
Abstract
Numerous genetic and epidemiologic studies have demonstrated an association between elevated levels of lipoprotein(a) (Lp[a]) and cardiovascular disease. As a result, lowering Lp(a) levels is widely recognized as a promising strategy for reducing the risk of new-onset coronary heart disease, stroke, and heart failure. Lp(a) consists of a low-density lipoprotein-like particle with covalently linked apolipoprotein A (apo[a]) and apolipoprotein B-100, which explains its pro-thrombotic, pro-inflammatory, and pro-atherogenic properties. Lp(a) serum concentrations are genetically determined by the apo(a) isoform, with shorter isoforms having a higher rate of particle synthesis. To date, there are no approved pharmacological therapies that effectively reduce Lp(a) levels. Promising treatment approaches targeting apo(a) expression include RNA-based drugs such as pelacarsen, olpasiran, SLN360, and lepodisiran, which are currently in clinical trials. In this comprehensive review, we provide a detailed overview of RNA-based therapeutic approaches and discuss the recent advances and challenges of RNA therapeutics specifically designed to reduce Lp(a) levels and thus the risk of cardiovascular disease.
Similar content being viewed by others
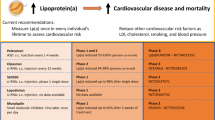

Elevated lipoprotein(a) (Lp[a]) levels are an independent risk factor for cardiovascular disease. |
Currently, there are no standard lipid-lowering therapies that sufficiently reduce Lp(a). |
Novel RNA-based drugs that lower serum Lp(a) concentrations, such as pelacarsen, olpasiran, SLN360, and lepodisiran, are in clinical trials. |
Long-term risks of Lp(a) depletion are difficult to assess due to the largely unknown physiological role of Lp(a). |
Introduction
The number of drugs that interfere with RNA transcription, maturation, and translation is rapidly growing, including antisense oligonucleotides (ASOs), small interfering RNAs (siRNAs), messenger RNAs (mRNAs), and microRNAs (miRNAs) [1]. Compared with established drugs such as recombinant proteins, RNA molecules have the advantage of recognizing a wide range of ligands [2, 3]. This property allows RNA-based therapeutics to reach previously inaccessible targets and enables them to act in a variety of ways, such as inhibiting protein translation or inducing protein coding [4]. However, challenges such as specific and efficient delivery to their intended target site remain to be resolved [5]. Nevertheless, nine ASOs and five siRNAs are currently approved by the Food and Drug Administration (FDA) for treatment of various diseases such as Duchenne muscular dystrophy and hereditary transthyretin-mediated amyloidosis [6,7,8]. In addition, two mRNAs have been approved for vaccination against the severe acute respiratory syndrome coronavirus type 2 (SARS-CoV-2) virus [9].
Notably, none of the approved RNA therapeutics directly target cardiovascular disease (CVD), but rather CVD-associated conditions such as familial hypercholesterolemia [10]. Given that CVD remains the leading cause of death worldwide, the development of new therapies and the discovery of novel approaches to disease prevention remain particularly important [11].
Among others, elevated serum levels of lipoprotein(a) (Lp[a]) are an established independent risk factor for CVD [12, 13]. Specifically, individuals with Lp(a) levels above 50 mg/dL have an increased risk of CVD and cardiovascular events including stroke, myocardial infarction, and cardiovascular death [14, 15]. Specifically, concentrations of Lp(a) within the range of 30–50 mg/dL indicate moderate risk, 51–180 mg/dL indicate high risk, and concentrations above 180 mg/dL are associated with very high cardiovascular risk [16]. Lp(a) levels are not influenced by lifestyle changes, but are genetically determined by the LPA gene, which encodes apolipoprotein(a) (apo[a]) [17, 18]. Therefore, modulation of apo(a) expression via LPA transcripts represents a promising target for RNA-based therapies, with numerous publications reporting on their beneficial effects in reducing Lp(a) levels and the associated CVD risk. This review aims to outline the current opportunities and challenges with RNA-based therapeutics targeting Lp(a). The review is based on previously published studies and does not include new human or animal studies conducted by any of the authors.
RNA-Based Therapeutics
Since the discovery of RNA in 1967 and its crucial role in translating genetic information, RNA-based therapeutics have emerged as an important approach to treat human diseases [19]. In general, four different classes of RNA drugs have evolved over time, which include ASOs, siRNAs, mRNAs, and miRNAs (Fig. 1).

Overview of gene-silencing mechanisms of RNA-based therapeutics. a ASOs bind to target mRNAs, resulting in RNase H1 cleavage or steric blockade of translation initiation and protein expression. Steric-blocking ASOs can also be used to express a different protein isoform by initiating alternative splicing. b The guide strand of the siRNA enters the RISC, and the activated complex cleaves the target mRNA. c Therapeutics based on mRNA are expressed by the cell's own synthesis machinery and encode a target protein. d MiRNA drugs act by binding and cleaving the target mRNA, thereby inhibiting protein translation, or by competing with endogenous miRNAs, preventing mRNA cleavage by blocking the RISC and allowing protein expression. ASOs antisense oligonucleotides, siRNA small interfering RNA, mRNA messenger RNA, miRNA microRNA, RISC RNA-induced silencing complex. Created with BioRender.com
ASOs
ASOs are 18–30 nucleotides long, single-stranded nucleic acids with a molecular weight of 6–9 kDa [5, 20]. They can be divided into two classes: RNase H-competent and steric-blocking ASOs [5]. RNase H-competent ASOs mostly use the gapmer approach, where a DNA-based gap is framed by RNA-based sequences for target mRNA binding [5]. Upon binding, RNase H1 is recruited, which recognizes the DNA–RNA duplex formed by the aforementioned DNA gap and the mRNA to cleave the bound target [5, 20, 21]. To date, four ASOs using this mechanism have been approved by the FDA: mipomersen, fomivirsen, inotersen, and tofersen [5, 22]. Inotersen and tofersen have also been approved by the European Medicines Agency (EMA) [23,24,25]. Table 1 lists FDA-approved ASOs and phase III RNA therapeutics for CVD. In contrast, steric-blocking ASOs bind their target mRNA without inducing degradation due to a lack of RNase H1 competence [5, 21]. Specifically, they work via two different mechanisms. First, they can promote and mediate alternative splicing by interacting with pre-mRNAs and thus interfering with the splicing complex [5, 20]. This can lead to an increase in protein expression rather than a silencing effect [20]. Due to the role of alternative splicing in the diversity of the proteome, ASOs could also be used to promote the switch to a different protein isoform and prevent the expression of pathological proteins [5]. There are currently five FDA-approved ASOs that use the splice-switching approach: eteplirsen, golodirsen, casimersen, viltolarsen, and nusinersen [5, 8, 22]. In addition, viltolarsen and nusinersen have been approved by the EMA [26, 27]. Second, ASOs can bind to the codon responsible for translation initiation, thereby inhibiting translation initiation and attenuating target protein expression [5, 20].
In summary, ASOs have significant therapeutic potential due to their ability to bind to mRNAs or pre-mRNAs, resulting in target cleavage, alteration of alternative splicing, or the steric inhibition of mRNA translation. This versatility has contributed to the development of several FDA-approved ASO drugs that provide effective treatments for a range of diseases.
SiRNAs
A second category of RNA-based drugs is siRNAs, which serve as effector molecules of RNA interference (RNAi) and have a very distinct and defined structure. They consist of double-stranded RNA of 20–25 base pairs in length, with the addition of two 3′-overhanging nucleotides [28]. An siRNA is composed of a guide strand complementary to the target mRNA sequence and a passenger strand [5]. Once inside the cell, the siRNA is incorporated into the RNA-induced silencing complex (RISC). The passenger strand is then removed and the activated complex cleaves the target mRNA upon binding of the guide strand to the target sequence [28, 29]. Current FDA- and EMA-approved siRNAs for the treatment of various diseases include vutrisiran, patisiran, givosiran, lumasiran, and inclisiran [30,31,32,33,34,35,36]. Table 1 lists FDA-approved siRNAs and phase III RNA therapeutics for CVD.
mRNAs
The third class of RNA-based therapeutics consists of mRNAs. These take advantage of the fact that exogenous mRNA can be translated into functional proteins [19]. Therapies using mRNAs can be divided into two classes: (i) exogenous mRNAs, which replace or supplement endogenous proteins, and (ii) vaccines against infectious diseases or cancer, where the mRNA encodes, for example, a viral protein [19]. Compared with DNA-based drugs, mRNAs have higher transfection efficiency and lower toxicity, since they function without cell nucleus entry and can be degraded in regular cell processes, thus eliminating the potential for infections or insertional mutagenesis [37, 38]. The two vaccines developed for SARS-CoV-2, BNT162b2 and mRNA-1273, are currently the only mRNAs approved by the FDA and the EMA [30, 39, 40].
MiRNAs
The fourth category of potential RNA-based drugs is miRNAs. These single-stranded RNAs are first expressed as longer transcripts and subsequently undergo modification and processing, resulting in the formation of a mature miRNA approximately 22 nucleotides in length [41, 42]. This mature miRNA then acts by specifically binding to a complementary sequence of the target mRNA [43]. In contrast to ASOs, which make the target mRNA recognizable for RNase H1, miRNAs recruit the RISC similarly to siRNAs [5, 44]. The main difference between siRNA and miRNA is that miRNAs target multiple mRNAs and thereby regulate their expression, whereas siRNAs target one specific mRNA [44]. In contrast to siRNAs, miRNAs require only partial pairing to recognize their target mRNA, enabling regulation of multiple mRNAs [44,45,46]. Consequently, miRNAs regulate mRNA silencing through mechanisms such as translational repression, decapping, deadenylation, or exonuclease activity [44]. Although miRNAs represent a promising field of drug development, miRNA drugs have not yet been approved by the FDA or the EMA [47, 48]. Currently, there are two main categories of miRNA drugs in clinical trials: (i) miRNA mimics and (ii) miRNA repressors [47]. MiRNA mimics are double-stranded RNA molecules designed to replicate the structure of mature miRNA duplexes, effectively mimicking endogenous miRNAs duplexes [42]. In contrast, miRNA repressors are RNA molecules that contain the reverse complement of a specific miRNA, allowing them to bind to endogenous miRNAs and consequently suppress their activity [42, 49].
Limitations of RNA-Based Therapeutics and Potential Solutions
Although RNA-based therapeutics hold immense potential for gene-silencing therapy approaches, there are multiple challenges to their application:
-
(i)
Stability. Short, unmodified oligonucleotides are rapidly degraded due to their phosphate-sugar backbone, which is recognized and cleaved by nucleases [50,51,52]. To counteract this, various chemical modifications are introduced to increase their resistance to enzymatic degradation [50, 52]. Early modifications include the substitution of an oxygen atom with a sulfur atom in the oligonucleotide backbone, known as phosphorothioate (PS) modifications [50, 52]. Additional changes such as the introduction of modified sugar molecules further improve oligonucleotide properties [29, 53] by preventing cellular defense mechanisms and degradation [50, 54]. For ASOs, siRNAs, and miRNAs, these sugar modifications include the conversion of the 2′-hydroxyl group to 2′-O-methyl (2′-O-Me), 2′-O-methoxyethyl (2′-MOE), and 2′-fluoro (2′-F) groups [54, 55]. In contrast, for mRNAs, modifying the cap and poly(a) structures of mRNA is utilized to improve the stability of mRNA therapeutics [56].
-
(ii)
Potency. Stability-improving modifications also contribute to potency, as the longer the oligonucleotides remain in their target tissue, the more pronounced their effect becomes [57]. PS modifications, for example, enhance stability but also potency by improving the cellular delivery of ASOs [51]. However, they can also reduce the binding affinity to the target nucleic acid [55, 58]. 2′-Sugar modifications can improve the effects of PS modifications on target binding affinity [55, 59] or even increase RNA binding affinity of ASOs [57]. Compared with ASOs, siRNAs have a certain inherent stability due to their double-stranded structure, but often exhibit reduced activity without appropriate modifications [60, 61]. To enhance the potency of siRNAs, the passenger strand is often heavily modified to prevent it from entering the RISC and to promote loading of the guide strand into the enzyme complex [28]. Since unintended cooperation of the passenger strand instead of the designated guide strand can lead to unwanted off-target effects and a reduction in target-directed potency [62], the 5′-end of the guide strand should have a relatively low pairing energy to ensure its selection as the strand loaded into the RISC [61, 63]. For mRNAs, changing the cap structure increases protein synthesis, while modifying the poly(a) tail enhances translation efficiency [64]. To increase the binding affinity of miRNA repressors similar to ASOs and siRNAs, 2′-MOE and 2′-F modifications are introduced [65]. In contrast, the potency of miRNA mimics is transient and largely limited by low transfection efficiency [66]. To overcome this, similar to siRNAs, nucleotides in the passenger strand are modified to enhance miRNA incorporation into the RISC [42].
-
(iii)
Toxicity. In general, the PS backbone is the most common cause of toxicity [50, 67], which tends to induce immune responses due to nonspecific protein binding of the oligonucleotide and subsequent complement activation [57, 58, 68]. In addition to the immunostimulatory effects of introduced modifications, the basic molecule itself can trigger the immune system. By binding double-stranded RNA (dsRNA)-activated protein kinase (PKR) or toll-like receptors (TLRs), dsRNA induces interferon responses that result in nonspecific degradation of mRNA and apoptosis [44, 63]. TLRs also recognize siRNAs, therefore triggering defense mechanisms and inducing inflammatory cytokine production [61, 69]. Depending on the cell type, limiting the siRNA sequence to less than 30 nucleotides can partially mitigate these effects [63]. Similar to siRNAs, mRNA drugs can trigger an immune response by TLRs, making cell delivery more challenging than with smaller therapeutic components [19, 37]. Since both innate and adaptive immune responses are required for effective immunization, which must induce a long-term stimulatory effect on effector and memory cell production, the immunostimulatory properties of native mRNA hold potential for adjuvant vaccine activity [56, 70]. To increase the safety of the mRNA by modulating this inherent immunogenicity and to enhance translatability, modifications of nucleotides, the 5′-cap, and the poly(a) tail are introduced [56, 71, 72]. In addition, off-target silencing can lead to an observable and toxic phenotype [73] affecting cell viability [63, 73], with off-target effects observed on numerous genes in siRNA-treated cells [63, 74]. Similar to siRNAs, ASOs induce toxicity by two mechanisms: RNase H1-dependent silencing of or hybridization to off-target nucleotides due to complete or partial complementarity [67, 75]. The functional groups of ASOs allow them to interact with proteins or other small-molecule drugs, potentially leading to targeting of unintended organs [75]. In addition, ASOs can be toxic through their accumulation in cytoplasmic granules and aptamer binding [67]. Similarly, the broad binding characteristics of miRNAs and their ability to regulate a wide range of mRNAs raise concerns that they may bind to yet unknown off-targets [76].
-
(iv)
Delivery. Emerging as an early transport approach, nanoparticles were used to protect siRNAs against degradation by RNases and to mask the charge of siRNA and to facilitate escape of the siRNA into the cytoplasm [54]. Today, N-acetylgalactosamine (GalNAc) modifications are a prototypical siRNA conjugate for delivery into hepatocytes [54]. This approach takes advantage of the presence of the asialoglycoprotein receptor (ASGPR) on hepatocytes, with approximately 500,000 receptor copies per cell [54, 77]. Upon binding of GalNAc by ASGPRs and internalization into endosomes, the ligand is released and the receptor undergoes rapid recycling [54, 77, 78]. The majority of FDA- and EMA-approved siRNAs utilize this mechanism of glycoprotein uptake to achieve liver-specific delivery [77, 79]. For miRNAs, several delivery approaches have been developed to facilitate cell membrane penetration, including liposomes, polymers, or exosomes [80]. Similarly, lipid nanoparticles, cationic nano-emulsions, or cationic peptides and polymers can be used for mRNA delivery [9].
-
(v)
Side effects. Common side effects of siRNA treatment include injection site reactions, infections of the upper respiratory tract, and in rare cases, more target organ-specific reactions such as elevated liver enzymes [81]. Similarly, ASO administration is commonly associated with injection site reactions, headache, fever, or nausea [75]. Common side effects of mRNA therapeutics that have been extensively studied in the context of SARS-CoV-2 vaccination include pain at the injection site, fatigue, and headache [82]. More serious complications include thrombosis, stroke, and myocarditis [83]. Most of the side effects are likely caused by the injection itself and the immune responses induced by the drug [75]. However, in some cases, more serious adverse effects such as hepatic or renal toxicity are reported, which may be explained by accumulation of the drug in the target tissue [75]. While mild effects such as headache or flu-like symptoms can be easily treated with nonprescription medications [75], more severe side effects require thorough investigation before approval and clinical use.
In summary, the major challenges in the development of RNA-based therapeutics include ensuring stability, efficacy in terms of silencing and induction of immune responses after administration, and specific delivery to the target site. While mild side effects, such as flu-like symptoms, can be easily managed, more serious side effects, such as liver toxicity, may pose the greatest challenge to clinical translation. However, the introduction of certain modifications and the development of new delivery systems are improving the properties of the oligonucleotide and helping to bring RNA-based drugs a step closer to clinical implementation.
RNA-Based Therapeutics and CVD
As CVD remains the leading cause of death worldwide, with an estimated 19 million deaths in 2020 [84], the potential of RNA-based therapeutics to prevent cardiovascular events such as myocardial infarction and to reduce potential risk factors associated with genetic predisposition has gained traction. CVD risk factors include high blood pressure, smoking, diabetes, and high blood cholesterol levels as well as age, family history, or sex [85]. Novel drugs marketed for the treatment of CVD have been predominantly limited to combinations of already existing and approved agents, including small molecules such as statins or beta-blockers [86]. However, recent advances, particularly in the field of nucleic acid-based therapies, have made it possible to target pathways, including those associated with CVD, for example through miRNA regulation [86]. The development and approval of RNA-based therapeutics for the prevention of CVD is mainly focused on hypertension and heart failure, angiogenesis, amyloidosis, and lipid management [86] (Table 1).
Elevated levels of low-density lipoprotein cholesterol (LDL-C) have been shown to be directly associated with CVD [41, 85]. After a meta-analysis demonstrated a 23% relative risk reduction of major cardiovascular events for every 38.7 mg/dL reduction in LDL-C [101], therapeutic lowering of LDL-C levels became a cornerstone for secondary CVD prevention [102]. While lifestyle changes can reduce high LDL-C levels, a subset of individuals require pharmacological interventions to achieve adequate reduction of lipoprotein concentrations [41]. In such cases, LDL-C levels are typically lowered with statins, which are hydroxymethylglutaryl-CoA reductase inhibitors [103] and the recommended drug for lowering LDL-C levels in patients with atherosclerotic cardiovascular disease (ASCVD) [104]. Nevertheless, certain patient populations such as those with familial hypercholesterolemia require additional or alternative treatment approaches to statins to achieve the predefined target [41, 105]. According to the European Society of Cardiology guidelines, the treatment goal for individuals with familial hypercholesterolemia and no concomitant risk factors is to achieve a reduction in LDL-C of ≥ 50% from baseline, along with LDL-C levels below 70 mg/dL [105, 106]. In the LDL metabolism, proprotein convertase subtilisin/kexin type 9 (PCKS9) plays a critical role by accelerating LDL receptor degradation and limiting receptor recycling [107]. In patients with an inadequate or no response to statin treatment, PCSK9 inhibitors offer a viable option [41]. These inhibitors increase the number of LDL receptors that bind LDL-C and subsequently facilitate its degradation [108] and include antibodies such as alirocumab or RNA-based drugs such as inclisiran, an FDA-approved siRNA targeting LDL-C for the treatment of ASCVD and familial hypercholesterolemia [109].
In addition to targeting elevated LDL-C levels, RNA-based drugs have been developed in the context of CVD-associated conditions such as hypertriglyceridemia, which has been associated with increased risk of CVD [110]. Advanced therapy medicinal products (ATMPs) developed and studied for their efficacy in hypertriglyceridemia include olezarsen, volanesorsen, and ARO-APO C3 [111,112,113]. Another cause of cardiac diseases is cardiac amyloidosis, which is caused by extracellular deposition of incorrectly folded proteins in the heart [114]. ATMPs that have been developed and studied for the treatment of amyloidosis, particularly hereditary transthyretin (hATTR) amyloidosis-associated polyneuropathy, include eplontersen, inotersen, patisiran, and vutrisiran [92, 115,116,117]. An additional CVD-related condition that has been the focus of RNA-based drug development is hemophilia, where CVD diagnoses and CVD-associated deaths in patients with hemophilia have increased in recent years [118, 119]. Fitusiran has been studied for its preventive effect on the rate of untreated bleeding in patients with hemophilia [120].
In summary, while LDL-C remains the primary target for CVD prevention, some individuals require additional treatment beyond standard statin therapy to effectively reduce CVD risk, such as targeting PCKS9, which has become a stepping stone in the development and clinical application of RNA-based drugs.
Lipoprotein(a) as an Independent Risk Factor for CVD
Despite receiving therapeutic interventions for lipid management, some patients may continue to experience cardiovascular events, suggesting the presence of additional risk factors beyond elevated LDL-C levels [121]. Additional risk factors include elevated levels of Lp(a), an LDL-like particle that contributes significantly to LDL-C levels [122]. Specifically, elevated Lp(a) serum concentrations have been described as a contributing factor to coronary artery disease and atherosclerosis-related disorders affecting overall life expectancy [12,13,14, 123,124,125,126]. Intriguingly, elevated Lp(a) levels are not uncommon, affecting an estimated 1.4 billion people, or approximately 20–30% of the general population [125, 126]. Serum concentrations are not influenced by lifestyle, but are primarily determined by genetic factors [12, 14]—for example, the dietary pattern recommended for blood lipid control had only a slight effect on Lp(a) levels [127].
Individuals with elevated Lp(a) levels have a 31% higher risk of CVD and are 41.5% more likely to have an ASCVD event [123]. Lp(a) values above 180 mg/dL are even associated with a CVD risk comparable to that observed in patients with familial hypercholesterolemia [12, 14, 126]. A randomized Mendelian study showed no difference between men and women with elevated Lp(a) levels in terms of associated CVD risk [124].
Structure, Genetics, and Function of Lp(a)
The Lp(a) particle, primarily synthesized in the liver, consists of covalently linked apolipoprotein A (apo[a]) and apolipoprotein B-100 (apo[b]) attached to an LDL-like particle [123, 125, 126] (Fig. 2). Almost 90% of the Lp(a) serum concentration can be explained by the LPA gene [12]. This gene encodes for apo(a), whereas apo(a) length is determined by the repetition of specific sequences called kringle IV factors (KIV) [12]. Individuals with fewer KIV repeats express a smaller apo(a) isoform and vice versa [12]. In addition to apo(a) isoform size, studies indicate other genetic variants that regulate Lp(a) serum levels [12], such as single-nucleotide polymorphisms rs10455872 or rs3798220 [128]. A comprehensive analysis of three placebo groups from the IONIS-APO(a)Rx and IONIS-APO(a)-LRx clinical trials revealed significant changes of up to −13.1 to 21.6% in Lp(a) levels within one individual without a significant change in mean absolute Lp(a) concentration in any group. This indicates a certain intra-individual variability and implies the existence of factors other than genetic influences [129]. Fluctuations in Lp(a) concentrations can also be attributed to metabolic and structural factors related to very low-density lipoprotein triglycerides, factors associated with thrombus formation, and indicators of acute-phase responses as reflected by changes in the erythrocyte sedimentation rate [130].

Structure of LDL-C and Lp(a) particle. a LDL-C consists of a lipid particle, associated with apoB-100. b Lp(a) consists of an LDL-like particle with apo(a). The length of the apo(a) isoform depends on the number of kringle IV (KIV) repeats. The shorter the apo(a) isoform, the higher the particle number. LDL-C low-density lipoprotein cholesterol, Lp(a) lipoprotein(a), Apo(a) apolipoprotein(a), ApoB-100 apolipoprotein B-100. Created with BioRender.com
In contrast to the well-investigated pathological effects, the physiological functions of Lp(a) are not well understood [12, 131]. Studies suggest a potential role in wound healing. Immunohistochemical analysis has revealed positive staining for apo(a)/apo(b) in wounds at various stages of the healing process, suggesting the involvement of Lp(a) in mediating wound healing [131, 132]. In addition, a proteomics study has identified a correlation between Lp(a) and many proteins involved in wound healing [131, 133].
Pathological Mechanism
The underlying pathological mechanisms of Lp(a) are well understood, supported by several studies demonstrating its pro-atherogenic and pro-thrombotic nature [134] (Fig. 3).

Pathological mechanism of Lp(a). Lp(a) has pro-thrombotic, pro-atherogenic, and pro-inflammatory properties. Lp(a) interferes with plasminogen activation and fibrin degradation by promoting prothrombin activation. OxPL on the particle surface makes Lp(a) pro-inflammatory and induces monocyte activation and transmigration. Lp(a) is associated with atherosclerosis by promoting foam cell formation, necrotic core formation, endothelial cell binding, and smooth muscle cell proliferation. As a result of plaque formation and rupture, individuals with elevated serum concentrations may experience myocardial infarction and ischemic stroke. Turbulent blood flow and calcification can lead to atherosclerotic stenosis and aortic valve stenosis [163, 206, 207]; SMC smooth muscle cell, EC endothelial cell, Lp(a) lipoprotein(a), Apo(a) apolipoprotein(a), ApoB-100 apolipoprotein B-100, OxPL oxidized phospholipids. Created with BioRender.com
A key factor is the structural resemblance of apo(a) to plasminogen, a pro-enzyme of the fibrinolytic system [125, 135], which is converted to its active protease plasmin, and plays a critical role in the degradation of fibrin and subsequent clots [136, 137]. Although the loop structures of Lp(a) allow binding of plasminogen activators, these regulators are unable to activate Lp(a), unlike plasminogen [138]. Consequently, Lp(a) has the potential to disrupt fibrinolysis and mediate intravascular thrombosis [135] by interfering with conversion of plasminogen to plasmin [139, 140] and by competing with plasminogen for fibrin binding, further reducing fibrinolysis [140]. These effects are more pronounced for smaller apo(a) isoforms due to their increased fibrin binding affinity [140].
Lp(a) also promotes prothrombin activation by binding to free or bound tissue factor pathway inhibitor [125, 141]. Upon activation, prothrombin is converted to thrombin, inducing the formation of insoluble fibrin [141], which can serve as a polymeric scaffold and potentially obstruct blood vessels, leading to pathological thrombosis [142]. In addition, Lp(a) impairs the fibrinolytic process by stimulating the expression of plasminogen activator inhibitor-1 (PAI-1), the primary inhibitor of fibrinolysis [140].
Lp(a) also plays a multifaceted role in the development of atherosclerotic plaques. The greater susceptibility of Lp(a) to oxidation than LDL potentially paves the way for Lp(a) uptake by macrophages, which then transform into foam cells and serve as precursors to atherosclerosis [135, 143]. This induces the expression of inflammatory cytokines and promotes the upregulation of endothelial cell adhesion molecules, both of which contribute to the formation and progression of atherosclerotic lesions [140].
Lp(a) is the most important carrier of oxidized phospholipids (OxPL), which serve as ligands for innate immune cell receptors, and, upon binding, initiate a cascade of diverse pro-inflammatory processes [144]. Activated monocytes play a particularly important role in these inflammatory processes that drive atherosclerosis and thus contribute significantly to its development [145]. Studies have shown increased arterial wall inflammation and mononuclear cell recruitment in individuals with elevated Lp(a) levels [144]. Moreover, Lp(a) was able to induce a pro-inflammatory response in monocytes from healthy individuals. However, when OxPLs are inactivated on the surface of Lp(a) particles, the pro-inflammatory effect is significantly attenuated, highlighting the important role of OxPLs in the pathophysiology of Lp(a), particularly through monocyte activation and consequent increase in CVD risk [144, 146].
In summary, based on the high sequence homology between apo(a) and plasminogen, which leads to the promotion of atherosclerosis and thrombosis, as well as the inflammatory properties of OxPL on the particle surface, Lp(a) may represent a functional link between thrombosis and atherosclerosis [12, 125, 143].
Lp(a) in the Context of Lipid-Lowering Approaches
Given the pathological effects of Lp(a) and its association with CVD, the reduction of elevated Lp(a) levels has emerged as a therapeutic target to reduce cardiovascular events [123]. Previously, it was estimated that a reduction in Lp(a) of more than 100 mg/dL was required to achieve a reduction in the associated risk of CVD [147]. However, more recent analyses have suggested a significantly lower value of 65.7 mg/dL to achieve a similar reduction in CVD risk as would be obtained by lowering LDL-C by 38.67 mg/dL [148]. The previous overestimation of the required reduction can be attributed to suboptimal standardization of Lp(a) measurement assays [148]. Apo(a) size polymorphism poses a major challenge to current assays. Other factors that may affect measurement accuracy include the difference in molar particle mass, poor purification properties of pure Lp(a), and mixed aggregates of Lp(a) and LDL, which cannot be fully dissociated [149].
Currently, there are no therapies specifically designed to target elevated Lp(a) serum concentrations [12, 14, 126, 150, 151]. The only method available to significantly reduce Lp(a) concentrations is a procedure known as lipoprotein apheresis [13, 152]. In this process, lipoprotein particles are physically removed from the blood [153], resulting in a significant reduction in both LDL and Lp(a) levels by 60–70% directly after the procedure [12]. However, Lp(a) levels tend to return to pre-therapy levels within approximately 1 week, necessitating weekly treatment to maintain low Lp(a) levels [154]. Lipoprotein apheresis has several additional drawbacks, including high cost, low capacity, poor accessibility, and its chronic nature [13, 152]. In addition, it is semi-invasive, time-consuming, and often associated with a decrease in quality of life and depression [152]. Furthermore, studies of lipoprotein apheresis have been criticized for poor randomization and blinding [12, 155], which may call into question the reported clinical benefit.
While Lp(a) has been recognized as a CVD risk factor, LDL-C–lowering therapies by statins remain the standard approach to reducing atherosclerotic cardiovascular risk in primary and secondary care populations [156]. In contrast to their efficacy in reducing LDL-C levels, studies have suggested that statins actually increase Lp(a) levels, a phenomenon first described in 1989 with lovastatin, which showed a dose-dependent increase in Lp(a) [157]. Meta-analyses have further confirmed this trend, reporting Lp(a) increases from 8.5 to 19.6% with statin treatment, compared with decreases of −0.4 to −2.3% in the placebo group. Therefore, it has been debated whether the increase in Lp(a) levels counteracts the risk reduction achieved by lowering LDL-C with statin therapy [12]. A meta-analysis showed a 47% increased risk of cardiovascular events in statin-treated patients with elevated Lp(a) levels relative to those with Lp(a) levels below 50 mg/dL (hazard ratio [HR] = 1.48, 95% CI 1.23–1.78). In the control group, subjects with Lp(a) levels above 50 mg/dL had an increased risk of 23% (HR = 1.23, 95% CI 1.04–1.45%) [12, 158]. This suggests that the Lp(a)-dependent risk of CVD is even more pronounced when LDL is reduced by statins [12, 158]. However, this study did not use either group as a reference, making direct risk comparisons difficult [12]. Nevertheless, studies by Tsimikas et al. show significant Lp(a) elevation under statin treatment, with variations among patient [156, 159]. Counteracting this increase by combining statins with Lp(a)-lowering therapeutics may hold great potential for maximizing statin-mediated CVD risk reduction.
While PCKS9 inhibitors primarily target LDL-C, they can also reduce Lp(a) levels by 25–30% [125]. The FDA-approved siRNA inclisiran, a PCKS9 inhibitor, showed no significant change but a trend toward Lp(a) reduction in 80% of patients at high risk for CVD due to elevated LDL-C levels [160]. In addition, two antibody-based PCKS9 inhibitors, evolocumab and alirocumab, have been shown to reduce Lp(a) levels [126]. In the FOURIER trial, evolocumab led to a 12 mg/dL reduction in Lp(a) levels, which was associated with a 15% relative risk reduction for CVD [161, 162]. Similarly, the ODYSSEY study showed a 5 mg/dL reduction in Lp(a) with alirocumab treatment, although these results were influenced by patients’ baseline serum levels [163]. Niacin, another PCKS9 inhibitor, showed a 30% reduction in Lp(a), but no CVD benefit [123, 126, 151]. Although PCSK9 inhibitors can lead to modest reductions in Lp(a) levels by 20% on average [160], they have no effect on arterial wall inflammation [146].
In summary, while established LDL-C-lowering statins appear to increase Lp(a) levels and the associated CVD risk in patients with elevated Lp(a) levels, PCSK9 inhibitors may moderately reduce both. Although lipid apheresis significantly reduces serum Lp(a) concentrations, its various drawbacks make its standardized use highly unlikely. Therefore, other approaches to directly target and reduce Lp(a) need to be developed and investigated.
Reduction of Lp(a) by RNA-Based Therapeutics
Given the limitations of existing drugs discussed above, there is an urgent need for innovative approaches for the reduction of Lp(a), and thus the risk of CVD warrants novel options. Due to its tight genetic control, Lp(a) has been a focal point for the development of RNA-based therapeutics. Most RNA-based approaches currently under development follow a general strategy of targeting the LPA gene in hepatocytes, ultimately leading to a reduction in apo(a) production and consequently Lp(a) particles [14, 123,124,125]. Another approach is to target apo(b), which is present not only in Lp(a), but also in LDL-C and other lipoproteins. Trials with the ASO mipomersen, which targets apo(b), resulted in moderate reductions in Lp(a) levels, but accompanied by hepatic steatosis during follow-up of up to 6 months. Thus, targeting apo(b) is less likely to be adopted for clinical practice [164]. Currently, there are only four RNA therapeutics in clinical trials that directly aim to prevent CVD by lowering Lp(a) levels: pelacarsen, olpasiran, SLN360, and lepodisiran (Table 2).
Pelacarsen
Pelacarsen [TQJ230], also known as AKCEA-APO[a]LRx, is an ASO and the most advanced RNA-based therapeutic designed to efficiently lower Lp(a) levels [173, 174]. Its predecessor, AKCEA-APO[a]Rx, is a second-generation ASO with 2′-O-(2-methoxyethyl) modifications and was initially tested in a phase 1 study [175]. In a single-dose regimen of AKCEA-APO[a]Rx administered to healthy male subjects with elevated Lp(a) levels, no reduction in Lp(a) or other lipid parameters was observed after 30 days [175]. However, with multiple doses of AKCEA-APO[a]Rx, there was a significant and sustained reduction in Lp(a), OxPL, and apo(b) levels when assessed at 84 days [175].
The phase 2 trial consisted of a randomized, double-blinded trial with placebo-controls. The primary objective of this study was to assess the reduction of Lp(a) levels in a cohort of 64 individuals with elevated plasma Lp(a) levels at day 85 or 99, respectively [176]. Cohort A included patients with Lp(a) concentrations between 125 and 437 nmol/L (62% men vs. 38% women); cohort B included patients with higher Lp(a) levels above 438 nmol/L (18% men vs. 82% women). Treatment with AKCEA-APO[a]Rx resulted in a mean reduction in Lp(a) levels of 66.8% in cohort A and 71.6% in cohort B, significantly different from the placebo control group. Similar beneficial effects were found for other parameters, including apo(b) and LDL-C levels [176].
As part of the same study, a phase 1/2a trial was conducted with a GalNAc-modified version of AKCEA-APO[a]Rx, referred to as AKCEA-APO[a]LRx, later known as pelacarsen. Fifty-eight healthy individuals were treated with either single or multiple doses of AKCEA-APO[a]LRx (single-dose: 71% men vs. 29% women; multi-dose: 47% men vs. 53% women). The single-dose regimen led to an 85.3% reduction at the highest dose of 80 mg. This reduction was sustained up to day 90, with a remaining reduction of 44%. In the multiple-dose cohort, a mean reduction of 92% was observed at day 36 for the 40-mg group. All participants successfully completed the study with no reported adverse events, validating the benefits of GalNAc modifications [176]. Moreover, the GalNAc modification improved the potency of AKCEA-APO[a]LRx, surpassing its predecessor by more than 30-fold [176].
A phase 2 study of AKCEA-APO[a]LRx recruited 286 individuals (66% men vs. 34% women) with established CVD and Lp(a) concentrations of at least 60 mg/dL [165]. Administration regimens included 20, 40, or 60 mg every 4 weeks, 20 mg every 2 weeks, and 20 mg weekly [165]. The primary endpoint of the study was the percent change in Lp(a) levels observed at 6 months after treatment initiation. All treatment regimens administered resulted in a significant reduction of Lp(a) levels at 6 months. The most significant reduction, reaching up to 80%, was observed when AKCEA-APO[a]LRx was administered weekly at a dose of 20 mg [165].
A phase 3 study is currently underway to determine the impact of Lp(a) lowering with pelacarsen on cardiovascular events in patients with established CVD and elevated Lp(a) levels (ClinicalTrials.gov: NCT04023552); the estimated study completion date is May 2025 [166].
In summary, phase 1 and 2 studies have shown promising results, with pelacarsen reducing Lp(a) concentrations by more than 80% after multiple administrations. A phase 3 study is now evaluating the Lp(a)-lowering potential of pelacarsen in CVD risk reduction.
Olpasiran
Another promising RNA-based drug currently in development is olpasiran, previously called AMG 890. Olpasiran is a GalNAc-modified siRNA designed to target the LPA gene and use RNAi to induce degradation of LPA mRNA, ultimately reducing Lp(a) levels [177]. Previous in vivo studies in transgenic mice and cynomolgus monkeys showed a reduction of more than 80% at a dose of 1 mg/kg, which was sustained for 5–8 weeks after administration [123, 177].
The primary outcome of a phase 1 trial of olpasiran was to assess its safety and tolerability. Secondary endpoints included assessment of Lp(a) reduction and pharmacokinetic analysis [123]. The study enrolled 64 individuals (cohorts 1–5: 70% men; cohorts 6–7 58% men) with elevated Lp(a) levels who were treated with one subcutaneous dose of olpasiran ranging from 3 to 225 mg. Olpasiran was generally well tolerated [123, 178]. Even 1 month after a single dose of olpasiran, Lp(a) levels remained below baseline. Treatment resulted in a maximum mean change of −71 to −97% in individuals with Lp(a) levels of 70–199 nM, and −76 to −91% in cohorts 6 (9 mg olpasiran, single dose) and 7 (75 mg olpasiran, single dose), which included participants with Lp(a) levels above 200 nM [123].
Based on these positive outcomes, a double-blind, placebo-controlled, randomized, dose-finding phase 2 study was conducted. The trial enrolled 281 patients diagnosed with established ASCVD and Lp(a) concentrations greater than 150 nmol/L [167, 179]. The primary objective of this study was to evaluate the efficacy of olpasiran treatment over 36 weeks using subcutaneous injections at doses of 10 mg, 75 mg, and 225 mg administered every 12 weeks or 225 mg every 24 weeks [167, 177]. The secondary endpoint was the percentage change in Lp(a) and other lipid parameters, at both 36 and 48 weeks [167, 179]. In the placebo group, Lp(a) concentration increased by 3.6% at 36 weeks (95% confidence interval [CI], −0.1 to 7.3), whereas Lp(a) concentration decreased significantly under olpasiran treatment [167]. Notably, Lp(a) concentrations were reduced in a dose-dependent manner by up to −101% (95% CI, −105.8 to −96.5) in subjects receiving the highest dose of olpasiran every 12 weeks [167]. At 48 weeks, Lp(a) concentrations were reduced by up to −100.9% (95% CI, −106.7 to −95.0) with the 225-mg dose every 12 weeks [167].
A phase 3 study evaluating the impact of olpasiran treatment on cardiovascular events including coronary heart disease death, myocardial infarction, and coronary revascularization in patients with established ASCVD and elevated Lp(a) levels is ongoing (ClinicalTrials.gov: NCT05581303), with an estimated end date of December 2026 [180].
In conclusion, a phase 2 study evaluating the effect of multiple doses of olpasiran in patients with ASCVD and elevated Lp(a) levels showed a strong decrease in Lp(a) concentrations of up to −101% at the highest dose administration. An ongoing phase 3 study is evaluating the impact of olpasiran on cardiovascular events.
SLN360
A more recent addition to the field of Lp(a)-targeting RNA therapeutics is SLN360. This innovative siRNA targets the LPA mRNA and consequently lowers apo(a) expression [14, 181]. It consists of a 19-mer siRNA modified with GalNAc [14, 181, 182].
Previous in vitro toxicology assessment studies have already shown a reduction of LPA expression in primary human hepatocytes without relevant off-target effects [182]. In preclinical studies, subcutaneous injections in rats resulted in specific delivery of SLN360 to the liver and kidneys, a promising sign for targeted therapy. Importantly, SLN360 administration was generally well tolerated in both rats and nonhuman primates. Liver LPA mRNA and serum Lp(a) levels were significantly reduced at 1 and 2 months after injection [181, 182]. Based on these promising results, this siRNA entered the first phase 1 trial. The primary objective of the phase 1 APOLLO study was to evaluate the tolerability of SLN360 after a single dose and to measure Lp(a) levels at a maximum of 150 days after administration [183]. The study enrolled 32 adults over the age of 18 with no history of ASCVD and Lp(a) levels greater than 60 mg/dL. Overall, SLN360 was well tolerated [14]. Although Lp(a) levels gradually increased after nadir, they did not return to baseline levels even 150 days after administration. Notably, median Lp(a) concentrations were reduced by 70% and > 80% after injection of the highest doses of 300 mg and 600 mg, respectively. Furthermore, SLN360 induced a dose-dependent 18% reduction in total cholesterol and a 26% reduction in LDL-C [14].
A multicenter, randomized, double-blind, placebo-controlled phase 2 trial will evaluate the efficacy, safety, and tolerability of SLN360 in 160 patients with elevated Lp(a) levels and at high risk for ASCVD [169]. The primary endpoint is the mean change in Lp(a) at 36 weeks. Secondary endpoints include assessment of Lp(a) levels at 48 and 60 weeks and changes in other lipid parameters such as LDL-C and apoB [169]. The study is expected to be completed in June 2024 [169].
In summary, a phase 1 trial demonstrated that treatment with the siRNA SLN360 reduced Lp(a) levels by more than 80% after single administration of 600 mg in healthy individuals with elevated Lp(a) levels. Based on these results, a phase 2 study is ongoing to evaluate the effect of SLN360 on Lp(a) levels in patients at high risk for ASCVD.
Lepodisiran (LY3819469)
The latest advances in Lp(a)-lowering therapies is the GalNAc conjugated and 2-O-Me, 2′-F, and unmodified Dicer siRNA lepodisiran or LY3819469, which targets the mRNA encoding apo(a) [184]. A single-ascending-dose, placebo-controlled phase 1 study enrolled 48 subjects without cardiovascular disease and with serum lipoprotein(a) concentrations ≥ 75 nmol/L (65% men vs. 35% women) [170]. The primary outcome was the assessment of tolerability and safety, pharmacokinetics, and effects on Lp(a) concentrations after administration of a single dose of lepodisiran [170]. Secondary outcomes included lepodisiran plasma levels 168 days after dosing and changes in serum Lp(a) concentrations after 336 days of follow-up (48 weeks) [170]. The maximum median change in Lp(a) concentration was −97% in the highest dose group (608 mg), which was maintained at day 337 post-injection (−94%) [170]. A placebo-controlled phase 2 study is ongoing to evaluate the efficacy of siRNAs in an estimated 254 participants with Lp(a) levels above 175 nmol/L over 20 months. The current study end date is October 18, 2024 [171]. In April 2023, a new phase 1 study focused on pharmacokinetics, safety, and tolerability was initiated in 28 patients with normal and impaired renal function [172].
Challenges in RNA-Based Therapies for Lp(a) Reduction
Due to its tight genetic control and various insights into its pathological mechanism, Lp(a) has been a target for the development of RNA therapeutics. However, as its physiological function remains unclear, it is uncertain whether the physiological or pathological role of Lp(a) is more significant in the human body. Therefore, achieving > 90% reduction in Lp(a) levels, already possible using ASO and siRNAs, may have unknown consequences, supported by the observation that very low Lp(a) levels are associated with increased risk of type 2 diabetes mellitus [185, 186]. In fact, a case–control study with 143,087 participants and a Mendelian randomized analysis revealed a causal relationship between very low Lp(a) levels (< 1.5 mg/dL) and type 2 diabetes mellitus [186]. In addition, depletion of apolipoprotein A1, a component of high-density lipoprotein particles, is associated with the development of atherosclerosis [187]. This illustrates how the reduction of an essential metabolic and structural component of lipid particles or excessive lowering of serum Lp(a) levels may induce new pathological conditions.
Cholesterol management is an established focus in CVD, for example by statin therapy. Cholesterol plays an important role in the physiology of the central nervous system, with particular emphasis on members of the LDL receptor family [188,189,190]. Apart from effects of statins on the mevalonate pathway, which is responsible for the majority of cholesterol production, emerging evidence suggests that statins may exert effects in tissues beyond the liver due to the importance of the mevalonate pathway in various cell types [190]. While some studies suggest a beneficial effect of statins on neurodegenerative diseases, a study in female mice showed an increase in plaques and β-amyloid production in the hippocampus. Therefore, low cholesterol levels may be a risk factor for Alzheimer’s disease in women [191, 192]. In addition, LDL-C deficiency has been associated with an increased risk of neurological disease [193]. However, other studies suggest that the responsible gene variants have no causal effects on Alzheimer’s disease risk or other neurological disorders [194]. A systematic review of randomized clinical trials and observational studies found no evidence of adverse cognitive effects with statin use [195]. The PROSPER study reported no difference in cognitive impairment in patients treated with pravastatin versus placebo [196, 197]. Although literature on the effect of low LDL-C levels on neurological disease is inconclusive, depletion of an LDL-like particle may have similar effects on the brain or other organs.
Additional safety concerns arise from the observed effect of immune responses induced by antisense therapy [28], such as the induction of systemic immune responses and flu-like symptoms after administration of an antisense phosphorothioate DNA oligonucleotide directed against the human immunodeficiency virus 1 in the mid-1990s [28, 198]. Since Lp(a) plays a crucial role in CVD initiation and development, Lp(a)-lowering drugs are likely to be used as a preventive measure for CVD. This implies that both patients with established CVD and healthy individuals would receive treatment for elevated Lp(a) levels. Although these concerns have been addressed in healthy donors during phase 1 trials, the potential for long-term complications remains, especially considering that phase 1 trials for pelacarsen were completed in 2016 [199]. Therefore, long-term effects that manifest later cannot yet be estimated.
In light of possible side effects, it is essential to carefully assess the safety and toxicity of newly developed RNA therapies. While phase 1 trials of RNA therapies targeting Lp(a) have generally reported positive results, adverse effects such as elevated liver enzymes have been reported following SLN360 administration [14]. Although the increase in liver enzymes was likely caused by SARS-CoV-2 vaccination, the possibility of elevated liver enzymes as a result of hepatic adverse events could not be definitively ruled out [14]. In phase 2 studies for pelacarsen, 90% of patients reported mild or moderate adverse effects, compared with 83% of patients in the placebo group [165]. Ten percent of patients treated with pelacarsen experienced severe adverse events, resulting in a total of 5% of patients discontinuing the trial due to arthralgia, myalgia, or malaise [165]. The most common adverse event was injection site reaction, reported in 27% of patients treated with pelacarsen [165]. Other common adverse events affecting more than 10% of patients were urinary tract infections, myalgia, and headaches [165]. In the phase 2 study of olpasiran, two patients receiving olpasiran (10 mg every 12 weeks, 225 mg every 24 weeks) experienced a cardiovascular event and underwent percutaneous coronary intervention [167]. Six percent of patients in the placebo group experienced cardiovascular events, including ischemic stroke and unstable angina, and one patient underwent coronary artery bypass grafting [167]. Twelve patients receiving olpasiran 225 mg every 12 weeks experienced injection site reactions compared with six patients in the placebo group [167]. Pain at the injection site led to discontinuation of the study in three cases in which other dermatologic conditions were described previously [167]. This underscores the fact that even when a drug is considered safe and well tolerated, certain adverse events, particularly those related to the injection site, may still occur.
Even after official approval, adverse events may still be found, as in the case of mipomersen, an ASO targeting apo(b) [41, 200]. Studies have shown that mipomersen can achieve a 30% reduction in LDL-C levels when administered weekly at a dose of 200 mg in various patient populations [41, 201, 202]. Therefore, mipomersen received FDA approval in 2013 for the treatment of heterozygous familial hypercholesterolemia [203]. In addition, mipomersen has been shown to significantly reduce Lp(a) levels in individuals with various lipid disorders and cardiovascular risk [204]. However, post-approval reports revealed several adverse effects including injection site reactions, elevations in hepatic alanine aminotransferases, and hepatic steatosis [41, 201]. Liver toxicity was generally reported after 6 months of treatment [41]. Cardiac events such as coronary artery disease and angina pectoris were also observed, with 12 events in the mipomersen group (n = 39) versus only one in the placebo group (n = 19) [41, 201]. These findings prompted the FDA to issue a boxed warning for mipomersen [41, 203], while the EMA had rejected the initial application for approval and a re-evaluation in 2013 [95]. Mipomersen is a compelling example of a drug that initially demonstrated the desired therapeutic effect, but later showed serious side effects, some of which occurred after a considerable time of treatment. This underscores the importance of conducting thorough studies of target protein reduction using siRNA or ASOs in a manner that allows for the detection of all potential adverse effects. However, the propensity for adverse effects can vary significantly between drugs and individuals, making them difficult to predict and estimate in advance.
Finally, it is important to consider the frequency of administration when evaluating the potential for clinical implementation of these drugs. Patients receiving the first approved siRNA drug patisiran are treated every 3 weeks. After pre-medication with antihistamines and corticosteroids to prevent infusion site reactions, the actual drug administration takes approximately 80 min; 60 min of pre-medication and 80 min of treatment results in a single treatment cycle of more than 2 h [205]. The phase 3 APOLLO study of patisiran showed adverse effects in 97% of patients, most of which were mild or moderate in severity [205]. Notably, the patisiran and placebo groups had similar rates of the most commonly reported adverse events [205]. In these cases, treatment with an RNA therapeutic involved multiple, prolonged treatment cycles with potentially serious adverse events for patients, perhaps for life. In particular in the context of Lp(a) reduction, which may serve as a preventive measure rather than a direct treatment for CVD, it is important to carefully weigh the benefits and burdens for the patient. In addition to considering the patient's perspective, particularly the need for inpatient care, RNA-based drug development requires an assessment of whether the potential benefits of the drug outweigh the need for repeated treatment cycles, typically every 3 to 4 weeks. It is important to determine whether the drug is designed for preventive use or primarily for the treatment of individuals with established disease.
Conclusions
RNA-based drugs represent an emerging class of drugs with potential applications in the treatment of CVD. An established risk factor associated with CVD and cardiovascular events is Lp(a) and its serum concentration. With both its pro-atherosclerotic and pro-thrombotic effects, Lp(a) contributes to the development and progression of cardiovascular disease. Based on its tightly regulated genetic control, Lp(a) has become an attractive target for the development of novel RNA-based therapeutics. To date, there are no standard lipid-lowering agents that exclusively reduce Lp(a). However, three promising RNA drugs (pelacarsen, olpasiran, and SLN360) are currently in development, each of which has demonstrated the ability to effectively reduce elevated serum Lp(a) levels. Another candidate, lepodisiran, is currently being investigated for its effect on Lp(a) serum levels. However, the physiological mechanisms of Lp(a) and the long-term consequences of Lp(a) reduction are still largely unknown. Although preliminary clinical data suggest that RNA-based therapeutics may offer a safe and effective option for achieving sustained Lp(a) reduction, several critical questions remain, including how they can be translated into clinical procedures and how effective the treatment is in terms of CVD risk reduction. In addition, the question remains as to whether excessive Lp(a) reduction is beneficial and what impact it would have on other organs and other pathologies, such as neurodegenerative diseases.
Data Availability
Data sharing is not applicable to this article, as no original datasets were generated or analyzed.
References
Stein CA, Castanotto D. FDA-approved oligonucleotide therapies in 2017. Mol Ther Elsevier Ltd. 2017;25:1069–75.
Kim CM, Smolke CD. Biomedical applications of RNA-based devices. Curr Opin Biomed Eng. 2017;4:106–15.
Kaczmarek JC, Kowalski PS, Anderson DG. Advances in the delivery of RNA therapeutics: from concept to clinical reality. Genome Med. 2017;9:60.
Mollocana-Lara EC, Ni M, Agathos SN, Gonzales-Zubiate FA. The infinite possibilities of RNA therapeutics. J Ind Microbiol Biotechnol. 2021;48: kuab063.
Roberts TC, Langer R, Wood MJA. Advances in oligonucleotide drug delivery. Nat Rev Drug Discov. 2020;19(10):673–94.
Igarashi J, Niwa Y, Sugiyama D. Research and development of oligonucleotide therapeutics in Japan for rare diseases. Futur Rare Dis. 2022;2:1–14.
Mullard A. FDA approves fifth RNAi drug - Alnylam’s next-gen hATTR treatment. Nat Rev Drug Discov. 2022;21(8):548–9.
Bireley JD, Morren JA. CNM-Au8: an experimental agent for the treatment of amyotrophic lateral sclerosis (ALS). Expert Opin Investig Drugs. 2023;32:677–83 (Taylor & Francis).
Ramachandran S, Satapathy SR, Dutta T. Delivery strategies for mRNA vaccines. Pharmaceut Med. 2022;36:11–20.
Kulkarni JA, Witzigmann D, Thomson SB, Chen S, Leavitt BR, Cullis PR, et al. The current landscape of nucleic acid therapeutics. Nat Nanotechnol. 2021;16:630–43.
Sinning D, Landmesser U. Low-density lipoprotein-cholesterol lowering strategies for prevention of atherosclerotic cardiovascular disease: focus on siRNA Treatment targeting PCSK9 (Inclisiran). Curr Cardiol Rep. 2020;22:176.
Kronenberg F. Lipoprotein(a). In: von Eckardstein A, Binder CJ, editors. Prev treat atheroscler improv state-of-the-art manag search nov targets. Cham: Springer International Publishing; 2022. p. 201–32.
Cegla J, France M, Marcovina SM, Neely RDG. Lp(a): When and how to measure it. Ann Clin Biochem. 2021;58:16–21.
Nissen SE, Wolski K, Balog C, Swerdlow DI, Scrimgeour AC, Rambaran C, et al. Single ascending dose study of a short interfering RNA targeting Lipoprotein(a) production in individuals with elevated plasma Lipoprotein(a) levels. J Am Med Assoc. 2022;327:1679–87.
Grundy SM. Correction. (Journal of the American College of Cardiology (2019) 73(24) (3168–3209), (S0735109718390338), (https://doi.org/10.1016/j.jacc.2018.11.002)). J Am Coll Cardiol. 2019;73:3234–7.
Banach M, Burchardt P, Chlebus K, Dobrowolski P, Dudek D, Dyrbuś K, et al. PoLA/CFPiP/PCS/PSLD/PSD/PSH guidelines on diagnosis and therapy of lipid disorders in Poland 2021. Arch Med Sci. 2021;17:1447–547.
Kronenberg F. Human genetics and the causal role of Lipoprotein(a) for various diseases. Cardiovasc Drugs Ther. 2016;30:87–100.
Li Y, Luke MM, Shiffman D, Devlin JJ. Genetic variants in the Apolipoprotein(a) gene and coronary heart disease. Circ Cardiovasc Genet. 2011;4:565–73 (American Heart Association).
Kim Y-K. RNA therapy: rich history, various applications and unlimited future prospects. Exp Mol Med. 2022;54:455–65.
Paunovska K, Loughrey D, Dahlman JE. Drug delivery systems for RNA therapeutics. Nat Rev Genet. 2022;23:265–80.
Bonham MA, Brown S, Boyd AL, Brown PH, Bruckenstein DA, Hanvey JC, et al. An assessment of the antisense properties of RNase H-competent and steric-blocking oligomers. Nucleic Acids Res. 1995;23:1197–203 (England).
Egli M, Manoharan M. Chemistry, structure and function of approved oligonucleotide therapeutics. Nucleic Acids Res. 2023;51:2529–73.
European Medicines Agency. Tegsedi. Inotersen. 2023. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/tegsedi
European Medicines Agency. EMEA-002403-PIP01–18 - paediatric investigation plan. Tofersen. 2018. Available from: https://www.ema.europa.eu/en/medicines/human/paediatric-investigation-plans/emea-002403-pip01-18
European Medicines Agency. EU/3/16/1732 - orphan designation for treatment of amyotrophic lateral sclerosis. Tofersen. 2019. Available from: https://www.ema.europa.eu/en/medicines/human/orphan-designations/eu-3-16-1732
European Medicines Agency. EU/3/20/2282 - orphan designation for treatment of Duchenne muscular dystrophy. Viltolarsen. 2023. Available from: https://www.ema.europa.eu/en/medicines/human/orphan-designations/eu-3-20-2282
European Medicines Agency. Spinraza. Nusinersen. 2023. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/spinraza
Gait MJ, Agrawal S. Introduction and history of the chemistry of nucleic acids therapeutics BT—antisense RNA design, delivery, and analysis. In: Arechavala-Gomeza V, Garanto A, editors. Methods Mol Biol. New York: Springer, US; 2022. p. 3–31.
Xu W, Jiang X, Huang L. RNA interference technology. Compr Biotechnol. Elsevier; 2019. p. 560–75.
Zogg H, Singh R, Ro S. Current advances in RNA therapeutics for human diseases. Int J Mol Sci. 2022.
Al Musaimi O, Al Shaer D, Albericio F, de la Torre BG. 2022 FDA TIDES (Peptides and Oligonucleotides) Harvest. Pharmaceuticals (Basel). Switzerland; 2023;16.
European Medicines Agency. Amvuttra. Vutrisiran. 2024. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/amvuttra
European Medicines Agency. Onpattro. Patisiran. 2023. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/tegsedi
European Medicines Agency. Givlaari. Givosiran. 2023. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/tegsedi
European Medicines Agency. Oxlumo. Lumasiran. 2023. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/tegsedi
European Medicines Agency. Leqvio. Inclisiran. 2023. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/tegsedi
Qin S, Tang X, Chen Y, Chen K, Fan N, Xiao W, et al. mRNA-based therapeutics: powerful and versatile tools to combat diseases. Signal Transduct Target Ther. 2022;7:166.
Duan Q, Hu T, Zhu Q, Jin X, Chi F, Chen X. How far are the new wave of mRNA drugs from us? mRNA product current perspective and future development. Front Immunol. 2022.
European Medicines Agency. Spikevax (previously COVID-19 Vaccine Moderna). 2023. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/spikevax-previously-covid-19-vaccine-moderna
European Medicines Agency. Comirnaty. 2023. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/comirnaty
Blom DJ, Marais AD, Moodley R, van der Merwe N, van Tonder A, Raal FJ. RNA-based therapy in the management of lipid disorders: a review. Lipids Health Dis. 2022. https://doi.org/10.1186/s12944-022-01649-3.
Jin HY, Gonzalez-Martin A, Miletic AV, Lai M, Knight S, Sabouri-Ghomi M, et al. Transfection of microRNA Mimics Should Be Used with Caution. Front Genet. 2015. https://doi.org/10.3389/fgene.2015.00340.
Rupaimoole R, Han H-D, Lopez-Berestein G, Sood AK. MicroRNA therapeutics: principles, expectations, and challenges. Chin J Cancer. 2011;30(6):368–70.
Lam JKW, Chow MYT, Zhang Y, Leung SWS. siRNA Versus miRNA as Therapeutics for Gene Silencing. Mol Ther Nucleic Acids. 2015;4: e252 (United States).
Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466:835–40.
Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet. 2008;9:102–14.
Hanna J, Hossain GS, Kocerha J. The Potential for microRNA Therapeutics and Clinical Research. Front Genet. 2019. https://doi.org/10.3389/fgene.2019.00478.
Guerriaud M, Kohli E. RNA-based drugs and regulation: toward a necessary evolution of the definitions issued from the European union legislation. Front Med. 2022.
Robertson B, Dalby AB, Karpilow J, Khvorova A, Leake D, Vermeulen A. Specificity and functionality of microRNA inhibitors. Silence. 2010;1:10.
Kuijper EC, Bergsma AJ, Pijnappel WWMP, Aartsma-Rus A. Opportunities and challenges for antisense oligonucleotide therapies. J Inherit Metab Dis. 2021;44:72–87 (John Wiley & Sons, Ltd).
Mansoor M, Melendez AJ. Advances in antisense oligonucleotide development for target identification, validation, and as novel therapeutics. Gene Regul Syst Bio. 2008;2: GRSB.S418 (SAGE Publications Ltd STM).
Meng M, Schmidtgall B, Ducho C. Enhanced stability of DNA oligonucleotides with partially Zwitterionic backbone structures in biological media. Molecules. 2018;23(11):2941 (Switzerland).
Irie A, Sato K, Hara RI, Wada T, Shibasaki F. An artificial cationic oligosaccharide combined with phosphorothioate linkages strongly improves siRNA stability. Sci Rep. 2020;10:14845.
Springer AD, Dowdy SF. GalNAc-siRNA conjugates: leading the way for delivery of RNAi therapeutics. Nucleic Acid Ther. 2018;28:109–18.
Yin W, Rogge M. Targeting RNA: a transformative therapeutic strategy. Clin Transl Sci. 2019;12:98–112 (John Wiley & Sons, Ltd).
Liu A, Wang X. The pivotal role of chemical modifications in mRNA therapeutics. Front Cell Dev Biol. 2022.
Geary RS, Norris D, Yu R, Bennett CF. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv Drug Deliv Rev. 2015;87:46–51.
Kurreck J, Wyszko E, Gillen C, Erdmann VA. Design of antisense oligonucleotides stabilized by locked nucleic acids. Nucleic Acids Res. 2002;30:1911–8.
Agrawal S, Jiang Z, Zhao Q, Shaw D, Cai Q, Roskey A, et al. Mixed-backbone oligonucleotides as second generation antisense oligonucleotides: in vitro and in vivo studies. Proc Natl Acad Sci USA. 1997;94:2620–5 (United States).
Gareri C, Polimeni A, Giordano S, Tammè L, Curcio A, Indolfi C. Antisense oligonucleotides and small interfering RNA for the treatment of dyslipidemias. J Clin Med. 2022.
Deleavey GF, Watts JK, Damha MJ. Chemical modification of siRNA. Curr Protoc Nucleic Acid Chem. 2009;39:16.3.1-16.3.22 (John Wiley & Sons, Ltd).
Varley AJ, Hammill ML, Salim L, Desaulniers JP. Effects of chemical modifications on siRNA strand selection in mammalian cells. Nucleic Acid Ther. 2020;30:229–36.
Aagaard L, Rossi JJ. RNAi therapeutics: principles, prospects and challenges. Adv Drug Deliv Rev. 2007;59:75–86 (Netherlands).
Kim SC, Sekhon SS, Shin W-R, Ahn G, Cho B-K, Ahn J-Y, et al. Modifications of mRNA vaccine structural elements for improving mRNA stability and translation efficiency. Mol Cell Toxicol. 2022;18:1–8.
Simonson B, Das S. MicroRNA therapeutics: the next magic bullet? Mini Rev Med Chem. 2015;15:467–74 (Netherlands).
Fan J, Feng Y, Zhang R, Zhang W, Shu Y, Zeng Z, et al. A simplified system for the effective expression and delivery of functional mature microRNAs in mammalian cells. Cancer Gene Ther. 2020;27:424–37.
Frazier KS. Antisense oligonucleotide therapies: the promise and the challenges from a toxicologic pathologist’s perspective. Toxicol Pathol. 2014;43:78–89 (SAGE Publications Inc).
Anderson BA, Freestone GC, Low A, De-Hoyos CL, Drury WJ III, Østergaard ME, et al. Towards next generation antisense oligonucleotides: mesylphosphoramidate modification improves therapeutic index and duration of effect of gapmer antisense oligonucleotides. Nucleic Acids Res. 2021;49:9026–41.
Kanasty RL, Whitehead KA, Vegas AJ, Anderson DG. Action and reaction: the biological response to siRNA and its delivery vehicles. Mol Ther. 2012;20:513–24 (United States).
Clem AS. Fundamentals of vaccine immunology. J Glob Infect Dis. 2011. https://doi.org/10.4103/0974-777X.77299.
Qin S, Tang X, Chen Y, Chen K, Fan N, Xiao W, et al. mRNA-based therapeutics: powerful and versatile tools to combat diseases. Signal Transduct Target Ther. 2022. https://doi.org/10.1038/s41392-022-01007-w.
Pardi N, Hogan MJ, Porter FW, Weissman D. mRNA vaccines—a new era in vaccinology. Nat Rev Drug Discov. 2018;17:261–79.
Fedorov Y, Anderson EM, Birmingham A, Reynolds A, Karpilow J, Robinson K, et al. Off-target effects by siRNA can induce toxic phenotype. RNA. 2006;12:1188–96.
Jackson AL, Bartz SR, Schelter J, Kobayashi SV, Burchard J, Mao M, et al. Expression profiling reveals off-target gene regulation by RNAi. Nat Biotechnol. 2003;21:635–7 (United States).
Alhamadani F, Zhang K, Parikh R, Wu H, Rasmussen TP, Bahal R, et al. Adverse drug reactions and toxicity of the food and drug administration-approved antisense oligonucleotide drugs. Drug Metab Dispos. 2022;50:879–87.
Bajan S, Hutvagner G. RNA-based therapeutics: from antisense oligonucleotides to miRNAs. Cells. 2020;9(1):137.
Debacker AJ, Voutila J, Catley M, Blakey D, Habib N. Delivery of oligonucleotides to the liver with GalNAc: from research to registered therapeutic drug. Mol Ther. 2020;28:1759–71.
Willoughby JLS, Chan A, Sehgal A, Butler JS, Nair JK, Racie T, et al. Evaluation of GalNAc-siRNA conjugate activity in pre-clinical animal models with reduced asialoglycoprotein receptor expression. Mol Ther. 2018;26:105–14.
Zhang M, Huang Y. siRNA modification and delivery for drug development. Trends Mol Med Elsevier. 2022;28:892–3.
Segal M, Slack FJ. Challenges identifying efficacious miRNA therapeutics for cancer. Expert Opin Drug Discov. 2020;15:987–91 (Taylor & Francis).
Ranasinghe P, Addison ML, Dear JW, Webb DJ. Small interfering RNA: discovery, pharmacology and clinical development—an introductory review. Br J Pharmacol. 2023;180:2697–720 (John Wiley & Sons, Ltd).
Riad A, Hocková B, Kantorová L, Slávik R, Spurná L, Stebel A, et al. Side effects of mRNA-Based COVID-19 vaccine: nationwide phase IV study among healthcare workers in Slovakia. Pharmaceuticals. 2021. https://doi.org/10.3390/ph14090873.
Yasmin F, Najeeb H, Naeem U, Moeed A, Atif AR, Asghar MS, et al. Adverse events following COVID-19 mRNA vaccines: a systematic review of cardiovascular complication, thrombosis, and thrombocytopenia. Immunity Inflamm Dis. 2023;11: e807 (John Wiley & Sons, Ltd).
Tsao CW, Aday AW, Almarzooq ZI, Alonso A, Beaton AZ, Bittencourt MS, et al. Heart disease and stroke statistics—2022 update: a report from the American Heart Association. Circu Am Heart Assoc. 2022;145:e153-639.
Hajar R. Risk factors for coronary artery disease: historical perspectives. Hear Views. 2017;18:109–14.
Robinson EL, Port JD. Utilization and potential of RNA-based therapies in cardiovascular disease. JACC Basic Transl Sci. 2022;7:956–69.
Bejar N, Tat TT, Kiss DL. RNA therapeutics: the next generation of drugs for cardiovascular diseases. Curr Atheroscler Rep. 2022;24:307–21.
Zhu Y, Zhu L, Wang X, Jin H. RNA-based therapeutics: an overview and prospectus. Cell Death Dis. 2022;13:644.
Tomasoni D, Bonfioli GB, Aimo A, Adamo M, Canepa M, Inciardi RM, et al. Treating amyloid transthyretin cardiomyopathy: lessons learned from clinical trials. Front Cardiovasc Med. 2023.
Severi D, Palumbo G, Spina E, Iovino A, Nolano M, Manganelli F, et al. A case of severe increase of liver enzymes in a ATTRv patient after one year of inotersen treatment. Neurol Sci. 2023;44:1419–22.
Tardif J-C, Karwatowska-Prokopczuk E, Amour ES, Ballantyne CM, Shapiro MD, Moriarty PM, et al. Apolipoprotein C-III reduction in subjects with moderate hypertriglyceridaemia and at high cardiovascular risk. Eur Heart J. 2022;43:1401–12.
Coelho T, Marques W Jr, Dasgupta NR, Chao C-C, Parman Y, França MC Jr, et al. Eplontersen for hereditary transthyretin amyloidosis with polyneuropathy. JAMA. 2023;330:1448–58.
Lazarte J, Hegele RA. Volanesorsen for treatment of familial chylomicronemia syndrome. Expert Rev Cardiovasc Ther. 2021;19:685–93 (Taylor & Francis).
Chambergo-Michilot D, Alur A, Kulkarni S, Agarwala A. Mipomersen in familial hypercholesterolemia: an update on health-related quality of life and patient-reported outcomes. Vasc Health Risk Manag. 2022;18:73–80 (Dove Medical Press).
European Medicines Agency. Kynamro. Mipomersen. 2013. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/kynamro
Merćep I, Friščić N, Strikić D, Reiner Ž. Advantages and disadvantages of inclisiran: a small interfering ribonucleic acid molecule targeting PCSK9—a narrative review. Tomlinson B, editor. Cardiovasc Ther. Hindawi; 2022;2022:8129513.
Yang J. Patisiran for the treatment of hereditary transthyretin-mediated amyloidosis. Expert Rev Clin Pharmacol. 2019;12:95–9 (Taylor & Francis).
Nie T, Heo Y-A, Shirley M. Vutrisiran: a review in polyneuropathy of hereditary transthyretin-mediated amyloidosis. Drugs. 2023;83:1425–32.
Boyce S, Rangarajan S. RNAi for the treatment of people with hemophilia: current evidence and patient selection. J Blood Med. 2023;14:317–27.
Wolska A, Yang Z-H, Remaley AT. Hypertriglyceridemia: new approaches in management and treatment. Curr Opin Lipidol. 2020;31(6):331–9.
Silverman MG, Ference BA, Im K, Wiviott SD, Giugliano RP, Grundy SM, et al. Association between lowering LDL-C and cardiovascular risk reduction among different therapeutic interventions: a systematic review and meta-analysis. JAMA. 2016;316:1289–97.
Expert Panel on Detection and Treatment of High Blood Cholesterol in Adults E. Executive Summary of the Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). JAMA. 2001;285:2486–97.
Sirtori CR. The pharmacology of statins. Pharmacol Res. 2014;88:3–11.
Ziaeian B, Fonarow GC. Statins and the prevention of heart disease. JAMA Cardiol. 2017;2:464.
Tomlinson B, Patil NG, Fok M, Lam CWK. Role of PCSK9 inhibitors in patients with familial hypercholesterolemia. Endocrinol Metab (Seoul, Korea). 2021;36:279–95 (Korea (South)).
Mach F, Baigent C, Catapano AL, Koskinas KC, Casula M, Badimon L, et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk: The Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS). Eur Heart J. 2020;41:111–88.
Pang J, Chan DC, Watts GF. The knowns and unknowns of contemporary statin therapy for familial hypercholesterolemia. Curr Atheroscler Rep. 2020;22:64 (United States).
Qiao YN, Zou YL, Guo SD. Low-density lipoprotein particles in atherosclerosis. Front Physiol. 2022;13:1–15.
Kosmas CE, Muñoz Estrella A, Skavdis A, Peña Genao E, Martinez I, Guzman E. Inclisiran for the treatment of cardiovascular disease: a short review on the emerging data and therapeutic potential. Ther Clin Risk Manag. 2020;16:1031–7.
Han SH, Nicholls SJ, Sakuma I, Zhao D, Koh KK. Hypertriglyceridemia and cardiovascular diseases: revisited. Korean Circ J. 2016;46:135–44.
Gouni-Berthold I, Schwarz J, Berthold HK. Updates in drug treatment of severe hypertriglyceridemia. Curr Atheroscler Rep. 2023;25:701–9.
Calcaterra I, Lupoli R, Di Minno A, Di Minno MND. Volanesorsen to treat severe hypertriglyceridaemia: a pooled analysis of randomized controlled trials. Eur J Clin Invest. 2022;52: e13841 (John Wiley & Sons, Ltd).
Akoumianakis I, Zvintzou E, Kypreos K, Filippatos TD. ANGPTL3 and apolipoprotein C-III as novel lipid-lowering targets. Curr Atheroscler Rep. 2021;23:20.
Garcia-Pavia P, Rapezzi C, Adler Y, Arad M, Basso C, Brucato A, et al. Diagnosis and treatment of cardiac amyloidosis: a position statement of the ESC Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2021;42:1554–68.
Robinson C, Pham C, Zamarripa AM, Dugay CS, Lee CA, Berger AA, et al. Inotersen to treat polyneuropathy associated with hereditary transthyretin (hATTR) amyloidosis. Heal Psychol Res. 2022;10:1–8.
Adams D, Polydefkis M, González-Duarte A, Wixner J, Kristen AV, Schmidt HH, et al. Long-term safety and efficacy of patisiran for hereditary transthyretin-mediated amyloidosis with polyneuropathy: 12-month results of an open-label extension study. Lancet Neurol. 2021;20:49–59.
Obici L, Ajroud-Driss S, Lin K-P, Berk JL, Gillmore JD, Kale P, et al. Impact of vutrisiran on quality of life and physical function in patients with hereditary transthyretin-mediated amyloidosis with polyneuropathy. Neurol Ther. 2023;12:1759–75.
Shapiro S, Benson G, Evans G, Harrison C, Mangles S, Makris M. Cardiovascular disease in hereditary haemophilia: the challenges of longevity. Br J Haematol. 2022;197:397–406 (John Wiley & Sons, Ltd).
Kamphuisen PW, ten Cate H. Cardiovascular risk in patients with hemophilia. Blood. 2014;123:1297–301.
Young G, Srivastava A, Kavakli K, Ross C, Sathar J, You C-W, et al. Efficacy and safety of fitusiran prophylaxis in people with haemophilia A or haemophilia B with inhibitors (ATLAS-INH): a multicentre, open-label, randomised phase 3 trial. Lancet. 2023;401:1427–37 (Elsevier).
Ahn CH, Choi SH. New drugs for treating dyslipidemia: beyond statins. Diabetes Metab J. 2015;39:87–94.
Fatica EM, Meeusen JW, Vasile VC, Jaffe AS, Donato LJ. Measuring the contribution of Lp(a) cholesterol towards LDL-C interpretation. Clin Biochem. 2020;86:45–51.
Koren MJ, Moriarty PM, Baum SJ, Neutel J, Hernandez-Illas M, Weintraub HS, et al. Preclinical development and phase 1 trial of a novel siRNA targeting lipoprotein(a). Nat Med. 2022;28:96–103 (Springer US).
Guertin J, Kaiser Y, Manikpurage H, Perrot N, Bourgeois R, Couture C, et al. Sex-specific associations of genetically predicted circulating Lp(a) (Lipoprotein(a)) and Hepatic LPA Gene expression levels with cardiovascular outcomes: Mendelian randomization and observational analyses. Circ Genomic Precis Med. 2021;14:E003271.
Vuorio A, Watts GF, Schneider WJ, Tsimikas S, Kovanen PT. Familial hypercholesterolemia and elevated lipoprotein(a): double heritable risk and new therapeutic opportunities. J Intern Med. 2020;287:2–18.
Hardy J, Niman S, Goldfaden RF, Ashchi M, Bisharat M, Huston J, et al. A review of the clinical pharmacology of pelacarsen: a Lipoprotein(a)-lowering agent. Am J Cardiovasc Drugs. 2022;22:47–54 (Springer International Publishing).
Mackinnon LT, Hubinger L, Lepre F. Effects of physical activity and diet on lipoprotein(a). Med Sci Sport Exerc. 1997;29:1429–36.
Clarke R, Peden JF, Hopewell JC, Kyriakou T, Goel A, Heath SC, et al. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N Engl J Med. 2009;361:2518–28 (Massachusetts Medical Society).
Marcovina SM, Viney NJ, Hughes SG, Xia S, Witztum JL, Tsimikas S. Temporal variability in lipoprotein(a) levels in patients enrolled in the placebo arms of IONIS-APO(a)Rx and IONIS-APO(a)-LRx antisense oligonucleotide clinical trials. J Clin Lipidol. 2018;12:122-129.e2.
Nakajima K, Hata Y. Intraindividual variations in Lipoprotein (a) levels and factors related to these changes. J Atheroscler Thromb. 1996;2:96–106.
Riches K, Porter KE. Lipoprotein(a): Cellular effects and molecular mechanisms. Cholesterol. 2012;2012.
Yano Y, Shimokawa K, Okada Y, Noma A. Immunolocalization of lipoprotein(a) in wounded tissues. J Histochem Cytochem. 1997;45:559–68.
von Zychlinski A, Kleffmann T, Williams MJA, McCormick SP. Proteomics of Lipoprotein(a) identifies a protein complement associated with response to wounding. J Proteomics. 2011;74:2881–91 (Elsevier B.V.).
Orsó E, Schmitz G. Lipoprotein(a) and its role in inflammation, atherosclerosis and malignancies. Clin Res Cardiol Suppl. 2017;12:31–7.
Maranhão RC, Carvalho PO, Strunz CC, Pileggi F. Lipoprotein (a): structure, pathophysiology and clinical implications. Arq Bras Cardiol. 2014;103:76–84.
Flick MJ, Bugge TH. Plasminogen–receptor KT: plasminogen activation and beyond. J Thromb Haemost. 2017;15:150–4 (John Wiley & Sons, Ltd).
Draxler DF, Sashindranath M, Medcalf RL. Plasmin: a modulator of immune function. Semin Thromb Hemost. 2017;43:143–53.
Edelberg JM, Pizzo SV. Lipoprotein (a) in the regulation of fibrinolysis. J Atheroscler Thromb. 1995;2 Suppl 1:S5–7 (Japan).
Jenkins AJ, Kostner KM, Kostner GM. Lipoprotein(a): structure, metabolism, and pathophysiology BT—Lipoproteins in diabetes Mellitus. In: Jenkins AJ, Toth PP, Lyons TJ, editors. New York. New York: Springer; 2014. p. 141–55.
Ugovšek S, Šebeštjen M. Lipoprotein(a)-the crossroads of atherosclerosis, atherothrombosis and inflammation. Biomolecules. 2021;12:26 (Switzerland).
Boffa MB, Koschinsky ML. Lipoprotein (a): truly a direct prothrombotic factor in cardiovascular disease? J Lipid Res. 2016;57:745–57.
Litvinov RI, Pieters M, de Lange-Loots Z, Weisel JW. Fibrinogen and Fibrin BT - Macromolecular Protein Complexes III: Structure and Function. In: Harris JR, Marles-Wright J, editors. Cham: Springer International Publishing; 2021. p. 471–501.
Boffa MB, Koschinsky ML. Oxidized phospholipids as a unifying theory for lipoprotein(a) and cardiovascular disease. Nat Rev Cardiol. 2019;16:305–18 (Springer US).
van der Valk FM, Bekkering S, Kroon J, Yeang C, Van den Bossche J, van Buul JD, et al. Oxidized phospholipids on Lipoprotein(a) elicit arterial wall inflammation and an inflammatory monocyte response in humans. Circulation. 2016;134:611–24.
Schnitzler JG, Poels K, Stiekema LCA, Yeang C, Tsimikas S, Kroon J, et al. Short-term regulation of hematopoiesis by lipoprotein(a) results in the production of pro-inflammatory monocytes. Int J Cardiol. 2020;315:81–5.
Stiekema LCA, Prange KHM, Hoogeveen RM, Verweij SL, Kroon J, Schnitzler JG, et al. Potent lipoprotein (a) lowering following apolipoprotein (a) antisense treatment reduces the pro-inflammatory activation of circulating monocytes in patients with elevated lipoprotein (a). Eur Heart J. 2020;41:2262–71.
Burgess S, Ference BA, Staley JR, Freitag DF, Mason AM, Nielsen SF, et al. Association of LPA variants with risk of coronary disease and the implications for lipoprotein(a)-lowering therapies: a mendelian randomization analysis. JAMA Cardiol. 2018;3:619–27.
Lamina C, Kronenberg F, Lp(a)-GWAS-Consortium. Estimation of the required lipoprotein(a)-lowering therapeutic effect size for reduction in coronary heart disease outcomes: a mendelian randomization analysis. JAMA Cardiol. 2019;4:575–9.
Scharnagl H, Stojakovic T, Dieplinger B, Dieplinger H, Erhart G, Kostner GM, et al. Comparison of lipoprotein (a) serum concentrations measured by six commercially available immunoassays. Atherosclerosis. 2019;289:206–13.
Tsimikas S. RNA-targeted therapeutics for lipid disorders. Curr Opin Lipidol. 2018;29:459–66.
Fernandez-Prado R, Perez-Gomez MV, Ortiz A. Pelacarsen for lowering lipoprotein(a): implications for patients with chronic kidney disease. Clin Kidney J. 2020;13:753–7.
Kayikcioglu M. LDL apheresis and Lp (a) apheresis: a clinician’s perspective. Curr Atheroscler Rep. 2021. https://doi.org/10.1007/s11883-021-00911-w.
Stefanutti C, Thompson GR. Lipoprotein apheresis in the management of familial hypercholesterolaemia: Historical perspective and recent advances. Curr Atheroscler Rep. 2015;17.
Julius U, Tselmin S, Schatz U, Fischer S, Birkenfeld AL, Bornstein SR. Actual situation of lipoprotein apheresis in patients with elevated lipoprotein(a) levels. Atheroscler Suppl. 2019;40:1–7.
Waldmann E, Parhofer KG. Apheresis for severe hypercholesterolaemia and elevated lipoprotein(a). Pathology. 2019;51:227–32.
Tsimikas S, Gordts PLSM, Nora C, Yeang C, Witztum JL. Statin therapy increases lipoprotein(a) levels. Eur Heart J. 2020;41:2275–84.
Kostner GM, Gavish D, Leopold B, Bolzano K, Weintraub MS, Breslow JL. HMG CoA reductase inhibitors lower LDL cholesterol without reducing Lp(a) levels. Circulation. 1989;80:1313–9.
Willeit P, Ridker PM, Nestel PJ, Simes J, Tonkin AM, Pedersen TR, et al. Baseline and on-statin treatment lipoprotein(a) levels for prediction of cardiovascular events: individual patient-data meta-analysis of statin outcome trials. Lancet. 2018;392:1211–320.
Tsimikas S, Gordts PLSM, Nora C, Yeang C, Witztum JL. Statins and increases in Lp(a): an inconvenient truth that needs attention. Eur Heart J. 2020;41:192–3.
Ruscica M, Greco MF, Ferri N, Corsini A. Lipoprotein(a) and PCSK9 inhibition: clinical evidence. Eur Hear J Suppl. 2020;22:L53–6.
Sabatine MS, Giugliano RP, Keech AC, Honarpour N, Wiviott SD, Murphy SA, et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. 2017;376:1713–22 (Massachusetts Medical Society).
O’Donoghue ML, Fazio S, Giugliano RP, Stroes ESG, Kanevsky E, Gouni-Berthold I, et al. Lipoprotein(a), PCSK9 inhibition, and cardiovascular risk. Circulation. 2019;139:1483–92 (American Heart Association).
Wilson DP, Jacobson TA, Jones PH, Koschinsky ML, McNeal CJ, Nordestgaard BG, et al. Use of Lipoprotein(a) in clinical practice: a biomarker whose time has come. A scientific statement from the National Lipid Association. J Clin Lipidol. 2019;13:374–92.
Langsted A, Nordestgaard BG. Antisense Oligonucleotides Targeting Lipoprotein(a). Curr Atheroscler Rep. 2019;21:1–7.
Tsimikas S, Karwatowska-Prokopczuk E, Gouni-Berthold I, Tardif J-C, Baum SJ, Steinhagen-Thiessen E, et al. Lipoprotein(a) reduction in persons with cardiovascular disease. N Engl J Med. 2020;382:244–55 (Massachusetts Medical Society).
ClinicalTrials.gov. A Randomized Double-blind, Placebo-controlled, Multicenter Trial Assessing the Impact of Lipoprotein (a) Lowering With Pelacarsen (TQJ230) on Major Cardiovascular Events in Patients With Established Cardiovascular Disease. NCT04023552. 2019. Available from: https://classic.clinicaltrials.gov/ct2/show/NCT04023552
O’Donoghue ML, Rosenson RS, Gencer B, López JAG, Lepor NE, Baum SJ, et al. Small interfering RNA to reduce Lipoprotein(a) in cardiovascular disease. N Engl J Med. 2022;387:1855–64 (Massachusetts Medical Society).
ClinicalTrials.gov. A Double-blind, Randomized, Placebo-controlled, Multicenter Study Assessing the Impact of Olpasiran on Major Cardiovascular Events in Participants With Atherosclerotic Cardiovascular Disease and Elevated Lipoprotein(a). NCT05581303. 2022. Available from: https://classic.clinicaltrials.gov/ct2/show/NCT05581303
ClinicalTrials.gov. A Multi-centre, Randomised, Double-blind, Placebo-controlled, Phase 2 Study to Investigate Efficacy, Safety and Tolerability of SLN360 in Participants With Elevated Lipoprotein(a) at High Risk of Atherosclerotic Cardiovascular Disease Events. NCT05537571. 2023. Available from: https://classic.clinicaltrials.gov/ct2/show/NCT05537571
Nissen SE, Linnebjerg H, Shen X, Wolski K, Ma X, Lim S, et al. Lepodisiran, an extended-duration short interfering RNA targeting Lipoprotein(a): a randomized dose-ascending clinical trial. JAMA. 2023;330:2075–83.
ClinicalTrials.gov. A Phase 2, Randomized, Double-Blind, Placebo-Controlled Study to Investigate the Efficacy and Safety of LY3819469 in Adults With Elevated Lipoprotein(a). NCT05565742. 2022. Available from: https://classic.clinicaltrials.gov/ct2/show/NCT05565742
ClinicalTrials.gov. Pharmacokinetics of LY3819469 Following Subcutaneous Dose in Participants With Renal Impairment Compared With Participants With Normal Renal Function. NCT05841277. 2023. Available from: https://classic.clinicaltrials.gov/ct2/show/NCT05841277
Yeang C, Karwatowska-Prokopczuk E, Su F, Dinh B, Xia S, Witztum JL, et al. Effect of pelacarsen on Lipoprotein(a) cholesterol and corrected low-density lipoprotein cholesterol. J Am Coll Cardiol. 2022;79:1035–46.
Ionis Pharmaceuticals. Pelacarsen. 2022. Available from: https://www.ionispharma.com/medicines/akcea-apoa-l/
Tsimikas S, Viney NJ, Hughes SG, Singleton W, Graham MJ, Baker BF, et al. Antisense therapy targeting apolipoprotein(a): a randomised, double-blind, placebo-controlled phase 1 study. Lancet. 2015;386:1472–83 (Elsevier Ltd).
Viney NJ, van Capelleveen JC, Geary RS, Xia S, Tami JA, Yu RZ, et al. Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet. 2016;388:2239–53 (Elsevier Ltd).
O’Donoghue ML, López JAG, Knusel B, Gencer B, Wang H, Wu Y, et al. Study design and rationale for the Olpasiran trials of Cardiovascular Events and lipoproteiN(a) reduction-DOSE finding study (OCEAN(a)-DOSE). Am Heart J. 2022;251:61–9 (Elsevier Inc.).
Koren MJ, Moriarty PM, Neutel J, Baum SJ, Hernandez-Illas M, Weintraub HS, et al. Abstract 13951: safety, tolerability and efficacy of Single-dose Amg 890, a Novel Sirna Targeting Lp(a), in Healthy Subjects and Subjects With Elevated Lp(a). Circulation. 2020;142:A13951–A13951 (American Heart Association).
ClinicalTrials.gov. A Double-blind, Randomized, Placebo-controlled Phase 2 Study to Evaluate Efficacy, Safety, and Tolerability of Olpasiran (AMG 890) in Subjects With Elevated Lipoprotein(a). NCT04270760. 2022. Available from: https://classic.clinicaltrials.gov/ct2/show/NCT04270760
ClinicalTrials.gov. A Double-blind, Randomized, Placebo-controlled, Multicenter Study Assessing the Impact of Olpasiran on Major Cardiovascular Events in Participants With Atherosclerotic Cardiovascular Disease and Elevated Lipoprotein(a). NCT05581303. 2022.
Rider DA, Eisermann M, Löffler K, Aleku M, Swerdlow DI, Dames S, et al. Pre-clinical assessment of SLN360, a novel siRNA targeting LPA, developed to address elevated lipoprotein (a) in cardiovascular disease. Atherosclerosis. 2022;349:240–7.
Rider D, Chivers S, Aretz J, Eisermann M, Löffler K, Hauptmann J, et al. Preclinical toxicological assessment of a novel siRNA, SLN360, targeting elevated Lipoprotein (a) in Cardiovascular Disease. Toxicol Sci. 2022;189:237–49.
ClinicalTrials.gov. A Randomised, Double-blind, Placebo Controlled, First-in-human Study to Investigate the Safety, Tolerability, Pharmacokinetic and Pharmacodynamic Response of SLN360 in Subjects With Elevated Lipoprotein(a). NCT04606602. 2020. Available from: https://clinicaltrials.gov/ct2/show/NCT04606602
Sheridan C. RNA drugs lower lipoprotein(a) and genetically driven cholesterol. Nat Biotechnol. 2022;40:983–5.
Mora S, Kamstrup PR, Rifai N, Nordestgaard BG, Buring JE, Ridker PM. Lipoprotein(a) and risk of type 2 diabetes. Clin Chem. 2010;56:1252–60.
Gudbjartsson DF, Thorgeirsson G, Sulem P, Helgadottir A, Gylfason A, Saemundsdottir J, et al. Lipoprotein(a) concentration and risks of cardiovascular disease and diabetes. J Am Coll Cardiol. 2019;74:2982–94.
Liu X, Wei W, Liu Z, Song E, Lou J, Feng L, et al. Serum apolipoprotein A-I depletion is causative to silica nanoparticles-induced cardiovascular damage. Proc Natl Acad Sci USA. 2021. https://doi.org/10.1073/pnas.2108131118.
Pfrieger FW. Outsourcing in the brain: do neurons depend on cholesterol delivery by astrocytes? BioEssays. 2003;25:72–8 (United States).
Segatto M, Di Giovanni A, Marino M, Pallottini V. Analysis of the protein network of cholesterol homeostasis in different brain regions: an age and sex dependent perspective. J Cell Physiol. 2013;228:1561–7 (United States).
Fracassi A, Marangoni M, Rosso P, Pallottini V, Fioramonti M, Siteni S, et al. Statins and the brain: more than lipid lowering agents? Curr Neuropharmacol. 2019;17:59–83.
Park I-H, Hwang EM, Hong HS, Boo JH, Oh SS, Lee J, et al. Lovastatin enhances Aβ production and senile plaque deposition in female Tg2576 mice. Neurobiol Aging. 2003;24:637–43.
Björkhem I, Meaney S, Fogelman AM. Brain cholesterol: long secret life behind a barrier. Arterioscler Thromb Vasc Biol. 2004;24:806–15.
Hussain G, Wang J, Rasul A, Anwar H, Imran A, Qasim M, et al. Role of cholesterol and sphingolipids in brain development and neurological diseases. Lipids Health Dis. 2019;18:26.
Benn M, Nordestgaard BG, Frikke-Schmidt R, Tybjærg-Hansen A. Low LDL cholesterol, PCSK9 and HMGCR genetic variation, and risk of Alzheimer’s disease and Parkinson’s disease: mendelian randomisation study. BMJ. 2017;357: j1648.
Adhikari A, Tripathy S, Chuzi S, Peterson J, Stone NJ. Association between statin use and cognitive function: a systematic review of randomized clinical trials and observational studies. J Clin Lipidol. 2021;15:22-32.e12.
Trompet S, van Vliet P, de Craen AJM, Jolles J, Buckley BM, Murphy MB, et al. Pravastatin and cognitive function in the elderly. Results of the PROSPER study. J Neurol. 2010;257:85–90.
Shepherd J, Blauw GJ, Murphy MB, Bollen ELEM, Buckley BM, Cobbe SM, et al. Pravastatin in elderly individuals at risk of vascular disease (PROSPER): a randomised controlled trial. Lancet. 2002;360:1623–30.
Agrawal S, Kandimalla ER. Role of Toll-like receptors in antisense and siRNA [corrected]. Nat Biotechnol. 2004;22:1533–7 (United States).
ClinicalTrials.gov. A Blinded, Placebo-Controlled, Dose-Escalation, Phase 1 Study to Assess the Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of Single and Multiple Doses of ISIS 681257 Administered Subcutaneously to Healthy Volunteers With Elevated Lipoprotein. NCT02414594. 2015. Available from: https://clinicaltrials.gov/ct2/show/NCT02414594
Raal FJ, Santos RD, Blom DJ, Marais AD, Charng M-J, Cromwell WC, et al. Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: a randomised, double-blind, placebo-controlled trial. Lancet (London, England). 2010;375:998–1006 (England).
McGowan MP, Tardif J-C, Ceska R, Burgess LJ, Soran H, Gouni-Berthold I, et al. Randomized, placebo-controlled trial of mipomersen in patients with severe hypercholesterolemia receiving maximally tolerated lipid-lowering therapy. PLoS One. 2012;7: e49006.
Kastelein JJP, Wedel MK, Baker BF, Su J, Bradley JD, Yu RZ, et al. Potent reduction of apolipoprotein B and low-density lipoprotein cholesterol by short-term administration of an antisense inhibitor of apolipoprotein B. Circulation. 2006;114:1729–35 (United States).
Wong E, Goldberg T. Mipomersen (kynamro): a novel antisense oligonucleotide inhibitor for the management of homozygous familial hypercholesterolemia. P T. 2014;39:119–22.
Santos RD, Raal FJ, Catapano AL, Witztum JL, Steinhagen-Thiessen E, Tsimikas S. Mipomersen, an antisense oligonucleotide to apolipoprotein B-100, reduces lipoprotein(a) in various populations with hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2015;35:689–99 (American Heart Association).
Urits I, Swanson D, Swett MC, Patel A, Berardino K, Amgalan A, et al. A review of patisiran (ONPATTRO®) for the treatment of polyneuropathy in people with hereditary transthyretin amyloidosis. Neurol Ther. 2020;9:301–15.
Liu T, Yoon W-S, Lee S-R. Recent updates of lipoprotein(a) and cardiovascular disease. Chonnam Med J. 2021;57:36–43.
Tsimikas S. A test in context: lipoprotein(a): Diagnosis, prognosis, controversies, and emerging therapies. J Am Coll Cardiol. 2017;69:692–711.
Funding
No funding or sponsorship was received for this study or publication of this article.
Author information
Authors and Affiliations
Contributions
All authors contributed to the conceptualization of this review. The first draft was written by Henriette Thau. Figures and tables were created by Henriette Thau. All versions were reviewed and edited by Sebastian Neuber, Maximilian Y. Emmert and Timo Z. Nazari-Shafti. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
Henriette Thau, Sebastian Neuber, Maximilian Y. Emmert and Timo Z. Nazari-Shafti declare no conflict of interest and no commercial or financial relationships.
Ethical Approval
This review is based on previously published studies and does not include new human or animal studies conducted by any of the authors.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Thau, H., Neuber, S., Emmert, M.Y. et al. Targeting Lipoprotein(a): Can RNA Therapeutics Provide the Next Step in the Prevention of Cardiovascular Disease?. Cardiol Ther 13, 39–67 (2024). https://doi.org/10.1007/s40119-024-00353-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40119-024-00353-w