethyl ether (yield 35percent). 1H NMR (CDCl3) δ 3.75-3.42 (m, 20 H, OCH2CH2O), 3.39 (s, 3H, MeO).
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2018, vol. 28, # 22, p. 3502 - 3505
[2] Bioorganic and Medicinal Chemistry Letters, 2018, vol. 28, # 22, p. 3502 - 3505
37
[ 112-60-7 ]
[ 74-88-4 ]
[ 23783-42-8 ]
Reference:
[1] Bioorganic and Medicinal Chemistry, 2008, vol. 16, # 24, p. 10295 - 10300
[2] Chemische Berichte, 1981, vol. 114, # 2, p. 477 - 487
[3] Journal of the American Chemical Society, 1981, vol. 103, # 25, p. 7484 - 7489
[4] Journal of Materials Chemistry, 1997, vol. 7, # 7, p. 1147 - 1154
38
[ 112-60-7 ]
[ 23783-42-8 ]
Reference:
[1] Helvetica Chimica Acta, 1984, vol. 67, p. 2128 - 2142
[2] Journal of the Chemical Society, Chemical Communications, 1990, # 13, p. 911 - 912
[3] Angewandte Chemie - International Edition, 2015, vol. 54, # 12, p. 3763 - 3767[4] Angew. Chem., 2015, vol. 127, # 12, p. 3834 - 3838,5
[5] Bioorganic and Medicinal Chemistry Letters, 2018, vol. 28, # 22, p. 3502 - 3505
39
[ 112-60-7 ]
[ 77-78-1 ]
[ 23783-42-8 ]
Reference:
[1] Molecular Crystals and Liquid Crystals, 2004, vol. 411, p. 421/[1463]-437/[1479]
[2] RSC Advances, 2014, vol. 4, # 22, p. 11064 - 11072
40
[ 112-60-7 ]
[ 100-44-7 ]
[ 55489-58-2 ]
[ 86259-87-2 ]
Reference:
[1] Angewandte Chemie - International Edition, 2009, vol. 48, # 7, p. 1248 - 1252
41
[ 112-60-7 ]
[ 62921-76-0 ]
Reference:
[1] Molecular Crystals and Liquid Crystals, 2004, vol. 411, p. 421/[1463]-437/[1479]
[2] Journal of the Chemical Society, Chemical Communications, 1990, # 13, p. 911 - 912
42
[ 112-60-7 ]
[ 86770-74-3 ]
Reference:
[1] Journal of the American Chemical Society, 2003, vol. 125, # 5, p. 1120 - 1121
[2] Journal of the American Chemical Society, 2001, vol. 123, # 31, p. 7560 - 7563
[3] Journal of Organic Chemistry, 2001, vol. 66, # 13, p. 4494 - 4503
[4] Helvetica Chimica Acta, 1991, vol. 74, # 8, p. 1697 - 1706
[5] Inorganica Chimica Acta, 2011, vol. 365, # 1, p. 38 - 48
[6] Chemistry - A European Journal, 2011, vol. 17, # 6, p. 1828 - 1836
[7] Journal of the American Chemical Society, 2011, vol. 133, # 24, p. 9242 - 9245
[8] Nucleosides, Nucleotides and Nucleic Acids, 2011, vol. 30, # 7-8, p. 490 - 502
[9] Patent: US2012/4423, 2012, A1,
[10] Angewandte Chemie - International Edition, 2012, vol. 51, # 9, p. 2151 - 2154
[11] Chemistry - A European Journal, 2012, vol. 18, # 21, p. 6548 - 6554
[12] Chemistry - A European Journal, 2012, vol. 18, # 33, p. 10419 - 10426
[13] Organic and Biomolecular Chemistry, 2013, vol. 11, # 1, p. 27 - 30
[14] Patent: WO2013/109859, 2013, A1,
[15] European Journal of Organic Chemistry, 2013, # 35, p. 7952 - 7959
[16] Angewandte Chemie - International Edition, 2014, vol. 53, # 3, p. 810 - 814[17] Angew. Chem., 2014, vol. 126, # 3, p. 829 - 833,5
[18] Macromolecular Bioscience, 2015, vol. 15, # 1, p. 63 - 73
[19] Angewandte Chemie - International Edition, 2015, vol. 54, # 35, p. 10327 - 10330[20] Angew. Chem., 2015, vol. 35, # 37, p. 10467 - 10471,5
[21] Chemical Communications, 2016, vol. 52, # 6, p. 1154 - 1157
[22] Patent: WO2017/15693, 2017, A1,
[23] Patent: WO2017/50979, 2017, A1,
[24] Bioconjugate Chemistry, 2017, vol. 28, # 9, p. 2284 - 2292
[25] Patent: US2018/44280, 2018, A1,
43
[ 112-60-7 ]
[ 86770-76-5 ]
Reference:
[1] European Journal of Medicinal Chemistry, 2015, vol. 102, p. 153 - 166
44
[ 112-60-7 ]
[ 106984-09-2 ]
Reference:
[1] Helvetica Chimica Acta, 1991, vol. 74, # 8, p. 1697 - 1706
[2] Inorganica Chimica Acta, 2011, vol. 365, # 1, p. 38 - 48
[3] Journal of the American Chemical Society, 2011, vol. 133, # 24, p. 9242 - 9245
[4] Patent: US2015/166595, 2015, A1,
[5] Angewandte Chemie - International Edition, 2015, vol. 54, # 35, p. 10327 - 10330[6] Angew. Chem., 2015, vol. 35, # 37, p. 10467 - 10471,5
45
[ 112-60-7 ]
[ 98627-22-6 ]
[ 105891-54-1 ]
Reference:
[1] Journal of Organic Chemistry, 2006, vol. 71, # 26, p. 9884 - 9886
With sodium hydride In dimethyl sulfoxide; mineral oil at 0 - 20℃; for 23 h; Inert atmosphere
To a cold (0 °C) stirred solution of tert-butyl 2-bromoacetate (5.4 mL, 36.6 mmol) and 2,2'-((oxybis(ethane-2,l-diyl))bis(oxy))diethanol (35.0 mL, 203 mmol) in DMSO (50 mL) was added NaH (1.6217 g, 40.6 mmol, 60percent in oil) in two portions. The reaction mixture was slowly warmed up to rt, stirred for 23 h, diluted with water (50 mL), saturated with NaCl (50 g), stirred for 5 min, extracted with Et20 (3 x 100 mL), and concentrated. The residue was taken in DCM (50 mL) and water (25 mL), stirred for 5 min. The organic solution was passed through a phase separator, concentrated and purified on a 80 g Si02 cartridge using a gradient of ethyl acetate in hexanes (50- 100percent) to afford the title compound (8.2 g, 72.7percent yield) as a pale yellow oil. Rf = 0.08 (EtOAc). NMR (400 MHz, CDCb) δ 4.02 (d, J = 1.0 Hz, 2H), 3.75 - 3.60 (m, 12H), 1.47 (s, 9H) ppm.LRMS (ESI) m/z: calculated for Ci4H2807 308.3679; found 330.95 (M+Na)+; Found 347.01 (M+K)+.
Reference:
[1] Patent: WO2018/200981, 2018, A1, . Location in patent: Paragraph 0209; 0294-0295
[2] Journal of Medicinal Chemistry, 2018, vol. 61, # 2, p. 583 - 598
[3] Patent: US2018/177750, 2018, A1, . Location in patent: Paragraph 1106
48
[ 112-60-7 ]
[ 169751-72-8 ]
Reference:
[1] Patent: WO2017/46036, 2017, A1,
49
[ 112-60-7 ]
[ 101187-40-0 ]
Reference:
[1] Journal of the American Chemical Society, 2012, vol. 134, # 1, p. 83 - 86
50
[ 112-60-7 ]
[ 462100-06-7 ]
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2004, vol. 14, # 5, p. 1105 - 1108
51
[ 112-60-7 ]
[ 1663-39-4 ]
[ 518044-32-1 ]
Yield
Reaction Conditions
Operation in experiment
82%
With sodium In tetrahydrofuran at 20℃;
To a solution of tetraethyleneglycol (40.61 mL; 45.64 g; 235 mmol) in abs. THF (125 mL) a piece of sodium (1/4 cm) was added. After the sodium had reacted completely, tert-butylacrylate (1 1.98 mL; 10.57 g; 82.5 mmol) was added dropwise over 20 min and the resulting solution was stirred at rt overnight. The pH was adjusted to 7-8 with NaOH solution (1 N) and the solvents were removed in vacuum. The residue was dissolved in sat. NaCl solution (75 mL) and extracted with EtOAc (3x100 mL). The combined organic layers were dried over MgSO4 and after evaporation of the solvent tert-butyl-15-hydroxy-4,7,10,13-tetraoxapentadecanoate (21.87 g; 67.8 mmol; 82percent based on tert-butylacrylate) was obtained as colorless oil. 1H-NMR (500 MHz, Chloroform-c/, TMS) δ [ppm] = 3.77 - 3.57 (m, 18H; OCH2); 3.01 (bs, 1 H, OH); 2.51 (t, 2H, J = 6.6 Hz, CH2COO'Bu); 1.45 (s, 9H, C(CH3)3); 13C{1H}-NMR (126 MHz, Chloroform-c/, TMS) δ [ppm] = 171.1 (COO); 80.7 (C(CH3)3); 72.6/70.8/70.7/70.6/70.5/67.0 (OCH2); 61.9 (HOCH2); 36.4 (CH2CO); 28.2 (C(CH3)3).
81%
Stage #1: With sodium In tetrahydrofuran at 20℃; Stage #2: at 20℃;
[00162] Synthesis of tert-butyl l-hydroxy-3,6,9, 12-tetraoxapentadecan-15-oate (2): To a solution of tetraethylene glycol 1 (58.3 g, 300 mmol) in dry THF (200 mL) was added sodium (115 mg), and the mixture was stirred until sodium was consumed. To the resulting solution was then added tert-butyl acrylate (12.8 g, 100 mmol) in dry THF (50 mL) dropwise, and the resulting mixture was stirred overnight. The reaction was quenched with AcOH (0.1 mL) and water (0.5 mL) and stirred for 0.5 h, and was then extracted with ethyl acetate (200 mL χ 3). The combined organics were worked up by a standard procedure to give a clear oil product 2 (26 g, 81percent). ESI m/z: 340 (M+18)+.
81%
With sodium In tetrahydrofuran at 20℃;
To a solution of tetraethylene glycol (VI-1, 58 g, 0.30 mol) in dry THF (200 mL) was added sodium (0.12 g), and the mixture was stirred until the sodium was consumed. To the resulting solution was then added tert-butyl acrylate (VI-2, 13 g, 0.10 mol) in dry THF (50 mL) dropwise, and the resulting mixture was stirred at RT overnight. The reaction was quenched with acetic acid (0.1 mL) first and then water (0.5 mL), and the resulting mixture was stirred at RT for half an hour, and subsequently was extracted with ethyl acetate (3 x 200 mL). The combined organic solution was washed with water (30 mL) and then brine (3 x 100 mL), dried over sodium sulfate, filtered and concentrated to yield product (VI-3, 26 g, 81percent yield) as colorless oil. ESI m/z: 340 (M + 18)+
80%
With sodium hydride In tetrahydrofuran; mineral oil at 20℃; for 20 h;
To a solution of tetraethylene glycol (20.0 g, 103 mol) in anhydrous THF (54.0 mL) were added NaH (60percent dispersion in mineral oil, 42 mg, 1.05 mmol) and tert-butyl acrylate (5.2 mL, 36.0 mol). The resulting solution was stirred for 20 h at room temperature. The solvent was removed under reduced pressure. The residue was purified by flash column chromatography (petroleum ether/EtOAc=1/1) to afford 16d-1 as a colorless oil (9.3 g, 80percent). IR (KBr) νmax 3445, 2873, 1729 cm-1. 1H NMR (400 MHz, CDCl3) δ (ppm) 3.65-3.53 (m, 18H), 2.43 (t, J=8.4 Hz, 2H), 1.37 (s, 9H). 13C NMR (100 MHz, CDCl3) δ (ppm) 170.7, 80.3, 72.6, 70.4 (2C), 70.3, 70.2, 70.1, 70.0, 66.7, 61.3, 36.0, 27.9 (3C). MS (ESI) m/z 340.2 [M+NH4]+. HRMS (ESI) m/z calcd for C15H30O7Na [M+Na]+: 345.1883; found, 345.1884.
