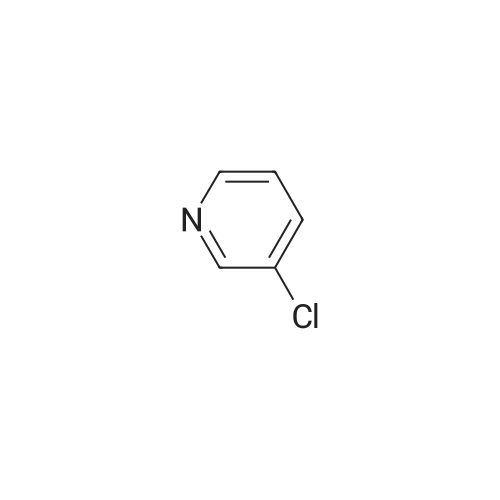
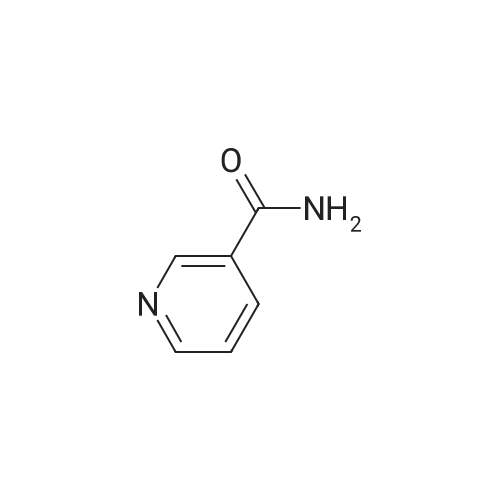
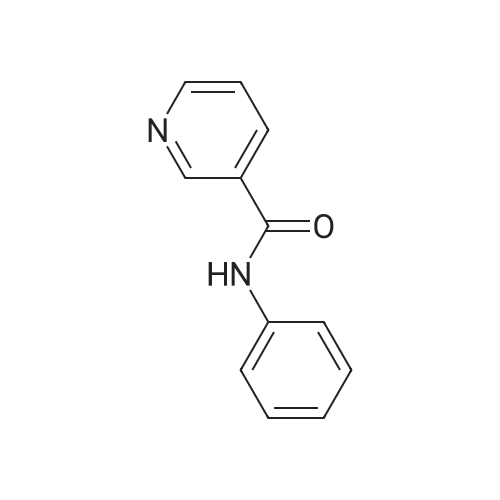
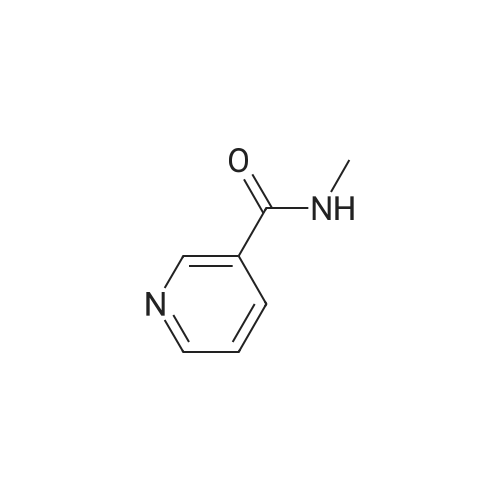
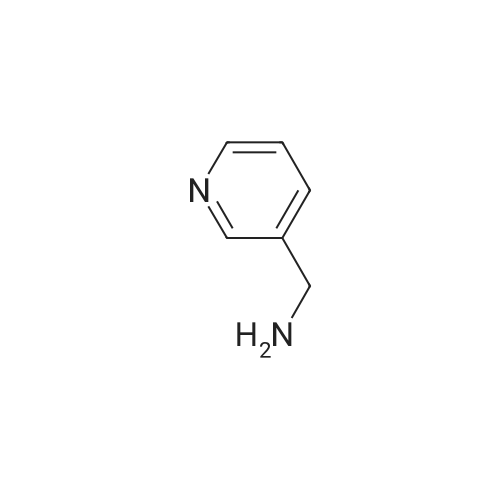
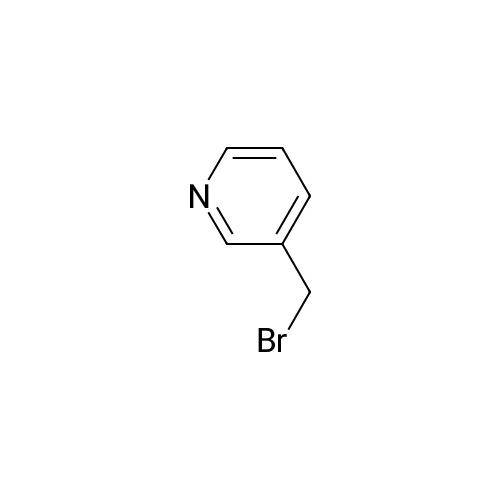
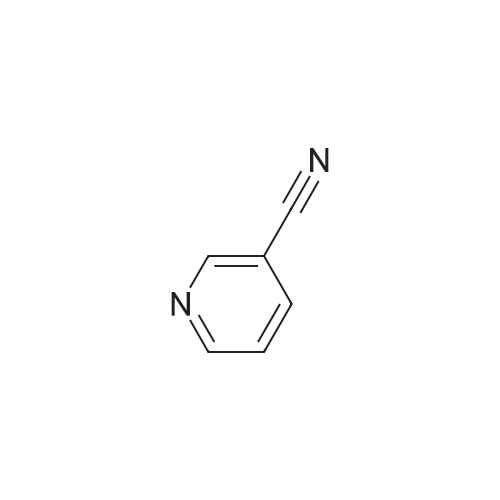
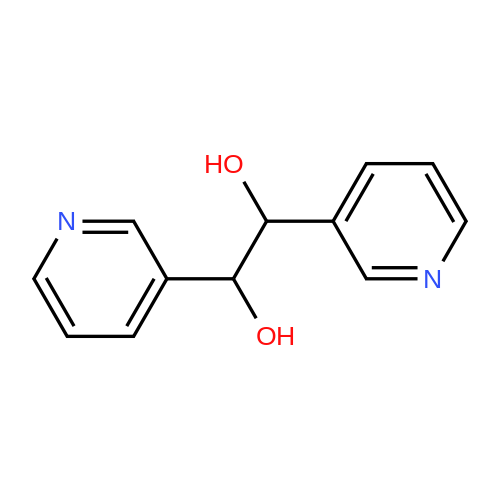
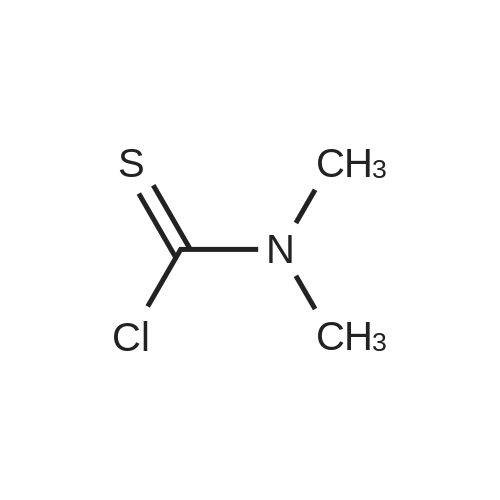
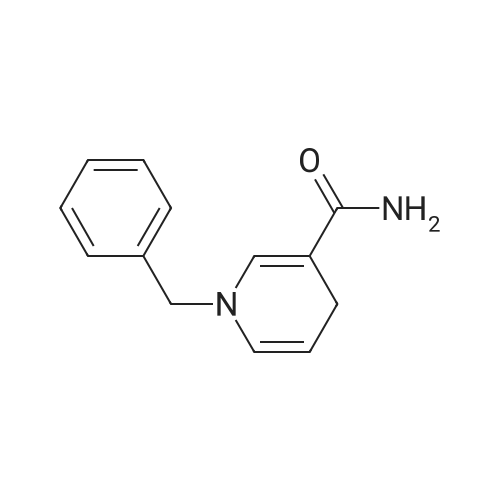
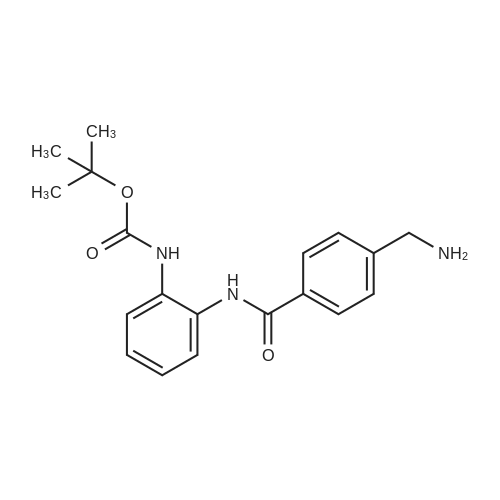
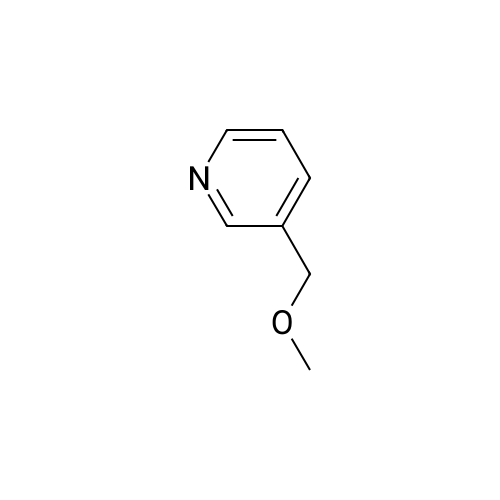
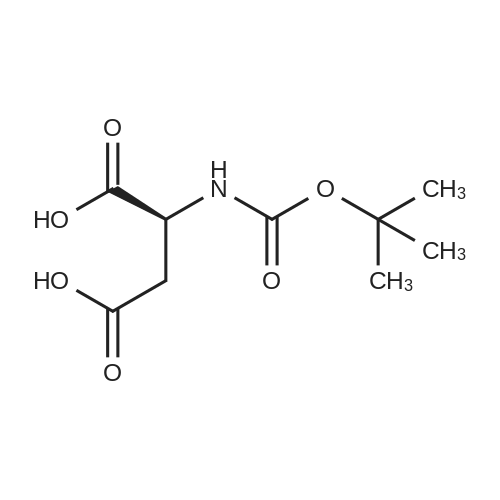
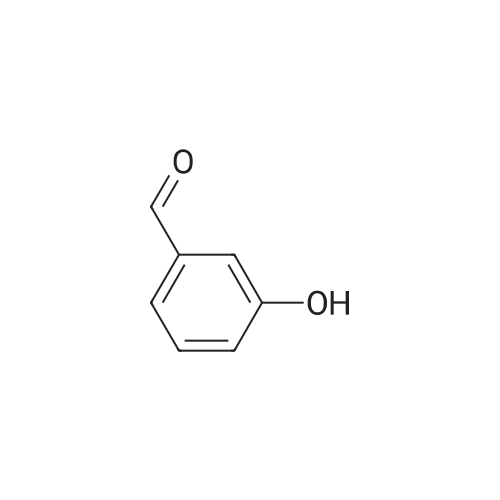
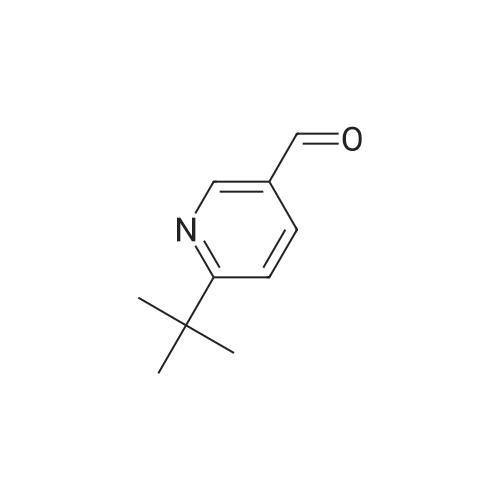
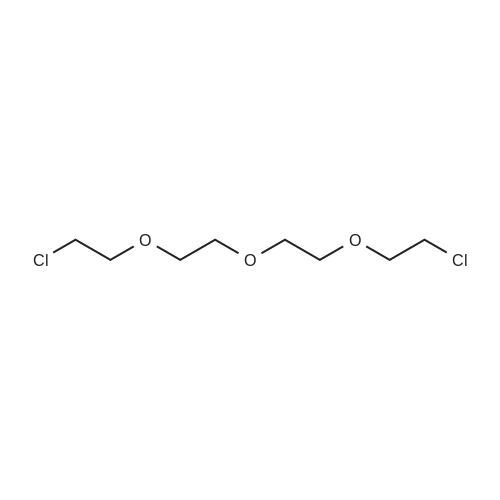
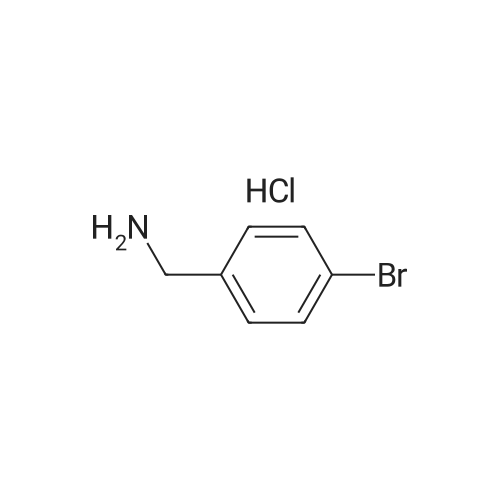
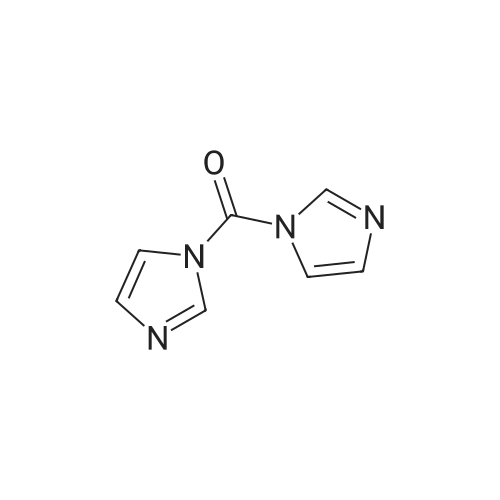
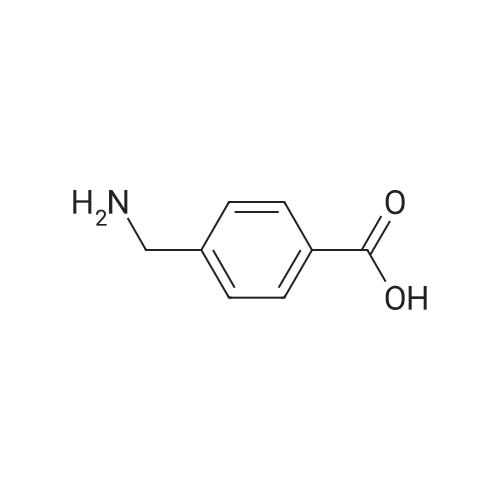
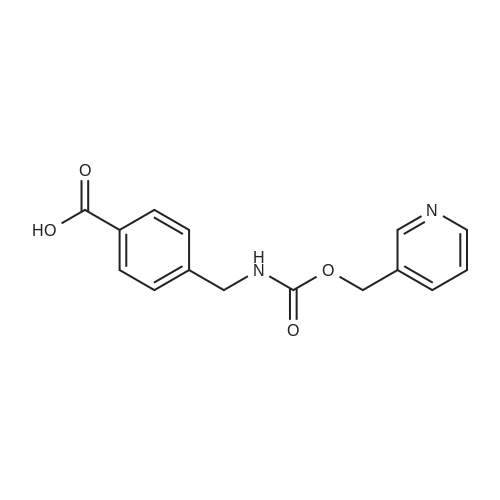
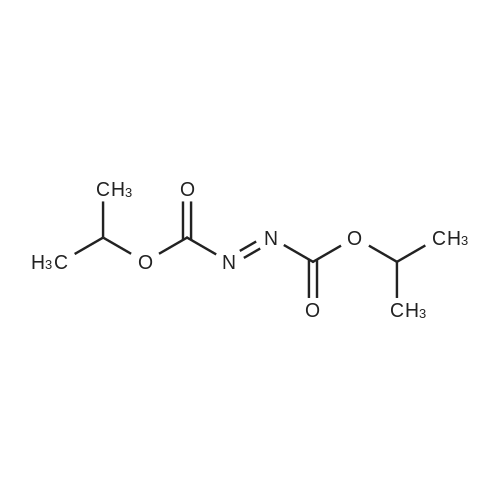
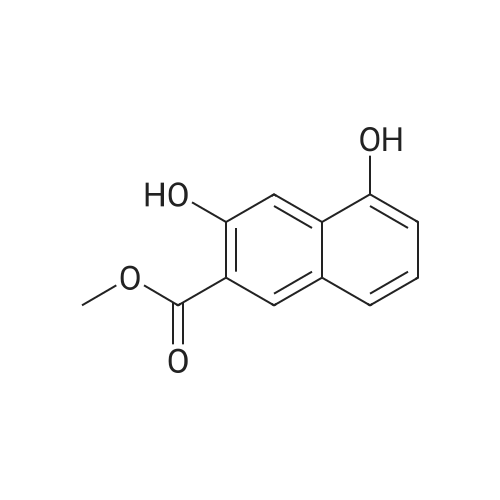
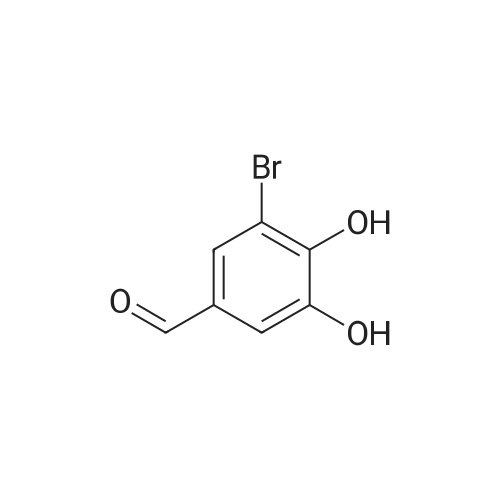
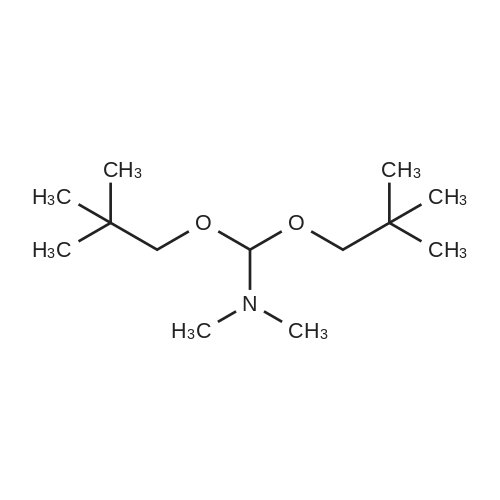
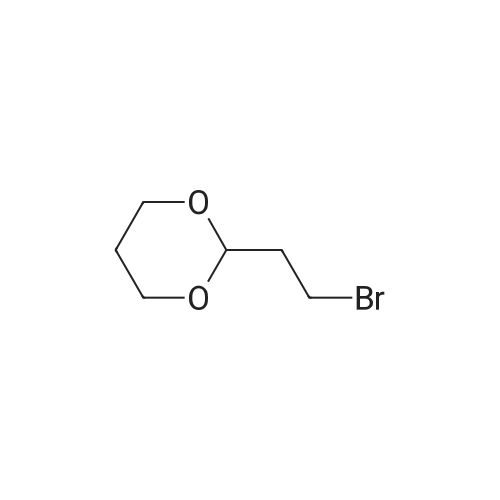
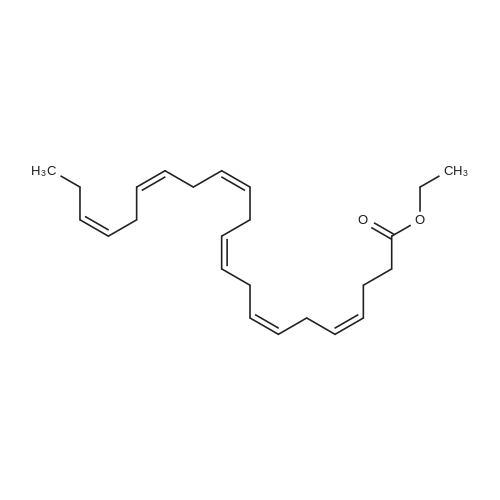
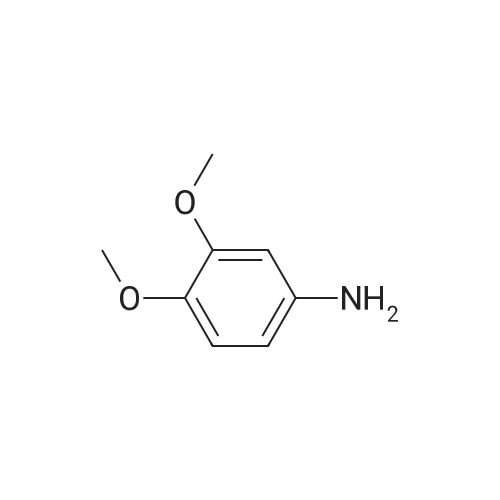
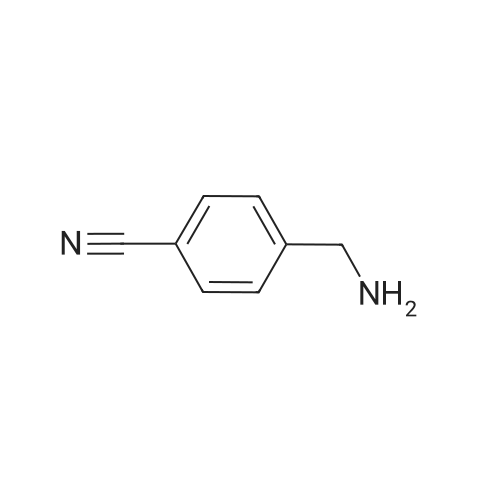
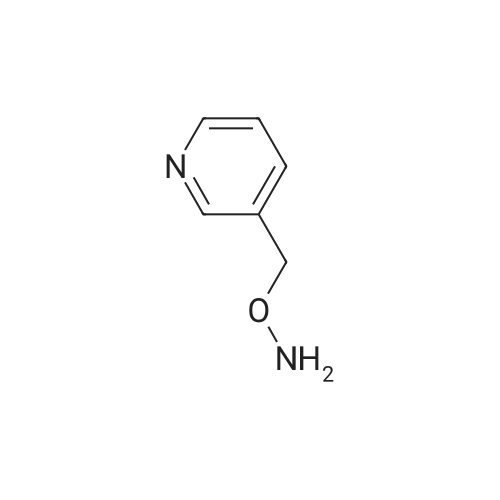
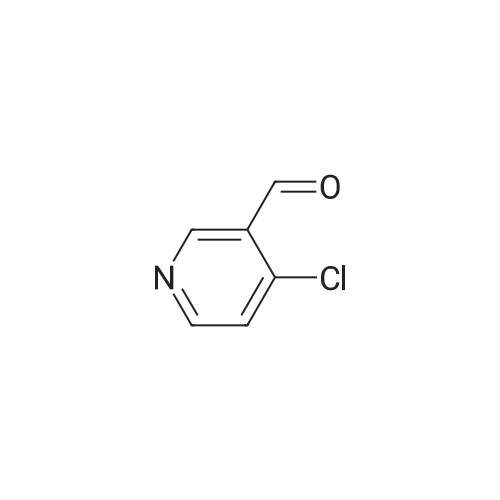
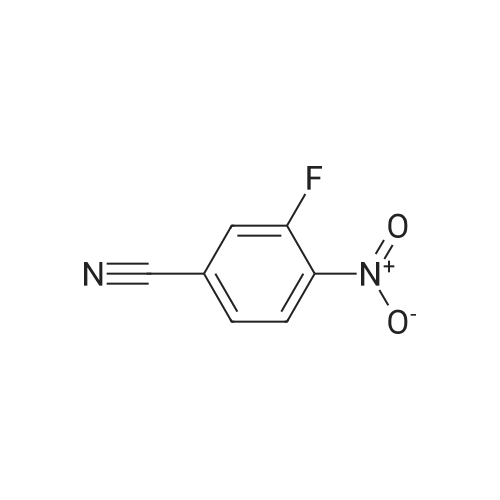
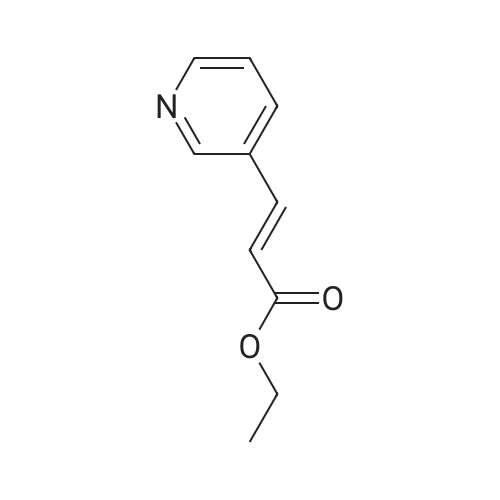
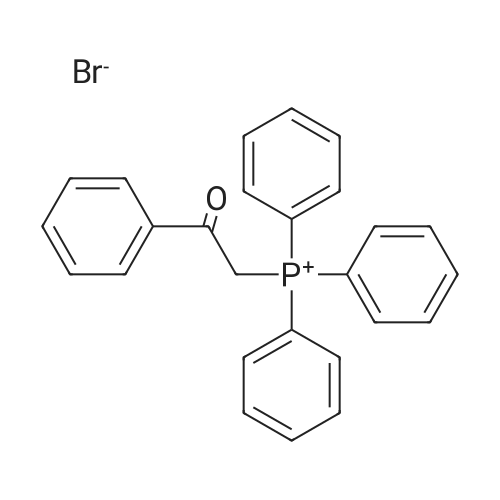
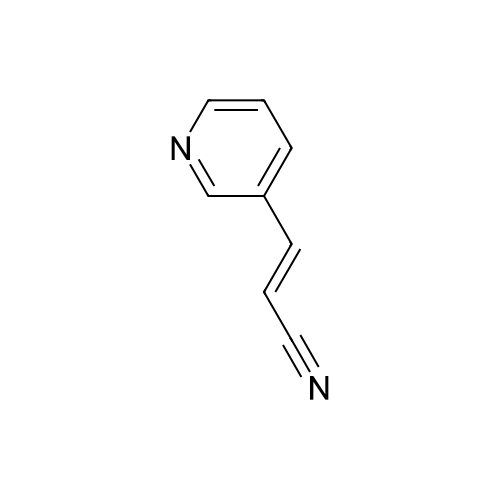
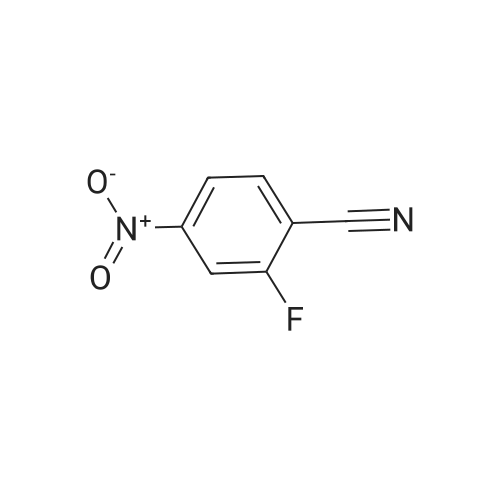
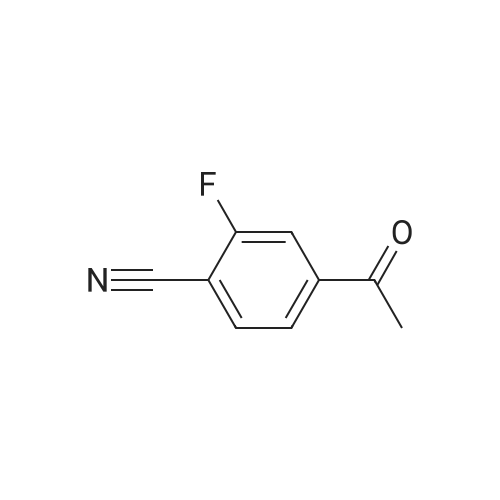
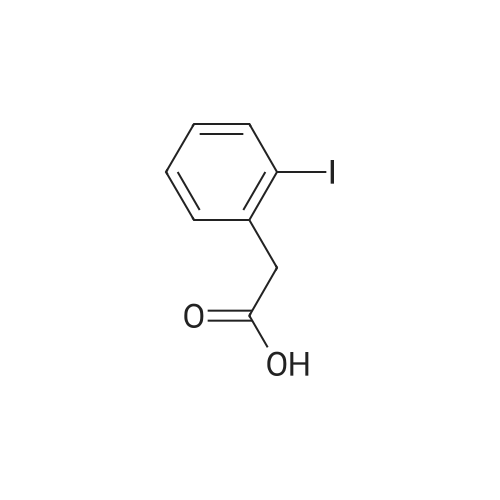
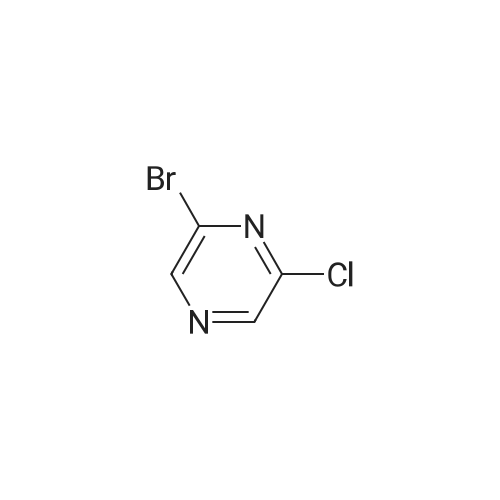
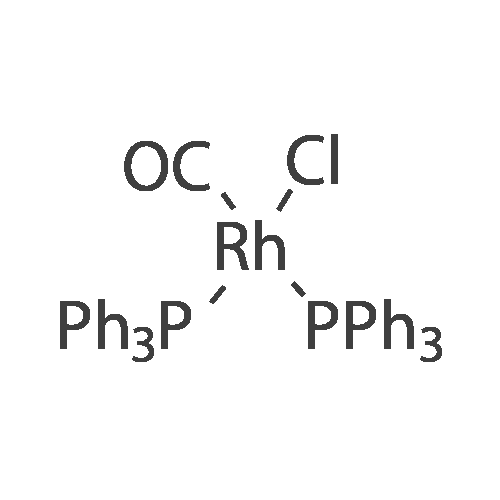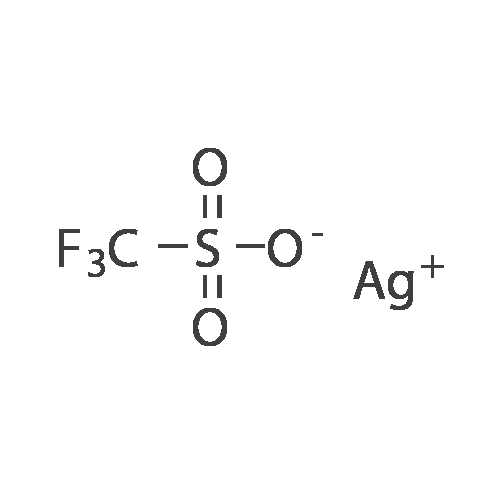| 99% |
With tert.-butylnitrite; oxygen; acetic acid In toluene at 50℃; for 1h; |
|
| 98% |
With 2,2,6,6-tetramethyl-1-piperidinyloxy free radical; bromine; NaNO2 In dichloromethane at 100℃; for 5h; |
|
| 97% |
With hydrogenchloride; tert.-butylnitrite; TEMPOL; oxygen; 2,3,5,6-tetrachlorocyclohexa-2,5-diene-1,4-dione; acetic acid In neat (no solvent) at 20℃; for 24h; Green chemistry; |
|
| 96% |
With sulfuric acid; dihydrogen peroxide; sodium bromide In 1,4-dioxane; water monomer at 70℃; Flow reactor; Green chemistry; |
|
| 96% |
With oxygen In 1,3,5-trimethyl-benzene at 60℃; for 12h; |
8.19 Example 8 Application of this method in the reaction of other alcohols to aldehydes and ketones
The typical reaction steps are as follows:1 mmol of the starting alcohol of the reactant column shown in Table 2,OH - Ni3In-LDH 14 mg,Mesitylene 5mL were added to the reactor,Into the oxygen,Atmospheric reaction,The reaction was stirred at 60 for a certain period of time.The solid catalyst was removed by filtration,Using gas chromatography internal standard method (chlorobenzene as internal standard) to analyze the content of liquid products,Calculate yield. |
| 95% |
With Amberlyst A-27 supported permanganate In dichloromethane |
|
| 95% |
With acetic anhydride; NaNO2 at 25℃; |
|
| 95% |
With 2,2,6,6-tetramethyl-1-piperidinyloxy free radical; tert.-butylnitrite; oxygen; acetic acid In 1,2-dichloro-ethane for 3h; Autoclave; Heating; |
|
| 95% |
With TGSE; Sodium hydrogenocarbonate; anhydrous sodium carbonate In water monomer for 8.33333h; Electrochemical reaction; |
|
| 95% |
With 3,3-dichloro-1,2-diphenylcyclopropene; dimethyl sulfoxide; triethylamine In dichloromethane at -78 - 20℃; Inert atmosphere; |
20 Example 20
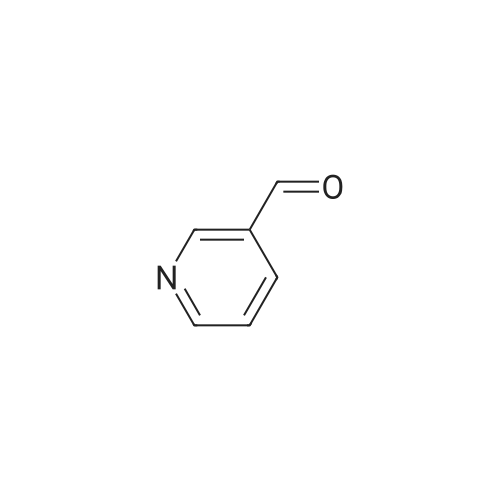
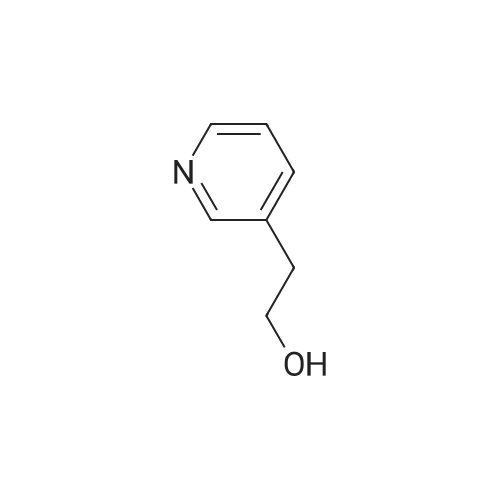
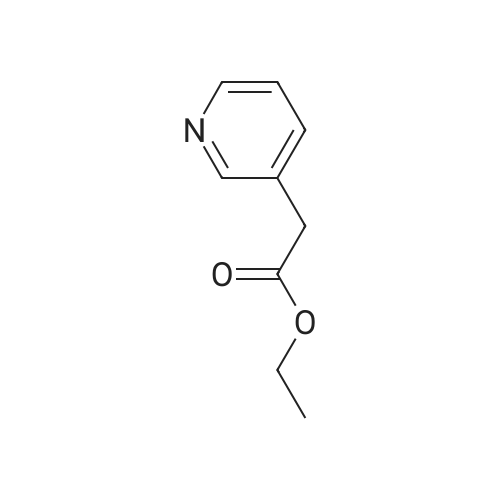
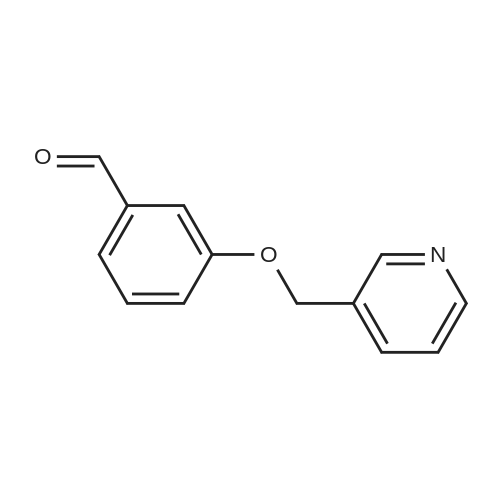
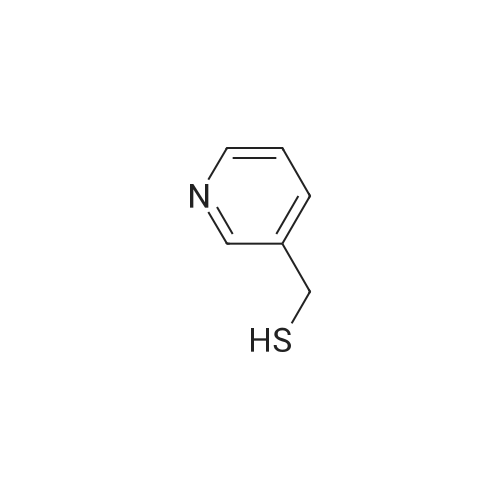
Place a round bottom flask in a cryogenic coolant circulation tank,Add 3,3-dichloro-1,2-diphenylcyclopropene (8) (522.30 mg, 2 mmol) to the bottle under Ar protection.And 10 ml of dichloromethane,Cool the inside of the bottle to -78 ° C,Dimethyl sulfoxide (312.52 mg, 4 mmol) was added dropwise.Keep the temperature below -70 ° C, continue to stir for 30 min after the dropwise addition.Keep the temperature inside the bottle at -78 ° C - 70 ° C,3-pyridinemethanol (13) (109.13 mg, 1 mmol) was added dropwise.The temperature of the dropping process does not exceed -65 ° C, and the stirring is continued for 30 min.Add triethylamine (404.76 mg, 4 mmol),Stirring was allowed to return to room temperature and the target product selectivity was 98%.After the reaction, a saturated ammonium chloride solution was poured into the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was combined, washed with saturated NaCI.Obtaining pure 4-heptanone,Colorless liquid, yield 95%. |
| 95% |
With potassium carbonate In n-heptane at 80℃; for 24h; |
S4. Procedure for the synthesis of aldehydes and ketones
General procedure: A magnetic stir bar, 0.5 mmol alcohol and 3 mL n-heptane solvent were added to 20 mL glass tube. Then, 35mg catalyst and 10 mol% of K2CO3 were added. The glass tube containing reaction mixture was f itted withseptum and connected to a balloon containing one bar of air. Then the glass tube was placed into a preheatedaluminum block at 85°C. Temperature inside the reaction tube was measured to be 80 oC and this temperaturehas been taken as the reaction temperature. The reaction was allowed to progress under continuous stirringfor the required time at 80 °C. Af ter completion of the reaction, the glass tube was cooled down to roomtemperature. Afterwards, the catalyst was f iltered-off and washed with ethyl acetate. The solvent f rom thef iltrate containing the reaction products was removed in vacuum and the corresponding aldehyde/ketone waspurif ied by column chromatography. All products were analyzed by GC-MS and NMR spectroscopy analysis.In the case of yields determined the by GC, 100 μL n-hexadecane was added to the reaction vial containingthe products and diluted with ethyl acetate. Then, the reaction mixture containing catalyst and products wasf iltered through a plug of silica and the filtrate containing product was analyzed by GC. |
| 93% |
With di-tert-butyl-diazodicarboxylate; potassium-t-butoxide; oxygen In fluorobenzene Heating; |
|
| 93% |
With 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical In dichloromethane at 20℃; for 2h; chemoselective reaction; |
|
| 93% |
With hydrogenchloride; NaBrO3; N-(1-oxo-2,2,6,6-tetramethylpiperidin-4-yl)-benzoylamide; acetic acid In dichloromethane; water monomer at 20℃; for 1h; |
|
| 93% |
With Cu<SUB>2</SUB>(ophen)<SUB>2</SUB>; oxygen In acetonitrile at 60℃; for 12h; Green chemistry; |
4.2 General procedure for the synthesis of 59 benzylaldehyde
General procedure: To a 10mL round bottom flask, the mixture of 12 benzylalcohol 1a (1.0mmol, 0.104mL) and Cu2(ophen)2 (0.05mmol, 26mg,) in 60 CH3CN (4mL), was stirred in oil bath at 60°C under oxygen atmosphere (oxygen ball). The reaction was monitored and conversions were determined by GC-MS. |
| 93% |
With 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; C110H202N8O47; oxygen; copper(II) bis(trifluoromethanesulfonate); potassium carbonate In water monomer at 20℃; for 12h; Green chemistry; |
General procedure for aerobic oxidation of alcohols in water
General procedure: To a 48 mL tube, were added Cu(II) or Cu(I) salt (0.05 mmol), PEG-PyTa (0.025 mmol) and H2O (3.0 mL). The mixture was stirred for 30 min at room temperature and a clear dark-blue solution was observed. Then alcohols (1.0 mmol), TEMPO (0.05 mmol), and K2CO3 (0.2 mmol) were sequentially added, followed by connecting a balloon of oxygen. The reaction mixture was stirred at room temperature until the reaction completed based on GC analysis. After that, the reaction mixture was extracted with MTBE (3 mL×3) and the extracts were combined, dried over anhydrous Na2SO4 and concentrated under vacuum. Finally, the residue was purified by flash chromatography on silica to afford the desired aldehydes. |
| 93% |
With sodium chlorine monoxide; Sodium hydrogenocarbonate; potassium bromide In dichloromethane at 0℃; for 0.5h; |
|
| 92% |
With 2,2,6,6-tetramethyl-1-piperidinyloxy free radical; air; NaNO2 for 6h; Heating; |
|
| 92% |
With hydrogen bromide; oxygen for 6h; Heating; |
|
| 92% |
With 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; anhydrous sodium carbonate; <i>L</i>-proline; copper(II) bromide In methanol at 20℃; for 5h; |
|
| 92% |
With tert.-butylhydroperoxide at 80℃; for 15h; Green chemistry; |
General procedure for the oxidation of alcohols tocarbonyl compounds
General procedure: The alcohol (1 mmol) was added to a mixture of TBHP(1 mmol) and VO(ephedrine)2MNPs (50 mg) in PEG(1 mL), and then the mixture was refluxed at 80 C for thetime specified. The progress was monitored by TLC (EtOAc/n-hexane, 1/2). After completion of the reaction, the catalystwas separated from the product by an external magnet(within 5 s), and the mixture was washed with EtOAc(25 mL) and decanted. The decanted mixturewas washedwith 30% NaOH (5 mL) and the organic layer was dried overanhydrous Na2SO4. The evaporation of EtOAc underreduced pressure gave the pure products in 85e98% yields. |
| 91% |
With C6H4MoNO7(1-)*C19H42N(1+); oxygen In water monomer at 100℃; for 18h; Green chemistry; chemoselective reaction; |
2.3. General procedure for the catalytic oxidation of alcohols toaldehydes
General procedure: A mixture of alcohol (0.75 mmol), and catalyst Mo1 (13 mg,3.0 mol%) taken in 0.5 mL of water was stirred at 100 ° C under oxygenatmosphere (O2 bladder) and the stirring was continued for16-24 h as per requirement. The progress of reaction was monitoredby TLC. After completion of the reaction, ethyl acetate was added to the mixture. The aqueous phase was extracted with ethyl acetate 2-3 times. Then the combined organic extracts were driedover anhydrous sodium sulfate and the solvent was removed under reduced pressure. The crude product so obtained was purified by column chromatography using hexane-ethyl acetate as eluent. While the known products were characterized by spectroscopic techniques and compared with reported data and the new products 22b and 36b were characterized completely. The characterization detail is provided in supporting information section. |
| 91% |
With sodium chlorine monoxide; C8H16NO3; tetrabutylammonium bromide; Sodium hydrogenocarbonate; potassium bromide In dichloromethane; water monomer at 0℃; for 0.5h; chemoselective reaction; |
General procedure for the oxidation using NaOCl
General procedure: A 50ml flask was charged with a solution of benzyl alcohol, morpholinone nitroxide 2 in CH2Cl2 and a sat. aqueous solution of NaHCO3 containing KBr and n-Bu4NBr. To this cooled to 0 °C by water-ice bath and well stirred mixture, a pre-mixed solution of aqueous NaOCl (6-14% Cl) and sat. aqueous solution of NaHCO3 was added dropwiseduring 10 min. The reaction was stirred for 30 min at 0 °C, then quenched with sat. aqueous solution of Na2S2O3. The aqueous layer was separated and extracted with CH2Cl2. The combined organic layers were washed with brine, dried over Na2SO4 and concentrated under reduced pressure. The crude material was purified by flash column chromatography to give benzaldehyde as a colorless oil. |
| 91% |
With double-atom catalyst FeCo-DAC In o-dimethylbenzene at 140℃; for 36h; Inert atmosphere; Sealed tube; |
|
| 90% |
With air In toluene at 110℃; for 4h; atmospheric pressure; |
|
| 90% |
With m-iodosylbenzoic acid; Sodium hydrogenocarbonate at 20℃; for 12h; |
|
| 90% |
With 1H-imidazole; [bis(acetoxy)iodo]benzene In dichloromethane at 20℃; for 1h; |
|
| 90% |
With tert.-butylhydroperoxide In acetonitrile at 80℃; for 10h; |
|
| 90% |
With chloramine-T; anhydrous zinc bromide In acetonitrile for 2h; Reflux; |
Experimental Procedures for oxidation of Various alcohols:
General procedure: A CH3CN solution of alcohol (1 mmol), ZnBr2 (45 mg, 0.2 mmol), and chloramine-T (282 mg, 1 mmol) was placed in a three necked flask with a reflux condenser. After the mixture was stirred under reflux for 1.5-5 h. After cooling to room temperature, the solution was quenched by adding water and the resulting mixture was extracted with AcOEt. Removal of the solvent under reduced pressure gave the crude product, which was purified by column chromatography on silica gel to give the corresponding carbonyls. |
| 90% |
With oxygen In benzene at 150℃; |
|
| 90% |
With 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; oxygen; rac-Ala-OH; iron(II) chloride In 1,4-dioxane for 12h; Reflux; Green chemistry; |
9 Example 9
Equipped with a magnetic stirrer in round bottom flask methyl benzyl alcohol (12. 22g, 100. Ommol, namely formula The R1 is 4-methyl, R2 is hydrogen, X is carbon, η is 1, m is 0), ferric chloride (0. 81g, 5mmol), L- isoleucineAcid (1.31g, 10mmol), TEMP0 (1.56g, 10mmol), toluene 300. OmL was added , then the reaction with oxygen in the air bottleReplacement, stirred and reflux for 6h. After completion of reaction, the reaction mixture was cooled to room temperature, filtered, the filtrate evaporated to give the crude product,The resultant crude product was purified by column chromatography, with n-hexane: Elution: (10 1 volume ratio) mixed liquid of ethyl acetate containing the desired collectionLabeled compound of the eluent, evaporation of the solvent and dried to give the product p-tolualdehyde 10. 93g, 91% yield.[0039]. 3-pyridyl methanol (1. 09g, 10. Ommol, i.e., of formula (I), R1 is hydrogen, R2 is hydrogen, X isN, [eta] is l, m is 0) is taken, experimental methods and procedures were the same as in Example 1, except that: ferrous chloride (0. 06g, 0 5mmol),DL- alanine (0. 09g, lmmol), TEMP0 (0. 156g, lmmol), dioxane 30. OmL, the reaction with oxygen bottleAfter stirring at reflux for air displacement 12h. The final product was obtained 0. 96g, yield 90%. |
