
Abstract
Close-coupled selective catalytic reduction (SCR) systems are one method to deal with tightening emission legislation for NOx in internal combustion engines. Due to smaller mixing sections and at unfavourable boundary conditions, however, urea-water solution (UWS) droplets can impact on the SCR catalyst itself. To investigate this phenomenon further, this work develops a modeling capability of this process. Established mechanism for NH3-SCR and HNCO hydrolysis from literature is integrated into DETCHEMCHANNEL and a 2D COMSOL model to simulate the influence in the SCR Channel. Simulations are validated against end-of-pipe experiments from literature and spatially resolved concentration profiles from a hot gas test rig with very good agreement. Finally, a channel simulation is coupled with a model to describe the catalytic decomposition of an urea droplet. The coupled simulation is able to simulate the influence of UWS droplet impact onto a catalyst channel. Fast droplet decomposition causes a peak in NH3 and HNCO in the single channel and thus increases NOx conversion. However, the overall uniformity and efficiency are decreased, which is why droplet impact on the catalyst should be strictly avoided.
Similar content being viewed by others


1 Introduction
Combustion processes of lean-burn engines, fueled with diesel, natural gas or hydrogen, inevitably lead to the formation of nitrogen oxides (NOx). These can have a severe impact on the environment and the human health [1], which makes the need for a reduction of NOx emissions apparent. Especially the emissions of diesel fueled internal combustion (IC) engines have become a matter of public interest in recent years. Despite significant advances in the optimisation of combustion systems, fuel injection equipment and turbocharging, catalytic exhaust gas aftertreatment is an indispensable tool to meet ever increasing regulatory standards for NOx emissions (Euro 7, US Tier 3) [2, 3]. In the case of lean burn engines, where excess oxygen is present, an additional reducing agent such as ammonia (NH3) is required to remove nitrogen oxides via selective catalytic reduction (SCR). In mobile applications, due to safety concerns, ammonia is generated on board by decomposition of urea. Urea is provided, by spraying 32.5 wt% urea-water solution (UWS), commercially available as AdBlue\(^{{{{\circledR }}}}\) in Europe or DEF in the USA, into the exhaust line in front of a catalytic honeycomb monolith. Urea undergoes thermolysis to form one equivalent of each NH3 and isocyanic acid HNCO. The latter then hydrolyses mainly on the catalyst into another equivalent of NH3 and CO2 [4, p. 247].
The actual reduction of NOx occurs on the SCR catalyst itself. Depending on the temperature and the ratio between NO and NO2 different reduction reactions are favoured: At lower operating temperatures and an equal ratio of NO2 and NO, the fast SCR reaction of one each with two equivalents of NH3 is predominant. At low conversion of NO to NO2 in the DOC, the standard SCR reaction of NO, NH3 and some parts of O2 to N2 and H2O is favoured. However, if the NO2/NO ratio exceeds 1 : 1, the slow SCR reaction takes place, which increases the NH3 consumption.
Extensive efforts have been made to model vanadium based SCR catalyst systems. Global kinetic [5, 6], as well as microkinetic models based on Langmuir Hinshelwood [7,8,9], Eley Rideal [10,11,12,13,14,15] and Mars van Krevelen [16, 17] mechanisms, has been proposed. Similarly, there were modeling studies for Cu- or Fe-Zeolithe SCR catalysts [18,19,20]. For monolithic catalyst structures, single channel representations [21,22,23] including washcoat models, as well as simulation of entire monoliths, have been conducted [24,25,26].
Close coupled systems, where the SCR catalyst is placed directly behind the engine, besides the obvious spatial benefit, have the advantage of a faster heat-up of the catalyst and therefore reach higher NOx conversion earlier, after the cold start of an engine [27]. However, depending on the operating conditions, the spatial limitations of the close-coupled system and incomplete evaporation can lead to spray-wall interactions and thus liquid film formation inside the dosing and mixing sections up to droplet impact on the catalyst itself, especially at lower temperatures [28, 29]. A recent study by [30] investigated the influence of SCR catalyst materials on the decomposition of urea deposits by TG experiments. Both pure TiO2 and a typical VWT powder have shown to accelerate the decomposition of the urea by-products. At the highest catalyst/sample ratio of about 0.5, it was possible to end the decomposition as early as 450∘C, leaving no further deposits. Based on the experiments, a surface mechanism was developed to simulate the catalytic influence on urea decomposition in addition to the non-catalytic mechanism by [31]. Nevertheless, at lower temperatures, urea by-products can remain on the catalyst surface, which can lead to deposit growth up to a blocking of catalyst channels. This deposit growth is a well-known and unwanted phenomena in urea-SCR exhaust systems as it increases the engine back pressure and decreases the system efficiency [32]. Deposits on the porous catalyst structure can furthermore decrease the available surface area and therefore decrease the catalytic DeNOx activity.
In this work, we set up a model to simulate the SCR reactions and the catalytic HNCO hydrolysis in a SCR catalyst and compare them with spatially resolved concentration measurements in a monolith channel. Furthermore, we study the effect of UWS droplets impacting on the SCR catalyst and the influence on the conversion of NOx inside a channel by combining the aforementioned catalyst channel model with a model to describe the catalytic decomposition of urea. To the best of our knowledge, this coupling of liquid and catalytic urea decomposition and the surface chemistry in SCR catalyst channels has not been studied before. As a result, it provides important insight on possible processes and phenomena in close-coupled SCR systems and can be used to investigate optimal boundary conditions.
2 Experimental Methods and Results
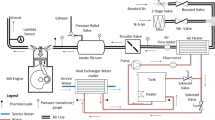
To obtain stationary spatially resolved concentrations of different gas phase species over catalytic honeycomb monoliths, a capillary-based in situ probe technique, previously described in [33,34,35,36], was employed. The experimental setup of the hot gas test rig allows for a wide range of operating conditions [37] (temperature, gas phase concentrations, UWS mass flow, gas volume flow) under which concentration and temperature measurements can be performed. A sketch of the hot gas test rig can be found in Fig. 1.

High gas flow rates of are generated by a side channel blower from ambient air. The flow rate is measured by a flow meter (Bronkhorst In-Flow F-106BI). An inline electrical gas heater (Leister LE 5000 DF) is used to heat the gas flow. NO and NH3 can be added in the heating section. The amount of NO and NH3 is measured and controlled by mass flow controllers (MFCs). Following the heating section, an inlet section is employed to generate defined flow conditions. First, the gas flow is passed through a mixing section to ensure a homogeneous distribution of the gas phase species. Subsequently, a cross-section expanse leads into a flow straightener with a honeycomb structure to reduce turbulence. The cross-section is reduced to the rectangular cross-section of the monolith (60 × 30 mm), which remains constant over the rest of the test rig.
The injection section is 200-mm long and features a commercial water-cooled three hole injector in a 33∘ inclination angle. UWS (AdBlue®;, EuroLub) can be dosed with an injection pressure of 5 bar. A commercial vanadia-titania-based catalyst monolith (Umicore) of 15-cm length and 600 cpsi was placed directly behind the dosing section. The honeycomb channels have rectangular cross-sections. A cordierite monolith without active substance was used for blank measurements. Following the catalyst is an outlet section of 100 cm.
A Fourier-transform infrared spectrometer (FTIR, MultiGas 2030, MKS) can be used to measure the gas phase concentrations downstream of the catalyst (end-of-pipe). To measure spatially resolved gas phase concentrations, a deactivated, fused silica capillary is connected to a motorised linear stage (OptoSigma). The capillary, with an outer diameter of 170 μm and an inner diameter of 100 μm, sucks in \({\sim 1}\) mL min − 1 and directs the small gas flow to a quadrupol mass spectrometer (HPR-20, Hiden Analytical). Before the measurement, the capillary is inserted against the direction of flow into a channel located in the center of the monolith until the open end of the capillary is flush with the beginning of the catalyst. During the measurement, the capillary is moved incrementally in the direction of the gas flow by the linear stage. At least four data points are recorded inside the catalyst for approx. 5 min at each position. The mass spectrometer is calibrated with two stationary concentration values of the FTIR (base value and operating condition). Temperatures were measured with type-K thermocouples, which were placed inside the inlet and dosing section, outside the dosing section and catalyst and moved along the capillary for spatially resolved measurements inside the catalyst channels.
The capillary can significantly increase the residence time of the gas phase species, depending on the ratio between channel and capillary diameter, as well as the axial and radial position of the probe [38]. Residence time corrections have been calculated by Hettel et al., by comparing CFD simulations and experimental results [38, 39]. For our setup, the volume flux is at most reduced by approx. 6 %, if the probe is inside the corner of a rectangular channel and 45 %, if the probe is in the center of the channel [39]. However, during experiments, the radial position of the probe is unknown. Therefore, due to the insecurity with regard to the quantitative influence of the capillary and the potential negligible change in volume flux, no residence time correction was applied to the experimental concentration profiles.
3 Model Description
In the following, the two applied simulation models are described. While the DETCHEM model convinces with its simplicity, the model in COMSOL offers the possibility of an extension to 3D CFD simulations.
3.1 DETCHEM
The DETCHEMCHANNEL package is a two-dimensional model for tubular reactors, simulating gas flow, as well as gas-phase and surface reactions. To reduce computational time, the steady-state boundary-layer approximation is employed [40]. The approximation yields similar results in comparison with a more detailed Navier-Stokes model, if axial diffusion can be neglected [21, 41]. This is the case for high fluid velocities and small channel diameters, meaning high Reynolds numbers Re, as long as the flow remains laminar [21]. For our experiments, Re numbers were calculated via Re = u ⋅ dchannel/ν(Tgas) and were between 70 (250∘C) and 119 (450∘C). Therefore, with the critical Reynolds number of 2030, laminar flow inside the channels is ensured and the boundary layer model should be ideally suited. Furthermore, several publications have shown that a single cylindrical channel can be representative of a monolith consisting of a multitude of rectangular channels [21, 41,42,43].A set of five governing equations are implemented into DETCHEM [44]:Total mass continuity:
Axial momentum:
Radial momentum:
Energy continuity:
Species continuity:
Because the code simulates the steady state of the channel, the total mass continuity equation (Eq. 3) does not require a sink term to consider adsorption and desorption reactions, as the net mass transfer between wall and gas phase is zero. The radial diffusion flux is calculated via
At the channel wall, the radial diffusion flux depends on the reaction source terms \(\dot {r}_{i}\), which are calculated based on the implemented reaction mechanism. Inside the channel ji is calculated based on the diffusivity Di of species i. After radial discretisation, a set of first-order differential equations is obtained. To solve the system, a finite volume method is applied, with the dependent variables being the mass flux, flux of axial momentum, enthalpy flux and species mass flux. Subsequently, the system of equations is transformed from using the mass flux instead of the radial coordinate, as independent variable. Finally, in order to solve the governing equations, the standard implicit LIMEX solver is used [40]. To model the surface and gas phase reaction source terms ji and \(\dot {\omega }_{i}\), microkinetic reaction mechanisms are implemented.
3.2 COMSOL
In addition to the simulations in DETCHEMCHANNEL in this work, CFD simulations were performed with COMSOL Mulitphysics Version 6.0 [45]. It is a commercial code which uses the finite-element-method (FEM) to solve multiphysical problems. Here a model for a two-dimensional channel was set up to simulate the experiments in Section 2 and to serve as an additional comparison to the simulations in DETCHEM. The simulation model of the channel consists of several submodels which are described in the following:The Geometry was created according to the experimental channel dimensions. Since a fully developed inlet flow was assumed in the channel inlet, comparable to the DETCHEM simulation, no additional inlet section upstream of the channel was added.It was meshed with the build-in mesher and element maximum and minimum sizes of 1.68 × 10− 4m and 7.71 × 10− 6m, respectively. Furthermore, four boundary layers were added to the top and bottom wall of the channel for a better and finer description of the wall effects. Overall the mesh consisted of 5955 elements over the 15-cm channel. A mesh independence study confirmed that a sufficiently fine mesh was used.In the Chemistry submodel, all gas and surface species and reactions were defined. The thermodynamic and transport data for the species were calculated based on the NASA polynomials and potential characteristic length, energy minimum and dipole moment, respectively, provided from the DETCHEM database. The reactions and respective reaction rates were implemented according to the developed mechanism described in Section 4.1.In addition, the Transport of Diluted Species model is used to compute the concentration field of the solute species in the solvent N2. Since most of the used species only have a concentration of a few hundred ppm in the solvent N2, this assumption is seen to be justified. The driving forces for the transport of the species are the diffusion by Fick’s law and convection through the fluid flow.The Laminar Flow model is used to calculate the velocity and pressure field of the laminar single-phase flow within the channel. Here the Navier-Stokes equations for conservation of momentum and the continuity equation for conservation of mass are solved. The fluid properties are calculated based on the thermodynamic and transport data provided within the chemistry module. At the inlet, a velocity distribution of a fully developed flow is set, while at the outlet, the pressure is set to ambient conditions.In the Heat Transfer in Fluids module, the boundary and initial thermal conditions are given. The channel inlet is set to the stationary or time-dependant inlet temperature, while all other boundaries are adiabatic. However, it is accounted for the surface heat of reaction.Lastly, the Surface Reactions module is used to define the surface properties and to simulate the different surface species and respective surface concentrations on the channel wall.
4 Simulation Results
The following sections are about the implementation of the used reaction mechanism and the simulation results of the experimental data. At first, the implementation of the NH3 SCR mechanism by [17] is validated against end-of-pipe experiments from [42]. Furthermore, a mechanism for HNCO hydrolysis from [46] is added. The combined model is compared to spatially resolved experiments from [36] and own measurements. Finally, the validated channel model is combined with a model for the urea decomposition to simulate the case of an impacting droplet onto the catalyst.
4.1 Implementation of Mechanism
For the modeling of the NH3 − SCRreactions, a dual-site Mars-van Krevelen mechanism, by Nova et al. [17], was implemented into DETCHEMCHANNEL and COMSOL. The mechanism consists of a set of ten reactions. These include the standard SCR, ammonia adsorption and desorption and ammonia oxidation, which becomes relevant at higher temperatures [47]. Furthermore, if NO2 is present in the gas phase, nitrate formation enables the fast SCR, NO2 SCR and nitrous oxide (NO2) formation, as well as nitrate desorption and adsorption. Since our experiments only involved NO, the reactions with NO2 were omitted and the NH3 mechanism reduced to 5 reactions.
A key part of the mechanism is the differentiation between two different adsorption sites. The S1 sites are associated with vanadium species, where NO can adsorb. Whereas the adsorption of ammonia and nitrates occurs on the S2 sites, which are associated with oxides of other metals, such as tungsten or titanium. Surface coverages of species i on the S1 and S2 sites are denoted as σi and 𝜃i, respectively (Table 1). The rate of adsorption of NH3 and HNO3 depends on a rate constant ki, the gas phase concentration Ci and the number of free adsorption sites 𝜃i, represented by the term \((1-\theta _{\text {NH}_{3}}-\theta _{\text {HNO}_{3}})\) (Eq. 1). Additionally, a so-called spillover effect is modeled, where NH3 adsorbed on S2 sites can migrate to the S1 sites and therefore block adsorption sites for NO (Eq. 1) [17]. In the reaction equation of the standard SCR, this is shown by the term (1 − σi). Thereby, the inhibition of DeNOx reactions by higher ammonia concentrations is represented [48]. The desorption reactions Eq. 1 depend on a rate constant and the surface coverage of the species (Eq. 1). In the case of NH3, the rate constant of desorption is also influenced by its surface coverage \(\theta _{{\text {NH}_{3}}}\). The relevant reaction parameters of reactions R1–R6 are listed in Table 2.
The implementation of the mechanism into DETCHEMCHANNEL was evaluated with experimental data taken from Rammelt et al. [42]. They investigated the catalytic activity of 1.9 wt% V2O5, 10 wt% WO3 and TiO2 over two different monoliths with 25 and 300 cpsi. Conversions of NH3 and NO at a GHSV of 65,000h− 1 were measured for the temperatures 573, 673 and 773 K at initial NO/NH3 concentrations of 500 and 1500 ppm. Furthermore, conversions at a GHSV of 150,000h− 1 were obtained for temperatures of 573 and 673 K and NO/NH3 concentrations of 300, 500 and 1500 ppm. The ratio between NH3 and NO concentrations was 1:1 for all experiments.
As can be seen from Fig. 2, where some exemplary results are shown, the simulated conversions are in very good agreement with the experimental values. The single channel model is able to accurately reproduce the trends of temperature, concentration and channel diameter, without having to take the entire monolith into account. The average relative deviation between simulated and experimental conversions is 5.2 % for the 300 cpsi and 12 % for the 25 cpsi monolith, while the absolute deviation was 4 % conversion for both. Predictions for NO are also moderately better than for NH3, likely because of the rate equation for the ammonia oxidation, which results in an underestimation of NH3 conversion at lower (573K) and overestimation at higher temperatures (673K). If NH3 is formed from UWS, the decomposition of urea has to be taken into account. Concentration measurements have revealed that thermolysis to NH3 and HNCO is completed in front of the catalyst and was therefore not modelled in this work. In contrary, significant conversion of HNCO was only observed inside the catalyst, which required the implementation of a mechanism to model axial concentration gradients, due to catalytic hydrolysis. Several publications have investigated the adsorption, chemistry [51, 52] and reaction kinetics of HNCO on VWT and other catalytic surfaces such as anatase (TiO2) [46, 53,54,55,56,57]. In some of these publications, the influence of other gas phase species (NO, NO2) is considered. However, to our knowledge, the only microkinetic model, derived from a set of five elementary steps, was developed by Hauck et al. for the hydrolysis on anatase [49].

Simulated and experimental NO/NH3 conversion over VWT catalyst monoliths. Experimental data taken from Rammelt et al. [42]
The algebraic solution was based on a quasi equilibrium approach, where the reaction of the surface species Ti-NCO and Ti-H2O is the rate-determining step (RDS). By substituting the surface concentrations of the two species by experimentally determined sorption isotherms, the only remaining variables are the partial pressures of HNCO, NH3 and H2O and the temperature. The remaining kinetic and thermodynamic parameters were determined by fitting the reaction rate of the model to the variation of the HNCO, H2O and NH3 concentration.
During the spatially resolved experiments with UWS on the hot gas test rig, complete urea thermolysis was achieved in front of the VWT catalyst. Accordingly, NO, NH3 and HNCO are the main SCR active species affecting the conversion on the VWT catalyst. To model the conversion of the three species, the two mechanisms by Nova et al. [17] and Hauck et al. [49] were combined. The NH3-SCR mechanism is implemented into the Channel model, according to the specifications of the original publication with some adjustments [17]. To add the hydrolysis reaction, several assumptions were made. As mentioned by Bernhard et al., TiO2 is highly active towards HNCO hydrolysis [54]. On the other hand, an increasing vanadium content has an inhibiting effect on the reaction rate [57]. For our system, we have therefore assumed that hydrolysis on vanadium species can be neglected. As a result, the model by Hauck et al. can be used, since it only models the hydrolysis on TiO2. Furthermore, the NH3-SCR mechanism distinguishes between the S1 (VOx) and S2 sites (other acidic metal oxides). In this case, we decided to let the modeled hydrolysis reaction take place on the S2 sites of the NH3-SCR mechanism. This assumption is supported by the fact, that NH3, as well as HNCO adsorption takes place on the acidic site of a VWT or TiO2 catalyst [17, 53].
4.2 Simulation of Spatially Resolved Experiments
Experimentally recorded spatially resolved concentration profiles for NO, NH3 and HNCO were modelled using DETCHEMCHANNEL and COMSOL Multiphysics. The amount of active sites was fitted manually, so that the highest agreement between experimental and simulated concentration profiles could be achieved, and was in the range for typical ammonia adsorption capacities of 110–200μmol g− 1 [28, 47, 57, 58]. The ratio between S1 and S2 sites was estimated from typical VOx surface coverages of 25–50 % [59] and was set to 50 %. To demonstrate the validity of the models, concentration profiles over the same catalytic VWT monolith were measured for different temperatures and inlet concentrations, and subsequently simulated. The gas velocity inside the channel was 8.3ms− 1
For the experiment in Fig. 3, NH3 and NO were dosed directly, without UWS at a temperature of 350∘C. The inlet concentrations were 650 ppm each. The NO concentration decreases rapidly at first, due to the standard SCR reaction. Over the length of the catalyst, the reaction rate then gradually decreases until the concentration approaches the final value of 43 ppm, amounting to a conversion of 93 %. The simulated NO concentrations (mixing cup average over the channel height) represent the axial development of the experimental values quite well. Especially at the end of the catalyst, where the deviation is < 1 ppm. In contrast, NH3 does not appear to react until two cm into the catalyst. After that, the concentration drops linearly to 120 ppm (\(X_{{\text {NH}_{3}}}=82 \%\)). This does not agree with neither the FTIR end-of-pipe measurement, nor the simulated concentration profile. Furthermore, the nitrogen balance would suggest an equivalent or increased NH3 consumption compared to the NO consumption. Especially because NH3 oxidation was observed at 350∘C during NH3-only experiments. However, this is not the case over the entire length of the catalyst. The end-of-pipe concentrations measured by FTIR also show that the NH3 concentration is higher than the NO concentration. However, the difference is significantly smaller and can possibly be attributed to an increased NH3 dosage. This deviation is attributed a low signal-noise ratio for NH3 from the MS, and an overlap in atomic masses for isotopologues of NH3 and H2O (m/z 17). As a result, faulty experimentally obtained ammonia concentration profiles are attributed to a measurement error and will not be further discussed for each experiment.

Experimental and simulated spatially resolved gas phase concentration of NH3 and NO inside a single channel of a stationary VWT monolith. T0 = 350∘C, \(\boldsymbol {C_{{\text {NH}_{3}},0}=650 \text {ppm}}\)
The simulated conversions of NH3 at the end of the catalyst are slightly overestimated, when compared to the FTIR measurement. This trend was already observable in Fig. 2 for higher temperatures. Furthermore, for simulations of NH3-only experiments at 350∘C, NH3 conversions are similarly overestimated. The DETCHEMCHANNEL model shows a steeper decline of gas phase concentrations, then the COMSOL model. As result, the COMSOL model is in better agreement with the experiment. However, both models approach the same final value at the end of the channel, further highlighting the validity of both a detailed CFD model, as well as a simplified boundary-layer approach. Nevertheless, the differences regarding the resolution, flow and transport modeling can explain the small differences between the two simulation results.
For experiments that mimic the conditions of real SCR exhaust gas aftertretament systems, HNCO is generated through the decomposition of UWS. An equimolar yield of HNCO and NH3 can be assumed according to Eq. 2 and was confirmed in front of the catalyst. In Fig. 4, the settings of the injector resulted in a concentration of 200 ppm each, which was further validated via FTIR measurements over an inactive cordierite monolith. Over the VWT catalyst, the HNCO concentration decreases rapidly within the channel, reaching 50 % conversion before 2cm. At the end of the catalyst, the concentration has decreased to 4 ppm, corresponding to a conversion of 98 %. The simulated HNCO values are in good agreement with the experimental values over the whole range of the channel. At the end of the channel, simulated HNCO conversions reach 99.8 %, which is higher than the experimental conversion. However, this deviation is within the measurement error of the mass spectrometer. The difference between the two models is negligible.

Experimental and simulated spatially resolved gas phase concentration of NH3 and HNCO inside a single channel of a stationary VWT monolith. T0 = 250∘C, \(\boldsymbol {C_{{\text {NH}_{3}},0}=C_{{\text {HNCO}},0}=200 \text {ppm}}\)
The ammonia concentration is expected to increase during the hydrolysis of HNCO to CO2 and NH3. Nonetheless, the experimental concentration profile of ammonia resembles that of HNCO, in some experiments decreasing to even negative values. To our knowledge, the hydrolysis reaction is the only reaction consuming HNCO. Therefore, the NH3 concentration should increase in equal parts as the HNCO concentration decreases, as shown by the simulated NH3 concentrations. Here an additional 200 ppm of NH3 are generated, amounting to a final concentration of 400 ppm, because no further ammonia conversion is observed, as the temperature is too low for a potential oxidation reaction.
If NO is dosed in addition to HNCO, a decrease of HNCO due to hydrolysis, as well as a decrease of NO due to the Standard SCR reaction should be visible throughout the catalyst. The spatially resolved gas phase concentrations of such an experiment with NO (CNO,0 = 400ppm), NH3 (\(C_{{\text {NH}_{3}},0}=200 \text {ppm}\)) and HNCO (CHNCO,0 = 200ppm) at 250∘C are depicted in Fig. 5. The NO concentration decreases gradually to 140 ppm at the end of the catalyst channel (XNO = 66% at 14cm), due to the standard SCR reaction with NH3. The models are in good agreement with the experimental values, but slightly overestimate the rate of the standard SCR reaction at the end of the catalyst channel. The HNCO concentration decreases rapidly and reaches almost complete conversion. Compared to the experiment performed at the same conditions without NO (Fig. 4), a faster HNCO hydrolysis is expected. The reason for this is the consumption of NH3 with NO via the standard SCR reaction. Due to competitive adsorption of ammonia with HNCO, an overall lower NH3 concentration leads to more adsorption sites being available for HNCO. This is evident from both experimental and modeled concentration profiles and end-of-pipe concentrations. Without NO, experimental end-of-pipe concentrations of 3.7 ppm HNCO are reached, while with additional NO, the HNCO concentration drops to 1.4 ppm. Furthermore, the simulated consumption of HNCO is faster with NO, but slightly overestimated. The simulated NH3 concentration undergoes a maximum early in the catalyst channel, due to the fast HNCO hydrolysis and subsequently converges towards the NO concentration, when almost no HNCO is present anymore. That the ammonia concentration does not fall significantly below the NO concentration indicates that ammonia oxidation does not take place at 250∘C. This is also supported by the previous experiments. In the COMSOL model, HNCO hydrolysis is faster than for the DETCHEM model. As a result, the ammonia concentration maximum is shifted towards the channel entrance. This further increases the reaction rate of the standard SCR averaged over the channel, causing an overall higher conversion of NO (\(X_{{\text {NO}}}^{\text {COMSOL}}=75 \%\) vs. \(X_{{\text {NO}}}^{\text {DETCHEM}}=72 \%\)). This maximum could be even more pronounced in reality, because in our model, HNCO adsorption is not modeled, which would potentially inhibit the SCR reaction in the front part of the channel. As a result, slower NH3 consumption would lead to a higher peak concentration. Further implications of the competitive adsorptions are discussed in the next section.A radial concentration profile associated with the experiment at 250∘C (Fig. 5) is shown in Fig. 6. Here the simulated concentrations of NH3, NO and HNCO at the axial position z= 2cm of the catalyst are presented over the total height of the channel.

Experimental and simulated spatially resolved gas phase concentration of NO, NH3 and HNCO inside a single channel of a stationary VWT monolith. \(\boldsymbol {T_{0}={250}^{{\circ }}}\)C, \(\boldsymbol {C_{{\text {NH}_{3}},0}=C_{{\text {HNCO}},0}=200 \text {ppm}, C_{{\text {NO}},0}=400 \text {ppm}}\)

Simulated radial gas phase concentration profile of NH3, NO and HNCO at z= 2cm inside a single channel of a stationary VWT monolith. \(\boldsymbol {T_{0}={250}^{\circ }}C\), \(\boldsymbol {C_{{\text {NH}_{3}},0}=C_{{\text {HNCO}},0}=200 \text {ppm}, C_{{\text {NO}},0}=400 \text {ppm}}\)
The profile of HNCO is parabolic and decreases towards the walls of the channel, where the hydrolysis reaction takes place. Both the profiles simulated with DETCHEM and COMSOL agree very well in the middle part of the channel. Slight differences arise directly at the wall, where the COMSOL simulation estimates a HNCO concentration of almost 0, whereas in the DETCHEM simulation still a concentration of about 15ppm is left. Nevertheless, both results suggest that the hydrolysis reaction at the channel wall is limited by the diffusion of HNCO from the inside towards the outside of the channel and not by the reaction at the catalyst surface. In the middle of the channel (r= 0.55mm), both simulation models predict a HNCO concentration of about 102ppm which is very close the experimental value of 107ppm of Fig. 5. The same observation can also be seen for NO. Here, both simulations estimate a concentration of about 357ppm which agrees with the value of 356ppm in the experiment. This might suggest that the position of the capillary in the experiment was more located in the middle of the channel than close to the wall, where the concentrations decrease due to the surface reaction. Slight differences between the two simulation models can be seen by the modeling of this concentration decrease for NO. COMSOL shows a higher conversion of NO at the catalyst surface and thus a lower concentration than the DETCHEM simulation. For NH3, an almost linear concentration profile over the height of the channel is estimated by both simulations, which is associated with the parallel formation of NH3 from the hydrolysis and its conversion by the NO reduction.
The axial concentration profile of the three SCR active species at 425∘ can be seen in Fig. 7. The increase in temperature from 250 to 425∘C led to an increase in NO conversion, from 66 to 81 %, and an almost complete conversion of HNCO.

Experimental and simulated spatially resolved gas phase concentration of NO, NH3 and HNCO inside a single channel of a stationary VWT monolith. \(\boldsymbol {T_{0}={425}^{\circ }}\), \(\boldsymbol {C_{{\text {NH}_{3}},0}=C_{{\text {HNCO}},0}=200 \text {ppm}, C_{{\text {NO}},0}=400 \text {ppm}}\)
In the case of the HNCO concentration profile, the experimental hydrolysis seems to be slower at 425∘C, compared to 250∘C. The reason for this is unknown, and this trend is not reflected by the model. One possibility is that the influence of dominant external mass transport limitations is incorrectly assessed.
Both the increase in the reaction rate of the standard SCR reaction and the hydrolysis reaction are represented by the modeled concentration profiles. However, the NO and HNCO conversions are slightly overestimated, especially in the front part of the channel between 2 and 8cm. The concentration profile modeled at 425∘C differs significantly from the profile at 250∘C. The concentration maximum is less pronounced and shifted about 2cm towards the catalyst inlet. This can be explained by the increased reaction rate of the standard SCR reaction. The increased rate of the modeled hydrolysis also contributes to shifting the concentration maximum in the negative z-direction. In contrast to NO, an almost complete conversion of ammonia can be observed at the end of the channel, due to its oxidation. The ammonia oxidation is relevant at these temperatures, which is consistent with the results from Fig. 2. It can therefore be assumed that NO conversion is limited by the availability of NH3. To increase DeNOx activity, it seems necessary to increase the NH3 to NO ratio α. Some differences between DETCHEM and COMSOL in the course of the simulated concentration profiles of NH3 and NO arise, while the two HNCO profiles are almost overlapping. As the NO conversion in the DETCHEM simulation seems to be a little bit faster than in COMSOL, also the NH3 profile is affected. Since NH3 is produced from the HNCO hydrolysis, a local maximum of NH3 can be seen at around 1cm in the COMSOL simulation, which is not that pronounced in the DETCHEM simulation in which the produced NH3 is equally consumed. Nevertheless, both simulations converge to almost the same concentrations of NH3 and NO at the end of the catalyst, which are in good agreement with the experimental value.
In Fig. 8, the simulated end-of-pipe concentrations (z = 15cm) are plotted over a temperature range from 100 to 600∘C. The inlet concentrations are 400 ppm NO and 200 ppm NH3 and HNCO. The onset of NO conversion is observed at \({\sim 100}^{\circ }\)C, followed by a fast increase in DeNOx activity up until TDETCHEM = 325∘C (\({X_{\text {NO}},{\max \limits }}=94 \%)\) for the DETCHEM model or TCOMSOL = 315∘C (\(X_{{\text {NO}},{\max \limits }}=91 \%\)) for the COMSOL model. Simultaneously the NH3 concentration decreases, parallel to the NO concentration. However, at 250∘C, the end-of-pipe concentrations of the two species start to diverge, with NH3 reaching full conversion between 350 and 400∘C. This is likely due to ammonia oxidation. Above 400∘C, low concentration of NH3 is observed again, because the equilibrium between adsorption and desorption is shifted towards the gas phase species. In the case of HNCO, almost all of it is hydrolysed, even at temperatures as low as 100∘C. In reality, HNCO would condensate and polymerise at lower temperatures [60]. However, the modelling of condensation was not part of this work, and the conclusion stands that the hydrolysis of HNCO is a very fast process, not just at high temperatures. When the DETCHEM and the COMSOL model are compared, the latter shows better results for temperatures above 300∘C, while the former is closer to the experimental value at 250∘C.

Simulated and experimental outlet concentrations of NH3, NO and HNCO with inlet concentrations of 400 ppm NO and 200 ppm NH3 and HNCO
4.3 Influence of UWS Droplet Impact on the Catalyst
A detailed 3D simulation of an impacting UWS droplet on a catalyst would be very computational expensive, since the length scales of the droplet (≈ 100μm) and the catalyst (≈ 10cm) are three orders of magnitude apart. Therefore, the two processes were simulated separately and important exchange variables were transferred from the one to the other simulation. For the simulation of catalytic and non-catalytic decomposition of urea, the DETCHEMMPTR model is successfully used for several years in our group [30, 31, 61]. The methodology of the model (detailed described in [30, 31] and in the manual [40]) is transferred into a COMSOL 0D simulation. The reactions in the catalyst channel are captured in a 2D simulation as described above. A scheme of the process with the separately considered urea decomposition and catalyst reactions, which are subsequently coupled, is shown in Fig. 9.

Scheme for the coupling of urea decomposition and channel reactions
The initial inlet flow V̇ini is split into one part which affects the impacted droplet and therefore flows into the COMSOL 0D simulation, and another part which directly flows into the channel V̇add. Inside of the 0D simulation an assumed droplet of urea with a diameter of 150μm has hit the catalyst surface, spreads out on the defined interface area and starts to decompose. A penetration of the liquid into the porous washcoat or secondary droplet generation from the droplet impingement could not be considered in the 0D model. The simulation covers the gas phase as well as the several liquid and solid phases of the urea decomposition by-products as described in [30]. The heat is supplied over the interface to the catalyst wall and the heated inlet flow. The volume flow into this simulation is constantly mixed with the existing gas phase with a constant volume and pressure. Therefore, produced gaseous species from the decomposition of urea leave the simulation in the outlet flow \(\Dot {V}^{out}_{MPTR}\), which is mixed with the remaining gas flow to enter the channel simulation as \(\Dot {V}^{in}_{Chan}\).
The different simulation steps of one example simulation can be seen in Fig. 10. Here it is assumed that a urea droplet with a diameter of 150μm impacts at the beginning of the catalyst with gas and wall temperatures of 200∘C, which is a typical low-temperature case for urea-SCR. Nevertheless, the urea droplet decomposes very fast, due to its low mass, and reaches a steady state after only 0.14s, as can be seen in Fig. 10a. The originated deposit consists of about 80% cyanuric acid and 20% ammelide, whose further decomposition is very slow and mainly takes places at higher temperatures. The resulting deposit mass is approx. 30% of the original droplet mass. This is not much for one single droplet, but over time, bigger deposit structures could accumulate.

Example of the different stages of UWS droplet impact on a VWT catalyst at 200 ∘C. In the first stage (a), a UWS droplet of 150 μm impacts at the beginning of the catalyst and decomposes. Resulting gas products from droplet impact at 1 s influence the inlet composition of the catalyst (b), especially for the species NH3 and HNCO. High NH3 concentrations in the inlet influence the reactions inside the catalyst channel, reduce the NO outlet and lead to a peak in NH3 outlet (c)
In addition to the deposits, gaseous products, like NH3 and HNCO, are also formed during decomposition. They mix with the gas flow and can react further downstream inside the channel. In fact, due to the concentrated gas evolution through the droplet in the single channel, instead of a uniform distribution over the whole catalyst, a very high concentration of NH3 and HNCO enters the channel as depicted in Fig. 10b. As the droplet hits the channel after 1 s, the NH3 concentration rises from the base level of 200ppm to a maximum of 3378ppm. Similarly, the HNCO concentration rises to a maximum of 1775ppm within the droplet decomposition time. Subsequently, the concentrations falls to the base level of 200ppm again. This peak of inlet concentration for NH3 and HNCO leads inside the channel to an increased NOx conversion. At a temperature of 200∘C and the boundary conditions described above usually a NO conversion of around 27 % is achieved, as can be seen in the temperature plot in Fig. 8. HNCO is almost completely hydrolised to NH3. This leads, for an inlet of 400ppm NO, to a base outlet signal of 292ppm as can be seen in Fig. 10c. The additional NH3 peak, which was caused by the droplet decomposition and HNCO hydrolysis, leads to a further drop of the NO concentration down to 259ppm. Higher conversion is however limited by the reaction rates at these low temperatures. The large inlet amounts of NH3 and HNCO could not be converted completely until the end of the channel. While HNCO hydrolysis is still very fast, resulting in a peak of 5.7ppm at 1.132s, a NH3 slip of up to 4317ppm was simulated, as can be seen in Fig. 10c. This also overloads possible subsequent ammonia storage or oxidation catalysts and would result in the emission of NH3.
5 Discussion
The experimental in situ probe technique on the hot gas test rig produced reliable concentration profiles for NO and HNCO. However, unrealistic NH3 profile were obtained and not further taken into account, as discussed in the previous section. The implementation of the dual site NH3-SCR mechanism by Nova et al. into DETCHEMCHANNEL and COMSOL allowed for reliable simulation of the concentration profiles with NO and NH3 present in the reacting system. Good agreements were achieved during validation of the implemented model with experimental conversions by Rammelt et al. [42]. However, ammonia conversions are slightly underestimated at lower and overestimated at higher temperatures, while no clear trend is observable for NO. Similar results are observed for the modeling of concentration profiles.
The combination of the NH3-SCR mechanism with the HNCO hydrolysis mechanism allowed for the simulation of the entire reacting system, when urea undergoes complete thermolysis in front of the catalyst. To evaluate the overall performance of the DETCHEM model in predicting the spatially resolved concentrations, absolute and relative deviations between experimental and simulated values were determined. This includes simulations at temperatures of 250, 350 and 425∘C. For NO, a mean relative deviation of 20% and a mean absolute deviation of 26 ppm were obtained. The model shows a higher agreement for the gas phase concentrations at the end of the catalyst. If only the deviation between the model and the last experimental value of each measurement is determined, the result improves to an absolute deviation of 10 ppm and a relative deviation of 16%. Most of the error occurs in the range between 2 and 6 cm, where simulated conversions are underestimated. One possible explanation is the influence of the probe. When it is entered from downstream, the increase in residence time is most significant in the front part of the catalyst channel [39]. This results in a erroneous reaction progress. When the probe is drawn closer to the exit of the channel, its influence continues to vanish. Therefore, these measured values would again agree with the simulations, which can be observed in Fig. 7, and further simulations which are not shown here.
Considering the implementation of the HNCO hydrolysis mechanism in combination with the SCR mechanism, an average absolute error of less than 2.2 ppm can be observed at the end of the channel, due the complete conversion. For all data points, the average absolute deviation is 16 ppm. As with the values of NO, this can possibly be explained by the influence of the in situ probe. Another reason may be the competitive adsorption, which was not considered for HNCO. Therefore, adsorbed HNCO cannot reduce the SCR reaction rate, and adsorbed NO cannot reduce the hydrolysis reaction rate. These effects would be more present in the front part of the catalyst, where the gas phase concentrations of the adsorbing species are higher. If the implemented model was to be used for simulating gas phases with NO2, then this effect would be even more pronounced, due to formed nitrates being known to block adsorption sites [55].
Furthermore, the implementation of the HNCO hydrolysis mechanism is based on simplifying assumptions to allow the application of the TiO2-based mechanism to a VWT catalyst. Ideally, a microkinetic mechanism developed for hydrolysis on VWT would be used, which to our knowledge is not available. However, the impact of an improved hydrolysis mechanism would be insignificant since the reaction is most likely limited by external mass transfer in our temperature range, which is consistent with the literature [46, 57, 62]. Accordingly, the HNCO concentration profile is mainly determined by the transport model. At lower temperatures below 160∘C, depending on the cpsi, internal mass transport may become significant, which would require the use of a washcoat model [46].
Even though both simulation models use the same reaction mechanism, some visible differences can be seen between the simulation results. While the agreement between DETCHEM and COMSOL for the spatial description of HNCO is very good, the differences for NH3 and NO are more pronounced, especially in the middle part of the catalyst channel. These differences can be attributed to the slightly different methods in the calculation and handling of the temperature-dependant diffusion, which could not be adjusted and equalised due to limited setting possibilities. The exact differences lie in the calculation of the diffusion coefficient and the associated reduced collision integral and can be found in the COMSOL and DETCHEM manual [40, 45]. For HNCO, the differences in diffusion coefficients are apparently smaller than for NH3 and NO. Since the concentrations of NH3 and NO are coupled through the SCR reaction, small errors furthermore amplify into a common larger error and the differences between the models. The simplifications in the DETCHEM model are also noticeable in the computational time. While a stationary channel simulation in DETCHEM took about 10s, the more detailed simulation in COMSOL required about 3min. The transient simulation for the overview of outlet concentrations from 100 to 600∘C (Fig. 8) took around 1 h in COMSOL.
Especially at low gas temperatures, incomplete evaporation and decomposition of UWS droplets can lead to spray/wall-impingement and film formation as it was detailed studied already in the literature [63,64,65,66]. Furthermore, this also gives the possibility for droplets to enter or hit the catalyst. Our example simulation of the coupled urea decomposition and channel simulation has shown the influence of UWS droplet impact on the SCR catalyst. The impacted droplet decomposes very fast, but leaves a significant amount of remaining deposits at the catalyst, which further decomposes only at higher temperatures. Nonetheless, the gaseous products from the droplet decomposition, mainly NH3 and HNCO, drastically increase the inlet into the channel. This increase in reducing agent in the inlet consequently leads to a decrease of NO at the outlet through higher conversion rates. Although this decrease turns out to be smaller than one might expect at first glance from the high NH3 peak. The catalyst surface takes some time to adsorb the increased amount of NH3 and adjust to the changed boundary conditions. Furthermore, the reaction rate of the SCR reaction is limited by the low temperature of 200∘.A detailed 3D CFD simulation including both the process of droplet impingement and decomposition, as well as the flow in the channel, would take several hours up to days of computational time even with some sort of simplification. The decoupling and further simplifications of the processes accelerates the computing time considerably. The transient 0D simulation of the droplet decomposition takes about 6s in COMSOL, while the more detailed 2D simulation of the catalyst channel takes about 5min for simulating 20s of real time. The acceleration of the decoupling method in the order of 2–3 magnitudes can justify the used simplifications. Nevertheless, a detailed description and simulation of the process in the future is essential for the validation of the method.
6 Conclusion
This study presented a model for the coupling of liquid and surface chemistry in Urea-SCR systems to investigate the influence of UWS droplets impinging on the channels of a VWT monolith. This required the modeling of both the decomposition of UWS and urea by-products, and the classical SCR reactions, as well as coupling of the two systems. Furthermore, the implemented reaction mechanisms allowed for the simulation of experiments with spatially resolved concentration profiles within a catalyst channel.
Experimental data was taken from [36] and additional experiments were performed on the same hot gas test rig equipped with a VWT monolith. Concentration profiles were measured along the catalyst channels using the SpaciPro technique. The analysed species included NH3, HNCO and NO over a temperature range from 200 to 425∘C. Whereas NO and NH3 could be dosed directly, in the experiments, HNCO was generated by thermolysis of injected UWS in front of the catalyst.
The implemented mechanism for NH3 SCR was first validated against end-of-pipe experiments from literature where a very good agreement with an average absolute deviation of 4 % between experiment and DETCHEMCHANNEL simulations was achieved. In the next step, the mechanism for HNCO hydrolysis was added and experiments with spatially resolved concentration profiles in a VWT catalyst were simulated with DETCHEMCHANNEL and COMSOL. Since experimental NH3 profiles could not be correctly quantified, due to the reasons described above, only NO and HNCO profiles were compared with the simulation results. Overall a good agreement between both simulation models and the experiments was achieved. Slight differences between the DETCHEM and the COMSOL model occur due to the differences in transport and diffusion model and the complexity of the models itself. For higher temperatures, it was shown that experiment and simulation have a higher deviation in the middle part of the catalyst than at its outlet for both simulation models. This could be due to errors from the measurement with the capillary in the channel itself, but also due to unrevealed or not covered phenomena by the mechanism. Therefore, we further emphasise the use of SpaciPro technique instead of only data from the outlet of the catalyst for validation and development of mechanism.
Finally, the SCR channel model in COMSOL was coupled with a simulation for the urea decomposition to model a UWS droplet impact onto a SCR catalyst. It was shown that the droplet decomposes very fast even at 200∘, but also leaves a significant amount of deposit behind, which would only further decompose at high temperatures. Thus, deposits could accumulate inside of the catalyst and increase the pressure drop or even block channels, which decreases the system efficiency. The gaseous products from urea droplet decomposition increase the NH3 concentration inside a SCR channel and therefore increase the NOx conversion. However, droplet impact and conversion increase in one channel also comes with a decrease in the overall uniformity and should therefore strongly be avoided.
Nevertheless, further tightening emission regulations require optimisation of the exhaust gas aftertreatment systems. Therefore, the interactions of physical and chemical processes have to be investigated. This work served to better understand spatially resolved concentration profiles, the process of impacting droplets onto SCR catalysts and gave a modeling capability. Since urea SCR is also in investigation for the aftertreatment of H2-engines, it is worth to continue developing and investigating the processes in order to better understand them in the future.
Data Availability
Please contact the corresponding author.
Abbreviations
- S1 :
-
VWT adsorption sites associated with vanadium species
- S2 :
-
VWT adsorption sites associated with associated with oxides of tungsten and titanium
- CO2 :
-
Carbon dioxide
- H2O:
-
Water
- HNCO:
-
Isocyanic acid
- NH3 :
-
Ammonia
- NO:
-
Nitric oxide
- O2 :
-
Oxygen
- DeNOx :
-
NOx removal
- FTIR:
-
Fourier-transform infrared spectrometer
- IC:
-
Internal combustion
- MFC:
-
Mass flow controller
- MFC:
-
Mass flow controller
- NO x :
-
Nitrogen oxides
- SCR:
-
Selective catalytic reduction
- TG:
-
Thermogracimetric analysis
- UWS:
-
Urea water solution
- VWT:
-
Vanadium-tungsten-titanium catalyst
- [Ti −OH2]Mono :
-
Monolayer sorption capacity of H2O on TiO2 mol L− 1
- α :
-
Parameter for surface coverage dependence
- β :
-
Pressure exponent
- \(\dot {\omega }_{i}\) :
-
Gas phase reaction rate of species i
- \(\dot {r}_{i}\) :
-
Surface-specific reaction rate of species i
- λ :
-
Thermal conductivity
- μ :
-
Dynamic viscosity
- ν :
-
Radial component of velocity
- \(\overline {M}\) :
-
Average molar mass of gas mixture
- ρ :
-
Volumetric mass density
- σ i :
-
Fraction of S1 surface sites occupied by species i
- \(\text {E}_{i}^{\circ }\) :
-
Activation energy of reaction j
- \(\text {k}_{\text {hyd}}^{\prime \prime }\) :
-
Combined rate constant of the hydrolysis reaction
- kj :
-
Rate constant of reaction j
- \(\text {k}_{j}^{\circ }\) :
-
Pre-exponential factor of reaction j
- 𝜃 i :
-
Fraction of S2 surface sites occupied by species i
- C i :
-
Concentration of species i
- cpsi :
-
Channels per square inch
- D i :
-
Diffusion coefficient of species i
- h :
-
Enthalpy density
- h i :
-
Enthalpy of species i J mol− 1
- j i :
-
Radial diffusion flux of species i
- K j :
-
Equilibrium constant of reaction
- M i :
-
Molar mass of species i
- p :
-
Pressure
- p i :
-
Partial pressure of species i
- \(p_{i}^{*}\) :
-
Normalised partial pressure of species i
- r :
-
Radial coordinate of catalyst channel
- r j :
-
Reaction rate of reaction j
- R e :
-
Reynolds number
- T :
-
Temperature
- u :
-
Axial component of velocity
- X i :
-
Mole Conversion of species i -
- Y i :
-
Mass fraction of species i
- z :
-
Axial coordinate of catalyst channel
- R:
-
Universal gas constant
- ∘C:
-
Temperature in degree Celsius
- J:
-
Energy in joule
- K:
-
Temperature in Kelvin
- L:
-
Volume in litres
- mol:
-
Amount of substance in mole
- m:
-
Length in meter
- s:
-
Time in seconds
- in:
-
Length in inch
- LN :
-
Gas quantity in standard liters
- ppm:
-
Concentration in parts per million
- wt%:
-
Mass fraction in weight percent
References
Brunekreef, B., Holgate, S.T.: Air pollution and health. Lancet 360, 1233–1242 (2002). https://doi.org/10.1016/S0140-6736(02)11274-8
European Comission: European vehicle emissions standards – Euro 7 for cars, vans, lorries and buses. https://ec.europa.eu/info/law/better-regulation/have-your-say/initiatives/12313-European-vehicle-emissions-standards-Euro-7-for-cars-vans-lorries-and-buses_en (2020)
Register Federal: Amendments related to: tier 3 motor vehicle emission and fuel standards, nonroad engine and equipment programs, and marpol annex vi implementation. https://www.epa.gov/regulations-emissions-vehicles-and-engines/amendments-related-tier-3-motor-vehicle-emission-and (2015)
Reif, K.: Dieselmotor-Management, 6th edn. Springer, Berlin (2020)
Roduit, B., Wokaun, A., Baiker, A.: Global kinetic modeling of reactions occurring during selective catalytic reduction of NO by NH3 over vanadia/titania-based catalysts. Ind. Eng. Chem. Res. 37, 4577–4590 (1998). https://doi.org/10.1021/ie980310e
Xiong, S., Xiao, X., Liao, Y., Dang, H., Shan, W., Yang, S.: Global kinetic study of no reduction by NH3 over V2O5 −WO3/TiO2 : relationship between the SCR performance and the key factors. Ind. Eng. Chem. Res. https://doi.org/10.1021/acs.iecr.5b03044
Odriozola, J.A., Heinemann, H., Somorjai, G.A., Garcia de la Banda, J.F., Pereira, P.: AES and TDS study of the adsorption of NH3 and NO on V2O5 and TiO2 surfaces: mechanistic implications. J. Catal. 119(1), 71–82 (1989). https://doi.org/10.1016/0021-9517(89)90135-8
Takagi, M., Kawai, T., Soma, M., Onishi, T., Tamaru, K.: The mechanism of the reaction between NOx and NH3 on V2O5 in the presence of oxygen. J. Catal. 50, 441–446 (1977). https://doi.org/10.1016/0021-9517(77)90056-2
Lintz, H.G., Turek, T.: Intrinsic kinetics of nitric oxide reduction by ammonia on a vanadia-titania catalyst. Appl. Catal. A Gen. 85, 13–25 (1992). https://doi.org/10.1016/0926-860X(92)80126-W
Willi, R., Roduit, B., Koeppel, R.A., Wokaun, A., Baiker, A.: Selective reduction of NO by NH3 over vanadia-based commercial catalyst: parametric sensitivity and kinetic modelling. Chem Eng Sci 51, 2897–2902 (1996). https://doi.org/10.1016/0009-2509(96)00171-6
Janssen, F., van den Kerkhof, F., Bosch, H., Ross, J.: Mechanism of the reaction of nitric oxide, ammonia, and oxygen over vanadia catalysts. 1. The role of oxygen studied by way of isotopic transients under dilute conditions. J. Phys. Chem. 91, 5921–5927 (1987). https://doi.org/10.1021/j100307a021
Miyamoto, A., Kobayashi, K., Inomata, M., Murakami, Y.: Nitrogen-15 tracer investigation of the mechanism of the reaction of NO with NH3 on vanadium oxide catalysts. J. Phys. Chem. 86, 2945–2950 (1982). https://doi.org/10.1021/j100212a027
Inomata, M., Miyamoto, A., Murakami, Y.: Mechanism of the reaction of NO and NH3 on vanadium oxide catalyst in the presence of oxygen under the dilute gas condition. J. Catal. 62, 140–148 (1980). https://doi.org/10.1016/0021-9517(80)90429-7
Ciardelli, C., Nova, I., Tronconi, E., Konrad, B., Chatterjee, D., Ecke, K., Weibel, M.: SCR-DeNOx for diesel engine exhaust aftertreatment: unsteady-state kinetic study and monolith reactor modelling. Chem. Eng. Sci. 59, 5301–5309 (2004). https://doi.org/10.1016/J.CES.2004.07.016
Tronconi, E., Nova, I., Ciardelli, C., Chatterjee, D., Bandl-Konrad, B., Burkhardt, T.: Modelling of an SCR catalytic converter for diesel exhaust after treatment: dynamic effects at low temperature. Catal. Today 105, 529–536 (2005). https://doi.org/10.1016/J.CATTOD.2005.06.043
Nova, I., Tronconi, E.: Kinetic study of the NO/NO2-NH3 SCR reactions over a V2O5–WO3/TiO2 commercial catalyst for the after treatment of Diesel engines exhausts. IFAC Proc. Vol. 42, 183–190 (2009). https://doi.org/10.3182/20091130-3-FR-4008.00025
Nova, I., Colombo, M., Tronconi, E.: Kinetic modeling of dynamic aspects of the standard NH3-SCR reaction over v2o5-WO3/tio2 and Fe-Zeolite commercial catalysts for the aftertreatment of diesel engines exhausts kinetic modeling of dynamic aspects of the standard NH 3-SCR reaction over v2o5-WO3/tio2 and Fe-Zeolite commercial catalysts for the aftertreatment of diesel engines exhausts. Oil Gas Sci. Technol. – Rev. IFP Energies Nouvelles 66, 681–691 (2011). https://doi.org/10.2516/ogst/2011132
Opitz, B., Bendrich, M., Drochner, A., Vogel, H., Hayes, R.E., Forbes, J.F., Votsmeier, M.: Simulation study of SCR catalysts with individually adjusted ammonia dosing strategies. Chem. Eng. J. 264, 936–944 (2015). https://doi.org/10.1016/j.cej.2014.11.114
Bendrich, M., Scheuer, A., E. Hayes, R., Votsmeier, M.: Unified mechanistic model for standard SCR, Fast SCR, and NO 2 SCR over a copper chabazite catalyst. Appl. Catal. B Environ. 222, 76–87 (2018). https://doi.org/10.1016/j.apcatb.2017.09.069
Bendrich, M., Opitz, B., Scheuer, A., Hayes, R.E., Votsmeier, M.: Selective catalytic reduction: adding an ammonia slip catalyst mitigates dosing errors. Can. J. Chem. Eng. 100, 1439–1447 (2022). https://doi.org/10.1002/cjce.24293
L. Raja, L., Kee, R.J., Deutschmann, O., Warnatz, J., Schmidt, L.D.: A critical evaluation of navier-stokes, boundary-layer, and plug-flow models of the flow and chemistry in a catalytic-combustion monolith. Catal. Today 59. https://doi.org/10.1016/S0920-5861(00)00271-6 (2000)
Shakor, Z., Al-Kareemawi, A., Al-Najar, J.: Modeling of single channel monolithic reactor with the single pass flow operation system. Eng. Technol. J. (28):2221–2236 (2010)
Hayes, R.E., Kolaczkowski, S.T., Thomas, W.J.: Finite-element model for a catalytic monolith reactor. Comput. Chem. Eng. 16, 645–657 (1992). https://doi.org/10.1016/0098-1354(92)80014-Z
Tischer, S., Correa, C., Deutschmann, O.: Transient three-dimensional simulations of a catalytic combustion monolith using detailed models for heterogeneous and homogeneous reactions and transport phenomena. Catal. Today 69, 57–62 (2001). https://doi.org/10.1016/S0920-5861(01)00355-8
Jahn, R., Šnita, D., Kubíček, M., Marek, M.: 3-D modeling of monolith reactors. Catal. Today 38, 39–46 (1997). https://doi.org/10.1016/S0920-5861(97)00037-0
Bertrand, F., Devals, C., Vidal, D., Séguineau De Préval, C., Hayes, R.E.: Towards the simulation of the catalytic monolith converter using discrete channel-scale models. Catal. Today 188, 80–86 (2012). https://doi.org/10.1016/J.CATTOD.2011.12.011
Neusser, H.-J., Kahrstedt, J., Dorenkamp, R., Jelden, H.: The euro 6 engines in the modular diesel engine system of volkswagen. MTZ Worldwide 74, 4–10 (2013). https://doi.org/10.1007/s38313-013-0057-9
Nova, I., Tronconi, E.: Urea-SCR Technology for deNOx After Treatment of Diesel Exhausts. Springer, Berlin (2014). https://doi.org/10.1007/978-1-4899-8071-7
Kojima, H., Fischer, M., Haga, H., Ohyay, N., Nishi, K., Mito, T., Fukushi, N.: Next generation all in one close-coupled urea-scr system. In: SAE Technical Paper Series, SAE Technical Paper Series, SAE International400 Commonwealth Drive. Warrendale. https://doi.org/10.4271/2015-01-0994 (2015)
Kuntz, C., Weickenmeiery, H., Börnhorst, M., Deutschmann, O.: Deposition and decomposition of urea and its by-products on TiO2 and VWT-SCR catalysts. Int. J. Heat Fluid Flow 95, 108969 (2022). https://doi.org/10.1016/j.ijheatfluidflow.2022.108969
Tischer, S., Börnhorst, M., Amsler, J., Schoch, G., Deutschmann, O.: Thermodynamics and reaction mechanism of urea decomposition. Phys. Chem. Chem. Phys. 21, 16785–16797 (2019). https://doi.org/10.1039/c9cp01529a
Brack, W., Heine, B., Birkhold, F., Kruse, M., Deutschmann, O.: Formation of urea-based deposits in an exhaust system: numerical predictions and experimental observations on a hot gas test bench. Emission Control Sci. Technol. 2, 115–123 (2016). https://doi.org/10.1007/s40825-016-0042-2
Sá, J., Fernandes, D.L.A., Aiouache, F., Goguet, A., Hardacre, C., Lundie, D., Naeem, W., Partridge, W.P.: SpaciMS: spatial and temporal operando resolution of reactions within catalytic monoliths. Analyst 135, 2260–2272 (2010). https://doi.org/10.1039/C0AN00303D
Livio, D., Diehm, C., Donazzi, A., Beretta, A., Deutschmann, O.: Catalytic partial oxidation of ethanol over Rh/Al2O3: spatially resolved temperature and concentration profiles. Appl. Catal. A Gen. 467, 530–541 (2013). https://doi.org/10.1016/j.apcata.2013.07.054
Donazzi, A., Livio, D., Maestri, M., Beretta, A., Groppi, G., Tronconi, E., Forzatti, P.: Synergy of homogeneous and heterogeneous chemistry probed by in situ spatially resolved measurements of temperature and composition. Angew. Chem. 123, 4029–4032 (2011). https://doi.org/10.1002/ange.201007346
Eck, M., Lott, P., Schweigert, D., Börnhorst, M., Deutschmann, O.: Spatially resolved measurements of HNCO hydrolysis over SCR catalysts. Chemie Ingenieur Technik. https://doi.org/10.1002/cite.202100192 (2022)
Börnhorst, M., Langheck, S., Weickenmeier, H., Dem, C., Suntz, R., Deutschmann, O.: Characterization of solid deposits from urea water solution injected into a hot gas test rig. Chem. Eng. J. 377. https://doi.org/10.1016/j.cej.2018.09.016
Hettel, M., Diehm, C., Torkashvand, B., Deutschmann, O.: Critical evaluation of in situ probe techniques for catalytic honeycomb monolithsy. Catal. Today 216, 2–10 (2013). https://doi.org/10.1016/j.cattod.2013.05.005
Hettel, M., Antinori, C., Deutschmann, O.: CFD evaluation of in situ probe techniques for catalytic honeycomb monoliths. Emission Control Sci. Technol. 2, 188–203 (2016). https://doi.org/10.1007/s40825-016-0043-1
Deutschmann, O., Tischer, S., Kleditzsch, S., Janardhanan, V., Correa, C., Chatterjee, D., Mladenov, N., Minh, H.D., Karadeniz, H., Hettel, M., Menon, V., Banerjee, A., Goßler, H., Detchem, D.E.: https://www.detchem.com (2020)
Mladenov, N., Koop, J., Tischer, S., Deutschmann, O.: Modeling of transport and chemistry in channel flows of automotive catalytic converters. Chem. Eng. Sci. 65, 812–826 (2010). https://doi.org/10.1016/j.ces.2009.09.034
Rammelt, T., Torkashvand, B., Hauck, C., Böhm, J., Gläser, R., Deutschmann, O.: Nitric oxide reduction of heavy-duty diesel off-gas by NH3-SCR in front of the turbocharger. Emission Control Sci. Technol. 3, 275–288 (2017). https://doi.org/10.1007/s40825-017-0078-y
Thormann, J., Maier, L., Pfeifer, P., Kunz, U., Deutschmann, O., Schubert, K.: Steam reforming of hexadecane over a Rh/CeO2 catalyst in microchannels: experimental and numerical investigation. Int. J. Hydrog. Energy 34, 5108–5120 (2009). https://doi.org/10.1016/j.ijhydene.2009.04.031
Deutschmann, O., Tischer, S., Kleditzsch, S., Janardhanan, V., Correa, C., Chatterjee, D., Mladenov, N., Minh, H.D., Karadeniz, H., Hettel, M., Menon, V., Banerjee, A., Goßler, H., Detchem D.E.: Manual version 2.8: chapter 14, 2022 (2022)
COMSOL: Comsol multiphysics reference manual: version 6.0, https://doc.comsol.com/6.0/doc/com.comsol.help.comsol/COMSOL_ReferenceManual.pdf (2021)
Hauck, P., Jentys, A., A. Lercher, J.: Surface chemistry and kinetics of the hydrolysis of isocyanic acid on anatase. Appl. Catal. B Environ. 70, 91–99 (2007a). https://doi.org/10.1016/j.apcatb.2005.12.025
Chatterjee, D., Burkhardt, T., Bandl-Konrad, B., Braun, T., Tronconi, E., Nova, I., Ciardelli, C.: Numerical simulation of ammonia scr-catalytic converters: model development and application. SAE Tech. Paper. https://doi.org/10.4271/2005-01-0965 (2005)
Nova, I., Ciardelli, C., Tronconi, E., Chatterjee, D., Bandl-Konrad, B.: NH3-scr of no over a v-based catalyst: low-t redox kinetics with NH3 inhibition. AIChE J. 52, 3222–3233 (2006). https://doi.org/10.1002/aic.10939
Hauck, P., Jentys, A., A. Lercher, J.: On the quantitative aspects of hydrolysis of isocyanic acid on TiO2. Catal. Today 127, 165–175 (2007b). https://doi.org/10.1016/j.cattod.2007.05.012
Nova, I., Ciardelli, C., Tronconi, E., Chatterjee, D., Weibel, M.: Unifying redox kinetics for standard and fast NH3-SCR over a V205-WO3/TiO2 catalyst. AIChE J. 55, 1514–1529 (2009). https://doi.org/10.1002/aic.11750
Czekaj, I., Kröcher, O.: Decomposition of urea in the SCR process: combination of DFT calculations and experimental results on the catalytic hydrolysis of isocyanic acid on TiO2 and Al2O3. Topics Catal. 52, 1740–1745 (2009). https://doi.org/10.1007/s11244-009-9344-8
Czekaj, I., Kröcher, O., Piazzesi, G.: DFT calculations, drift spectroscopy and kinetic studies on the hydrolysis of isocyanic acid on the TiO2-anatase (1 0 1) surface. J. Mol. Catal. A Chem. 280, 68–80 (2008). https://doi.org/10.1016/j.molcata.2007.10.027
Piazzesi, G., Kröcher, O., Elsener, M., Wokaun, A.: Adsorption and hydrolysis of isocyanic acid on TiO2. Appl. Catal. B Environ 65, 55–61 (2006a). https://doi.org/10.1016/j.apcatb.2005.12.018
Bernhard, A.M., Peitz, D., Elsener, M., Schildhauer, T., Kröcher, O.: Catalytic urea hydrolysis in the selective catalytic reduction of NOx: catalyst screening and kinetics on anatase TiO2 and ZrO2. Catal. Sci. Technol. 3, 942–951 (2013). https://doi.org/10.1039/c2cy20668d
Piazzesi, G., Elsener, M., Kröcher, O., Wokaun, A.: Influence of NO2 on the hydrolysis of isocyanic acid over TiO2. Appl. Catal. B Environ. 65, 169–174 (2006b). https://doi.org/10.1016/j.apcatb.2006.01.002
Wu, Y.-J., Wang, F., Tang, W., Kakwani, R., Hou, Y., Feng, G.: Urea decomposition and implication for NOx reduction with cu-zeolite and vanadia-selective catalytic reduction. Chem. Eng. Technol. 43, 1758–1764 (2020). https://doi.org/10.1002/ceat.202000036
Kleemann, M., Elsener, M., Koebel, M., Wokaun, A.: Hydrolysis of isocyanic acid on SCR catalysts. Ind. Eng. Chem. Res. 39, 4120–4126 (2000). https://doi.org/10.1021/ie9906161
Gabrielsson, P.L.T.: Urea-SCR in automotive applications. Topics Catal. 28, 177–184 (2004). https://doi.org/10.1023/B:TOCA.0000024348.34477.4c
Zhang, J., Li, X., Chen, P., Zhu, B.: Research status and prospect on vanadium-based catalysts for NH3-SCR denitration. Materials 11, 1632 (2018). https://doi.org/10.3390/MA11091632
Hantzsch, A.: Ueber das cyamelid. Ber. Dtsch. Chem. Ges 38, 1013–1021 (1905). https://doi.org/10.1002/cber.190503801189
Kuntz, C., Kuhn, C., Weickenmeier, H., Tischer, S., Börnhorst, M., Deutschmann, O.: Kinetic modeling and simulation of high-temperature by-product formation from urea decomposition. Chem. Eng. Sci. 246. https://doi.org/10.1016/j.ces.2021.116876
Tronconi, E., Beretta, A.: The role of inter- and intra-phase mass transfer in the SCR-DeNOx reaction over catalysts of different shapes. Catal. Today 52, 249–258 (1999). https://doi.org/10.1016/S0920-5861(99)00079-6
Birkhold, F., Meingast, U., Wassermann, P., Deutschmann, O.: Analysis of the injection of urea-water-solution for automotive SCR DeNOx-systems: modeling of two-phase flow and spray/wall-interaction. In: SAE Technical Paper Series, SAE Technical Paper Series, SAE International400 Commonwealth Drive. Warrendale. https://doi.org/10.4271/2006-01-0643 (2006)
Börnhorst, M., Deutschmann, O.: Single droplet impingement of urea water solution on a heated substrate. Int. J. Heat Fluid Flow 69, 55–61 (2018). https://doi.org/10.1016/j.ijheatfluidflow.2017.10.007
Quissek, M., Lauer, T., García-Afonso, O., Fowles, S.: Identification of film breakup for a liquid urea-water-solution and application to CFD. In: SAE Technical Paper Series, SAE Technical Paper Series, SAE International400 Commonwealth Drive. Warrendale. https://doi.org/10.4271/2019-01-0983 (2019)
Kuhn, C., Schweigert, D., Kuntz, C., Börnhorst, M.: Single droplet impingement of urea water solution on heated porous surfaces. Int. J. Heat Mass Trans. 181. https://doi.org/10.1016/j.ijheatmasstransfer.2021.121836
Acknowledgements
Steinbeis GmbH & Co. KG für Technologietransfer (STZ 240 Reaktive Strömung) is acknowledged for a cost-free license of DETCHEM. Furthermore, we acknowledge support by the state of Baden-Württemberg through bwHPC. We would like to thank the Abgaszentrum Karlsruhe, in particular Mario Eck, for conducting experiments and using its facilities.
Funding
Open Access funding enabled and organized by Projekt DEAL. The authors kindly acknowledge the financial support from the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG) through project 237267381 – TRR 150. C.K. acknowledges Karlsruhe House of Young Scientist (KHYS) for funding the stay at UofA through a Research Travel Grant.
Author information
Authors and Affiliations
Contributions
Christian Kuntz: conceptualisation, methodology, formal analysis, investigation, writing — original draft, writing — review and editing, visualisation. Paul Jakob Jägerfeld: formal analysis, investigation, writing — original draft, writing — review and editing, visualisation. Joe Mmbaga: supervision, project administration. Robert E. Hayes: writing — review and editing, supervision, project administration. Olaf Deutschmann: writing — review and editing, supervision, project administration, funding acquisition.
Corresponding author
Ethics declarations
Ethics Approval
On behalf of all the co-authors, the corresponding author states there is not any violations or inconsistencies to the ethics standards.
Consent to Participate
On behalf of all the co-authors, the corresponding author confirms the consent to participate.
Consent for Publication
On behalf of all the co-authors, the corresponding author confirms the consent for publication.
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kuntz, C., Jägerfeld, P.J., Mmbaga, J. et al. Coupling of Liquid and Surface Chemistry in Urea SCR Systems. Emiss. Control Sci. Technol. 9, 77–92 (2023). https://doi.org/10.1007/s40825-023-00224-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40825-023-00224-1