77%
Stage #1: With sodium In tetrahydrofuran at 20℃; for 2 h; Stage #2: at 20℃; for 24 h;
To dry tetraethylene glycol (17.2 mL, 0.10 mol) in dry tetrahydrofuran(100 mL), sodium (0.02 g, 0.87 mmol) was added. After 2 h, the sodium had dissolved and tert-butyl acrylate (4.35 mL, 0.03 mol) was added. The solution was stirred under exclusion of moisture for 24 h. After neutralisation with 1 M HCl (0.8 mL), the solvent was evaporated under reduced pressure. The residue was dissolved in brine(100 mL) and extracted three times with ethyl acetate (150 mL). The combined organic layers were washed with water (50 mL) and dried with MgSO4. The solvent was evaporated under reduced pressure to afford 2 as clear, pale yellow liquid; 4.3 g, 77percent; 1H NMR (300 MHz,CDCl3): 3.77–3.52 (m, 18H), 3.06 (s, 1H), 2.41 (t, J = 5.8 Hz, 2H), 1.40(s, 9H); ESI-MS m/z: [M + H]+ 323.20252.
36%
With N-benzyl-trimethylammonium hydroxide In methanol at 20℃;
To a flask containing PEG4 (3.88 g, 20 mmole) was added triton B (40percent solution in methanol, 1.08 mL, 0.25 mmole) and tert-butyl acrylate (3.62 mL, 24 mmole) followed after 15 min. The mixture was stirred at room temperature overnight. The mixture was concentrated in vacuo and the residue was purified by flash chromatography on silica gel with 1 percent methanol in dichloromethane as eluent to give the title compound as an colorless oil (2.35 g, 36percent). 1H NMR No. 1.45 (s, 9H), 2.5 (t, 2H), 3.65 (m, 18H).
23%
With sodium In tetrahydrofuran at 20℃; for 24 h;
To a solution of tetra (ethylene glycol) 11-1 (40.6 mL, 235 mmol) in 100 mL of tetrahedrofuran was added 47 mg of sodium. 12 mL of tert-butylacrylate was added after sodium was dissolved. The reaction mixture was stirred at room temperature for 24 hours. The reaction mixture was concentrated in vacuo and quenched with 2 mL of 1 N HC1. The residue was suspended in brine and extracted with ethyl acetate (100 rriLXl, 50 mL X2). The organic layer was combined and washed with brine, dried over sodium sulfate and concentrated in vacuo to give 6.4 g (23percent) of compound 11-3.
23%
at 20℃; for 24 h;
Compound 2a: To a solution of tetra (ethylene glycol) (40.6 mL, 235 mmol) in 100 mL of tetrahydrofuran is added 47 mg of sodium. 12 mL of tert-butylacrylate is added after sodium is dissolved. The reaction mixture is stirred at room temperature for 24 hours. The reaction mixture is concentrated in vacuo and quenched with 2 mL of 1 N HCI. The residue is suspended in brine and extracted with ethyl acetate (100 mLX1 , 50 mL X2). The organic layer is combined and washed with brine, dried over sodium sulfate and concentrated in vacuo to give 6.4 g (23percent) of compound 2a.
23%
With sodium In tetrahydrofuran at 20℃; for 24 h;
Compound 11-3: To a solution of tetra (ethylene glycol) 11-1 (40.6 mL,235 mmol) in 100 mL of tetrahedrofuran was added 47 mg of sodium. 12 mL of tertbutylacrylatewas added after sodium was dissolved. The reaction mixture was stirred at roomtemperature for 24 hours. The reaction mixture was concentrated in vacuo and quenched with mL of 1 N HCI. The residue was suspended in brine and extracted with ethyl acetate (1 00mLXl, 50 mL X2). The organic layer was combined and washed with brine, dried oversodium sulfate and concentrated in vacuo to give 6.4 g (23percent) of compound 11-3.
23%
With sodium In tetrahydrofuran at 20℃; for 24 h;
To a solution of tetra (ethylene glycol) 11-1 (40.6 mL, 235 mmol) in100 mL oftetrahedrofuran was added 47 mg of sodium. 12 mL oftert-butylacrylate was added aftersodium was dissolved. The reaction mixture was stirred at room temperature for 24 hours. Thereaction mixture was concentrated in vacuo and quenched with 2 mL of 1 N HCl. The residue wassuspended in brine and extracted with ethyl acetate (100 mLXl, 50 mL X2). The organic layer was10 combined and washed with brine, dried over sodium sulfate and concentrated in vacuo to give 6.4 g(23percent) of compound 11-3.
Reference:
[1] Synthetic Communications, 2004, vol. 34, # 13, p. 2425 - 2432
[2] Patent: WO2016/146638, 2016, A1, . Location in patent: Page/Page column 20
[3] Patent: WO2017/147542, 2017, A2, . Location in patent: Paragraph 00162
[4] Patent: WO2018/89373, 2018, A2, . Location in patent: Paragraph 0638-0639
[5] Tetrahedron, 2011, vol. 67, # 12, p. 2251 - 2259
[6] Journal of Medicinal Chemistry, 2004, vol. 47, # 20, p. 4802 - 4805
[7] Carbohydrate Research, 2007, vol. 342, # 3-4, p. 541 - 557
[8] Journal of Chemical Research, 2016, vol. 40, # 6, p. 368 - 370
[9] Advanced Synthesis and Catalysis, 2012, vol. 354, # 17, p. 3259 - 3264
[10] Patent: WO2005/112919, 2005, A2, . Location in patent: Page/Page column 150
[11] Patent: WO2012/166560, 2012, A1, . Location in patent: Page/Page column 163; 168
[12] Patent: WO2013/68874, 2013, A1, . Location in patent: Page/Page column 20
[13] Patent: WO2013/192360, 2013, A1, . Location in patent: Paragraph 00458; 00479
[14] Patent: WO2013/185117, 2013, A1, . Location in patent: Paragraph 00444
[15] Synthetic Communications, 2007, vol. 37, # 11, p. 1899 - 1915
[16] Patent: CN107235848, 2017, A, . Location in patent: Paragraph 0021
52
[ 112-60-7 ]
[ 1663-39-4 ]
[ 518044-32-1 ]
Yield
Reaction Conditions
Operation in experiment
73%
With hydrogenchloride In tetrahydrofuran
15-Hydroxy-4,7,10,13-tetraoxapentadecanoic acid tert-butyl ester (9a) To 300 mL of anhydrous THF was added 80 mg (0.0025 mol) of sodium metal and 128 mL of tetraethylene glycol 4a (0.94 mol) with stirring (Seitz and Kunz, J. Org. Chem., 62:813-826 (1997)). After the sodium had completely dissolved, tert-butyl acrylate (24 mL, 0.33 mol) was added. The solution was stirred for 20 hrs at room temperature and neutralized with 8 mL of 1.0 M HCl. The solvent was removed in vacuo and the residue was suspended in brine (250 mL) and extracted with ethyl acetate (3*125 mL). The combined organic layers were washed with brine (100 mL) then water (100 mL), dried over sodium sulfate, and the solvent was removed. The resulting colorless oil was dried under vacuum to give 77.13 g (73percent yield) of product 9a (). 1H NMR: 1.40 (s, 9H), 2.49 (t, 2 H, J=6.4 Hz), 3.59-3.73 (m, 18 H).
Reference:
[1] Patent: US2004/1838, 2004, A1,
53
[ 112-60-7 ]
[ 124-63-0 ]
[ 477781-69-4 ]
Reference:
[1] Journal of Organic Chemistry, 2008, vol. 73, # 4, p. 1371 - 1378
54
[ 112-60-7 ]
[ 477781-69-4 ]
Reference:
[1] Journal of Organic Chemistry, 2006, vol. 71, # 26, p. 9884 - 9886
[2] Chemical Communications, 2014, vol. 50, # 58, p. 7800 - 7802
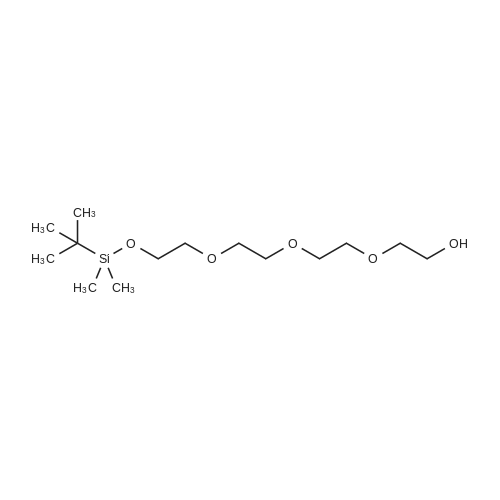
With 1H-imidazole; In dichloromethane; at 0℃; for 0.666667h;Inert atmosphere;
Example 25 Tetraethylene glycol mono(tert-butyl dimethylsilyl)ether CH2Cl2 (100 mL) was added to a 250 mL round bottom flask and flushed with nitrogen. Tetraethylene glycol (4.00 mL, 23.17 mmol) and imidazole (4.73 g, 69.51 mmol) were added and stirred to dissolve. The stirred solution was cooled to 0 C. and tert-butyl dimethylsilyl chloride (TBSCl, 4.32 g, 27.80 mmol) was added in a single portion. After stirring for 40 min at 0 C., TLC (EtOAc) revealed the di-silylated product (Rf=0.6) and the mono-silyl ether (Rf=0.3). The reaction was quenched with 250 mL of H2O and the organic layer separated. The aqueous layer was then extracted with CH2Cl2 (2*100 mL). The combined organics were washed with brine (200 mL), dried (Na2SO4), filtered, and concentrated under reduced pressure. Purification by column chromatography (EtOAc) afforded the title compound as a clear oil (2.92 g, 41%). 1H NMR (400 MHz, CDCl3): δH=0.01 (6H, s, TBS), 0.83 (9H, s, TBS), 2.97 (1H, br. s, OH), 3.48-3.72 (16H, m, -OCH2CH2O-).
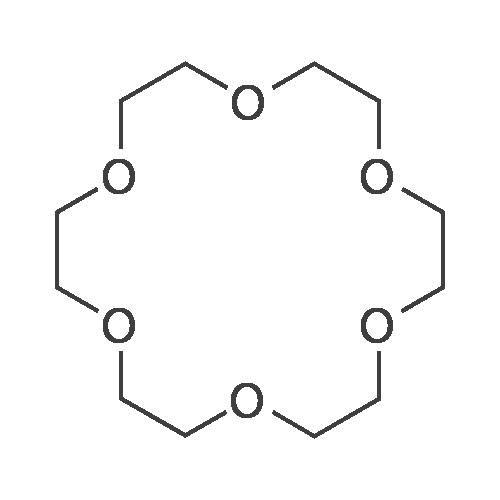
With sodium hydroxide; In 1,4-dioxane; at 27℃; for 0.5h;
1 part by mole of diethylene glycol bis-p-toluenesulfonate, 1.3 parts by mole of tetraethylene glycol, 4 parts by mole of sodium hydroxide, and 2 parts by mole of 1,4-dioxane are sequentially added to an industrial microwave reactor. The reaction was carried out at 27 C for 30 minutes. After the completion of the reaction, the mixture was separated by demineralization, extracted, and the solvent was evaporated. The crude product was distilled under reduced pressure to give 18-crown ether-6, yield 58%.
With 1H-imidazole; dmap; In dichloromethane; N,N-dimethyl-formamide; at 0℃;
To a stirred mixture of tetraethylene glycol 6 (66.0 g, 340 mmol), imidazole(33.9 g, 498 mmol) and 4-dimethylaminopyridine (5 g, 41 mmol) in dichloromethane (1000 mL) and dimethylformamide (100 mL) at 0 0C was slowly added a solution of tert- Butylchlorodimethylsilane (25.1 g, 166 mrnol) in dichloromethane (100 mL) over 4 hours. The resulted mixture was stirred overnight. The reaction mixture was quenched with IN HCl (300 mL), extracted with ethyl acetate. The combined organic phase was dried over magnesium sulfate, concentrated to dryness, and purified by flash chromatography to afford compound 7 (44.5 g, 87%) as clear oil. 1H NMR (400 MHz7 CDCl3) δ 0.00 (s, 6H), 0.83 (s, 9H), 1.79 (s, IH), 3.45-3.70 (m, 16H).
82.6%
With dmap; In dichloromethane; at 0 - 20℃; for 5h;
To the reaction flask was added tetraethylene glycol (8g, 41.18mmol, 5.0eq.), dichloromethane (25mL), DMAP (201mg, 8.24mmol, 0.2eq.) and triethylamine (833.1mg, 8.24mmol, 1.0 eq.), and the mixture was cooled to 0C in an ice-water bath. Then, a dichloromethane solution of TBSCl (1.24 g dissolved in 5 mL dichloromethane, 8.24 mmol, 1.0 eq.) was added dropwise to the reaction system. The reaction system was warmed to room temperature and stirred for 5 hours. The organic layer was washed twice with saturated ammonium chloride aqueous solution (30 mL each), and the organic phase was separated. The organic phase was concentrated to obtain 3,6,9,12-tetraoxa-13-sila-13,13,14,14-tetramethylpentadecanol (2.1 g, 82.6%).
82.6%
With dmap; triethylamine; In dichloromethane; at 0 - 20℃; for 5h;
[00166] Tetraethylene glycol (8 g, 41.18 mmol, 5.0 eq.), DCM (25 mL), DMAP (201 mg, 8.24 mmol, 0.2 eq.), and triethylamine (833.1 mg, 8.24 mmol, 1.0 eq.) were mixed in a flask and cooled to 0 C in an ice-water bath, followed by addition of a solution of TBSC1 (1.24 g, 8.24 mmol, 1.0 eq.) in DCM (5 mL). The temperature of the reaction was raised to rt; and the mixture was stirred at rt for 5 h. The mixture was washed with saturated ammonium chloride aqueous solution (2 x 30 mL); and the organic layer was separated and concentrated to dryness, providing 13,13,14,14-tetramethyl-3,6,9,12- tetraoxa-13-silapentadecan-1-ol (2.1 g, 82.6%).
78%
With 1-methyl-1H-imidazole; In dichloromethane; for 3h;
Example 1 (0117) FIG. 1 shows four pentagol-containing (Eg5) protected monomeric building blocks (tetragol=(ethylene glycol)4) with different reactive side chain precursor groups (R2). The monomeric building blocks (BBs) have the general structure TsOEg4OCH(CH2R2)CH2OPG [R2=-N3 (10a), -CC-Tbdps (10b), -SPmb (10c) and -OCH2C(O3C6H9) (10d)], which are obtained according to the synthetic route described in FIG. 1. Different protecting groups [PG=-Dmtr, -Mip, -Cyc, -Mthp, -Thp and -SO3-] are used for simple protection and effective deprotection, with a range of stability/controllable rate of acid-labile deprotection during each chain-extension cycle. (0118) Three hetero-functionalized tetragol derivatives with different reactive side-groups, namely TsO-EG4(R)-OThp (R=-OBn, -N3 and -SPmb), were synthesised as building blocks according to the procedure shown in FIG. 2. Notably, the coupling reaction between compound 2 and compound 5 generates some side products. Thus, it is preferable to completely dry the starting materials using acetonitrile before adding NaH catalyst to initiate the reaction. The hydrogenolysis of compound 8 to produce compound 11 was found to work best in acetonitrile and ethyl acetate. Here, the acid-labile Thp group was selected as a protecting group due to its acid-sensitivity and its small size, which means that it can be both effectively deprotected after each chain-extension cycle and, also easily removed by OSN diafiltration. The Tosyl group has high reactivity with OH group using NaH as a catalyst, making it suitable for chain extension of PEGs. The three different side-groups including BnO-, N3- and PmbS- on these building blocks can be readily converted into highly reactive groups after deprotection procedure, such as -OH, -NH2 and -SH, respectively. (0119) The chemical structure and molecular weight of the resulting building blocks including BnO-BB, N3-BB and PmbS-BB, have been confirmed by NMR spectroscopy and mass spectroscopy in FIGS. 3 and 4. The clearly observed m/z peaks at 614.2, 549.3 and 660.3 are assigned to BnO-BB, N3-BB and PmbS-BB, respectively.
73%
With pyridine; dmap; at 20℃;
General procedure: To a solution of ethylene glycol (19, 20, 21 or 22) (8-10 equiv) and DMAP (cat.) in dry pyridine (5 mL) was added TBDMS-Cl (1 equiv). The reaction mixture was stirred at room temperature overnight and then diluted with CH2Cl2 before the organic phase was washed twice with 1 M HCl, once with H2O and then satd NaHCO3. The organic phase was dried over MgSO4, filtered and concentrated under reduced pressure. The TBDMS-protected ethylene glycols (23, 24, 25 or 26) were used without further purification.
48%
With sodium hydride; In tetrahydrofuran; mineral oil; at 0 - 25℃; for 1.75h;
NaH (60% oil dispersion, 10 mmol) was slowly added to solution of tetraethylene glycol (10 mmol) in dry THF (20 mL) at 25 C. and stirred the suspension for 45 minutes at same temperature. The reaction mixture was cooled to 0 C. with ice-bath and tert-Butyldimethylsilyl chloride (10 mmol) was added dropwise and ice-bath was removed and reaction was continued at 25 C. After 60 minutes, H2O (5 mL) was added to quenched the reaction and extracted with ethyl acetate (3×50 mL). The combined organic layer was washed with brine and dried over anhydrous sodium sulphate, filtered and evaporated to get oil. The crude oily product was purified by silica gel column chromatography using 2% Methanol/Dichloromethane to provide mono-TBDMS protected tetraethylene glycol, 3 (4.8 mmol, yield 48%) as a colorless oil. 1H NMR (CDCl3, 400 MHz): δ 3.79 (t, J=4.0 Hz, 2H), 3.75 (t, J=4.0 Hz, 2H), 3.71-3.67 (m, 8H), 3.63 (t, J=4.0 Hz, 2H), 3.57 (t, J=4.0 Hz, 2H), 2.51 (s, 1H), 0.91 (s, 9H), 0.09 (s, 6H), ESI-LC/MS: Expected [M+H]+ for C14H32O5Si is 309.20, found m/z: 309.25 [M+H]+.
42%
With triethylamine; In dichloromethane; at 20℃; for 2h;
Tetraethylene glycol, 4c (1.12 g, 5.77 mmol) and TBDMSCl (0.87 g,5.77 mmol) were dissolved in dichloromethane (25 ml) followed by triethyl amine (1.46 g, 14.4 mmol). The solution was stirred at room temperature for 2 hours. After standard workup with dichloromethane, the residue was purified by silica gel column chromatography (50 % ethyl acetate in hexane) to afford 5c (744 mg, 42 %): 1H NMR δ 3.66 (m, 16 H), 2.51 (t, IH, J = 5.86 Hz), 0.89 (s, 9H), 0.07 (s, 6H).
41%
A solution of sodium hydride (60% in mineral oil, 2.94g, 73.5mmol) in anhydrous THF (135mL) was stirred at room temperature for 5min under argon. Dried tetraethylene glycol (8.80mL, 51.0mmol) was added dropwise at 0C and the reaction mixture was stirred for 5 more min before addition of tert-butyldimethylsilyl chloride (TBDMSCl, 12.00g, 79.6mmol). The resulting mixture was stirred at room temperature for 1h and the reaction was stopped by addition of water (150mL). The mixture was extracted with ethyl acetate (3×100mL). The organic layers were combined, washed successively with a saturated aqueous ammonium chloride solution (150mL) and brine (150mL), dried on magnesium sulphate, filtered and evaporated under vacuum. The crude product was purified by chromatography (SiO2 pad, EtOAc/pentane, 75/25, v/v then 50/50, v/v and finally CH2Cl2/EtOH, 98/2, v/v) to yield compound 21 (6.42g, 20.8mmol) as a yellow oil. Yield 41%; Rf (SiO2, EtOAc/pentane, 75/25, v/v) 0.16; IR (ATR diamond accessory) ν 778, 837, 1109, 1255, 1472, 2859, 2930, 3300-3600cm-1; 1H NMR (200MHz, CDCl3) δ 0.05 (s, 6H, (CH3)2Si), 0.88 (s, 9H, (CH3)3C), 2.63 (br s, 1H, OH), 3.65 (m, 16H, 8CH2O); 13C NMR (50MHz, CDCl3) δ-5.2 (2C), 18.4, 26.0 (3C), 61.8, 62.8, 70.4-70.7 (4C), 72.6, 72.7; MS m/z 251 (M-57, 9), 163 (26), 119 (46), 103 (55), 89 (100), 75 (93).
41%
A solution of sodium hydride (60% in mineral oil, 2.94 g, 73.5 mmol) in anhydrous tetrahydroiuran (135 mL) was stirred at room temperature for 5 min under argon. Dried tetraethylene glycol (8.80 mL, 51.0 mmol) was added dropwise at 0C and the reaction mixture was stirred for 5 min before addition of teri-butyldimethylsilyl chloride (12.00 g, 79.6 mmol). After return back to room temperature, the resulting mixture was stirred for 1 h and the reaction was quenched by addition of water (150 mL). The mixture was extracted with ethyl acetate (3x100 mL). The organic layers were combined, washed successively with a saturated aqueous ammonium chloride solution (150 mL) and brine (150 mL), dried on magnesium sulfate, filtered and evaporated under vacuum. The crude product was purified by chromatography (Si02 pad, EtOAc/pentane, 75/25, v/v then 50/50, v/v and finally CH2Cl2/EtOH, 98/2, v/v) to yield the expected compound (6.42 g, 20.8 mmol) as a yellow oil. Yield 41%. Rf (Si02, EtOAc/pentane, 75/25, v/v) 0.16. IR (ATR diamond accessory) v 778, 837, 1109, 1255, 1472, 2859, 2930, 3300-3600 cm"1. 1H NMR (200 MHz, CDC13) δ (ppm) 0.05 (s, 6H), 0.88 (s, 9H), 2.63 (brs, 1H), 3.65 (m, 16H). 13C NMR (50 MHz, CDC13) δ (ppm) -5.2 (2C), 26.0 (3C), 61.8, 62.8, 70.4-70.7 (4C), 72.6, 72.7. MS m/z 309 (1, [M+H]+), 163 (26), 147 (22), 133 (23), 119 (46), 103 (55), 89 (100), 87 (9), 75 (93), 73 (61), 59 (29).
41%
A solution of sodium hydride (60% in mineral oil, 2.94 g, 73.5 mmol) in anhydrous tetrahydroiuran (135 mL) was stirred at room temperature for 5 min under argon. Dried tetraethylene glycol (8.80 mL, 51.0 mmol) was added dropwise at 0C and the reaction mixture was stirred for 5 min before addition of teri-butyldimethylsilyl chloride (12.00 g, 79.6 mmol). After return back to room temperature, the resulting mixture was stirred for 1 h and the reaction was quenched by addition of water (150 mL). The mixture was extracted with ethyl acetate (3x100 mL). The organic layers were combined, washed successively with a saturated aqueous ammonium chloride solution (150 mL) and brine (150 mL), dried on magnesium sulfate, filtered and evaporated under vacuum. The crude product was purified by chromatography (Si02 pad, EtOAc/pentane, 75/25, v/v then 50/50, v/v and finally CH2Cl2/EtOH, 98/2, v/v) to yield the expected compound (6.42 g, 20.8 mmol) as a yellow oil. Yield 41%. Rf (Si02, EtOAc/pentane, 75/25, v/v) 0.16. IR (ATR diamond accessory) v 778, 837, 1109, 1255, 1472, 2859, 2930, 3300-3600 cm"1. 1H NMR (200 MHz, CDC13) δ (ppm) 0.05 (s, 6H), 0.88 (s, 9H), 2.63 (brs, 1H), 3.65 (m, 16H). 13C NMR (50 MHz, CDC13) δ (ppm) -5.2 (2C), 26.0 (3C), 61.8, 62.8, 70.4-70.7 (4C), 72.6, 72.7. MS m/z 309 (1, [M+H]+), 163 (26), 147 (22), 133 (23), 119 (46), 103 (55), 89 (100), 87 (9), 75 (93), 73 (61), 59 (29).
22%
With 1H-imidazole; In dichloromethane; at 0 - 20℃; for 6h;
To a stirred solution of 2,2'-(2,2'-oxybis(ethane-2,l-diyl)bis(oxy))diethanol 4-1 (5 g, 25.7 mmol) in DCM (100 mL) was added imidazole (2.1 g, 30.9 mmol) and teri-butyldimethylsilyl chloride (TBDMSC1; 3.48 g, 23.1 mmol) at 0 C and stirred to RT for 6 hr. The reaction mixture was diluted with DCM (100 mL) and washed with water (2 X 100 mL) and brine (50 mL). The organic layer was concentrated under reduced pressure and the crude residue was purified by flash column chromatography (100-200 silica) using 30% EtOAc/pet. ether gave 4-2 (1.8 g, 5.8 mmol, 22% yield) as an off-white solid.
5.7 g
Compound 20-2: To a solution of tetra (ethylene glycol) 20-1 (8.0 g, 41.2 mmol)in 100 mL of tetrahedrofuran was added 1.65 g of sodium hydride at 0C. The reaction mixture was stirred at room temperature for 30min. 6.21 g of TBS-Cl was added to thissolution. The reaction mixture was stirred at room temperatme overnight. The reactionmixture was concentrated in vacuo and quenched with 2 mL of 1 N HCl. The residue wassuspended in brine and extracted with ethyl acetate (1 00 mLXl, 50 mL X2). The organic5 layer was combined and washed with brine, dried over sodium sulfate and concentrated invacuo. The residue was purified by flash column chromatography to give 5.7g of compound20-2.
5.7 g
With sodium hydride; In tetrahydrofuran; at 0 - 20℃;
To a solution of tetra (ethylene glycol) 20-1 (8.0 g, 41.2 mmol) in 100mL of tetrahedrofuran was added 1.65 g of sodium hydride at 0C. The reaction mixture was stirred15 at room temperature for 30min. 6.21 g of TBS-Cl was added to this solution. The reaction mixture-was stirred at room temperature overnight. The reaction mixture was concentrated in vacuo andquenched with 2 mL of 1 N HCl. The residue was suspended in brine and extracted with ethylacetate (100 mLXl, 50 mL X2). The organic layer was combined and washed with brine, dried oversodium sulfate and concentrated in vacuo. The residue was purified by flash column5 chromatography to give 5.7g of compound 20-2.
polymer, product of random copolymerization at 100 deg C for 24 h, ε-caprolactone/trimethyl carbonate/tetra(ethylene glycol) units molar ratio 19:18:1; monomer(s): ε-caprolactone; trimethylene carbonate; tetra(ethylene glycol)[ No CAS ]
polymer, product of random copolymerization at 120 deg C for 24 h, ε-caprolactone/trimethyl carbonate/tetra(ethylene glycol) units molar ratio 19:19:1; monomer(s): ε-caprolactone; trimethylene carbonate; tetra(ethylene glycol)[ No CAS ]
polymer, product of random copolymerization at 100 deg C for 48 h, ε-caprolactone/trimethyl carbonate/tetra(ethylene glycol) units molar ratio 20:19:1; monomer(s): ε-caprolactone; trimethylene carbonate; tetra(ethylene glycol)[ No CAS ]
polymer, product of random copolymerization at 120 deg C for 48 h, ε-caprolactone/trimethyl carbonate/tetra(ethylene glycol) units molar ratio 20:17:1; monomer(s): ε-caprolactone; trimethylene carbonate; tetra(ethylene glycol)[ No CAS ]
polymer, A-B-A triblock copolymer, Mn 13000 Da, Mw 15000 Da by SEC, Mn 9800 Da by NMR, inherent viscosity at 20 deg C in CH2Cl2 (2g/l) 0.180 dl/g; monomer(s): ε-caprolactone; trimethylene carbonate; tetra(ethylene glycol); L-lactide[ No CAS ]
polymer, A-B-A triblock copolymer, Mn 21000 Da, Mw 26000 Da by SEC, Mn 15800 Da by NMR, inherent viscosity at 20 deg C in CH2Cl2 (2g/l) 0.255 dl/g; monomer(s): ε-caprolactone; trimethylene carbonate; tetra(ethylene glycol); L-lactide[ No CAS ]
polymer, A-B-A triblock copolymer, Mn 13000 Da, Mw 16000 Da by SEC, Mn 11000 Da by NMR, inherent viscosity at 20 deg C in CH2Cl2 (2g/l) 0.205 dl/g; monomer(s): ε-caprolactone; trimethylene carbonate; tetra(ethylene glycol); L-lactide[ No CAS ]
polymer, A-B-A triblock copolymer, Mn 22000 Da, Mw 27000 Da by SEC, Mn 18000 Da by NMR, inherent viscosity at 20 deg C in CH2Cl2 (2g/l) 0.260 dl/g; monomer(s): ε-caprolactone; trimethylene carbonate; tetra(ethylene glycol); L-lactide[ No CAS ]
With (carbonyl)chloro(hydrido)tris(triphenylphosphine)ruthenium(II); ammonia; 4,5-bis(diphenylphos4,5-bis(diphenylphosphino)-9,9-dimethylxanthenephino)-9,9-dimethylxanthene; In tert-Amyl alcohol; at 140℃; for 20h;Inert atmosphere; Cooling;
Example 6Direct Single-Stage Amination of Alcohols andHydroxy Acids by Means of Ammonia Over aHomogeneous Ruthenium Catalyst and Xantphos ata high V7J Vgas (according to the invention)Under an argon atmosphere, m g of starting material, mRU g of [carbonylchlorohydridotris(triphenylphosphane)ruthenium(II)] and mp g of 9,9-dimethyl-4,5-bis (diphenylphosphino)xanthene as catalyst and V07 ml of 2-methyl-2-butanol as solvent were introduced into a 50 mlsteel tube. The vessel was closed, pressurized three times with 20 bar of argon and depressurized each time. The vessel was then cooled by means of dry ice and m g of ammonia were condensed in. The reactor is heated to T C. and maintained at this temperature for 20 hours. Afier cooling to room temperature, the reactor was depressurized and opened, the solvent was removed on a rotary evaporator and the residue was dissolved in methanol and then analysed by gas chromatography. Reaction parameters and conversions and selectivities to the desired reaction product are shown in Tab. 5. The results show that many different hydroxy-thnctionalized substrates can be aminated by the method described.
Multi-step reaction with 2 steps
1.1: toluene-4-sulfonic acid / dichloromethane / 0.5 h / 20 °C
1.2: 10 h / 80 °C
2.1: potassium hydroxide / dichloromethane / 0 - 20 °C
2.2: 10 h / 20 °C
Multi-step reaction with 2 steps
1: toluene-4-sulfonic acid / dichloromethane / 4 h / 0 - 20 °C
2: triethylamine / dichloromethane / 3 h / 0 - 20 °C
Multi-step reaction with 2 steps
1: p-toluenesulfonyl chloride / dichloromethane / 4 h / 0 - 20 °C
2: triethylamine / dichloromethane / 3 h / 0 - 20 °C
potassium t-butoxide (Aldrich, 95%, 39.0 g) was added to a solution of tetraethylene glycol (Aldrich, 412.7 g) and methyl hexaethylene glycol chloride from above (95.8 g) at 18 C. over a 25 min period (ice/acetone bath). The reaction mixture was then stirred at 120 C. overnight. The pH was adjusted to 7 by the addition of hydrochloric acid (12 N, 11.7 ml), and volatiles were removed (64 Pa (0.48 Torr), up to 185 C.). The residual dark oil was diluted with toluene (250 ml), and treated with calcium chloride (38.1 g). After stirring for 18 h, the mixture was filtered through Celite and concentrated to a dark oil (102 g), which was fractionally distilled to provide decaethylene glycol monomethyl ether as an amber oil (64.4 g, 45%). An analytical sample was obtained via chromatography on silica gel, eluding with 4% methanol in chloroform to provide a colorless oil; 1H-NMR (DMSO-d6) delta: 4.58 (t, J=5.5 Hz, 1, OH), 3.58-3.38 (m, 40, 20 OCH2), 3.24 (s, 3, OCH3). Anal. Calcd for C21H44O11: C, 53.37; H, 9.38. Found: C, 53.09; H, 9.47.
mesylate of triethylene glycol monomethyl ether[ No CAS ]
[ 112-60-7 ]
[ 4437-01-8 ]
Yield
Reaction Conditions
Operation in experiment
41%
With sodium chloride; In tetrahydrofuran; hydrogenchloride; dichloromethane; ethyl acetate;
Example 20 Heptaethylene glycol monomethyl ether (25) To a solution of non-polydispersed tetraethylene glycol (51.5 g, 0.27 mol) in THF (1L) was added potassium t-butoxide (14.8 g, 0.13 mol, small portions over -30 min). The reaction mixture was then stirred for lh and then 24 (29.15 g, 0.12 mol) dissolved in THF (90 mL) was added dropwise and the reaction mixture was stirred overnight. The crude reaction mixture was filtered through Celite (washed CH2Cl2, ~200 mL) and evaporated to dryness. The oil was then dissolved in HCl (250 mL, 1 N) and washed with ethyl acetate (250 mL) to remove excess 24. Additional washings of ethyl acetate (125 mL) may be required to remove remaining 24. The aqueous phase was washed repetitively with CH2Cl2 (125 mL volumes) until most of the 25 has been removed from the aqueous phase. The first extraction will contain 24, 25, and dicoupled side product and should be back extracted with HCl (125 mL, 1N). The organic layers were combined and evaporated to dryness. The resultant oil was then dissolved in CH2Cl2 (100 mL) and washed repetitively with H2O (50 mL volumes) until 25 was removed. The aqueous fractions were combined, total volume 500 mL, and NaCl was added until the solution became cloudy and then was washed with CH2Cl2 (2*500 mL). The organic layers were combined, dried MgSO4, and evaporated to dryness to afford a the non-polydispersed title compound as an oil (16.9 g, 41% yield). It may be desirable to repeat one or more steps of the purification procedure to ensure high purity.
41%
With sodium chloride; In tetrahydrofuran; hydrogenchloride; dichloromethane; ethyl acetate;
Example 20 Heptaethylene Glycol Monomethyl Ether (25) To a solution of non-polydispersed tetraethylene glycol (51.5 g, 0.27 mol) in THF (1L) was added potassium t-butoxide (14.8 g, 0.13 mol, small portions over ~30 min). The reaction mixture was then stirred for lh and then 24 (29.15 g, 0.12 mol) dissolved in THF (90 mL) was added dropwise and the reaction mixture was stirred overnight. The crude reaction mixture was filtered through Celite (washed CH2Cl2,~200 mL) and evaporated to dryness. The oil was then dissolved in HCl (250 mL, 1 N) and washed with ethyl acetate (250 mL) to remove excess 24. Additional washings of ethyl acetate (125 mL) may be required to remove remaining 24. The aqueous phase was washed repetitively with CH2Cl2 (125 mL volumes) until most of the 25 has been removed from the aqueous phase. The first extraction will contain 24, 25, and dicoupled side product and should be back extracted with HCl (125 mL, 1N). The organic layers were combined and evaporated to dryness. The resultant oil was then dissolved in CH2Cl2 (100 mL) and washed repetitively with H2O (50 mL volumes) until 25 was removed. The aqueous fractions were combined, total volume 500 mL, and NaCl was added until the solution became cloudy and then was washed with CH2Cl2 (2*500 mL). The organic layers were combined, dried MgSO4, and evaporated to dryness to afford a the non-polydispersed title compound as an oil (16.9 g, 41% yield). It may be desirable to repeat one or more steps of the purification procedure to ensure high purity.
Heptaethylene glycol mono methyl ether (30). To a stirred solution of monodispersed tetraethylene glycol (35.7 mmol) in dry DMF (25.7 mL), under N2 was added in portion a 60% dispersion of NaH in mineral oil, and the mixture was stirred at room temperature for 1 hour. To the resulting sodium salt of the tetraethylene glycol was added a solution of monodispersed mesylate 29 (23.36) in dry DMF (4 ml) in a single portion, and the mixture was stirred at room temperature for 3.5 hours.
With NaH; In N,N-dimethyl-formamide; mineral oil;
Heptaethylene glycol mono methyl ether (30). To a stirred solution of monodispersed tetraethylene glycol (35.7 mmol) in dry DMF (25.7 mL), under N2 was added in portion a 60% dispersion of NaH in mineral oil, and the mixture was stirred at room temperature for 1 hour. To the resulting sodium salt of the tetraethylene glycol was added a solution of monodispersed mesylate 29 (23.36) in dry DMF (4 ml) in a single portion, and the mixture was stirred at room temperature for 3.5 hours.
With NaH; In N,N-dimethyl-formamide; mineral oil;
Heptaethylene glycol mono methyl ether (30). To a stirred solution of monodispersed tetraethylene glycol (35.7 mmol) in dry DMF (25.7 mL), under N2 was added in portion a 60% dispersion of NaH in mineral oil, and the mixture was stirred at room temperature for 1 hour. To the resulting sodium salt of the tetraethylene glycol was added a solution of monodispersed mesylate 29 (23.36) in dry DMF (4 ml) in a single portion, and the mixture was stirred at room temperature for 3.5 hours.
EXAMPLE 3 Tetraethylene glycol bis{3-(3,5-di-tert.-butyl-4-hydroxyphenyl)propionate} 4.1 grams of sodium methylate is added to 48.5 grams of tetraethylene glycol at 60C in a dry nitrogen atmosphere and stirred until dissolved. 100 grams of methyl 3-(3,5-di-tert.-butyl-4-hydroxyphenyl)propionate is added and the reaction mixture heated at 150-154C. 10.2 ml of methanol is collected in a Dean Stark trap over a period of 2 hours. The reaction mixture is then heated at 20 mm Hg. pressure for an additional 25 hours during which an additional 9.5 ml of methanol is collected. The reaction mixture is dissolved in a solvent mixture of about 700 ml of benzene and ether, and neutralized with glacial acetic acid. The solution is then washed with water, sodium carbonate solution, water, and finally dried over sodium sulfate. The crude product is recovered by removing the benzene and ether solvent under reduced pressure. Unreacted methyl 3-(3,5-di-tert.-butyl-4-hydroxyphenyl) propionate and other volatile impurities are removed by distillation with a wiped film evaporator, the wall temperature being first 180 to 200C, then 255C, the pressure being in the range of 1 to 5 microns. The product thus isolated is a syrupy liquid.
To a stirred solution of non-polydispersed compound 11 (35.7 mmol) in dry DMF (25.7 ML), under N2 was added in portion a 60% dispersion of NaH in mineral oil, and the mixture was stirred at room temperature for 1 hour.To this salt 12 was added a solution of non-polydispersed mesylate 9 (23.36) in dry DMF (4 ml) in a single portion, and the mixture was stirred at room temperature for 3.5 hours.Progress of the reaction was monitored by TLC (12% CH3OH-CHCl3).The reaction mixture was diluted with an equal amount of 1N HCl, and extracted with ethyl acetate (2*20 ml) and discarded.Extraction of aqueous solution and work-up gave non-polydispersed polymer 10 (82-84% yield).
41%
To a solution of non-polydispersed tetraethylene glycol (51.5 g, 0.27 mol) in THF (1L) was added potassium t-butoxide (14.8 g, 0.13 mol, small portions over 30 min). The reaction mixture was then stirred for 1 h and then 24 (29.15 g, 0.12 mol) dissolved in THF (90 mL) was added dropwise and the reaction mixture was stirred overnight. The crude reaction mixture was filtered through Celite (washed CH2Cl2, 200 mL) and evaporated to dryness. The oil was then dissolved in HCl (250 mL, 1 N) and washed with ethyl acetate (250 mL) to remove excess 24. Additional washings of ethyl acetate (125 mL) may be required to remove remaining 24. The aqueous phase was washed repetitively with CH2Cl2 (125 mL volumes) until most of the 25 has been removed from the aqueous phase. The first extraction will contain 24, 25, and dicoupled side product and should be back extracted with HCl (125 mL, 1N). The organic layers were combined and evaporated to dryness. The resultant oil was then dissolved in CH2Cl2 (100 mL) and washed repetitively with H20 (50 mL volumes) until 25 was removed. The aqueous fractions were combined, total volume 500 mL, and NaCl was added until the solution became cloudy and then was washed with CH2Cl2 (2×500 mL). The organic layers were combined, dried MgSO4, and evaporated to dryness to afford a the non-polydispersed title compound as an oil (16.9 g, 41% yield). It may be desirable to repeat one or more steps of the purification procedure to ensure high purity.
41%
To a solution of monodispersed tetraethylene glycol (51.5 g, 0.27 mol) in THF (1L) was added potassium t-butoxide (14.8 g, 0.13 mol, small portions over 30 min). The reaction mixture was then stirred for 1 h and then 9 (29.15 g, 0.12 mol) dissolved in THF (90 mL) was added dropwise and the reaction mixture was stirred overnight. The crude reaction mixture was filtered through Celite (washed CH2Cl2, 200 mL) and evaporated to dryness. The oil was then dissolved in HCl (250 mL, 1 N) and washed with ethyl acetate (250 mL) to remove excess 9. Additional washings of ethyl acetate (125 mL) may be required to remove remaining 9. The aqueous phase was washed repetitively with CH2Cl2 (125 mL volumes) until most of the compound 18 has been removed from the aqueous phase. The first extraction will contain 9, 10, and dicoupled side product and should be back extracted with HCl (125 mL, 1N). The organic layers were combined and evaporated to dryness. The resultant oil was then dissolved in CH2Cl2 (100 mL) and washed repetitively with H2O (50 mL volumes) until 10 was removed. The aqueous fractions were combined, total volume 500 mL, and NaCl was added until the solution became cloudy and then was washed with CH2Cl2 (2×500 mL). The organic layers were combined, dried MgSO4, and evaporated to dryness to afford the monodispersed compound 10 as an oil (16.9 g, 41% yield). It may be desirable to repeat one or more steps of the purification procedure to ensure high purity.
30 g
The solution of the compound of formula-9 (54.5 gms) in 34 ml of tetrahydrofuran was slowly added to pre-cooled mixture of sodium hydride (5.6 gms) and tetrahydrofuran (170 ml) at 0-5 C under nitrogen atmosphere and stirred the reaction mixture for 45 minutes at the same temperature. To this reaction mixture, the solution of the compound of formula-8a (34 gms) in tetrahydrofuran (34 ml) was added. Heated the reaction mixture to 40-45 C and stirred for 12 hours at the same temperature. Cooled the reaction mixture to 25-30C and acidified the reaction mixture using aqueous hydrochloric acid solution. Distilled off the solvent completely from the reaction mixture. To this reaction mixture aqueous sodium chloride solution was added and washed with ethyl acetate. Extracted the compound into dichloromethane from the reaction mixture. Combined the dichloromethane layers and washed with aqueous sodium solution and distilled off the solvent completely from the organic layer to get the title compound. Yield: 30 gms.
With NaH; In N,N-dimethyl-formamide; mineral oil;
Heptaethylene glycol mono methyl ether (30). To a stirred solution of monodispersed tetraethylene glycol (35.7 mmol) in dry DMF (25.7 mL), under N2 was added in portion a 60% dispersion of NaH in mineral oil, and the mixture was stirred at room temperature for 1 hour. To the resulting sodium salt of the tetraethylene glycol was added a solution of monodispersed mesylate 29 (23.36) in dry DMF (4 ml) in a single portion, and the mixture was stirred at room temperature for 3.5 hours.
To a solution of tetrahydrofiran (140 ML) containing sodium hydride (0.43 g, 18 mmol) was added dropwise a solution of non-polydispersed tetraethylene glycol (3.5 g, 18 mmol) in tetrahydrofuran (10 ML) and the reaction mixture was stirred for 1 h.Then mesylate of non-polydispersed tetraethylene glycol monobenzylether 40 (6.0 g, 16.5 mmol) dissolved in tetrahydrofuran (10 ML) was added dropwise and the reaction mixture was stirred overnight.The crude reaction mixture was filtered through Celite (washed, CH2Cl2, 250 ML) and the filtrate was washed H2O, dried MgSO4, and evaporated to dryness.The resultant oil was chromatographed (silica, ethyl acetate/methanol, 10:1) and chromatographed (silica, chloroform/methanol, 25:1) to afford the non-polydispersed title compound as a clear oil (2.62 g, 34% yield).
34%
To a solution of tetrahydrofuran (140 ML) containing sodium hydride (0.43 g, 18 mmol) was added dropwise a solution of monodispersed tetraethylene glycol (3.5 g, 18 mmol) in tetrahydrofuran (10 ML) and the reaction mixture was stirred for 1 h.Then mesylate of monodispersed tetraethylene glycol monobenzylether 24 (6.0 g, 16.5 mmol) dissolved in tetrahydrofuran (10 ML) was added dropwise and the reaction mixture was stirred overnight.The crude reaction mixture was filtered through Celite (washed, CH2Cl2, 250 ML) and the filtrate was washed H2O, dried MgSO4, and evaporated to dryness.The resultant oil was chromatographed (silica, ethyl acetate/methanol, 10:1) and chromatographed (silica, chloroform/methanol, 25:1) to afford the monodispersed compound 25 as a clear oil (2.62 g, 34% yield).
Tetra(ethylene glycol) (55 mmol, 10.7 g) was dissolved in 100 mL of tetrahydrofuran (?THF?) and to this solution was added KOtBu (55 mL, 1.0 M in THF) at room temperature. The resulting solution was stirred at room temperature for 30 minutes, followed by dropwise addition of CH3OCH2CH2Br (55 mmol, 5.17 mL in 50 mL THF). The reaction was stirred at room temperature overnight, followed by extraction with H2O (300 mL)/CH2Cl2 (3×300 mL). The organic extracts were combined and then dried over anhydrous Na2SO4. After filtering off the solid drying agent and removing the solvent by evaporation, the recovered crude residue was purified by column chromatography using a silica gel column (CH2Cl2:CH3OH=60:140:1) to give pure penta(ethylene glycol) monomethyl ether (yield 35%). 1H NMR (CDCl3) delta 3.75-3.42 (m, 20 H, OCH2CH2O), 3.39 (s, 3H, MeO).
Into a solution of 17.7 g (126 mmol) of phosphorus trichloride in 100 g of toluene, a solution of 41.0 g (250 mmol) of 2-te/f-butyl-6-methylphenol and 20.6 g (250 mmol) of 1- methylimidazole (MIM) in 70 g of toluene is added dropwise at 20-300C over 2 hours while under a nitrogen purge. The resulting mixture is held at 20-250C for an additional 30 minutes. A solution of 12.3 g (63 mmol) of tetraethyleneglycol and 10.3 g (122 mmol) of MIM is added dropwise over 30 minutes. The mixture is held for an additional hour. The reaction mixture is filtered and the filtrate is concentrated in vaccuo at 70-750C to give a pale yellow, viscous syrup.31P NMR (400MHz) (Benzene-d6) (ppm): 160
A synthesis of Undec-1-en-11-yltetra (ethylene glycol). A mixture of 0.34 mL of 50% aqueous sodium hydroxide (4.3 mmol) and 4.08 g of tetra (ethylene glycol) (21 mmol) was stirred for about 0.5 h in an oil bath at 100 C. under an atmosphere of argon, and then 1.0 g of 11-bromoundec-1-ene (4.3 mmol) was added. After 24 h, the reaction mixture was cooled and extracted six times with hexane. Concentration of the combined hexane portions by rotary evaporation at reduced pressure gave yellow oil containing a mixture of mono- and diethers, according to analysis by 1HNMR spectroscopy. Purification of the oil by chromatography on silica gel (eluant: ethyl acetate) gave 0.98 g of monoether: 76% yield; 1H NMR (400 MHz, CDCl,) 1.22-1.27 (m, 10H), 1.29-1.34 (m, 2H), 1.49-1.56 (m, 2H), 1.96-2.02 (m, 2H), 2.73-2.76 (t, 1H), 3.38-3.42 (t, 2H, J=7 Hz), 3.52-3.69 (m, 16H), 4.86-4.97 (m, 2H), 5.71-5.82 (m, 1H). MS (ESI-MS) calcd for C19H38O5 346.50, found 347.2 [M+H]+.
74%
General procedure: 4.1. O-(Penten-4-yl)-tetra ethyleneglycol (27a) To a solution of tetraethyleneglycol (17 mL, 100 mmol) in THF (10 mL) was added a solution of 50% NaOH (2 mL). The reaction mixture was reflux for 45 min. Then, alkyl bromide or mesylate (20 mmol) was added and the mixture was refluxed further for 16 h. A solution of 10% HCl was added to the mixture and the aqueous layer was extracted with EtOAc (3x). The organic layer was dried over MgSO4, concentrated in vacuum and purified by flash chromatography on silica gel (DCM / MeOH (18:2)) to furnish 27a (75% yield) as a translucent oil.
60%
A mixture of 1 ml of 50% aqueous sodium hydroxide (0.012 mol) and 23.4 g of tetra(ethylene glycol) (0.12 mol) was stirred for 0.5 h in an oil bath at 100 C. under Ar, and 2.8 g of 11-bromoundec-1-ene (0.012 mol) was then added. At the completion of the reaction as indicated by the TLC analysis, the reaction mixture was cooled and extracted several times with hexane. The combined organic layers were washed with brine and dried over sodium sulfate. After evaporation of the solvent in vacuo, the crude product was purified by chromatography column on silica gel (hexane/ethyl acetate, 1:1) to give pure product 2.5 g (yield: 60%). 1H NMR (CDCl3) delta 5.82-5.72 (m, 1H), 4.97-4.87 (m, 2H), 3.68-3.64 (t, 2H, J=4.8 Hz), 3.62-3.54 (m, 14H), 3.42-3.39 (t, 2H, J=7.2 Hz), 2.01-1.97 (m, 3H), 1.55-1.50 (m, 2H), 1.35-1.24 (m, 12H). 13C NMR (CDCl3) delta 139.5, 114.4, 72.8, 71.8, 70.8, 70.7, 70.5, 70.2, 34.0, 29.8, 29.7, 29.6, 29.3, 29.1, 26.3. MS ES+ m/z 369.28 (M+Na).
To 7.28 g (37.5 mmol) of tetraethylene glycol dissolved in 20 mL of DMF was added 0.900 g (37.5 mmol) of NaH and the resulting suspension was stirred for 10 minutes at rt. Then 2.0 g (12.5 mmol) of <strong>[5734-64-5]2-amino-4-chloro-6-methoxypyrimidine</strong> was added and the reaction mixture heated at 80 0C for 3 h. Solvent was removed under reduced pressure and the oily residue was purified by column chromatography (EtOAc:MeOH, 100:0 to 95:5) to give 3.30 g (83 %) of an oil 38. TLC (EtOAc:MeOH, 95:5 v/v): Rf = 0.24; 1U NMR (500 MHz, CDCl3): delta 5.48 (s, IH), 5.08 (br s, 2H), 4.39 (t, J = 4.8 Hz, 2H), 3.83 (s, 3H), 3.79 (t, J = 4.8 Hz, 2H), 3.60-3.73 (m, 12H), 3.21 (br s, IH); 13C NMR (166 MHz, CDCl3): delta 172.6, 172.0, 162.4, 80.2, 72.8, 70.86, 70.80, 70.77, 70.57, 69.7, 65.6, 61.8, 53.9; MS {mlz): [M+H]+ 318.1.
To a solution of tetraethylene glycol (8.66 g, 44.6 mmol) in dry THF (35 mL) was added sodium hydride (60% dispersion in mineral oil) in portions. The mixture was stirred 15 - 20 min under argon before adding a solution of tetraethylene glycol monobenzyl ether tosylate in THF (15 mL) dropwise over 5 h. The reaction mixture was then allowed to stir overnight at room temperature. The reaction was quenched with brine (100 mL) and then THF was removed in vacuo. The aqueous mixture was extracted with dichloromethane (9 x 100 mL), dried over sodium sulfate and then concentrated in vacuo to afford a yellow oil. The crude product was purified by flash chromatography on silica gel (methanol:chloroform, step gradient from 1:99 to 2:98 to 5:95 to 7:93) to yield 7.89 g of 3 as a colorless oil (65%). 1H NMR and 13C NMR spectra of this material agrees with previously published data.2
Multi-step reaction with 2 steps
1.1: 3,4-dihydro-2<i>H</i>-pyran; toluene-4-sulfonic acid / dichloromethane / 24 h / 20 °C / Inert atmosphere
1.2: 0 - 20 °C / Inert atmosphere
1.3: 36 h / 20 °C / Inert atmosphere
2.1: toluene-4-sulfonic acid / tetrahydrofuran / 36 h / 20 °C / Inert atmosphere
Multi-step reaction with 2 steps
1: triethylamine / acetonitrile / 24 h
2: toluene-4-sulfonic acid / dichloromethane; water / 2 h / 20 °C / Cooling with ice
Multi-step reaction with 2 steps
1: triethylamine / dichloromethane / 0 °C
2: pyridinium p-toluenesulfonate / dichloromethane / 5 h / 20 °C
Stage #1: 3,4-dihydro-2<i>H</i>-pyran; Tetraethylene glycol With toluene-4-sulfonic acid In dichloromethane at 20℃; for 24h; Inert atmosphere;
Stage #2: p-toluenesulfonyl chloride With potassium hydroxide In tetrahydrofuran; water at 0 - 20℃; Inert atmosphere; Overall yield = 285 g;
Synthesis of a mixtureof monotetrahydropyran-protected tetraethylene glycol monotosylate (9a) and di-tetrahydropyran-protectedtetraethylene glycol (8a) from a mixture of 7a and 8a.
To a 5-L round bottom flask charged with tetraethylene glycol (6a, 1552 g, 8.000 mol), CH2Cl2(3.0 L) and p-toluenesulfonic acidmonohydrate (6.0 g, 32 mmol) was added dihydropyran (174 mL, 1.84 mol) dropwiseand the resulting reaction mixture was stirred for 24 h at room temperature. Abouthalf of solvent was evaporated, and the reaction mixture was washed with water(1 x 3 L), aqueous NaCl solution (6 x 1 L) and dried over anhydrous sodiumsulfate. The organic layer was concentrated and further dried under vacuum toafford a mixture of 7a and 8a as a colorless liquid which was usedfor subsequent reaction without further purification (442 g, 86.3%). The molar ratioof 7a to 8a was found to be 8:1 estimated by 1H NMR analysis. A mixture of 7aand 8a (190 g, ~0.680 mol) preparedabove and THF (500 mL) were added to a 2-L round bottom flask, and theresulting reaction mixture was cooled in an ice-water bath. A solution of KOH(150 g, 2.70 mol) in water (140 mL) was rapidly added and the resulting mixturewas treated with a solution of TsCl (196 g, 1.03 mol) in 300 mL of THF dropwiseat 0 oC while vigorous stirring. The mixture was allowed to warm to roomtemperature and stirred overnight. The resulting mixture was poured into ice-water(500 mL), and the product was extracted with dichloromethane (5 x 200 mL). Theorganic layer was rinsed with aqueous NaCl solution (5 x 500 mL), dried overanhydrous sodium sulfate and concentrated invacuo to afford the required product (a mixture of 9a and 8a) as a lightyellow viscous liquid (285 g, 97.0%). The molar ratio of 9a to 8a was found to be8:1 estimated by 1H NMR analysis. 1H NMR (mixture, 500MHz, CDCl3) δ 7.82 (d, 2H, J= 10 Hz), 7.36 (d, 2H, J = 10Hz), 4.64 (m, 1H), 4.18 (t, 2H, J = 5 Hz), 3.83-3.91 (m, 2H), 3.56-3.74(m, 15H), 3.46-3.53 (m, 1H), 2.47 (s, 3H), 1.80-1.90 (m, 1H), 1.68-1.77 (m,1H), 1.48-1.66 (m, 4H).
2-(2-(2-(2-hydroxyethoxy)ethoxy)ethoxy)ethyl 2-(2-chlorophenoxy)acetate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
65%
With iodine; In toluene;Reflux;
General procedure: A mixture of phenoxy carboxylic acid 1 (2 mmol), ethylene glycol (10 mmol), I2 (0.025g, 0.1 mmol) in toluene (10 mL) was heated under reflux for 2-3 h. When the reaction was completed (TLC), the mixture was washed with 10% Na2S2O3 (10 mL × 3) and H2O (10 mL × 2). The organic layer was dried over anhydrous Na2SO4. The crude was purified by column chromatography to afford the desired products 3a-3p.
2-(2-(2-(2-hydroxyethoxy)ethoxy)ethoxy)ethyl 2-(2-chloro-4-methylphenoxy)acetate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
63%
With iodine; In toluene;Reflux;
General procedure: A mixture of phenoxy carboxylic acid 1 (2 mmol), ethylene glycol (10 mmol), I2 (0.025g, 0.1 mmol) in toluene (10 mL) was heated under reflux for 2-3 h. When the reaction was completed (TLC), the mixture was washed with 10% Na2S2O3 (10 mL × 3) and H2O (10 mL × 2). The organic layer was dried over anhydrous Na2SO4. The crude was purified by column chromatography to afford the desired products 3a-3p.
tetra(ethylene glycol) mono-butyraldehyde diethyl acetal[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
20.3 g
With potassium tert-butylate; In toluene; tert-butyl alcohol; at 90℃;Inert atmosphere;
A mixture of tetra(ethylene glycol) (97.1 g, 0.500 moles) and toluene (200 ml) was azeotropically dried by distilling off toluene under reduced pressure. The dried tetra(ethylene glycol) was dissolved in anhydrous toluene (180 ml), and a 1.0 M solution of potassium tert-butoxide in tert-butanol (120.0 ml, 0.120 moles) and <strong>[6139-83-9]4-chlorobutyraldehyde diethyl acetal</strong> (18.1 g, 0.100 moles) were added. The mixture was stirred at 90 C. overnight under argon atmosphere. After cooling to room temperature, the mixture was filtered and the solvents were distilled off under reduced pressure. The crude product was dissolved in 1000 ml deionized water and the resulting solution was filtered through activated carbon. Sodium chloride (10 g) was added and the product was extracted with dichloromethane (250, 200, and 150 ml). The extract was dried (MgSO4) and the solvent was distilled off under reduced pressure. Yield: 20.3 g. NMR (d6-DMSO): 1.10 ppm (t, CH3-C-) 1.51 ppm (m, C-CH2-CH2-), 3.49 ppm (bm, -OCH2CH2O-), 4.46 ppm (t, -CH, acetal), 4.58 ppm (t, -OH). Purity: 100%.
20.3 g
With potassium tert-butylate; In toluene; tert-butyl alcohol; at 95 - 100℃;Inert atmosphere;
A mixture of tetra(ethylene glycol) (97.1 g, 0.500 moles) and toluene (200 ml) was azeotropically dried by distilling off toluene under reduced pressure (rotary evaporator). The dried tetra(ethylene glycol) was dissolved in anhydrous toluene (180 ml) and 1.0 M solution of potassium tert-butoxide in tert-butanol (120.0 ml, 0.120 moles) and <strong>[6139-83-9]4-chlorobutyraldehyde diethyl acetal</strong> (18.1 g, 0.100 moles) (Alfa Aesar, Ward Hill, Mass.) were added. The mixture was stirred at 95-100 C. overnight under argon atmosphere. After cooling to room temperature, the mixture was filtered and the solvents were distilled off under reduced pressure. The crude product was dissolved in 1000 ml deionized water and the resulting solution was filtered through active carbon. Sodium chloride (100 g) was added and the product was extracted with dichloromethane (250, 200, and 150 ml). The extract was dried (over MgSO4) and the solvent was distilled off under reduced pressure (by rotary evaporation). The crude product was dissolved in 300 ml 10% phosphate buffer (pH=7.5) and impurities were extracted with ethyl acetate (2*50 ml). The resulting product was extracted with dichloromethane (200, 150, and 100 ml). The extract was dried (over MgSO4) and the solvent was distilled off under reduced pressure (by rotary evaporation). Yield: 20.3 g. NMR (d6-DMSO): 1.10 ppm (t, CH3-C-) 1.51 ppm (m, C-CH2-CH2-), 3.49 ppm (bm, -OCH2CH2O-), 4.46 ppm (t, -CH, acetal), 4.58 ppm (t, -OH). Purity: ?100% (no signs of unreacted starting materials).
20.3 g
With potassium tert-butylate; sodium chloride; In toluene; tert-butyl alcohol; at 95 - 100℃;Inert atmosphere;
A mixture of tetra(ethylene glycol) (97.1 g, 0.500 moles) and toluene (200 ml) was azeotropically dried by distilling off toluene under reduced pressure (rotary evaporator). The dried tetra(ethylene glycol) was dissolved in anhydrous toluene (180 ml) and 1.0 M solution of potassium tert-butoxide in tert-butanol (120.0 ml, 0.120 moles) and <strong>[6139-83-9]4-chlorobutyraldehyde diethyl acetal</strong> (18.1 g, 0.100 moles) (Alfa Aesar, Ward Hill, Mass.) were added. The mixture was stirred at 95-100 C. overnight under argon atmosphere. After cooling to room temperature, the mixture was filtered and the solvents were distilled off under reduced pressure. The crude product was dissolved in 1000 ml deionized water and the resulting solution was filtered through active carbon. Sodium chloride (100 g) was added and the product was extracted with dichloromethane (250, 200, and 150 ml). The extract was dried (over MgSO4) and the solvent was distilled off under reduced pressure (by rotary evaporation). The crude product was dissolved in 300 ml 10% phosphate buffer (pH=7.5) and impurities were extracted with ethyl acetate (2*50 ml). The resulting product was extracted with dichloromethane (200, 150, and 100 ml). The extract was dried (over MgSO4) and the solvent was distilled off under reduced pressure (by rotary evaporation). Yield: 20.3 g. NMR (d6-DMSO): 1.10 ppm (t, CH3-C-) 1.51 ppm (m, C-CH2-CH2-), 3.49 ppm (bm, -OCH2CH2O-), 4.46 ppm (t, -CH, acetal), 4.58 ppm (t, -OH). Purity: ?100% (no signs of unreacted starting materials).
[2-(4-chlorophenyl)-6,6-dimethyl-1-phenyl-6,7-dihydro-5H-pyrrolizin-3-yl]-acetic acid 2-{2-[2-(2-hydroxy-ethoxy)-ethoxy]-ethoxy}-ethyl ester[ No CAS ]
[2-(4-chlorophenyl)-6,6-dimethyl-1-phenyl-6,7-dihydro-5H-pyrrolizin-3-yl]-acetic acid 2-{2-[2-(2-{2-[2-(4-chlorophenyl)-6,6-dimethyl-1-phenyl-6,7-dihydro-5H-pyrrolizin-3-yl]-acetoxy}-ethoxy)-ethoxy]-ethoxy}-ethyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
1.06 g; 0.76 g
With dmap; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; In tetrahydrofuran; for 6h;
Example 4a: [2-(4-Chloro-phenyl)-6,6-dimethyl-l-phenyl-6,7-dihydro-5H- pyrrolizin-3-yl] -acetic acid 2-{2-[2-(2-hydroxy-ethoxy)-ethoxy]-ethoxy}-ethyl ester (4a) and Reference Example 4b: [2-(4-Chloro-phenyl)-6,6-dimethyl-l-phenyl-6,7-dihydro- 5H-pyrrolizin-3-yl]-acetic acid 2-{2-[2-(2-{2-[2-(4-chloro-phenyl)-6,6-dimethyl-l- phenyl-6,7-dihydro-5H-pyrrolizin-3-yl]-acetoxy}-ethoxy)-ethoxy]-ethoxy}-ethyl ester (4b) (0141) (0142) 1.53 g (7.9 mmol) tetraethylenglykol, 1.21 g (6.3 mmol) EDCxHCl, 30 mg (0.3 mmol) DMAP and 2 g (5.3 mmol) 6-(4-chlorophenyl)-2,2-dimethyl-7-phenyl-2,3-dihydro- lH-pyrrolizin-5-yl] acetic acid (<strong>[156897-06-2]licofelone</strong>) were dissolved in 40 ml THF. The reaction mixture was stirred for 6 hours. Stirring was stopped (bright yellow solution with a syrupy white precipitate) and the organic layer was decanted off from the syrupy product. To the residue 40 ml THF were added and the mixture was stirred for 1 minute before being decanted off again. The solvent of the combined THF layers was evaporated, yielding a yellow syrup. The crude product contained two major products, 4a and 4b. After flash chromatography (gradient: ethyl acetate/n-heptane 1 :2 (v/v) -> ethyl acetate/n-heptane 2: 1 -> ethyl acetate (100%) to yield the above products: (0143) 4b (0.76 g (0.8 mmol)) was obtained as a colorless solid (Rf (ethyl acetate/n-heptane 2: 1): 0.63). The product was dried at 7x10" mbar at RT for 2 hours. (0144) 1H NMR (400 MHz, acetone- d6) delta [ppm] : 1.29 (s, 12 H) 2.80 - 2.82 (m, 5 H) 3.55 (s, 4 H) 3.58 (s, 8 H) 3.66 - 3.70 (m, 4 H) 3.81 (s, 4 H) 4.20 - 4.26 (m, 4 H) 7.02 -7.07 (m, 6 H) 7.14 - 7.19 (m, 8 H) 7.29 (d, J=8.21 Hz, 4 H) LC-MS: tr: 15.0 min.; m/z: 917 [M+H]+ (method D) (0145) 4a (Rf EtOAc/H 2/1 : 0.12) was obtained as slightly brown to yellow oil. After a second chromatography (silica, 100% ethyl acetate), 4a was obtained as a yellow oil (1.06 g; 0.8 mmol). The product was dried at 7x10" mbar at RT for 2 hours. (0146) 1H NMR (400 MHz, acetone- d6) delta [ppm] : 1.30 (s, 6 H) 2.79 - 2.84 (m, 3 H) 3.44 - 3.56 (m, 3 H) 3.57 (s, 6 H) 3.59 - 3.61 (m, 5 H) 3.69 - 3.72 (m, 2 H) 3.83 (s, 2 H) 4.23 - 4.27 (m, 2 H) 7.03 - 7.08 (m, 3 H) 7.14 - 7.20 (m, 4 H) 7.31 (d, J=8.60 Hz, 2 H) (0147) LC-MS: tr: 10.7 min.; m/z: 556 [M+H]+ (method D)
With sodium hydride; In dimethyl sulfoxide; mineral oil; at 0 - 20℃; for 23h;Inert atmosphere;
To a cold (0 C) stirred solution of tert-butyl 2-bromoacetate (5.4 mL, 36.6 mmol) and 2,2'-((oxybis(ethane-2,l-diyl))bis(oxy))diethanol (35.0 mL, 203 mmol) in DMSO (50 mL) was added NaH (1.6217 g, 40.6 mmol, 60% in oil) in two portions. The reaction mixture was slowly warmed up to rt, stirred for 23 h, diluted with water (50 mL), saturated with NaCl (50 g), stirred for 5 min, extracted with Et20 (3 x 100 mL), and concentrated. The residue was taken in DCM (50 mL) and water (25 mL), stirred for 5 min. The organic solution was passed through a phase separator, concentrated and purified on a 80 g Si02 cartridge using a gradient of ethyl acetate in hexanes (50- 100%) to afford the title compound (8.2 g, 72.7% yield) as a pale yellow oil. Rf = 0.08 (EtOAc). NMR (400 MHz, CDCb) delta 4.02 (d, J = 1.0 Hz, 2H), 3.75 - 3.60 (m, 12H), 1.47 (s, 9H) ppm.LRMS (ESI) m/z: calculated for Ci4H2807 308.3679; found 330.95 (M+Na)+; Found 347.01 (M+K)+.
With sodium chloride; In tetrahydrofuran; hydrogenchloride; dichloromethane; ethyl acetate;
Example 20 Heptaethylene Glycol Monomethyl Ether (25) To a solution of non-polydispersed tetraethylene glycol (51.5 g, 0.27 mol) in THF (1L) was added potassium t-butoxide (14.8 g, 0.13 mol, small portions over ~30 min). The reaction mixture was then stirred for lh and then 24 (29.15 g, 0.12 mol) dissolved in THF (90 mL) was added dropwise and the reaction mixture was stirred overnight. The crude reaction mixture was filtered through Celite (washed CH2Cl2,~200 mL) and evaporated to dryness. The oil was then dissolved in HCl (250 mL, 1 N) and washed with ethyl acetate (250 mL) to remove excess 24. Additional washings of ethyl acetate (125 mL) may be required to remove remaining 24. The aqueous phase was washed repetitively with CH2Cl2 (125 mL volumes) until most of the 25 has been removed from the aqueous phase. The first extraction will contain 24, 25, and dicoupled side product and should be back extracted with HCl (125 mL, 1N). The organic layers were combined and evaporated to dryness. The resultant oil was then dissolved in CH2Cl2 (100 mL) and washed repetitively with H2O (50 mL volumes) until 25 was removed. The aqueous fractions were combined, total volume 500 mL, and NaCl was added until the solution became cloudy and then was washed with CH2Cl2 (2*500 mL). The organic layers were combined, dried MgSO4, and evaporated to dryness to afford a the non-polydispersed title compound as an oil (16.9 g, 41% yield). It may be desirable to repeat one or more steps of the purification procedure to ensure high purity.
41%
With sodium chloride; In tetrahydrofuran; hydrogenchloride; dichloromethane; ethyl acetate;
Example 20 Heptaethylene glycol monomethyl ether (25) To a solution of non-polydispersed tetraethylene glycol (51.5 g, 0.27 mol) in THF (1L) was added potassium t-butoxide (14.8 g, 0.13 mol, small portions over -30 min). The reaction mixture was then stirred for lh and then 24 (29.15 g, 0.12 mol) dissolved in THF (90 mL) was added dropwise and the reaction mixture was stirred overnight. The crude reaction mixture was filtered through Celite (washed CH2Cl2, ~200 mL) and evaporated to dryness. The oil was then dissolved in HCl (250 mL, 1 N) and washed with ethyl acetate (250 mL) to remove excess 24. Additional washings of ethyl acetate (125 mL) may be required to remove remaining 24. The aqueous phase was washed repetitively with CH2Cl2 (125 mL volumes) until most of the 25 has been removed from the aqueous phase. The first extraction will contain 24, 25, and dicoupled side product and should be back extracted with HCl (125 mL, 1N). The organic layers were combined and evaporated to dryness. The resultant oil was then dissolved in CH2Cl2 (100 mL) and washed repetitively with H2O (50 mL volumes) until 25 was removed. The aqueous fractions were combined, total volume 500 mL, and NaCl was added until the solution became cloudy and then was washed with CH2Cl2 (2*500 mL). The organic layers were combined, dried MgSO4, and evaporated to dryness to afford a the non-polydispersed title compound as an oil (16.9 g, 41% yield). It may be desirable to repeat one or more steps of the purification procedure to ensure high purity.
(21E,23E,25E,26E,38R,39S,40R,41R,43S,45S,48S,49R,50R,59R)-49,59-dihydroxy-47-[2-[2-[2-(2-hydroxyethoxy)ethoxy]ethoxy]ethoxy]-48-[(1R)-2-[(1S,3R,4R)-4-(2-hydroxyethoxy)-3-methoxy-cyclohexyl]-1-methyl-ethyl]-50-methoxy-38,39,40,41,51,52-hexamethyl-70,71-dioxa-60-azatricyclohexatriaconta-21,23,25(51),26(52)-tetraene-53,54,55,56,57-pentone[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
17%
With toluene-4-sulfonic acid; In tetrahydrofuran; water; at 20℃; for 2h;
A mixture of <strong>[159351-69-6]everolimus</strong> (0.5 g, 0.52 mmol), 2-[2-[2-(2- hydroxyethoxy)ethoxy]ethoxy]ethanol (2.03 g, 10.44 mmol) and p-toluenesulfonic acid hydrate (0.54 g, 3.13 mmol) in THF (6 mL) was stirred at 20 C for 2 h. The mixture was poured into ice cold saturated NaHCO3 (30 mL) and extracted with EtOAc (50 mL*3). The combined organic layers were washed with water, brine, then concentrated in vacuo. The residue was purified by reversed phase chromatography (CH3CN/pure water = 1:1) to afford (21E,23E,25E,26E,38R,39S,40R,41R,42S,45S,47S,49R,50R,59R)-49,59-dihydroxy-46-[2-[2- [2-(2-hydroxyethoxy)ethoxy]ethoxy]ethoxy]-47-[(1R)-2-[(1S,2R,3R)-3-(2-hydroxyethoxy)- 2-methoxy-cyclohexyl]-1-methyl-ethyl]-50-methoxy-38,39,40,41,51,52-hexamethyl-70,71- dioxa-60-azatricyclohexatriaconta-21,23,25(51),26(52)-tetraene-53,54,55,56,57-pentone (I- 22: 0.1 g, 17% yield) as a white solid. ESI-MS (EI+, m/z): 1043.4 [M+Na]+. 1H NMR (500 MHz, CDCl3) d 6.71- 5.78 (m, 4H), 5.77- 5.01 (m, 4H), 4.65- 3.88 (m, 3H), 3.87- 3.50 (m, 20H), 3.50- 3.00 (m, 14H), 2.77- 1.95 (m, 11H), 1.89- 1.53 (m, 13H), 1.53- 0.79 (m, 27H), 0.76- 0.62 (m, 1H).
(21E,23E,25E,26E,38R,39S,40R,41R,43S,45S,47R,48S,49R,50R,59R)-49,59-dihydroxy-47-[2-[2-[2-(2-hydroxyethoxy)ethoxy]ethoxy]ethoxy]-48-[(1R)-2-[(1S,3R,4R)-4-(2-hydroxyethoxy)-3-methoxy-cyclohexyl]-1-methyl-ethyl]-50-methoxy-38,39,40,41,51,52-hexamethyl-70,71-dioxa-60-azatricyclohexatriaconta-21,23,25(51),26(52)-tetraene-53,54,55,56,57-pentone[ No CAS ]
(21E,23E,25E,26E,38R,39S,40R,41R,43S,45S,47S,48S,49R,50R,59R)-49,59-dihydroxy-47-[2-[2-[2-(2-hydroxyethoxy)ethoxy]ethoxy]ethoxy]-48-[(1R)-2-[(1S,3R,4R)-4-(2-hydroxyethoxy)-3-methoxy-cyclohexyl]-1-methyl-ethyl]-50-methoxy-38,39,40,41,51,52-hexamethyl-70,71-dioxa-60-azatricyclohexatriaconta-21,23,25(51),26(52)-tetraene-53,54,55,56,57-pentone[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
300 mg; 563 mg
With toluene-4-sulfonic acid; In tetrahydrofuran; at 0 - 25℃; for 3h;
3'-O-(tert-butyl)dimethylsilyl-5-[(4-decyl-1,2,3-triazol-1-yl)methyl]-2'-deoxyuridine[ No CAS ]
3'-O-(tert-butyl)dimethylsilyl-5'-O-carbonyltetraethylene glycol-5-[(4-decyl-1,2,3-triazol-1-yl)methyl]-2'-deoxyuridine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
58%
General procedure: A nucleoside (0.28mmol) was dissolved in dry dimethylformamide(1ml) and CDT was added (137mg, 0.84mmol).The mixture was heated for 24 h at 37 C. Then the anhydroustri- or tetraethylene glycol (1.65 g for triethylene glycol, 2.1 gfor tetraethylene glycol, 11mol) and dioxane (0.5ml) wereadded. The mixture was heated for 24 h at 37 C and thenevaporated to obtain crude oil with constant volume. Theproduct was extracted in the chloroform-water system, theorganic layer was evaporated. The product was isolated bycolumn chromatography in the chloroform:ethyl acetate:ethanol (5:5:0.1 v/v) eluting system.
5'-O-(tert-butyl)dimethylsilyl-5-azidomethyl-2'-deoxyuridine[ No CAS ]
C25H43N5O11Si[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
66%
General procedure: A nucleoside (0.28mmol) was dissolved in dry dimethylformamide(1ml) and CDT was added (137mg, 0.84mmol).The mixture was heated for 24 h at 37 C. Then the anhydroustri- or tetraethylene glycol (1.65 g for triethylene glycol, 2.1 gfor tetraethylene glycol, 11mol) and dioxane (0.5ml) wereadded. The mixture was heated for 24 h at 37 C and thenevaporated to obtain crude oil with constant volume. Theproduct was extracted in the chloroform-water system, theorganic layer was evaporated. The product was isolated bycolumn chromatography in the chloroform:ethyl acetate:ethanol (5:5:0.1 v/v) eluting system.
5'-O-(tert-butyl)dimethylsilyl-5-[(4-decyl-1,2,3-triazol-1-yl)methyl]-2'-deoxyuridine[ No CAS ]
3'-O-carbonyltetraethylene glycol-5'-O-(tert-butyl)dimethylsilyl-5-[(4-decyl-1,2,3-triazol-1-yl)methyl]-2'-deoxyuridine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
56%
General procedure: A nucleoside (0.28mmol) was dissolved in dry dimethylformamide(1ml) and CDT was added (137mg, 0.84mmol).The mixture was heated for 24 h at 37 C. Then the anhydrous tri- or tetraethylene glycol (1.65 g for triethylene glycol, 2.1 g for tetraethylene glycol, 11mol) and dioxane (0.5ml) wereadded. The mixture was heated for 24 h at 37 C and thenevaporated to obtain crude oil with constant volume. Theproduct was extracted in the chloroform-water system, theorganic layer was evaporated. The product was isolated bycolumn chromatography in the chloroform:ethyl acetate:ethanol (5:5:0.1 v/v) eluting system.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping