
Plastome sequences help to improve the systematic position of trinerved Lindera species in the family Lauraceae
- Published
- Accepted
- Received
- Academic Editor
- Marcial Escudero
- Subject Areas
- Bioinformatics, Conservation Biology, Genetics, Plant Science, Forestry
- Keywords
- Lauraceae, Complete plastid genome, Lindera, Phylogenetic relationship, Comparative genomics
- Copyright
- © 2019 Tian et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ) and either DOI or URL of the article must be cited.
- Cite this article
- 2019. Plastome sequences help to improve the systematic position of trinerved Lindera species in the family Lauraceae. PeerJ 7:e7662 https://doi.org/10.7717/peerj.7662
Abstract
Lindera is a genus (c. 100 spp.) of trees belonging to the “core Laureae” group in the family Lauraceae. It is often confused with Litsea, and the systematics of the genus is unclear. Here, total 10 complete plastomes from nine trinerved Lindera species and another species Lindera obtusiloba (sect. Palminerviae Meissn.) were sequenced. Nine highly variable regions, trnH-GUG/psbA, psbM/trnD-GUC, petA/psbL, ndhF, trnL-UAG/ndhD, and ycf1, were identified among the 10 Lindera species. In addition, a total of 1,836 mutation events including six micro-inversions, 156 indels, and 1,674 substitutions, were also summarized. Comparing our sequences with other available plastomes in the “core Laureae,” we put forward that six hypervariable loci, trnH-GUG/psbA, ndhF, ndhF/rpl32, trl32/trnL-UAG, ndhD, and ycf1, could potentially be used as plastid barcode candidates for species identification. Further phylogenetic analyses were conducted using 49 complete Lauraceae plastomes. The results supported a close relationship among trinerved Lindera species and suggested an improved trinerved group comprising species of trinerved Lindera species and Iteadaphne caudate.
Introduction
The “core Lauraceae” group, originally defined by Rohwer & Rudolph (2005), consists of the Laureae-Cinnamomeae group and Persea group. Accumulating evidence shows a number of problems in the systematics of the intra- or inter-genus. The genus Lindera Thunb., a member of the “core Laureae” is widely distributed in the tropical to temperate regions of Asia, North America, and even Australia (Hyland, 1989), with 38 of the 100 known species in China alone (Li et al., 2008b). It has a close relationship with Litsea, Neolitsea, Laurus, Actinodaphne, etc., as shown in Chanderbali, van der Werff & Renner (2001). It appears to be polyphyletic according to its current constitution, despite the precise generic boundaries and species delineations based on morphological characters being currently unclear. Lindera umbellata Thunb., a deciduous shrubby tree with pinnately compound leaves, is the type species. Following Hooker’s (1890) criteria and Li’s (1985) concept, Tsui (1987b) divided Lindera into eight sections using characteristics such as being evergreen or deciduous, having penninerved or trinerved leaves, well-developed or non-developing terminal buds, non-enlarged or enlarged perianth tubes, and normal or shortened brachyblasts. However, these divisions were not explicitly phylogenetic (Chanderbali, van der Werff & Renner, 2001; Li et al., 2008a; Rohwer, 2000).
Using the plastid marker matK, the first molecular study on Lindera suggested that Lindera benzoin (L.) Blume together with other “core Laureae” represents a weakly supported paraphyletic group (Rohwer, 2000). Another phylogenetic study comparing different combinations of plastid sequences (trnL-trnF, psbA-trnH, trnT-trnL, and rpl16) and nuclear barcoding markers (26S rDNA and ITS rDNA), showed a well-supported “core Laureae” group comprising three Lindera species, Lindera benzoin, Lindera erythrocarpa Makino, and Lindera umbellata Thunb., as well as other species in their ITS analysis (Chanderbali, van der Werff & Renner, 2001). However, in their trnL-trnF + psbA-trnH analysis, the “core Laureae” group had no support from the paraphyletic relationships among these species. Although the reports using ITS or the single-copy nuclear marker rpb2 (including different plastid markers) also confirmed that the “core Laureae” group and Lindera were polyphyletic (Fijridiyanto & Murakami, 2009; Li et al., 2008a; Liu et al., 2017; Nie, Wen & Sun, 2007), the question still remained: were there any useful sequences for the phylogenetic classification of the “core Laureae” group in plastid genomes?
Trinerved and pinninerved-leaf Lindera species were merged into a single genus: 13 species of Lindera with trinerved leaves in sect. Daphnidium (Nees) Benth., and one species in sect. Uniumbellae H.P. Tsui and Subg. Iteodaphne (Bl.) Kosterm (Li et al., 2008b). The complex relationships between the 34 Lauraceae taxa had been well resolved based on plastome sequence data, and included five Lindera species: Lindera aggregata, Lindera obtusiloba Blume, Lindera megaphylla, Lindera communis Hemsl. and Lindera glauca (Siebold & Zucc.) Blume (Song et al., 2017b). In this study, one of the trinerved leaf species, Lindera aggregate, was found to be more closely related to the Actinodaphne-Neolitsea clade than to other species of Lindera and Litsea, similar to trees inferred from rpb2 (Fijridiyanto & Murakami, 2009; Song et al., 2017b). Most recently, another phylogenomic analysis using four complete plastomes of Lindera latifolia Hook.f., Lindera robusta (C.K. Allen) H.P. Cui, Lindera metcalfiana C.K. Allen and Lindera nacusua (D. Don) Merr. confirmed that nine Lindera species grouped into two strongly supported clades. One clade consisting of five species of Lindera formed a sister group to the second, which included Laurus nobilis, Litsea glutinosa, and four other species of Lindera (Zhao et al., 2018). However, the scarcity of complete plastid genomes of trinerved species in Lindera seriously impedes species identification and phylogenetic relationship determination. Thus, the phylogenetic relationships and genome information of trinerved Lindera truly needs further study.
Typical plastid genomes in most angiosperm species, which have maternal inheritance and haploidy, show high resolution in land plants with complex relationships (Barrett et al., 2016; Daniell et al., 2016; Ma et al., 2014; Wolfe & Randle, 2004; Zhang et al., 2017). Until now, over 43 (38 species, 19 genera) Lauraceae accessions with complete plastid genomes have been assembled and submitted to GenBank at the National Center for Biotechnology Information (data: April 11, 2018). The lengths of these complete genomes range from 114,622 bp (Cassytha filiformis Meisn, accession: MF939338) to 158,530 bp (Beilschmiedia tungfangensis S.K. Lee & L.F. Lau, accession: MF939348) (Song et al., 2017b, 2019). Among these plastids, eight intergenic spacers (accD-psaI, ndhC-trnV, ndhF-rpl32, psbM-trnD, rpl32-trnL, rpl14-rps8, rps3-rps19, and rps16-trnQ), three gene segments (rbcL, ycf1, and ycf2), and three introns (clpP, ndhA, and trnG-UCC) showed promising levels of variation for application in DNA-barcoding or intrageneric studies in Lauraceae (Hinsinger & Strijk, 2017; Song et al., 2015, 2016, 2017b, 2018; Zhao et al., 2018).
The genera Laurus, Lindera, and Litsea are closely related—they all share similar morphological traits such as being dioecious, having umbellate inflorescences subtended by large involucral bracts, having all introrse anthers, and having two stipitate glands at the third whorl (Li et al., 2004; Li & Christophel, 2000). Divisions based on only one characteristic may result in “twin species” (Ng, 2005), such as Lindera/Litsea, Parasassafras D.G. Long/Sinosassafras H.W. Li, Dodecadenia Nees/Iteadaphne Blume, differing only in 2- or 4-locules. Curiously, phylogenetic analysis of “core Laureae” based on single copy nuclear rpb2 gene sequences and complete plastid genomes have found that Lindera aggregata was found to be more closely related to the Actinodaphne-Neolitsea group than to other species of Lindera and Litsea (Fijridiyanto & Murakami, 2009; Song et al., 2017b). This was unanticipated because of the different numbers of celled anthers between Lindera and the Actinodaphne-Neolitsea group. Additionally, trinerved leaf Lindera species when merged with pinninerved ones into one genus would be morphologically homoplasious. Therefore, the study of the phylogenetic relationships and genome information of trinerved Lindera still needs improvement.
In this study, we further sequenced nine samples of seven trinerved Lindera species, plus one from Lindera obtusiloba, using next-generation sequencing technology. In our assessment of plastid genomes of the nine morphologically similar trinerved Lindera species with one trinerved or pentanerved species, we will address the following objectives: (1) investigate the size range of complete plastid genomes; (2) examine the type of mutation events and variation regions; and (3) analyze the phylogenetic relationships with other “core Laureae” species in the family Lauraceae. The results will provide further information for Lindera species identification and taxonomic revision.
Materials and Methods
Material preparation and DNA sequencing
Dry leaves of all Lindera species were deposited in the Herbarium of the Xishuangbanna Tropical Botanical Garden (HITBC), Chinese Academy of Sciences (CAS) (Table 1). High-quality genomic DNA was isolated from ca. 6 cm2 sections of dry leaf using an improved CTAB method (Yang, Li & Li, 2014). Total DNA (0.5 μg) was checked on the Agilent BioAnalyzer 2100. Libraries with short-inserts of 500 bp in length were generated with an Illumina Paired-End DNA library Kit according to the manufacturer’s protocol (Illumina, San Diego, CA, USA). Genomic DNA samples from each species were indexed with tags and pooled together in one lane of Illumina Genome Analyzer Hiseq 2000 for sequencing by Beijing Genomics Institute (Shenzhen, China).
| Taxa | L. aggregata | L. chunii | L. limprichtii | L. pulcherrima | L. obtusiloba | L. supracostata | L. thomsonii BOP | L. thomsonii SY | L. thomsonii var. vernayana | L. fragrans |
|---|---|---|---|---|---|---|---|---|---|---|
| Voucher | SY01432 | SY34518 | SY34167 | SY34019 | SY35538 | SY34832 | SY34836 | SY34818 | SY35026 | SY34828 |
| Geographic origin | Yunnan, China | Guangdong, China | Gansu, China | Guizhou, China | Shanxi, China | Yunnan, China | Yunnan, China | Yunnan, China | Yunnan, China | Yunnan, China |
| Coverage | 180× | 174× | 137× | 136× | 168× | 105× | 135× | 146× | 567× | 123× |
| Total length (bp) | 152,717 | 152,734 | 152,696 | 152,693 | 152,717 | 152,662 | 152,673 | 152,649 | 152,626 | 152,644 |
| LSC (bp) | 93,733 | 93,716 | 93,748 | 93,726 | 93,637 | 93,698 | 93,711 | 93,702 | 93,669 | 93,681 |
| SSC (bp) | 18,822 | 18,838 | 18,816 | 18,837 | 18,912 | 18,824 | 18,830 | 18,815 | 18,825 | 18,823 |
| IR (bp) | 20,081 | 20,090 | 20,066 | 20,065 | 20,084 | 20,070 | 20,066 | 20,066 | 20,066 | 20,070 |
| GC content (%) | 39.1 | 39.2 | 39.1 | 39.1 | 39.2 | 39.2 | 39.2 | 39.1 | 39.2 | 39.2 |
| Total number of genes (unique) | 127 (113) | 127 (113) | 127 (113) | 127 (113) | 127 (113) | 127 (113) | 127 (113) | 127 (113) | 127 (113) | 127 (113) |
| Protein encoding | 83 | 83 | 83 | 83 | 83 | 83 | 83 | 83 | 83 | 83 |
| tRNA | 36 | 36 | 36 | 36 | 36 | 36 | 36 | 36 | 36 | 36 |
| rRNA | 8 | 8 | 8 | 8 | 8 | 8 | 8 | 8 | 8 | 8 |
Genome assembly and annotation
The low-quality score paired raw reads were filtered and trimmed, and de novo assembled to form contigs using GetOrganelle software (Jin et al., 2018). The assembled contigs were searched against the referenced plastid genome sequence of Laurus nobilis L. (accession: KY085912) using a BLASTn (ncbi-blast-2.6.0) (Hinsinger & Strijk, 2017). The high similarity contigs were created using a consensus mapping sequence in Geneious R11 (Kearse et al., 2012). The generated consensus sequence was used as a reference to map high-quality trimmed reads to correct ambiguous nucleotides in Geneious R11 and to extract a new consensus sequence as the final circular plastid genomic sequence. The plastid genomes were annotated using a web-based annotation program GeSeq (Tillich et al., 2017) with an uploaded reference of Laurus nobilis L. Protein, rRNA, and tRNA were annotated with BLASTn, CDS, and rRNA using HMMER search (Finn, Clements & Eddy, 2011). tRNA was checked with tRNAscan-SE v1.3.1 (Lowe & Eddy, 1997), which is part of the GeSeq programs. The output GenBank files were inspected and edited manually, and plastid genome circle maps were drawn using OGDraw v1.2 (Lohse et al., 2013).
Whole genome comparison and divergence hotspot identification
Repetitive genomic sequences were identified using the REPuter web-service program (Kurtz et al., 2001) to distinguish forward, reverse, complemented, and palindromic repeats, with a 30 bp minimal repeat size and more than 90% sequence identity (hamming distance equal to 3). Simple sequence repeats (SSRs) were detected using MISA-web (http://webblast.ipk-gatersleben.de/misa/) with the following parameters: 10, five, four repeat units for mono-, di-, tri-nucleotides and three repeat units for tetra-, penta-, hexa-, 7-, 8-, 9-, 10-nucleotides (Beier et al., 2017). Indel events were counted after aligning the nine trinerved Lindera species plastid genome sequences using the MAFFT program (Katoh et al., 2002). Nucleotide diversity (Pi) and total number of mutations (Eta) of plastid genome sequences were conducted through a sliding window with a window length of 600 bp and step size of 200 bp using DnaSP 6.10.03 (Rozas et al., 2017).
Phylogenetic analysis
Phylogenetic analyses were performed with 47 species from the “core Lauraceae” group (Appendix S1), and two species of Endiandra R.Br. were treated as outgroup based on previous studies (Rohwer & Rudolph, 2005; Song et al., 2017b). After sequences were aligned by MAFFT 7.395 with auto strategy (Katoh et al., 2002), all 49 complete plastid genome sequences were modified manually (SSRs and indel events regions may not have aligned) in BioEdit 7.2.5 (Hall, 1999). Maximum-likelihood (ML) phylogenetic relationships were conducted in RaxML 8.2.10 tool (Stamatakis, 2014) using the CIPRES Science Gateway web (Miller, Pfeiffer & Schwartz, 2010) using the substitution model (GTRGAMMA) determined from jModelTest 2.1.9 (Darriba et al., 2012). Individual branches were assessed using the nonparametric bootstrap (BS) with 1,000 fast BS search performed in each analysis to obtain confidence support. Bayesian inference (BI) was performed with BEAST 1.8.3 for 50,000,000 generations using default parameters (Drummond et al., 2012). The best nucleotide substitution model, GTR+I+G, was estimated from jModelTest 2.1.9 (Darriba et al., 2012) using the Akaike information criterion methods. Uncorrelated relaxed clocks (Drummond et al., 2006) and the Yule process (Gernhard, 2008) were used in phylogenetic reconstruction. All parameter effective sample sizes were more than 200 according to Tracer v.1.6 (Rambaut, Drummond & Suchard, 2013), with the first 25% of sampled generations discarded as burn-in. Results of all phylogenetic analysis are displayed at Figtree 1.4.3 (http://tree.bio.ed.ac.uk/publications/).
Results
Complete plastid genome sequences
After sequencing the plastid genome, we obtained more than six Gb of raw data from each Lindera species. The assembled plastid genome of the 10 Lindera species, with sizes ranging from 152,626 bp to 152,734 bp, was within the range of the other Lauraceae species (Song et al., 2017b). The total length of the Lindera obtusiloba plastid genome sequence was only 17 bp shorter than that of Lindera chunii, but it was longer than the other species (Table 1). All 10 Lindera plastid genomes showed a typical quadripartite circular structure (Fig. 1), consisting of a pair of inverted repeat (IR) regions divided by a small single copy (SSC) region and a large single copy (LSC) region. The entire ycf1 crossed the SSC/IRb boundary, while 1,370 bp of 3′-ycf1 were truncated at the boundary of the SSC/IRa. The entire ycf2 crossed the LSC/IRa boundary, while the region 3,161 to 3,165 bp of 5′-ycf2 was truncated at the boundary of the LSC/IRb (Fig. 2). The general G+C content of the full plastid genome was in the range of 39.1% to 39.2%, and the G+C content was 38%, 33.9%, and 44.4% in LSC, SSC, and IR regions, respectively. There was a total of 127 functional genes in the whole plastid genomes, including 83 protein coding genes, 36 tRNA genes, and eight rRNA genes (Table 1). Additionally, among these 10 plastid genomic sequences, 13 genes possessed only a single intron, and two genes (ycf3 and clpP) possessed two introns.
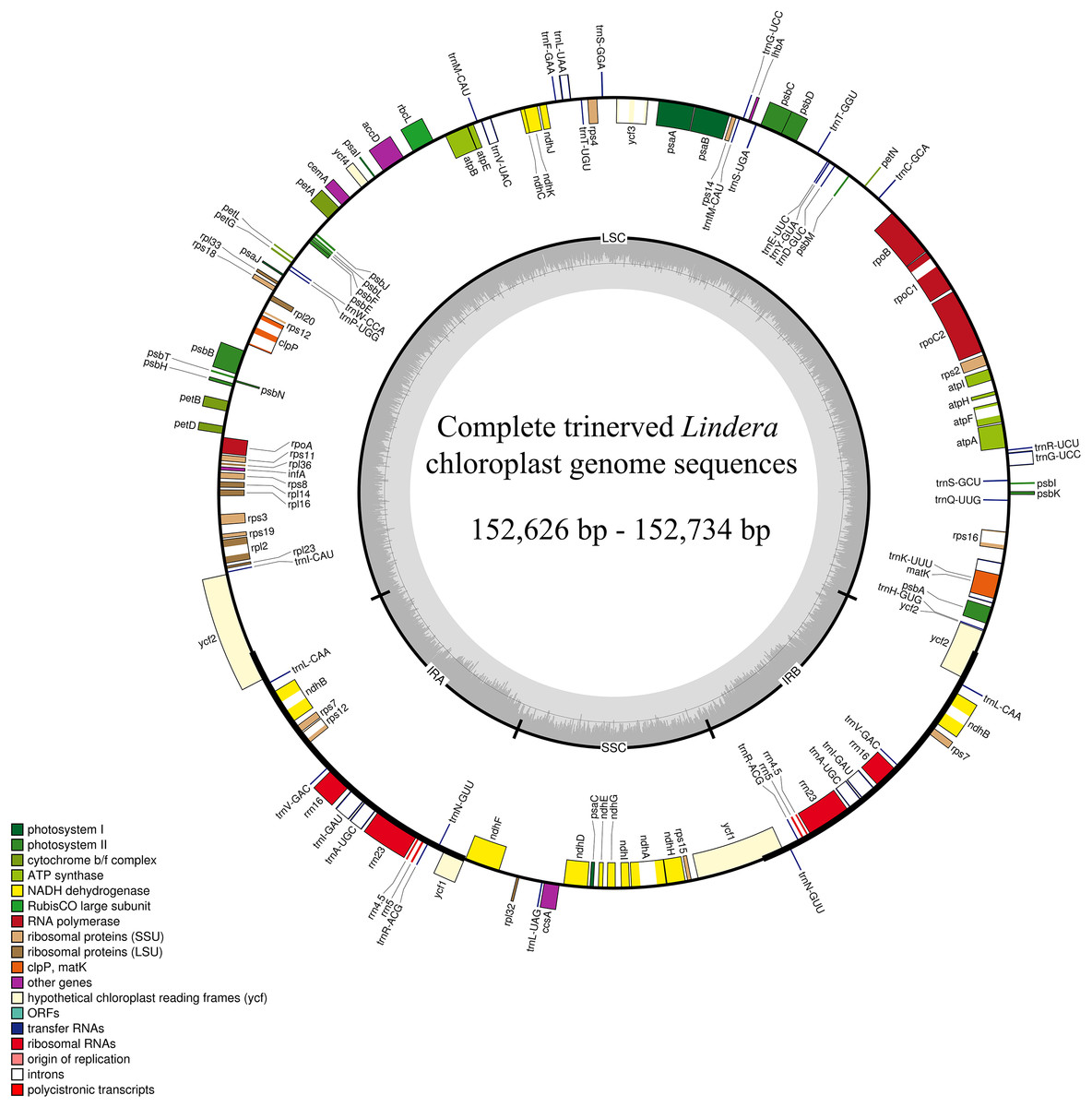
Figure 1: The plastid genomes map of 10 Lindera species.
Genes shown inside the circle are transcribed clockwise, and those outside the circle are transcribed counter clockwise.{kind=link}
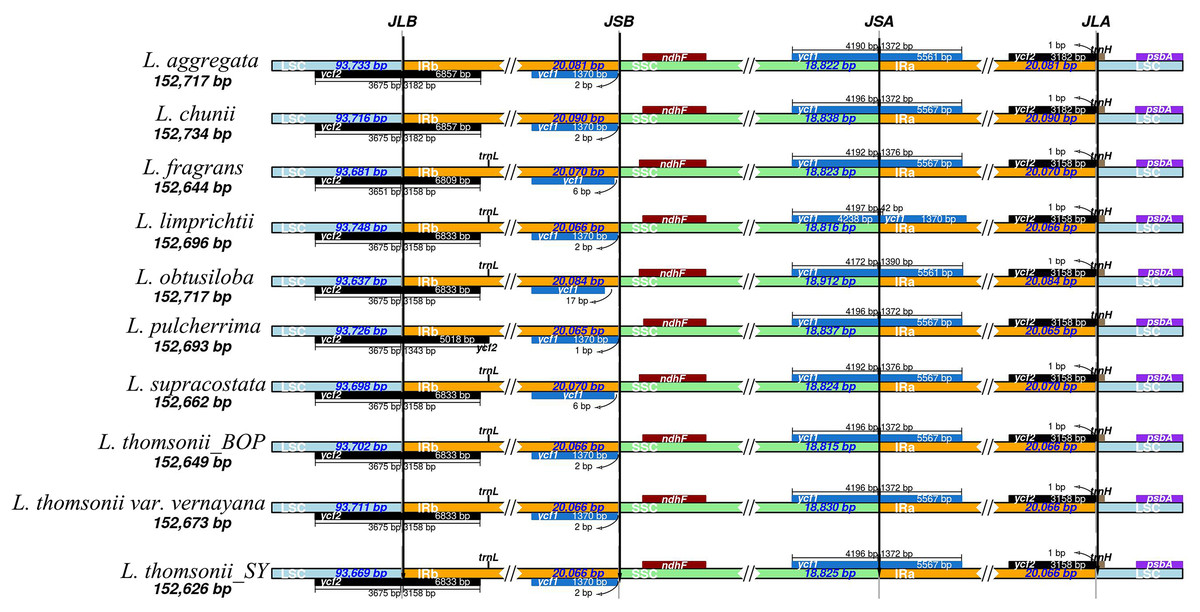
Figure 2: Comparison of the borders of the LSC, SSC, and IR regions among 10 Lindera plastid genome sequences.
LSC, large single-copy; SSC, small single-copy; IR, inverted repeat.{kind=link}
Number and forms of microstructural mutations
When being tested as potential mutation sites, 30–43 repeats and 74–92 SSRs were located in the 10 plastome sequences, 31 repeats and 74 SSRs in the plastome of Lindera obtusiloba, 33 repeats and 92 SSRs in the plastome of Lindera limprichtii, and 43 repeats and 91 SSRs were in the plastome of Lindera pilcherrima, with other species within those ranges (Fig. 3A; Appendix S2). Most repeats were forward and palindromic matches, except Lindera pulcherrima had 13 repeats that were reverse matches. In 67.57% of Lindera obtusiloba and 73.81% of Lindera chunii, the majority of SSR motifs were mononucleotides. Two adjacent repeat units with mononucleotides A/G were in the psbM/trnD-GUC, and dinucleotide TC and mononucleotide T were in ndhC/trnV-UAC among all species. Nevertheless, eight Lindera species contained 10-nucleotide SSR within trnL-UAA, except for Lindera aggregata and Lindera obtusiloba (Appendix S3). In these SSR sites, we found 89 different ones among the four plastomes. There were 79 single nucleotide repeats of A/T ranging from 4 to 16 bp, seven single nucleotide repeats of G/C ranging from 8 to 27 bp (Fig. 3B), two 10-nucleotide repeats in the atpH-atpI and trnT- trnL gene spacer regions, and one 18-nucleotide repeat in the trnF-ndhJ gene spacer region (Appendix S3). Besides these SSR indels, 48 non-SSR indels were identified in these plastomes. The size of most non-SSR indels ranged from 1 to 26 bp, while that of the psbM-trnD gene spacer sequence was 41 bp (Appendix S3). Additionally, 20 motifs, including seven forward, 10 palindromic, two reverse, and one complementary repeat, were shared by all plastomes (Appendix S2). In addition, we manually identified six micro-inversion events in the regions of rpl16 gene, ccs-ndhD, petA-psbJ, rps3-rps19, and trnH-psbA (Appendix S3). Two micro-inversions with three and eight bp were detected in the trnH-psbA region (Appendix S3). In the plastomes of all trinerved Lindera species, we detected 1,836 mutation events, including 156 indels and 1,674 substitutions, of which there were 24 double varieties, 181 parsimony informative sites, and 778 SNPs. Six hundred and eighty-four variable sites were in the LSC region, 226 in the SSC region, and 73 in the IR regions. There were 170 SNPs, including 591 transition (Ts) and 368 transversion (Tv) events in the plastomes.
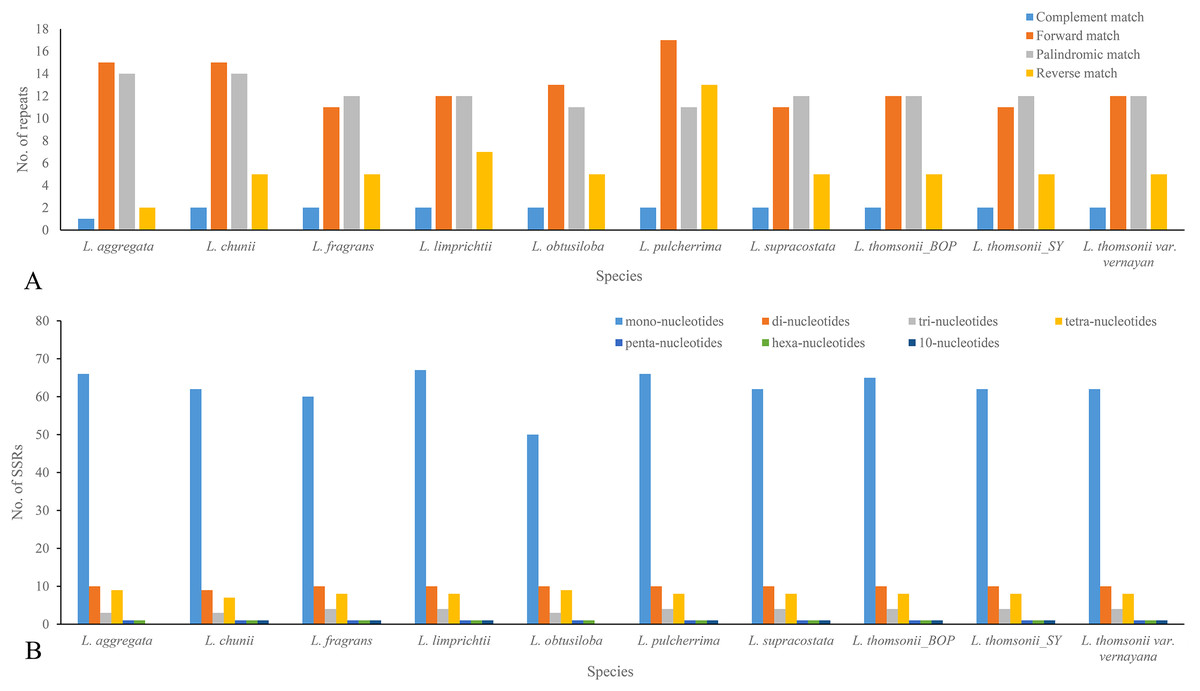
Figure 3: Repeats (A) and SSRs (B) in 10 Lindera species plastid genomic sequences.
(A) Frequency of forward, palindrome, reverse, complement type repeats in the Lindera plastome sequences. (B) Number of SSRs in the Lindera plastome sequences.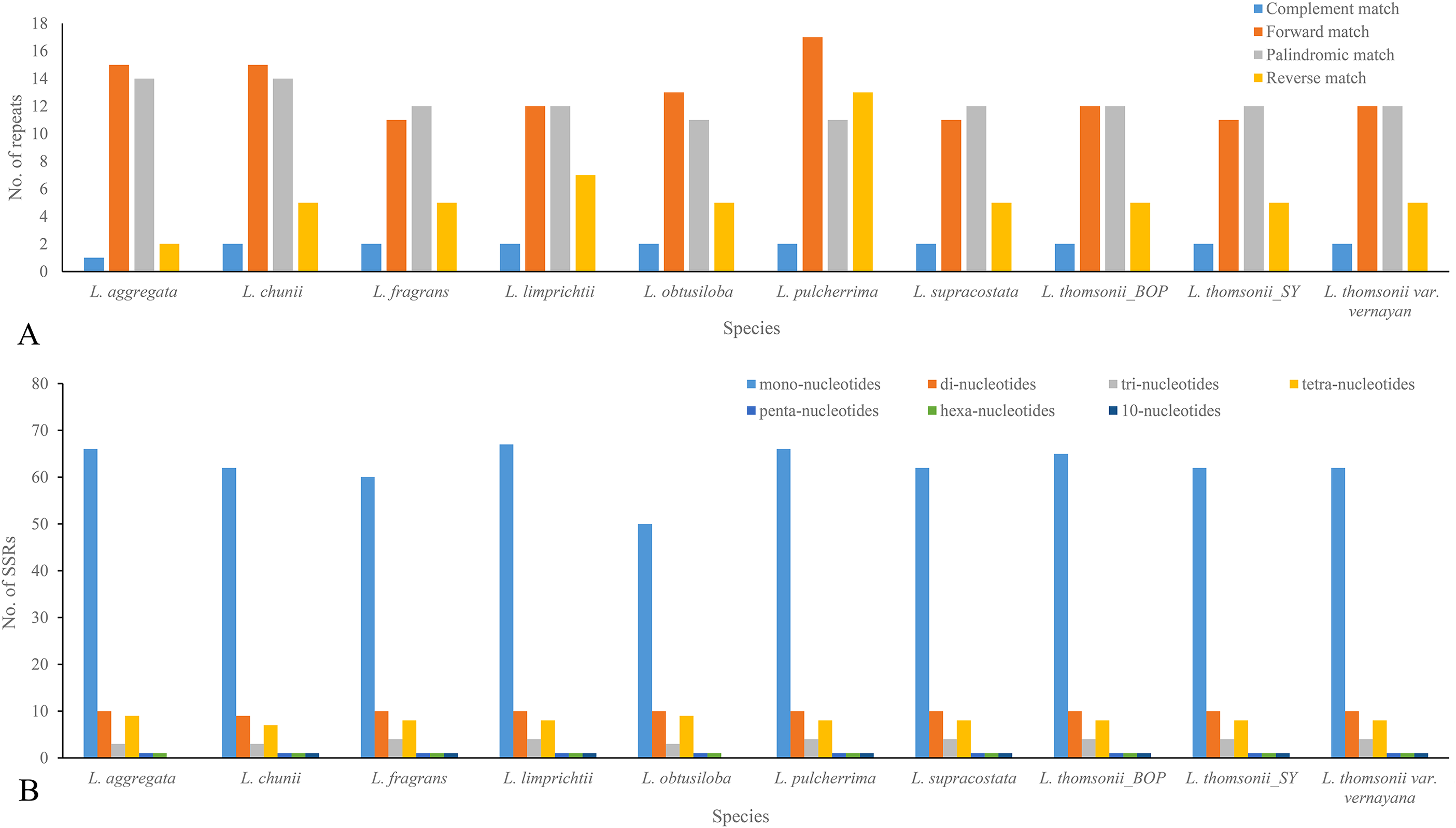{kind=link}
Genome sequence mutations and divergence
Both indel and SNP markers were not randomly distributed in the plastomes, and we calculated the genetic divergence among plastomes of the four trinerved Lindera and all of the “core Laureae” species. The nucleotide diversity (Pi) values within 600 bp among the trinerved Lindera varied from 0 to 0.01248 with a mean of 0.00154, while among the 23 plastomes of the “core Laureae” species the values varied from 0 to 0.01989 with a mean of 0.00418 (Fig. 4A). Six highly variable loci (Pi >0.006) were identified in the plastomes of the trinerved Lindera. They were trnH-GUG/psbA, psbM/trnD-GUC, petA/psbJ, ndhF, trnL-UAG/ndhD, and ycf1. Different hypervariable regions including ndhD, rpl32/trnL-UAG, and three fragments of ycf1 were precisely located in the plastomes of the “core Laureae” species (Fig. 4B). However, only five divergence hotspots were detected in the “core Lauraceae” plastome sequences of 41 species within the Persea group and Laureae-Cinnamomeae group, with values ranging from 0 to 0.01922 with a mean of 0.00447 (Fig. 4C), suggesting that two fragments of ycf2, ndhD, ndhF/rpl32, ndhH, and three fragments of ycf1 may serve as useful DNA barcodes for Lauraceae. Therefore, these regions may be suitable prospective markers to reconstruct the phylogenetic relationship within Lindera, or the intergeneric relationship of Lauraceae.

Figure 4: Sliding window analysis of the complete plastid genome sequences of trinerved Lindera species (A), 23 “core Laureae” species (B) and 47 “Core Lauraceae” species (C).
Note: Window length: 600 bp; step size: 200 bp. Y-axis means nucleotide diversity.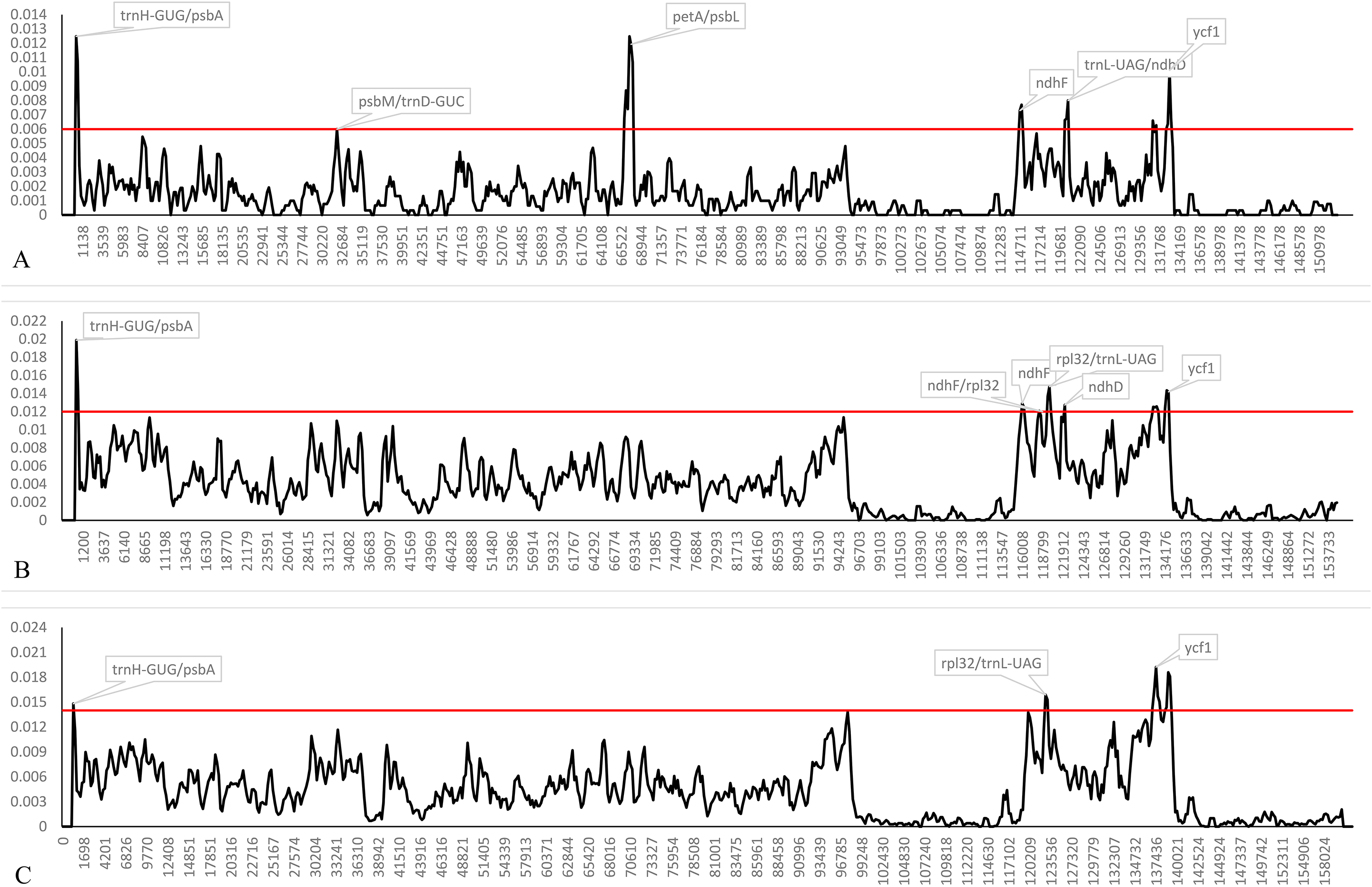{kind=link}
Phylogenetic analysis
The matrix of complete plastomes was used to reconstruct a phylogenetic tree of the 47 species from the “core Lauraceae” group. The topologies using ML and BI methods were similar. All species were divided into three groups: the Laureae group, the Cinnamomeae group, and the Perseae group. The Laureae group taxa were divided into six clades based on node support (Fig. 5). The posterior probability (PP) of BI analysis at most of the nodes was more than 0.90, and the BS values of the nodes were more than 90 in ML inference. Clade I contained the trinerved Lindera species, and Iteadaphne caudata with BS 100 and PP of 1.00. Clade II contained Actinodaphne trichocarpa C.K. Allen and Neolitsea sericea (Blume) Koidz. species with BS 100 and PP 1.00. Clade III contained Lindera benzoin, Lindera robusta, Lindera latifolia, and Lindera metcalfiana species with PP 0.99. Clade IV contained two Lindera obtusiloba species. Clade V contained L. communis, Lindera glauca, and Lindera nacusua with BS 100 and PP 1.00. Clade VI contained Laurus nobilis, three Litsea species, and Lindera megaphylla with BS 100 and PP 1.00 (Fig. 5). Among these clades, eight trinerved Lindera species formed a monophyletic lineage which nested with the trinerved Iteadaphne caudata, and Actinodaphne trichocarpa and Neolitsea sericea formed the sister clade with BS 100 and PP 1.00 (Clades I and II). Clade III was supportive in a sister position to Clades I and II in ML and BI trees.
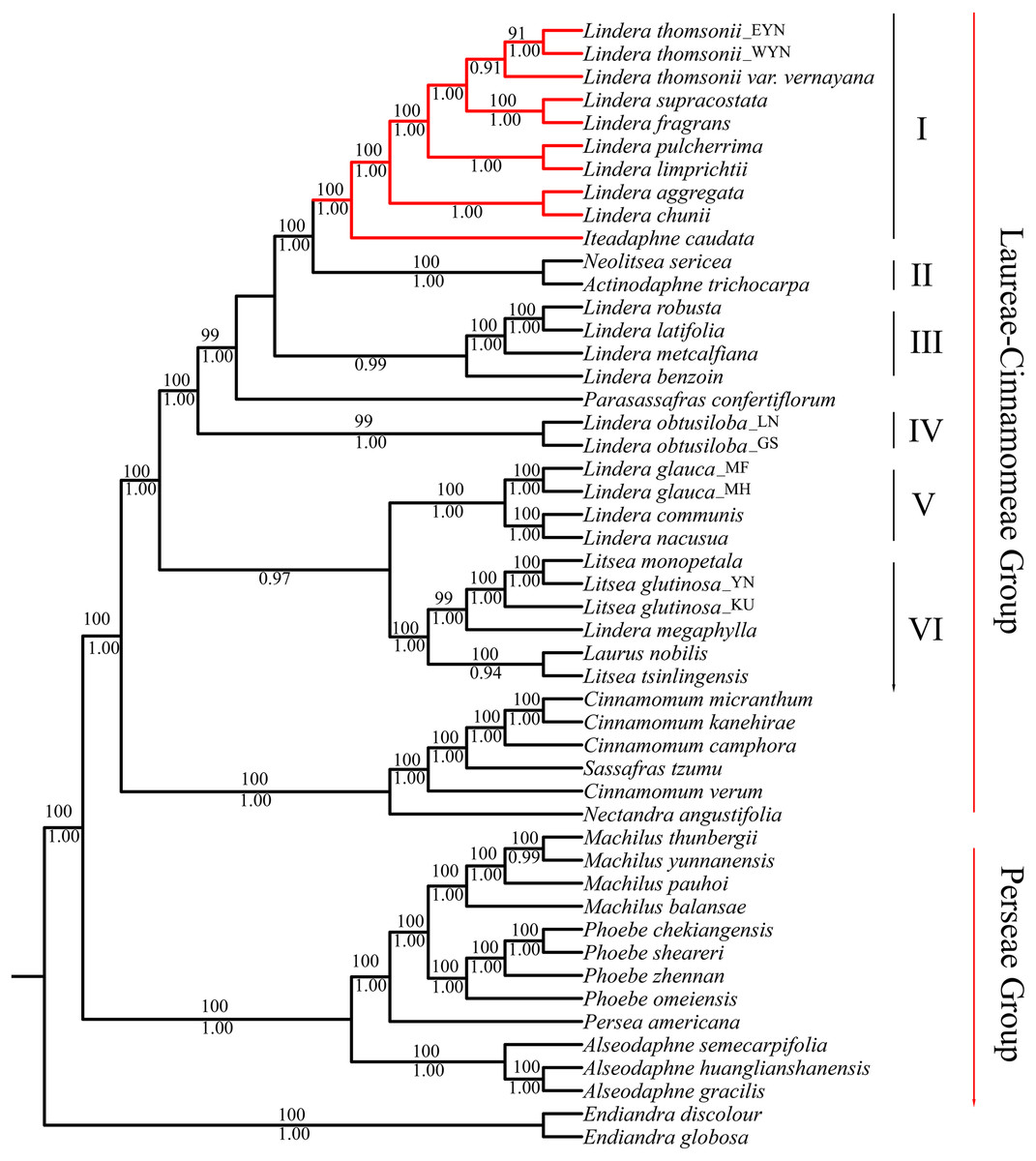
Figure 5: Complete plastid genome phylogenetic tree of 49 species based on maximum likelihood analysis and Bayesian inference.
Branches with red color means trinerved Lindera species in this study. Numbers above branches are posterior probabilities and bellow branches are bootstrap values. EYN means East of Yunnan; WYN means West od Yunnan; LN means Liaoning; GS means Gansu; YN means Yunnan; MF, MH and KU mean sequence from Genbank.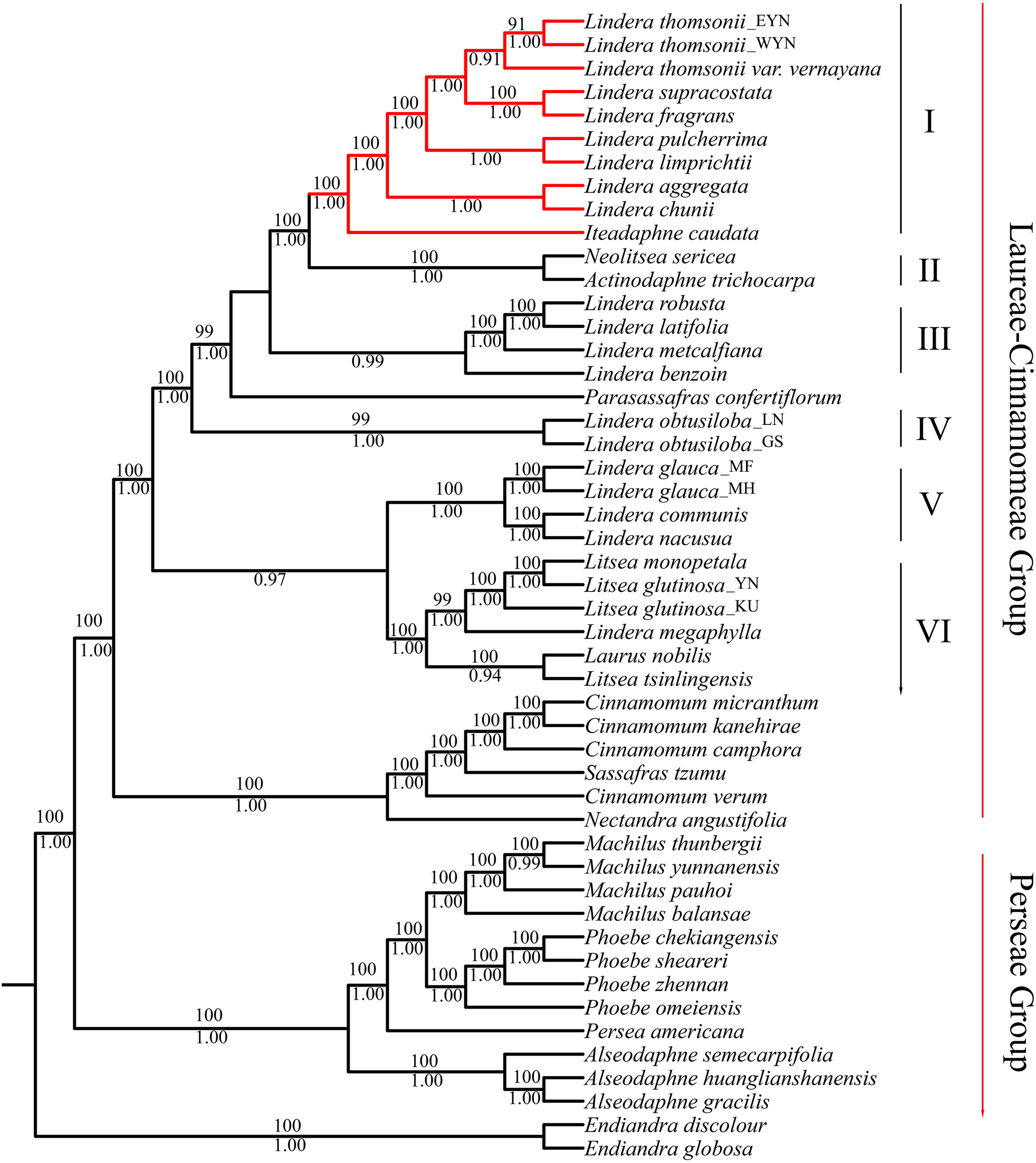{kind=link}
Discussion
In this study, we accomplished 10 complete plastid genomes of trinerved Lindera species using Illumina high-throughput sequencing technology (Fig. 1). The sizes of the trinerved Lindera plastomes were very similar to each other—longer than Lindera benzoin (152,478 bp) and Lindera nacusua (152,211 bp), but smaller than Lindera metcalfiana (152,968 bp) and Lindera robusta (152,852 bp) (Zhao et al., 2018). All species shared a typical angiosperm quadripartite structure, and the IR regions were more conservative than the LSC and SSC regions (Figs. 2 and 4; Appendix S4). Further analysis on sequence conservation between the 10 trinerved Lindera species and 18 previously sequenced “core Laureae” species has been carried out by using the BLAST tool, with results showing high similarities in both conserved structure and gene content (≥99%) (Chen et al., 2017; Li et al., 2017; Song et al., 2015, 2016, 2017b, 2018; Wu et al., 2017; Wu, Ho & Chang, 2016). In addition, the nucleotide variable value of the whole plastomes among the trinerved Lindera species was 0.14%. This value is approximate to the value of two Machilus species (0.15%) (Song et al., 2015), two Phoebe species (0.10%) (Song et al., 2017a), and three Alseodaphone species (0.12%) (Song et al., 2018), which indicates that the genetic divergence within the genera in the family Lauraceae is low (Appendix S4).
Microstructural mutations including perfect SSRs with a minimum size of 10 bp were detected in the plastid genomes of the trinerved Lindera species. Similarly, 83, 88, 81, 82, 86, 65, and 66 SSRs were recognized among the plastid genomes of Cinnamomum camphora (L.) T. Nees & C.H. Eberm, Cinnamomum micranthum Hayata, Litsea glutinosa, Machilus yunnanensis Lecomte, Persea americana Mill., Phoebe bournei (Hemsl.) Yang, and Phoebe chekiangensis P.T. Li, respectively (Chen et al., 2017; Li et al., 2017). Among the trinerved Lindera plastomes, mononucleotide repeats (A/T) were found to be abundant among SSRs (about 68–74%), followed by dinucleotide repeats (AT/AT and AG/CT, 10–13%) among the plastome sequences (Fig. 3B). This suggested that mononucleotide repeats may contribute more phylogenetic signal information in comparison to other SSRs (Appendix S3). Also, repeat motifs are generally found in the intergenic regions of trinerved Lindera plastid genomes, which shows the great variable nucleotide regions in the plastid genomes (Appendices S2 and S3). Repeat numbers were similar to Cinnamomum camphora, Cinnamomum micranthum, Litsea glutinosa, Persea americana, Phoebe bournei, and Phoebe chekiangensis, abundant palindromic repeats (Chen et al., 2017; Li et al., 2017; Song et al., 2016; Wu et al., 2017; Wu, Ho & Chang, 2016), which may provide some secondary structures or duplicate sequences in the whole genome (Kurtz et al., 2001).
All phylogenetic trees supported the relationships among the “core Lauraceae” described by Rohwer & Rudolph (2005) as a sister group of the Laureae-Cinnamomeae group and Persea group in accordance with other previous studies (Chanderbali, van der Werff & Renner, 2001; Huang et al., 2016; Li et al., 2011; Rohwer, 2000; Rohwer et al., 2009; Rohwer & Rudolph, 2005; Song et al., 2017b). The BI and ML analyses with complete plastid genomes yielded 1.00 Bayesian PPs and 100 BS values at each node. The relationships among the 20 Lindera species and 13 representatives of eight other genera species in the Laureae-Cinnamomeae group were phylogenetic trees divided into six clades, as described by Rohwer & Rudolph (2005). All trinerved Lindera species belonging to sect. Daphnidium were monophyletic. The phylogenetic trees also showed that the trinerved Lindera species had a sister relationship with Iteadaphne caudata, and they grouped with Neolitsea and Actinodaphne with higher variables among ndhF, rpl32/trnL-UAG, and ycf1, similarly to how Fijridiyanto & Murakami (2009) constructed phylogenetics with a low copy nuclear rpb2 gene, and how Song et al. (2017b, 2019) did the same with plastid genomes. Consequently, the systematics of Iteadaphne were recombined from Lindera caudata (Nees) Hook. f. by a single floret per umbellule, while the type specimen of Parasassafras, based on Actinodaphne confertiflora Meisn., was appropriated (Li, 1985; Ng, 2005; Tsui, 1987a, 1987b). Our results support that the number of anther cells is not a valuable characteristic, and the hypothesis of Neolitsea and Actinodaphne being more closely related to the trinerved Lindera species and Iteadaphne needs further studies based on intensive sampling.
Conclusions
Two highly variable regions, ndhF and psbA-trnH, were identified among the trinerved Lindera species, and their highly conserved structures and gene content have been found to be very similar. This similarity may be useful when studying phylogenetic information and in species identification. Our results also supported that the sect. Daphnidium species, as an independent sub-clade with Iteadaphne caudata, formed a sister clade to Actinodaphne and Neolitsea. The results also show that the systematics of Iteadaphne were recombined from Lindera caudata (Nees) Hook. f. when a single floret per umbellule was misappropriated, which indicated a close relationship with the trinerved Lindera species. Altogether, this study will help improve our understanding of phylogenetics and the evolution of the Lindera species.
Supplemental Information
Raw data of 10 Lindera species.
Complete plastid genome sequences and annotations of Lindera aggregata, Lindera chunii, Lindera limprichtii, Lindera pulcherrima, Lindera obtusiloba, Lindera supracostata, Lindera thomsonii BOP, Lindera thomsonii SY, Lindera thomsonii var. vernayana, Lindera fragrans.
Characteristic of 10 Lindera plastome sequences.
Appendix S1 List and accession number of 49 taxa for complete plastid genome phylogenetic analysis
Appendix S2 The repeats distribution in the plastid genome sequences of Lindera species (Note: F, P, R, and C represent forward, palindrome, reverse, and complement type repeat sequences in the plastome.)
Appendix S3 The mutations and Indels distribution in the plastid genome sequences of Lindera species
VISTA-based identity plots showed sequence identity of 10 Lindera plastid genome sequences with Laurus nobilis as a reference.
Appendix S4 VISTA-based identity plots showed sequence identity of 10 Lindera plastid genome sequences with Laurus nobilis as a reference.
Tree flies of 10 Lindera species.
Appendix S5 tree file of Bayesian inference based on BEAST 2.4.8. Appendix S6 tree file of maximum likelihood based on RAxML 8.2.10.