| 90% |
With C6H12N2*2ClCrO3(1-)*2H(1+) In dichloromethane at 20℃; for 1.5h; |
Typical Procedure for the Oxidation of Alcohols
General procedure: For oxidation of alcohols with DABCO-bis-CC, the procedureapplied was similar to that reported [4a] for oxidationof alcohols with PCC. DABCO-bis-CC (1.0 mmol) was suspendedin CH2Cl2 (2 ml), and the alcohol (1 mmol in 0.5 to1.5 ml of CH2Cl2) was rapidly added to the system at roomtemperature. After completion (TLC) of the reaction theproduct was isolated by filtration of the organic extractsthrough Celite and further evaporation of the solvent underreduced pressure. The product was finally purified by columnchromatography over silica gel using n-hexane:ethylacetate (9:1 v/v) to get the desired oxidized product. Thepurity of each carbonyl product was established by comparativeTLC study against the authentic sample. Furthermore,the IR spectrum of each carbonyl compound was recordedwhich displayed a strong C=O stretching peak in the rangefrom 1670 cm-1 to 1720 cm-1 depending upon the nature ofthe carbonyl product. |
| 89% |
With oxygen; Azobenzene; sodium bromide In 1,4-dioxane at 80℃; for 36h; |
|
| 88% |
With mesoporous silica; 4-acetylamino-2,2,6,6-tetramethylpiperidine-1-oxoammonium perchlorate In dichloromethane Ambient temperature; |
|
| 88% |
With ammonium nitrate; hydrogenchloride; 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; oxygen In water monomer; acetonitrile at 60℃; for 19h; Green chemistry; |
|
| 88% |
With 2,2'-azobis(isobutyronitrile); oxygen; sodium bromide In 1,4-dioxane at 70℃; |
|
| 88% |
With tert.-butylhydroperoxide In water monomer; acetonitrile at 80℃; |
|
| 88% |
With ammonium hydroxide; copper (I) iodide; 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical at 100℃; for 9h; |
12 Example 1:
General procedure: In a 150 mL thick-walled pressure tube equipped with a magnetic stirrer,In an air atmosphere,To the system was added benzyl alcohol (i.e., R1 in formula (I) H) 1.0 mmol (108.1 mg)Ammonia (1.6 x 10-2 mol / L) 5.0 mL,5 mol% (9.5 mg) of cuprous iodide,TEMPO 5 mol% (7.8 mg),100 & lt; 0 & gt; C for 12 h,After the reaction is over,The reaction solution was cooled to room temperature,And extracted with ethyl acetate (3 x 5.0 mL). The organic layers were combined and concentrated in vacuo to remove ethyl acetate to give the crude product. The crude product was purified by column chromatography(Petroleum ether: ethyl acetate = 10: 1) to give the pure desired product.The yield of 97.6 mg was 92%. |
| 88% |
With dihydrogen peroxide In neat (no solvent) at 25℃; for 12h; Green chemistry; |
Typical experimental procedure for oxidation reaction
General procedure: To a mixture of Pd-BIL (0.1 mol%) and benzyl alcohol(5 mmol) was slowly added H2O2 (10 ml, 30%). The resulting reaction mixture was stirred at 25°C until the disappearance of starting material (TLC). After completionof the reaction, ethyl acetate was added (3 mL × 5 mL) to separate product from catalyst and concentrated. The results shown in Table 4 demonstrated that this oxidative system was readily recyclable for seven runs without any significant loss of catalytic activity. |
| 87% |
With ferric(III) chloride; 4-acetylamino-2,2,6,6-tetramethyl-1-piperidinoxy; oxygen; acetic acid; NaNO2 In 1,2-dichloro-ethane at 50℃; for 8h; Autoclave; |
|
| 87% |
With 1-tosyloxy-1-oxo-1H-1λ5-benzo[d][1,2]iodoxol-3-one In acetonitrile at 20℃; |
|
| 87% |
With sodium chlorine monoxide; Sodium hydrogenocarbonate; potassium bromide In dichloromethane; water monomer at 0℃; for 0.166667h; Schlenk technique; |
|
| 86% |
With 5% ruthenium on carbon; oxygen In water monomer at 75℃; for 24h; |
|
| 86% |
With 2-ethyl-2-(2-iodylphenyl)butanoic acid In dichloromethane at 25 - 30℃; |
|
| 86% |
With 1-acetoxy-5-nitro-1,2-benziodoxol-3(1H)-one In N,N-dimethyl-formamide at 65℃; for 24h; |
|
| 86% |
With 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; copper (II) acetate In water monomer; acetonitrile at 20℃; for 6h; Green chemistry; |
General procedure: A mixture of alcohol (5.0 mmol), Cu(OAc)2 (9.1 mg, 0.05 mmol), and TEMPO (7.8 mg, 0.05 mmol) in CH3CN/H2O (5/10 mL) was stirred at room temperature for specified time. After completion of the reaction (monitored by TLC, eluents: petroleum ether/ethyl acetate = 4/1), dichloromethane (10 mL) was added to the resulting mixture. The dichloromethane phase was separated, and the aqueous phase was further extracted with dichloromethane (10 mL × 2). The combined organic layers were dried over anhydrous sodium sulfate and concentrated to give a residue, which was purified by column chromatography (eluents: petroleum ether/ethyl acetate = 10/1) to provide the desired product. |
| 85% |
With potassium permanganate In various solvent(s) at 20℃; for 2h; |
|
| 85% |
With tripotassium phosphate tribasic; 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; Cu2(phenanthroline)2(μ-Cl)2Cl2; oxygen In acetonitrile at 20℃; for 2h; |
|
| 85% |
With 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; calcium methanolate In acetonitrile at 0 - 20℃; |
|
| 85% |
With 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; Sodium hydrogenocarbonate In dichloromethane; water monomer at 20℃; for 6h; Green chemistry; chemoselective reaction; |
The procedure for the oxidation of benzyl alcohol is as follows:
General procedure: A mixture of H2O (5 mL) and CH2Cl2 (5 mL) wasadded to the chlorinated polymeric beads (2 g). (The unchlorinated beads were obtained from HaloSource, Inc. The unchlorinated beads were chlorinated by soaking the beads in bleach solution, with pH adjusted to 7 by additionof AcOH, for an hour. Then, the chlorinated beads were filtered and dried in air.) After addition of TEMPO (10 mg,0.064 mmol) and benzyl alcohol (0.2 g, 1.8 mmol), NaHCO3(0.5 g) was added to the mixture. The mixture was stirred at r.t. for 3 h and filtered. The residue on the filter paper was washed with H2O (20 mL) and CH2Cl2 (10 mL), and the organic phase of the filtrate was separated, dried over MgSO4, and filtered. The solvent was removed under vacuum to obtain benzaldehyde. |
| 85% |
With 2,2,6,6-tetramethyl-1-piperidinyloxy free radical; [{Cu(NO3)}(μ-3-(6-(1H-pyrazol-1-yl)pyridin-2-yl)pyrazol-1-ide)]2; oxygen In water monomer at 30℃; for 24h; |
|
| 85% |
With 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; [{(MeOH)Cu(OAc)}(μ-k2:k1-2-(3-(pyridin-2-yl)-1H-pyrazol-1-yl)acetic acid(-H))]2*0.5H2O; tetraethylammonium iodide; oxygen; potassium carbonate In water monomer at 40℃; for 24h; |
|
| 85% |
With ammonium hydroxide; copper (I) iodide; 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical In water monomer for 24h; Reflux; Green chemistry; |
|
| 84% |
With 3-(tert-butoxycarbonyl amino)-9-azabicyclo[3.3.1]nonane N-oxyl; oxygen; NaNO2 In acetic acid at 25℃; for 0.8h; |
Typical procedure for the aerobic oxidation of p-methylbenzyl alcohol to p-methyl benzaldehyde (Table 2, entry 2):
General procedure: A 25-mL tube equipped with a magnetic stirrer bar was added p-methylbenzyl alcohol (0.122 g, 1 mmol), sodium nitrite (5.5 mg, 8 mol%) and 3-(tert-butoxycarbonyl amino)-9-azabicyclo[3.3.1]nonane N-oxyl (3-BocNH-ABNO) (7.7 mg, 3 mol%). After the air in the tube was replaced with O2, 1 mL of acetic acid was added with syringe. Then the mixture was stirred under dioxygen atmosphere (balloon) at room temperature until the reaction was completed. After the reaction was finished, to the reaction mixture was added 8 mL of diethyl ether. Then the mixture was transferred into a separation funnel, and washed with saturated sodium bicarbonate solution (10 mL×3). The aqueous phase was extracted with 8 mL of ether. The combined organic phases was concentrated on a rotary evaporator and the residue was purified by column chromatography on silica gel using petroleumether/diethyl ether as eluent to afford p-methyl benzaldehyde as a colorless liquid; yield: 0.108 g (90%). |
| 83% |
With chromium(VI) oxide; N-hexadecyl-N,N,N-trimethylammonium bromide In water monomer at 80℃; for 5h; |
|
| 83% |
With 1-(benzoylamino)-3-methylimidazolium chlorochromate In dichloromethane for 8h; Heating; |
|
| 83% |
With [RuCl(1-ethyl-3-(N-mesitylimidazoliumylmethyl)-5-methylpyrazole)(p-cymene)]*(PF6); dihydrogen peroxide In acetonitrile at 60℃; for 5h; |
|
| 83% |
With 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; [Cu2(1,2-benzenedicarboxylate)2(1,4-bis(1,2,4-triazol-1-ylmethyl)-2,3,5,6-tetrafluorobenzene)2]·3H2O}n; anhydrous sodium carbonate In acetonitrile at 75℃; for 16h; |
2.2 Catalytic reaction
General procedure: In a typical experiment, 4-methoxybenzyl alcohol (12μL, 0.1mmol), Cu-FMOF (11.3mg, 10mmol %), TEMPO (7.8mg, 0.05mmol) and NaCO3 (10.6mg, 0.1mmol) in 1mL of air saturated acetonitrile were taken in a 15-mL three-necked round-bottom flask. The solution was magnetically stirred for 16h at 75°C under air atmosphere. The progress of the reaction was monitored via gas chromatography (Shimadzu GC-2010AF) involving a Chromopak capillary column and flame ionization detector. The products were further confirmed by using gas chromatography-mass spectroscopy (GC-MS) (Shimadzu GCMS-2010). The concentrations of 4-methoxybenzyl alcohol and 4-methoxybenzylaldehyde were calibrated by external standard method with standard samples (see Fig. S1). |
| 82% |
With orthoperiodic acid In acetonitrile at 20℃; for 6h; |
|
| 82% |
With iodic acid In N,N-dimethyl-formamide at 60℃; for 8h; Inert atmosphere; |
4.1.20 4.1.1 Typical experimental procedure with HIO3 (Method A)
General procedure: To a solution of p-bromobenzyl alcohol I-1 (187 mg, 1.0 mmol) in DMF (2.0 mL) was added HIO3 (194 mg, 1.1 mmol). The mixture was stirred at 60 °C for 2 h under an Ar atmosphere. After the reaction, the reaction mixture was poured into aq Na2S2O3, and extracted with a mixture of Et2O: hexane=1:1 (3*10 mL). The organic layer was dried over Na2SO4. After being filtration and removal of the solvent under reduced pressure, the residue was purified by flash short column chromatography on silica gel (EtOAc-hexane, 1:4) to give p-bromobenzaldehyde II-1 in 95% yield. |
| 81% |
With Phenanthroline; oxygen; potassium carbonate; copper chloride (I) In toluene at 90℃; |
|
| 81% |
With 3-(6-(3,5-dimethyl-1H-pyrazol-1-yl)pyridin-2-yl)-1-(2-((1-oxyl-2,2,6,6-tetramethylpiperidin-4-yl)oxy)-2-oxoethyl)-1H-imidazol-3-ium iodide; oxygen; anhydrous sodium carbonate; copper(II) bromide at 50℃; for 24h; Ionic liquid; |
|
| 80% |
With aluminium oxyhydroxide; ruthenium In toluene at 110℃; for 36h; |
|
| 80% |
With NaBrO3; sodium hydrogen sulphate In hexane for 10h; Heating; |
|
| 80% |
With 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; anhydrous sodium carbonate; N-Phenylglycine; copper (II) bromide In water monomer for 16h; Reflux; Schlenk technique; |
General Procedures for the Copper-Catalyzed Primary BenzylicAlcohol Oxidation under Air in Water (p-MethylbenzylAlcohol)
General procedure: A mixture of p-methylbenzyl alcohol (1.0 mmol), N-phenylglycine(0.0076 g, 0.05 mmol), CuBr2 (0.0112 g, 0.05 mmol),Na2CO3 (0.1060 g, 1.0 mmol), TEMPO (0.0078 g, 0.05 mmol),H2O (3.0 mL) were added to a 100 mL Schlenk tube, which wasvigorously stirred in air under reflux for 0.5 h. After the reaction,the product was extracted with CH2Cl2 (3 × 2.0 mL). Thecombined organic phase was washed with H2O (3.0 mL) anddried over anhydrous MgSO4. After concentration undervacuum, the residue was purified by column chromatography toafford p-methylbenzaldehyde.Isolated yield: 0.1080 g (90%). |
| 79% |
With IBX; 10-camphorsufonic acid In 1,4-dioxane; dichloromethane at 20℃; Inert atmosphere; |
General procedure
General procedure: Under nitrogen atmosphere, 1.1-1.5 mmol IBX and 10 to 20 mol% (±)-CSA monohydrate was added in round bottom flask already charged with magnetic bar and 2 mL DCM:1,4-Dioxane. Stirred the mixture for 10 minutes at room temperature and added the solution of alcohol dropwise for 5 minutes. Stirred the solution at room temperature till complete consumption of alcohol. Strip off the solvent and dilute the reaction mass with DCM. Filter the suspension through sintered funnel and wash the residue properly with DCM. This residue (white powdered solid, reduced part of IBX) was successfully used for preparation of IBX. Concentrate the filtrate on rotavapor and purify the product by column chromatography. |
| 78% |
With tert.-butylhydroperoxide In acetonitrile at 80℃; for 6h; |
|
| 78% |
Stage #1: 3-pyridil carbinol With 1,1-dichlorocycloheptatriene In dichloromethane; dimethyl sulfoxide at -30℃; for 0.333333h; Inert atmosphere;
Stage #2: With triethylamine In dichloromethane; dimethyl sulfoxide at -30 - 20℃; for 0.333333h; Inert atmosphere; |
General procedure for the Swern-type oxidation reaction
General procedure: A solution of DMSO (3.0 mmol) in CH2Cl2 (2.0 mL) was added toa solution of 1 (1.2 mmol) in CH2Cl2 (5.0 mL) at 30 C, and themixture was stirred for 20 min at the same temperature. The alcoholsubstrate (1.0 mmol) was added, and the mixture was stirredfor another 20 min before the dropwise addition of Et3N(3.0 mmol). The mixture was subsequently left to warm to room temperature (20 min) and concentrated under reduced pressure.The product was isolated by flash column chromatography. |
| 78% |
With ammonium hydroxide; 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; oxygen In ethanol at 50℃; for 36h; |
23 4.5. General procedure for the aerobic alcohol oxidation
General procedure: Under an air atmosphere, a Schlenk tube was charged with MCM-41-bpy-CuI (40 mg, 0.025 mmol), alcohol (0.5 mmol), TEMPO (4 mg, 0.025 mmol), aqueous ammonia (0.5 mmol, 25e28%, w/w) and EtOH (1.0 mL). The mixture was stirred at 50 °C for 18-48 h. After completion of the reaction, the reaction mixture was cooled to room temperature, diluted with ethyl acetate (10 mL), and filtered. The MCM-41-bpy-CuI complex was washed with EtOH (2*5 mL), and Et2O (5 mL) and reused in the next run. The filtrate was concentrated under reduced pressure and the residue was purified by flash column chromatography on silica gel (petroleum/ethyl acetate=15:1 to 10:1) to provide the desired product. |
| 78% |
In neat (no solvent) at 100℃; for 6h; |
Condition B
General procedure: A mixture of the substrate (250 μmol) and10% Ru/C (12.6 mg, 12.5 μmol) was stirred at 100 °C using atest tube equipped with air balloon. After the correspondingreaction time, the mixture was filtered through a membranefilter (pore size: 0.2 μm). The catalyst on the filter was washedwith H2O and CH2Cl2 and extracted with CH2Cl2 (5 mL × 3).The combined organic layers were dried over Na2SO4 andconcentrated in vacuo. The residue was further purified bysilica-gel column chromatography |
| 76% |
With potassium carbonate at 100℃; for 8h; Neat (no solvent); |
|
| 76% |
With tert.-butylnitrite; oxygen; 3,6-di(2'-pyridyl)-1,2,4,5-tetrazine; acetic acid In acetonitrile at 20℃; for 4.5h; Irradiation; |
|
| 75% |
With 2,2,6,6-tetramethyl-1-piperidinyloxy free radical; oxygen; copper chloride (I) In various solvent(s) at 65℃; for 36h; |
|
| 75% |
With air; potassium carbonate at 100℃; for 7h; |
|
| 75% |
With sodium chlorine monoxide; 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; potassium bromide In dichloromethane; water monomer at -10 - 15℃; |
|
| 73% |
With 1-hydroxy-3H-benz[d][1,2]iodoxole-1,3-dione In dimethyl sulfoxide at 20℃; for 2h; |
|
| 73% |
With dmap; 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; copper trifluoromethanesulfonate; (S)-(-)-5-(2-pyrrolidinyl)-1H-tetrazole In acetonitrile at 25℃; for 12h; chemoselective reaction; |
4 4.1.2. The oxidation of primary alcohols
General procedure: A round-bottom flask was charged with alcohol (2 mmol), CuOTf (0.1 mmol, 0.05 equiv) (S)-5-(pyrrolidin-2-yl)-1H-tetrazole (0.1 mmol, 0.05 equiv), TEMPO (0.1 mmol, 0.05 equiv), DMAP (0.15 mmol, 0.075 equiv) and CH3CN (5 ml). The reaction mixture was stirred at 25 °C open to air until the completion of the reaction, as monitored by TLC. After completion, CH3CN was evaporated under vacuum. The residue was then diluted with CH2Cl2 (5 ml) and filtered through a plug of silica gel to afford the desired product. |
| 73% |
With 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; C40H54CuN4O5(2+)*2I(1-); dihydrogen peroxide; potassium carbonate In water monomer at 60℃; for 12h; |
Typical procedure for the formation of aldehydes (3a-q)
General procedure: A test tube equipped with a magnetic stirring bar was charged with 1 (5 mg, 0.005 mmol, 0.25 mol%), TEMPO (16 mg, 0.10 mmol, 5 mol%), and K2CO3 (69 mg, 0.50 mmol, 25 mol%). Alcohols (2a-q, 2.0 mmol), H2O (5 mL) and H2O2 (30%, 1 mL) were added. The mixed light green solution was stirred at 60°C for 12 h, cooled to room temperature, and then extracted by Et2O (3 × 5mL). The organic layers were combined, washed with brine (20 mL), dried over anhydrous Na2SO4 and concentrated under reduced pressure. On elution with petroleum ether and ethyl acetate, the crude product was further purified by column chromatography on silica. All aldehyde products synthesized in this work are known and confirmed by 1H NMR spectra. |
| 72% |
With dmap; 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; copper chloride (I) In water monomer at 55℃; for 18h; Green chemistry; |
|
| 68% |
With 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; [Cu(BTPMPA)(OH2)(OTf)]OTf}; oxygen; potassium carbonate In toluene; acetonitrile at 27℃; for 20h; |
|
| 67% |
With [Cu6(pyridine-2-thiolate)6]; potassium hydroxide In toluene at 70℃; for 24h; Schlenk technique; Glovebox; Inert atmosphere; |
|
| 65% |
With quinolinium fluorochromate In hexane for 3h; Heating; |
|
| 65% |
With oxygen; potassium carbonate In toluene at 100℃; for 20h; |
|
| 65% |
With oxygen; hydroquinone In tetrahydrofuran; aq. phosphate buffer at 20℃; for 24h; Green chemistry; |
2.8. General procedure for the aerobic oxidation of alcohols
General procedure: A 25 mL round-bottomed flask equipped with a magnetic stirrerwas charged with alcohol (1 mmol), Pd-Laccase(at)MCF (0.2 g,0.27 mmol Pd), hydroquinone (HQ, 0.27 mmol) and NaPBS/THF(5 mL, 4/1 v/v). The reaction mixture was stirred under the O2 (balloon balloon)or in an open-air round-bottom flask at room temperature forthe time specified (Scheme 2). After completion of the reaction(monitored by TLC), the catalyst was separated using filtrationand washed with CH2Cl2 and the product was extracted with CH2-Cl2 (2 x 5 mL). The oganic phase was dried over anhydrousMgSO4. After evaporation of the solvent under reduced pressure,the crude product was purified by recrystallization from ethanolor chromatography on silica gel (n-hexane-EtOAc = 3:1). |
| 65% |
With amine-functionalized zinc-containing metal organic framework-based palladium nanoparticles; air In toluene at 80℃; for 8.5h; |
|
| 61% |
With 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical In acetonitrile at 70℃; for 11.5h; |
|
| 58% |
With aluminum(III) oxide In N,N-dimethyl-formamide at 120℃; for 8h; Inert atmosphere; |
|
| 58% |
With tert.-butylhydroperoxide; C56H62Cl2Mn2N6O10P2 In water monomer; acetonitrile at 80℃; for 4h; Green chemistry; |
|
| 55% |
With Bi(NO3)3·5H2O; cellulose supported copper(0); oxygen In acetonitrile at 60℃; for 2h; |
|
| 50% |
With 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical In <i>tert</i>-butyl alcohol at 25℃; aq. sodium carbonate/bicarbonate buffer; Electrolysis; |
|
| 46% |
With carbonylchlorohydridotris(triphenylphosphine)ruthenium(II); oxygen In toluene at 110℃; for 24h; Molecular sieve; Sealed tube; |
|
| 37% |
With oxygen; Cs2CO3 In N,N-dimethyl-formamide at 90℃; for 18h; Sonication; Schlenk technique; |
|
| 36% |
With K<SUB>2</SUB>OsO<SUB>4</SUB>.2H<SUB>2</SUB>O; potassium carbonate; potassium hexacyanoferrate(III) In water monomer; acetonitrile at 60℃; for 6h; chemoselective reaction; |
|
| 33% |
With nitric acid; ytterbium(III) tris(trifluoromethanesulfonate) In 1,2-dichloro-ethane for 24h; Heating; |
|
| 27% |
With sodium (meta)periodate; manganese(II) 5,10,15,20-tetrakis(N-ethylpyridinium-4-yl)porphyrin In water monomer; toluene at 30℃; for 3h; |
|
| 21% |
With Fe2(SO4)3; TEMPOL; oxygen; NaNO2 In water monomer; acetonitrile at 20℃; for 24h; |
The oxidation of alcohols was carried out under O2 in a 50-mL two-necked, round-bottom flask equipped with a magnetic stirrer. Typically, Fe2(SO4)3 (0.25 mmol) and TMHPO (0.25 mmol) were added to the flask, followed by 15 mL of a CH3CN/H2O (1:2) solvent mixture. After stirring for 5 min, the alcohol (5 mmol) was added, followed by NaNO2 (0.25 mmol). The resulting mixture was stirred at room temperature and 1 atm pressure of oxygen. When the reactions were completed, the reaction mixture was transferred to a separating funnel and extracted with dichloromethane. The organic layer was dried over anhydrous Na2SO4 and concentrated and further purified by flash chromatography to give the desired product. |
| 7% |
With [Mn(bp)(NCS)0.9(NO3)0.1(CH3OH)]*CH3OH; dihydrogen peroxide In acetonitrile at 60℃; for 4h; |
|






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping