
Abstract
Toll-like receptors (TLRs), which serve as a bridge between innate and adaptive immunity, may be viable treatment targets. TLRs are the first line of defense against microbes and activate signaling cascades that induce immune and inflammatory responses. Patients with “hot” versus “cold” tumors may respond more favorably to immune checkpoint inhibition, and through their downstream effects, TLR agonists have the potential to convert “cold tumors” into “hot tumors” making TLRs in combination with immune checkpoint inhibitors, potential targets for cancer therapies. Imiquimod is a topical TLR7 agonist, approved by the FDA for antiviral and skin cancer treatments. Other TLR adjuvants are used in several vaccines including Nu Thrax, Heplisav, T-VEC, and Cervarix. Many TLR agonists are currently in development as both monotherapy and in combination with immune checkpoint inhibitors. In this review, we describe the TLR agonists that are being evaluated clinically as new therapies for solid tumors.
Similar content being viewed by others
Introduction
Tumors evade host immune surveillance through a variety of mechanisms, including selecting less immunogenic clones and exploiting immune checkpoints (e.g. cytotoxic T-lymphocyte-associated protein 4 [CTLA4], programmed cell death protein-1 [PD-1]) to promote immunologic tolerance1,2. Tumor cell engagement of either CTLA4 or PD-1 leads to the downregulation of effector T-cell responses by blocking T-cell receptor co-stimulation and driving T-cell anergy, respectively. Immunotherapy, a treatment approach that involves harnessing and augmenting the host immune system to respond to and eliminate malignant cells, seeks to circumvent these tumor defenses.
Immunotherapy has revolutionized the treatment of cancer and includes vaccines, monoclonal antibodies, adoptive cell therapies, and immune checkpoint inhibitors (ICIs). ICIs targeting CTLA4 (ipilimumab), PD-1 (nivolumab, pembrolizumab, cemiplimab), and the PD-1 ligand programmed death-ligand 1 (PD-L1; atezolizumab, avelumab, durvalumab) are approved for the treatment of various solid tumors, including melanoma, non-small-cell lung cancer, and urothelial carcinoma3,4. Although ICIs represent a significant advance in the treatment of cancer, not all patients respond to available agents, and there have been reports of serious immune-related adverse events (AEs) and delayed toxicity3,5,6. Consequently, efforts are underway to identify biomarkers of response/safety to currently available agents, to develop immunotherapies that are effective in a broader range of patients, and to evaluate the potential of combination regimens to enhance the antitumor activity of ICIs3,7,8. As part of these efforts, Toll-like receptors (TLRs), which serve as a bridge between innate and adaptive immune responses, have been proposed as viable treatment targets, both as single therapies and in combination with ICIs. TLR agonists induce cytokine secretion, leading to activation of cytotoxic T lymphocytes (CTLs), resulting in an immune response that mediates inflammation and can reduce tumor burden9,10. In a melanoma mouse model, tumor volume was reduced by resiquimod (TLR7 agonist) and enhanced when mice were treated with resiquimod in combination with a PD-L1 blocker11. Similarly, MBS8 (TLR7/8 agonist) demonstrated anti-cancer activity, leading to the elimination of tumors in syngeneic mouse models12. Guretolimod (DSP-0509), a TLR 7 agonist under evaluation in a clinical study (NCT03416335), showed significant tumor reduction in mice13. In these mouse models, the adaptive immune response was initiated as evidenced by the generation of tumor specific CD8 + T cells. There has been a progressive interest to explore the role of TLRs in the treatment of cancer. This review provides an overview of the trials and TLR compounds in development.
TLRS: an overview
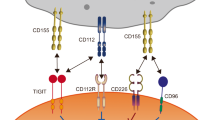
TLRs are a family of transmembrane receptors expressed by various immune (e.g. macrophages, dendritic cells, lymphocytes) and non-immune (e.g. epithelial cells, fibroblasts) cells14,15. TLRs recognize conserved exogenous and endogenous danger signals known as pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), respectively14,16. PAMPs include the bacterial endotoxin lipopolysaccharide (LPS) and viral and bacterial nucleic acids14,15, while DAMPs are released by dead or dying host cells during programmed cell death processes15,16. Of the 10 TLRs expressed in humans, six are found on cell surfaces (TLR1, 2, 4, 5, 6, and 10), and four are localized to endosomes (TLR3, 7, 8, and 9; Fig. 1)14,17. The former recognizes proteins and lipids, whereas the latter engages nucleic acids18.

The diagram illustrates the location of the TLRs. Dimerization of TLRs is required to activate downstream signaling but has not been shown.
The binding of PAMPs or DAMPs to TLRs triggers the maturation and activation of antigen-presenting cells14,19. Once mature, macrophages and dendritic cells (1) secrete cytokines that stimulate pro-inflammatory responses and (2) present antigen to naive T lymphocytes, prompting their differentiation into effector T cells17,20. Through their critical roles in both innate and adaptive immunity, TLRs defend against invading pathogens and function in immune surveillance.
Role of TLRs in cancer
TLRs have been shown to be overexpressed in different cancers, such as TLR7 and TLR8 in pancreatic cancer; TLR3, TLR4, TLR7, and TLR9 in esophageal cancer; TLR4, TLR5, and TLR7–9 in lung cancer; and TLR2–5 in ovarian cancer18,21. In a meta-analysis, overexpression of TLR1–5 or TLR9 was found to negatively correlate with clinical outcomes in patients with squamous cell carcinoma of the head and neck (SCCHN)22. In addition to overexpression, the localization of individual TLRs can become perturbed in malignant cells23. As mentioned, TLR2, TLR4, and TLR5 are normally localized to the plasma membrane, but can be found in the cytoplasm of colorectal cancer (CRC) cells. Similarly, TLR5 exhibits diffuse intracellular expression in esophageal carcinoma.
As a family, TLRs have been implicated in both cancer progression and suppression, with the effects of individual receptors varying by tumor histology18,21,24,25,26. For example, TLR4, whose principal ligand is LPS, promotes antitumor responses in hepatoblastoma, but pro-tumor responses in hepatocellular carcinoma and cervical cancer25. The antitumor effects of TLRs are mediated by the secretion of pro-inflammatory cytokines and the induction of tumor cell death, whereas their pro-tumor effects include facilitating cancer cell proliferation, survival, and metastasis, as well as immunosuppression18,24,26. TLRs can also stimulate regulatory T cells, which further contribute to the creation of a tumor-permissive immune environment21,26. The antithetical effects of TLRs have been attributed to variations in the response and expression of individual receptors by tumor cells and cells in the tumor microenvironment18,19. As a consequence, it is not possible to regard all TLRs and tumor types as equal; rather, it is necessary to parse out the role of a particular TLR in a given treatment setting, as some patients may derive greater clinical benefit from a TLR antagonist and others from a TLR agonist.
Rationale for targeting TLRs in cancer
Pre-clinical and early clinical studies in solid tumors using TLRs therapies in development have shown antitumor activity. In healthy human donor whole blood, the TLR7/8 agonist, MBS8 showed induction of the cytokine, IFN-inducible protein-10 (IP-10), with low levels of tumor necrosis factor [TNF]-alpha (TNF-α), and interferon [IFN]-gamma (INF-γ) and demonstrated antitumor activity as a monotherapy and rescued anti-PD-1 resistance in mouse models12. Similar anti-tumor activity and resistance rescue results were found with the TLR7 agonists, resiquimod and guretolimod (DSP-0509)11,13.
To date, no TLR antagonist has received regulatory approval for the treatment of cancer. Two vaccines with adjuvants that contain TLR agonist components have been approved by the United States Food and Drug Administration (FDA)14,18. One is monophosphoryl lipid A (MPLA), a TLR4 ligand processed from the LPS of Salmonella minnesota14,18. MPLA is employed as an adjuvant in a prophylactic vaccine against human papillomavirus types 16 and 18, common causes of cervical cancer. As an adjuvant, MPLA enhances the antigen-presenting capabilities of macrophages and B cells, primes naive T cells, induces the maturation of dendritic cells, and stimulates antibody production27. The second TLR agonist approved for human use is bacillus Calmette-Guérin (BCG), a TLR2/4 agonist derived from a live attenuated variant of Mycobacterium bovis14. BCG was originally designed for use as a tuberculosis vaccine14, but as immunotherapy, BCG is administered intravesically to patients with non-muscle-invasive bladder cancer14,18. A third TLR agonist, imiquimod, has also been approved by the FDA. Imiquimod is a nucleoside analog that is applied topically to the skin of patients with superficial basal cell carcinoma. Imiquimod acts as ligand for TLR714,18 which, in contrast to TLR2 and TLR4, is located intracellularly14,17. Upon engaging their respective receptors, BCG and imiquimod are believed to stimulate antitumor responses by stabilizing the antigen-presenting machinery of macrophages and dendritic cells28,29,30,31. These actions, in turn, lead to the secretion of pro-inflammatory cytokines (e.g. interleukin [IL]-2, TNF-α, INF-γ), and the activation of effector T cells that subsequently infiltrate tumors.
Therefore, through their downstream effects, TLR agonists have the potential to convert “cold tumors” into “hot tumors” characterized by intense immunologic activity. It has been suggested that patients with “hot tumors” respond more favorably to immune checkpoint inhibition than those with “cold tumors”32.
Considering the clinical success of MPLA, BCG, and imiquimod and the potential to augment the clinical efficacy of existing immunotherapeutic agents, novel TLR agonists are being explored as monotherapy, as part of combination therapy, and as vaccine adjuvants in patients with a variety of solid tumors, as described in detail below.
TLR agonists in clinical development
agonists of cell surface-expressed TLRs
Most TLR agonists in clinical development for the treatment of solid tumors target intracellularly expressed receptors. However, synthetic ligands for cell surface-expressed TLR4 and TLR5 are being evaluated.
TLR4
A phase 1 study of the intravenously administered TLR4 agonist GSK1795091 in combination with other immunotherapies including pembrolizumab, in adults with advanced solid tumors, has been completed (NCT03447314), however due to changes in manufacturing during the study, differences in biological activity occurred33. In the study, GSK1795091 was used with a combination partner of immunotherapy; 54 patients were treated. Most patients (51/54 [94%]) experienced at least 1 Grade 1/2 TEAE. Events included chills 41% (n = 22), nausea 37% (n = 20), fatigue 35% (n = 19), anemia 26% (n = 14), vomiting 22% (n = 12), decreased appetite 20% (n = 11), pyrexia and headache, each 15% (n = 8), weight decreased, dizziness and hypertension each 13% (n = 7) and constipation, diarrhea, arthralgia, and back pain, each 11% (n = 6). Events ≥3 were experienced by 44% (n = 24) patients. These included: anemia 11% (n = 6) fatigue, back pain and hypertension, each 4% (n = 2) and constipation 2% (n = 1). Although the data are limited, 3 patients had a PR including 2 patients treated with GSK1795091 in combination with pembrolizumab, and 13 patients achieved stable disease (SD) including 1 patient treated with GSK1795091 in combination with pembrolizumab. A pharmacodynamic response was observed for IP-10, IL-10, IL-Ra, IL-6 and monocyte chemoattractant protein-1 (MCP-1) but was not observed for TNF-α33.
TLR5
Entolimod is a subcutaneously administered TLR5 agonist derived from Salmonella flagellin. In a phase 1 study (NCT01527136) of 26 patients with advanced malignancies who received entolimod daily for 2 weeks Grade 1/2 adverse events included: transient hypotension 62% (n = 16), hyperglycemia 54% (n = 14) and fever 50% (n = 13). There were 3 adverse events of grade ≥3: rigors and pyrexia 4% (n = 1), transaminitis 4% (n = 1), and hypotension 4% (n = 1)34. Entolimod induced the secretion of IL-6, IL-8, and IL-10 and decreased the numbers of immunosuppressive cells and cytokines. No tumor responses were observed. A randomized phase 2 study (NCT02715882) of entolimod in patients with colorectal cancer (CRC) was initiated in Russia in 201635, but the status of this trial is unknown. Currently, there is no active clinical study of entolimod as an anticancer agent.
Agonists of intracellular TLRs
Agents specific to TLR3 (polyinosinic-polycytidylic [poly-ICLC], rintatolimod, BO-112) and TLR8 (motolimod, SBT6050) are being actively investigated. In this article, our discussion focuses on novel agents targeting TLR7, TLR 7/8 and TLR9 (Table 1) as a comprehensive review regarding the studies for TLR 3 products has already been published36.
TLR7
TLR7 primarily recognizes viral genetic material, specifically single-stranded RNA17. TQ-A3334 is an oral TLR7 agonist that has been shown to induce the production of pro-inflammatory cytokines, such as IFN-α and IP-1037. Although the status is unknown, there was a phase 1/2 study of TQ-A3334 being conducted (NCT04273815) for the treatment of non-small-cell lung cancer. In this study, TQ-A3334 tablets were being administered weekly either alone or in combination with the anti-vascular endothelial growth factor receptor inhibitor anlotinib. In a dose-ascending, phase 1a study of 42 healthy Chinese volunteers, a single dose of TQ-A3334 was tolerable, with no grade 4–5 AEs, serious AEs, or treatment-related discontinuations reported37. AEs occurred in 67% (28/42) of clinical trial participants administered TQ-A3334, with decreased lymphocyte 50% (n = 21), neutrophil 29% (n = 12), and white blood cell counts 26% (n = 11) and headache (6% (n = 11) being the most frequent. The rates of the most common AEs were dose-dependent and generally resolved without intervention within 72 h of dosing. Nine (21%) grade 3 AEs were reported (decreased neutrophil count, 5% (n = 2); decreased lymphocyte count, 14% (n = 6); hypertriglyceridemia, 2% (n = 1)). Treatment-induced changes in cytokine expression also returned to baseline within 72 hours.
Intratumoral LHC165 (NCT03301896), was being explored as a single agent (n = 20) and in combination with the investigational PD-1 inhibitor spartalizumab (PDR001) (n = 19) in patients with advanced solid tumors. Treatment-emergent AEs were reported in 56% (22/39) of patients (all grades) and 5% of patients (n = 2) experienced ≥ grade 3 event38. The most frequent events in both the monotherapy and combination arms were pyrexia 10% (n = 2), and 26% (n = 5), injection site reaction 15% (n = 3) and 10.5% (n = 2), chills 5% (n = 1) and 10.5% (n = 2) and decreased appetite 10% (n = 2) and 5% (n = 1), respectively. In the combination arm 16% of patients (n = 3) reported pruritus and 10.5% (n = 2 each) reported asthenia, malaise, and vitiligo. One patient in the monotherapy arm reported 2 grade 3 events: neutropenia and lymphopenia and another patient in the combination reported a grade 3 event of pancreatitis. The best overall response for LHC165 showed that in the monotherapy arm 10% (n = 2) of patients had a partial response (PR) and in the LHC165 with spartalizumab arm, 5% (n = 1) had a PR and 21% (n = 4) had stable disease. Recently (Aug. 2022), the Sponsor terminated the study.
Intravenous BNT411 (NCT04101357) is being evaluated as monotherapy in patients with solid tumors and in combination with atezolizumab, carboplatin, and etoposide in those with chemotherapy-naïve, extensive-stage small-cell lung cancer. In 11 patients treated with monotherapy, drug-related Grade 1/2 AEs included: pyrexia 18% (n = 1), and anemia 18% (n = 2), and one patient reported a Grade 3 event of pyrexia. No dose limiting toxicities, serious AEs, or drug related grades 4 and 5 AEs reported. At the highest dose level tested, cytokine movement was observed with increases in IP10. Preliminary efficacy data included 1 patient who had a best response of SD for 5 months39.
Intravenous NJH395 is an immune-stimulating antibody conjugate (ISAC) that combines an unspecified TLR7 agonist with a monoclonal antibody against human epidermal growth factor receptor-2 (HER2)40. Despite its systemic distribution, the HER2 component directs the TLR7 agonist specifically to HER2-expressing cells, enabling it to act locally and theoretically limit its off-target effects. A phase 1 dose escalation study of NJH395 has been completed (NCT03696771) in individuals with non-breast, HER2-positive advanced malignancies. In an interim analysis performed on 18 patients who received a single infusion of NJH395, cytokine release syndrome 56% (n = 10), pyrexia 44% (n = 8), and nausea 44% (n = 8) were the most common AEs40. Grade ≥3 AEs included decreased lymphocyte counts 28% (n = 5) and increased aspartate aminotransferase 11% (n = 2). The authors described these toxicities as “significant, but manageable”. NJH395 was also shown to trigger an increase in the number of tumor-infiltrating cytotoxic T cells, and stable disease (SD) was the best observed tumor response in 50% (n = 9) patients at 3 weeks post-dose.
A phase 1 study (NCT04645797) of an orally administered TLR7 agonist, APR003, administered to patients with advanced CRC and metastatic liver lesions is recruiting. APR003 was designed to concentrate in the gastrointestinal tract and liver; this targeted distribution is anticipated to lead to a more favorable safety and tolerability profile relative to other agonists with a more systemic distribution41.
Several other first-in-human studies of TLR7 agonists are currently underway. These include oral SHR2150 (NCT04588324), which is being tested in combination with chemotherapy plus anti–PD-1 or anti-CD47 in patients with advanced solid tumors; oral RO7119929 (NCT04338685), which is being evaluated as a single agent in patients with advanced hepatocellular carcinoma, biliary tract cancer, or solid tumors with liver metastases. DSP-0509 (NCT03416335) administered intravenously, is being studied alone and in combination with pembrolizumab in adults with advanced solid tumors; this study is ongoing. Although clinical data is not yet available for these studies several other TLR7 agonists under study have reported some of the clinical data.
TLR7/8
Because of their shared phylogeny, it is not uncommon for agents that bind to TLR7 to also engage TLR842. NKTR-262 is a TLR7/8 agonist in clinical development for the treatment of relapsed/refractory advanced solid tumors43. In patients with melanoma, intratumoral injection of NKTR-262 stimulated the up-regulation of IFN-inducible genes and IP-10 in a dose-dependent manner and increased the density of CD11c-positive cells; CD11c is expressed primarily on the surfaces of dendritic cells. In preclinical models, the antitumor activity of NKTR-262, which targets the innate immune system, was found to synergize with the investigational IL-2 pathway agonist bempegaldesleukin, which promotes the expansion of tumor-infiltrating cytotoxic T cells44. In the terminated phase 1/2 REVEAL study (NCT03435640), NKTR-262 was evaluated in combination with bempegaldesleukin ± nivolumab. In an interim analysis of phase 1, 97% (35/36) of patients receiving NKTR-262 plus bempegaldesleukin experienced ≥1 treatment-related AE, most commonly flu-like symptoms 78% (n = 8), fatigue 44% (n = 16), nausea 42% (n = 15), and pruritus 42% (n = )43. In 31% (n = 11) patients, Grade ≥3 AEs were reported in 6% (n = 2) patients each: elevated ALT, hypotension, leukocytosis, rash and syncope. A disease control rate of 41% (7/17 patients) including 2 with a partial response was also reported however, the Sponsor has terminated the study due to the overall Phase 1 results: but not due to safety reasons.
The intravenously administered TLR7/8 agonist BDB001 is being investigated in two ongoing phase 1 studies of patients with advanced solid tumors. In both, BDB001 will be administered as monotherapy and in combination with an immune checkpoint inhibitor, one with pembrolizumab (NCT03486301) and the other with atezolizumab (NCT04196530). Based on preliminary data from 36 patients participating in NCT03486301, the most common AEs following weekly administration of single-agent BDB001 were chills/rigor 19% (n = 7), fever 19% (n = 7), fatigue 11% (n = 4), nausea 11% (n =n = 4), and pruritus 11% (n = 4)45. Treatment-related AEs occurred in 70% (n = 25) patients. Two of the three patients with grade 3 AEs experienced cytokine release syndrome; no grade 4–5 AEs. BDB001 stimulated IFN-inducible genes, IFN-gamma, and IP-10 and the maturation of antigen-presenting cells. Efficacy data were available for 32 clinical trial participants, of whom 6% (n = 2) had a partial response (PR) and 56% (n = 20) had SD. A total of 23 patients with advanced solid tumors received BDB001 plus pembrolizumab in NCT0348630146. The safety profile of BDB001 when combined with pembrolizumab was largely similar to that observed with single-agent BDB001, with the most common treatment-related AEs being fever 39% (n = 9), fatigue 39% (n = 9)), chills/rigor 35% (n = 8), pruritus/rash 22% (n = 5), and nausea 13% (n = 3). Three treatment-related AEs were grade 3 (fatigue, rash, stomatitis, and alkaline phosphatase elevation); no grade 4–5 treatment-related AEs were reported. Among the 14 patients evaluated for efficacy, the disease control rate was 57% (n = 8). Two phase 2 studies of BDB001 are underway in combination with atezolizumab plus immunogenic radiotherapy in patients with solid tumors (NCT03915678; AGADIR) and as monotherapy in patients with solid tumors.
Like the TLR7 agonist NJH395, BDC-1001 is an ISAC. Although BDC-1001 also bears a HER2 monoclonal antibody, its TLR-binding component engages both TLR7 and TLR847. In an ongoing, first-in-human, phase 1/2 study (NCT04278144), BDC-1001 is being studied as monotherapy and in combination with pembrolizumab in patients with HER2-positive advanced solid tumors. Interim data from the monotherapy cohort in this ongoing study demonstrated a favorable safety profile. In 57 patients the most frequent reported treatment-emergent AEs included: Grade 1 and 2 infusion-related reactions 19% (n = 11), pyrexia and diarrhea 8.8% (n = 5), fatigue 7% (n = 4), nausea and arthralgia 5.3% (n = 3), abdominal pain, anemia, and vomiting 1.8% (n = 1), and Grade ≥3 anemia 1.8% (n = 1)47.
CV8102 is a non-coding RNA sequence that activates innate immunity via TLR7/8 and the retinoic acid-inducible gene I pathway48. CV8102 is being explored as a single agent, administered via intratumoral injection, and in combination with a PD-1 antibody in a phase 1 study (NCT03291002) of patients with advanced melanoma, cutaneous squamous cell carcinoma (cSCC), SCCHN, or adenoid cystic carcinoma. In an interim analysis performed on 23 patients receiving CV8102 monotherapy and 13 patients receiving the doublet regimen, the most common AEs were fatigue, fever, chills, and headache. Grade 3 drug-related events (monotherapy cohort 17% (n = 4); and combination cohort 23% (n = 3)) occurred: increases in liver enzymes (n = 3), abscess at injection site (n = 1), hypotension (n = 1) and asymptomatic elevation of pancreatic enzymes (n = 2)48. In terms of antitumor efficacy, two objective responses, one complete and one partial, were observed in patients with melanoma receiving CV8102 monotherapy. Preliminary efficacy data included 3 patients treated with single agent CV8102 with SD for >6 months and regression of both injected and non-injected tumors, and 1 patient with a partial response (PR). In 2/25 patients treated in combination with an anti-PD-1 therapy 1 patient had a PR in the injected lesion but developed additional lesions and the other had a mixed response with regression in both injected and non-injected lesions but progression in other non-injected lesions49.
Intratumoral TransCon TLR7/8 Agonist is a prodrug of resiquimod50,51, a more potent derivative of the approved TLR7 agonist imiquimod52. To limit systemic effects, TransCon TLR7/8 Agonist was designed for intratumoral retention50,51. It is currently being evaluated as monotherapy and in combination with pembrolizumab in a first-in-human phase 1/2 study (NCT04799054) of patients with advanced solid tumors. Based on an interim analysis of eight clinical participants, TransCon TLR7/8 Agonist has yet to be associated with dose-limiting toxicities or treatment-related systemic AEs53. The only treatment-related AE reported to date has been transient, grade 1–2 injection site reactions.
TLR9
TLR9 recognizes unmethylated DNA sequences that are rich in phosphate-linked cytosine-guanine (CpG) dinucleotides54. Unmethylated CpG is common in bacterial and viral genomes, but rare in mammals, as their genomes are predominately methylated54,55. There are three classes of oligonucleotides56. Class A promotes the production and release of IFN-alpha by plasmacytoid dendritic cells and is a poor activator of B cells. The effects of Class B are converse to those of Class A, and Class C stimulates both plasmacytoid dendritic cells and B cells (ie, mix of Class A and Class B).
Lefitolimod (MGN1703), a double-stranded TLR9 agonist with a dumbbell-shaped structure55, is being evaluated in conjunction with ipilimumab in a dose-finding, phase 1 study (NCT02668770) of patients with advanced solid tumors. Per a company press release57, the decision to assess subcutaneous lefitolimod in combination with immunotherapy was made following disappointing topline results from the phase 3 IMPALA study (NCT02077868). In IMPALA, patients with metastatic CRC who responded to first-line chemotherapy were randomized to receive maintenance treatment of either twice-weekly lefitolimod or standard of care. In the primary analysis, median overall survival (OS) was 22.0 months among lefitolimod-treated patients versus 21.9 months among those receiving standard of care (hazard ratio [HR], 1.12; 95% confidence interval [CI], 0.91–1.38; p = 0.28), and median progression-free survival (PFS) was poorer for lefitolimod versus standard of care (data values not reported). Regarding safety, the press release only stated that “no new safety signals were detected”. In the phase 2 IMPACT study (NCT01208194), which compared maintenance lefitolimod (n = 43) with placebo (n = 13) in the same patient population, the most common treatment-related AEs were flu-like symptoms (lefitolimod 14% (n = 6); placebo 8% (n = 1)) and injection site reactions (lefitolimod 5% (n = 2); placebo 8% (n = 1)); the only grade 3–4 AE to occur in more than two patients was ileus 9% (n = 4), and no treatment-related serious AEs were reported58. Of note, in pre-planned biomarker analyses, higher (≥3.08%) versus lower (<3.08%) baseline levels of activated natural killer T cells were associated with longer PFS in lefitolimod-treated patients58.
The intratumoral TLR9 agonist tilsotolimod (IMO-2125) induces increases in IFN-γ and IFN-responsive genes within 24 hours of dosing59. Tilsotolimod plus ipilimumab was evaluated in the phase 1/2 ILLUMINATE-204 study (NCT02644967), where it was shown to exhibit promising efficacy in the same patient population. Among the 49 patients who received the recommended phase 2 dose (8 mg) and who were evaluable for efficacy in ILLUMINATE-204, the overall response rate (ORR) was 22%, including two CRs, with regression seen in injected and non-injected lesions60. Although the combination regimen was tolerable, with no AEs leading to treatment discontinuation or death, almost half (48% [30/62)) of all patients experienced grade 3–4 AEs, most commonly liver enzyme increases and colitis. In the phase 1b, ILLUMINATE-101 study (NCT03052205), which enrolled patients with solid tumors, the most frequent treatment-related AEs associated with single-agent tilsotolimod were pyrexia, fatigue, chills, nausea, and vomiting61. The phase 3 ILLUMINATE-301 study (NCT03445533), evaluated tilsotolimod plus ipilimumab versus ipilimumab alone in patients with PD-1 inhibitor-refractory advanced melanoma, however the study was terminated due to lack of efficacy. Tilsotolimod is also being evaluated in combination with ipilimumab plus nivolumab in patients with solid tumors, including microsatellite-stable CRC and melanoma (NCT03865082 [ILLUMINATE-206], NCT04270864 [PRIMO]); in combination with investigational immune checkpoint inhibitors in patients with recurrent/metastatic SCCHN (NCT04196283); and as monotherapy in patients with melanoma (NCT04126876; INTRIM).
Vidutolimod/CMP-001 is a virus-like particle containing a TLR9 agonist that induces the up-regulation of IFN-alpha in plasmacytoid dendritic cells62,63 and the production of IFN-inducible genes in T cells and natural killer cells62. It is a Class A oligonucleotide61. Intratumoral CMP-001 is being investigated as monotherapy and in combination with pembrolizumab in patients with PD-(L)1 inhibitor-refractory advanced melanoma in an ongoing, dose-escalation, phase 1b study (NCT02680184). In an interim analysis, 7 of the 40 patients administered single-agent CMP-001 for 7 weeks developed PRs, corresponding to an ORR of 18%64. Among the 98 patients who received CMP-001 (at a polysorbate-20 concentration of 0.01%) plus pembrolizumab for 7 weeks, a total of 23% (n = 23) responded (complete response [CR], n = 7; PR, n = 16). Notably, in patients who had previously progressed on anti-PD(L)1, CMP-001 plus pembrolizumab reduced the size of injected and non-injected tumors by ~50%. The most common treatment-related AEs in both treatment arms were flu-like symptoms; injection site reactions were also reported in patients receiving the combination regimen. In patients administered CMP-001 alone or in combination with pembrolizumab, the most common treatment-related grade 3 or 4 AE was hypotension (5% (n = 2) and 7% (n = 7), respectively). Importantly, no patient died due to a treatment-related AE.
In a recently completed, investigator-initiated, phase 2 study (NCT03618641), neoadjuvant treatment with CMP-001 plus nivolumab was assessed in 30 patients with stage IIIB–D melanoma. After 7 weekly doses, pathologic responses were observed in 70% (21/30) of clinical trial participants (pathologic CR [pCR], 50% (n = 15); major pathologic response [MPR], 10% (n = 3); pathologic PR, 10% (n = 3))65. In biomarker analyses, pCR/MPR was shown to be associated with a greater influx of intratumoral plasmacytoid dendritic cells and CD8-positive T cells. Infusion-related grade 3 or 4 AEs occurred in three patients, two of whom discontinued CMP-001.
Additional studies of CMP-001 in combination with approved or investigational immune checkpoint inhibitors are planned or underway in patients with melanoma (NCT04698187, NCT04695977, NCT04401995, NCT04708418, NCT03618641, NCT04401995), recurrent/metastatic SCCHN (NCT04633278), metastatic CRC (NCT03507699), metastatic pancreatic cancer and non-melanoma advanced solid tumors (NCT04387071), patients with castration resistant prostate cancer (NCT05445609), and Merkel cell carcinoma (MCC), cSCC and triple negative breast cancer (NCT04916002). A subcutaneous formulation of CMP-001 is also being explored. A study using the subcutaneous administration of 5 mg, once for 2 weeks followed by intratumoral injections once weekly for 3 weeks followed by every 3 weeks thereafter, in combination with atezolizumab in 29 patients with NSCLC who had progressed s following PD-1 treatment was stopped due to lack of response66.
Cavrotolimod (AST-008) is a spherical synthetic oligonucleotide undergoing evaluation in a phase 1b/2 study (NCT03684785) of patients with advanced solid tumors. Although this study was terminated (due to administrative reasons), the phase 1b portion of the study completed enrollment. Patients received single agent cavrotolimod or cavrotolimod plus pembrolizumab. ORR in the overall cohort was 21% (4/19), with responses observed in two patients with MCC and two with melanoma67. Three of the four responders had experienced disease progression on anti–PD-(L)1 therapy before study enrollment. Like tilsotolimod and CMP-001, cavrotolimod induced the regression of injected and non-injected tumors. The most frequently reported AEs were injection site reactions and flu-like symptoms. With regard to pharmacodynamics, cavrotolimod (alone and in combination with pembrolizumab) was associated with dose-dependent increases in various cytokines (eg, IP-10) and in the numbers of T cells infiltrating injected and non-injected lesions68.
The development of SD-101, a Class C oligonucleotide61, was discontinued by its Sponsor in 2019 following a restructuring event69. The decision was strategic, as SD-101 in combination with pembrolizumab exhibited promising anti-tumor activity in the phase 1b/2 SYNERGY-001/KEYNOTE-184 study (NCT02521870) of PD-(L)1 inhibitor-naive patients with metastatic melanoma or recurrent/metastatic SCCHN. In the subgroup of patients with metastatic melanoma, an ORR of 76% was observed in those receiving SD-101 2 mg and 49% in those receiving SD-101 8 mg70,71. Responses were seen in injected and non-injected lesions, as well as in PD-L1–positive and PD-L1–negative tumors. Among patients with recurrent/metastatic SCCHN, an ORR of 22% was reported in those administered SD-101 2 mg and 26% in those administered SD-101 8 mg72,73. Treatment with SD101 and pembrolizumab precipitated an influx of T cells in both tumor types, and the most common SD-101–associated AEs were injection site reactions and flu-like symptoms71,73.
Clinical development of SD-101 continues in trials and is currently being evaluated in combination with nivolumab and radiation therapy in patients with chemotherapy-refractory metastatic pancreatic cancer (NCT04050085); in combination with pembrolizumab, intermittent androgen deprivation therapy, and stereotactic body radiation therapy in patients with newly diagnosed, hormone-naive, oligometastatic prostate cancer (NCT03007732); as neoadjuvant therapy in combination with pembrolizumab in patients with breast cancer (NCT01042379; I-SPY); in combination with nivolumab and ipilimumab in patients with uveal melanoma (NCT04935229); in combination with pembrolizumab or nivolumab and ipilimumab in patients with liver tumors (NCT05220722); in combination with BMS986178 in patients with solid tumors (NCT03831295).
Conclusion
Given the potential to both stimulate and enhance anti-tumor immunity, as well as the number of planned/active clinical studies, it is apparent that clinicians see promise in the use of TLR agonists to treat cancer. Based on the preliminary data available, TLR7 and TLR9 agonists currently in development suggest anti-tumor activity when used as monotherapy or in combination with approved immune checkpoint inhibitors. Notably, objective responses have been reported in patients with PD-(L)1 inhibitor-resistant disease treated with CMP-001 ± pembrolizumab or cavrotolimod ± pembrolizumab and in those with PD-L1–negative tumors treated with SD-101 plus pembrolizumab. Given the role of TLRs in both innate and adaptive immunity, the anti-tumor effects of TLR agonists are likely attributable to their ability to convert “cold tumors” into “hot tumors”, as pharmacodynamic analyses have demonstrated the ability of these agents to induce the expression of pro-inflammatory cytokines and to stimulate the influx of effector T cells into tumor tissue (Fig. 2). In terms of safety, the most common AEs associated with TLR agonists appear to be flu-like symptoms, injection site reactions, fatigue, and decreased leukocyte counts (e.g., lymphocytes, neutrophils). Importantly, when used as part of a combination regimen, investigational TLR7 and TLR9 agonists, such as CV8102, tilsotolimod, and CMP-001, do not appear to increase the toxicity of approved immune checkpoint inhibitors. This is notable as other TLR agonists, such as IMO-2055, have been discontinued owing to an increased risk of AEs when used as part of combination therapy74.

TLRs are involved in both innate and adaptive immunity. It is hypothesized that the anti-tumor effects of TLR agonists are likely attributable to their ability to convert “cold tumors” into “hot tumors”, as pharmacodynamic analyses have demonstrated the ability of these agents to induce the expression of pro-inflammatory cytokines and to stimulate the influx of effector T cells into tumor tissue.
Given the conflicting roles of different TLRs and of the same TLR in different tumor types, novel clinical study designs, namely adaptive designs, may be of value in efficiently evaluating the efficacy and safety of novel TLR agonists. Adaptive study designs allow for smaller numbers of patients to be assessed and/or for shorter evaluation periods relative to traditional clinical trial designs75. Once preliminary data become available, informed decisions can be made on whether assessment of a specific tumor cohort and/or treatment regimen should be abandoned or expanded. Biomarkers, such as those identified in the translational research setting, tend to underpin adaptive study designs, as they can inform patient selection (e.g., those with HER2-positive tumors) or be used to measure treatment response.
There is precedent for the use of adaptive study designs in the oncology setting, including MyPathway (NCT02091141), a multiple-basket, phase 2a study evaluating the clinical potential of approved targeted agents in non-approved tumor types; the ongoing phase 1/2 study (NCT03416335) of DSP-0509 ± pembrolizumab in those with advanced solid tumors; the phase 2 AGADIR study (NCT03915678), which is assessing atezolizumab plus BDB001 and radiotherapy in various tumor types, including pancreatic cancer and PD-(L)1 inhibitor-refractory NSCLC and bladder cancer; and the phase 1 I-SPY study (NCT01042379), which is studying approximately 20 different regimens for the neoadjuvant treatment of breast cancer. As the anti-tumor activity of TLR agonists has been correlated to the proliferation of dendritic cells and lymphocytes, this may serve as a suitable biomarker of clinical activity in adaptive studies. Given the positive signals stemming from preliminary analyses, more robust efficacy and safety data are eagerly anticipated from ongoing studies of TLR agonists in development for the treatment of solid tumors.
Data availability
No datasets were generated or analyzed for this article.
References
Velcheti, V. & Schalper, K. Basic overview of current immunotherapy approaches in cancer. Am. Soc. Clin. Oncol. Educ. Book 35, 298–308 (2016).
Vinay, D. S. et al. Immune evasion in cancer: mechanistic basis and therapeutic strategies. Semin. Cancer Biol. 35, S185–S198 (2015).
Stephen, B. & Hajjar, J. Overview of basic immunology and clinical application. (eds. Naing A, Hajjar J) Immunotherapy. 3rd edn. Vol 1244. p. 1–36 (Springer Nature Switzerland, 2020).
Russo, A. et al. Immunotherapy in lung cancer: from a minor god to the Olympus. (eds. Naing, A., & Hajjar, J.) Immunotherapy. 3rd edn. Vol 1244. p. 1–36. (Springer Nature Switzerland, 2020).
Albittar, A. A. Alhalabi, O., Glitza Oliva, I.C. Immunotherapy for melanoma. (eds. Naing, A., & Hajjar, J.) Immunotherapy. 3rd edn. Vol 1244. p. 51–68. (Springer Nature Switzerland, 2020).
Nigro, O. et al. Late immune-related adverse events in long-term responders to PD-1/PD-L1 checkpoint inhibitors: a multicentre study. Eur. J. Cancer 134, 19–28 (2020).
Naing, A. Being realistic and optimistic in curing cancer. J. Immunother. Precis. Oncol. 1, 53–55 (2018).
Fujii, T., Naing, A., Rolfo, C. & Hajjar, J. Biomarkers of response to immune checkpoint blockade in cancer treatment. Crit. Rev. Oncol. Hematol. 130, 108–120 (2018).
Anwar, M. A., Shah, M., Kim, J. & Choi, S. Recent clinical trends in Toll-like receptor targeting therapeutics. Med Res Rev. 39, 1053–1090 (2019).
Chi et al. Anti-tumor Activity of Toll-Like Receptor 7 Agonists. Front. Pharmacol. https://doi.org/10.3389/fphar.2017.00304 (2017).
Nishii, N. et al. Systemic administration of a TLR7 agonist attenuates regulatory T cells by dendritic cell modification and overcomes resistance to PD-L1 blockade therapy. Oncotarget. 9, 13301–13312 (2018).
Jensen, S. S. et al. Systemic TLR7/8 micelles trigger a novel and potent anti-tumor response by strong recruitment of neutrophils leading to massive tumor cell killing. J. Clin. Oncol. 40, 2576–2576 (2022).
Ota Y. et al. Novel intravenous injectable TLR7 agonist, DSP-0509, synergistically enhanced antitumor immune responses in combination with anti-PD-1 antibody [abstract]. In: Proceedings of the American Association for Cancer Research Annual Meeting 2018; 2018 Apr 14-18; Chicago, IL. Philadelphia (PA): AACR; Cancer Res 2018;78: Abstract nr 4726.
Smith, M. et al. Trial Watch: Toll-like receptor agonists in cancer immunotherapy. Oncoimmunology 7, e1526250 (2018).
Aranda, F. et al. Trial Watch: Toll-like receptor agonists in oncological indications. Oncoimmunology 3, e29179 (2014).
Amarante-Mendes, G. P. et al. Pattern recognition receptors and the host cell death molecular machinery. Front. Immunol. 9, 2379 (2018).
Tran, T. H. Toll-like receptor-targeted particles: a paradigm to manipulate the tumor microenvironment for cancer immunotherapy. Acta Biomater. 94, 82–96 (2019).
Urban-Wojciuk, Z. et al. The role of TLRs in anti-cancer immunity and tumor rejection. Front. Immunol. 10, 2388 (2019).
Huang, L., Xu, H. & Peng, G. TLR-mediated metabolic reprogramming in the tumor microenvironment: potential novel strategies for cancer immunotherapy. Cell. Mol. Immunol. 15, 428–437 (2018).
Suresh, R. & Mosser, D. M. Pattern recognition receptors in innate immunity, host defense, and immunopathology. Adv. Physiol. Educ. 37, 284–291 (2013).
Javaid, N. & Choi, S. Toll-like receptors from the perspective of cancer treatment. Cancers (Basel) 12, 297 (2020).
Hasnat, S. et al. The prognostic value of Toll-Like receptors in head and neck squamous cell carcinoma: a systematic review and meta-analysis. Int. J. Mol. Sci. 21, 7255 (2020).
Jouhi, L. et al. Different Toll-like receptor expression patterns in progression toward cancer. Front. Immunol. 5, 638 (2014).
Dajon, M., Iribarren, K. & Cremer, I. Toll-like receptor stimulation in cancer: a pro- and anti-tumor double-edged sword. Immunobiology 222, 89–100 (2017).
Shi, M. et al. Application potential of toll-like receptors in cancer immunotherapy: systematic review. Medicine (Baltimore) 95, e3951 (2016).
Huang, B. et al. TLR signaling by tumor and immune cells: a double-edged sword. Oncogene 27, 218–224 (2008).
De Becker, G. et al. The adjuvant monophosphoryl lipid A increases the function of antigen-presenting cells. Int. Immunol. 12, 807–815 (2000).
Fuge, O., Vasdev, N., Allchorne, P. & Green, J. S. Immunotherapy for bladder cancer. Res. Rep. Urol. 7, 65–79 (2015).
Kawai, K. et al. Bacillus Calmette-Guerin (BCG) immunotherapy for bladder cancer: current understanding and perspectives on engineered BCG vaccine. Cancer Sci. 104, 22–27 (2013).
Miller, R. L. et al. Imiquimod applied topically: a novel immune response modifier and new class of drug. Int. J. Immunopharmacol. 21, 1–14 (1999).
Sauder, D. N. Immunomodulatory and pharmacologic properties of imiquimod. J. Am. Acad. Dermatol. 43, S6–S11 (2000).
Duan, Q., Zhang, H., Zheng, J. & Zhang, L. Turning cold into hot: firing up the tumor microenvironment. Trends Cancer 6, 605–618 (2020).
Steeghs, N. et al. Manufacturing-dependent change in biological activity of the TLR4 agonist GSK1795091 and implications for lipid A analog development. Clin. Transl. Sci. 00, 1–15 (2022).
Bakhribah, H. et al. A phase I study of the Toll-like receptor 5 (TLR5) agonist, entolimod in patients (pts) with advanced cancers. J. Clin. Oncol. 33, 3063 (2015).
Cleveland BioLabs. Cleveland BioLabs announces start of colorectal cancer study of entolimod in Russian Federation [press release]. Available at: http://irdirect.net/prviewer/release_only/id/1718254. Accessed 1 April 2022 (2016).
Le Naour, J., Galluzzi, L., Zitvogel, L., Kroemer, G. & Vacchelli, E. Trial watch: TLR3 agonists in cancer therapy. Oncoimmunology. 9, 1771143 (2020).
Hu, Y. et al. Safety, pharmacokinetics and pharmacodynamics of TQ-A3334, an oral toll-like receptor 7 agonist in healthy individuals. Expert Opin. Investig. Drugs 30, 1–7 (2021).
Curigliano et al. Phase I study of LHC165 ± spartalizumab (PDR001) in patients (pts) with advanced solid tumors. Cancer Res. 81, CT103, https://doi.org/10.1158/1538-7445.AM2021-CT103 (2021).
Symeonides S. et al. Preliminary safety, PK/PD and efficacy results from a first-in-human phase I/IIa clinical trial of BNT411, a systemic Toll-like receptor 7 agonist in patients with solid tumors. J. ImmunoTher. Cancer https://doi.org/10.1136/jitc-2021-SITC2021.525 (2021).
Janku, F. et al. Preclinical characterization and phase I study of an anti–HER2-TLR7 immune-stimulator antibody conjugate in patients with HER2+ malignancies. Caner Immunol. Res. https://doi.org/10.1158/2326-6066.CIR-21-0722 (2022).
Miller, A. T. et al. Gastrointestinal/liver-targeted TLR7 agonist for treatment of colorectal and liver cancers. Cancer Res. 80, Abstract nr 684 (2020).
Cervantes, J. L., Weinerman, B., Basole, C. & Salazar, J. C. TLR8: the forgotten relative revindicated. Cell Mol Immunol 9, 434–438 (2012).
Diab, A. et al. REVEAL: phase 1 dose-escalation study of NKTR-262, a novel TLR7/8 agonist, plus bempegaldesleukin: local innate immune activation and systemic adaptive immune expansion for treating solid tumors. Presented at the 35th annual meeting of the Society for Immunotherapy of Cancer; November 9–14, 2020; Abstract 368 (2020).
Kivimae, S. et al. Harnessing the innate and adaptive immune system to eradicate treated and distant untreated solid tumors. J Immunother. Cancer 5, P275 (2017).
Patel, M. et al. BDB001, a Toll-Like receptor 7 and 8 (TLR7/8) agonist, can be safely administered intravenously and shows clinical responses in advanced solid tumors. J Immunother. Cancer 8, A199 (2020).
Patel, M. R. et al. BDB001, an intravenously administered toll-like receptor 7 and 8 (TLR7/8) agonist, in combination with pembrolizumab in advanced solid tumors: Phase 1 safety and efficacy results. J. Clin. Oncol. 39, 2512 (2021).
Sharma, M. et al. Phase 1/2 study of novel HER2-targeting, TLR7/8 immune-stimulating antibody conjugate (ISAC) BDC-1001 with or without immune checkpoint inhibitor in patients with advanced HER2-expressing solid tumors. J. Immunother. Cancer 8, A224 (2020).
Eigentler, T. et al. A phase I dose-escalation and expansion study of intratumoral CV8102 as single-agent or in combination with anti-PD-1 antibodies in patients with advanced solid tumors. J. Clin. Oncol. 38, 3096 (2020).
Eigentler, T. et al. Intratumorally administered CV8102 in patients with advanced solid tumors: preliminary results from completed dose escalation in study 008. Ann. Oncol. 32, S829–S866 (2021).
Zúñiga, L. A. et al. Intratumoral delivery of TransCon™ TLR7/8 Agonist promotes sustained anti-tumor activity and local immune cell activation while minimizing systemic cytokine induction. Cancer Cell Int 22, 286. https://doi.org/10.1186/s12935-022-02708-6 (2022).
Mirza A. et al. 16 Tumor growth inhibition mediated by a single dose of intratumoral TransCon™ TLR7/8 agonist was associated with activated circulating T and B cells and sustained low levels of systemic cytokines. J. Immunother. Cancer 9, https://doi.org/10.1136/jitc-2021-SITC2021.016 (2021).
Burns, R. P. Jr, Ferbel, B., Tomai, M., Miller, R. & Gaspari, A. A. The imidazoquinolines, imiquimod and R-848, induce functional, but not phenotypic, maturation of human epidermal Langerhans’ cells. Clin. Immunol. 94, 13–23 (2000).
Ascendis Pharma. Ascendis Pharma A/S virtual R&D program update highlights continued development across R&D pipeline [press release]. Available at: https://investors.ascendispharma.com/node/11076/pdf. Accessed 1 April 2022 (2021).
Krieg, A. M. Toll-like receptor 9 (TLR9) agonists in the treatment of cancer. Oncogene 27, 161–167 (2008).
Wittig, B., Schmidt, M., Scheithauer, W. & Schmoll, H. J. MGN1703, an immunomodulator and toll-like receptor 9 (TLR-9) agonist: From bench to bedside. Crit. Rev. Oncol. Hematol. 94, 31–44 (2015).
Vollmer, J. et al. Characterization of three CpG oligodeoxynucleotide classes with distinct immunostimulatory activities. Eur. J. Immunol. 34, 251–262 (2004).
Mologen A. G. MOLOGEN AG announces top line data of pivotal IMPALA study in metastatic colorectal cancer [press release]. 2019. Available at:. Accessed 1 April 2022.
Schmoll, H. J. et al. Maintenance treatment with the immunomodulator MGN1703, a Toll-like receptor 9 (TLR9) agonist, in patients with metastatic colorectal carcinoma and disease control after chemotherapy: a randomised, double-blind, placebo-controlled trial. J. Cancer Res. Clin. Oncol. 140, 1615–1624 (2014).
Babiker, H. M. et al. Tilsotolimod engages the TLR9 pathway to promote antigen presentation and type-I IFN signaling in solid tumors. Cancer Res. 80, Abstract nr CT134 (2020).
Haymaker, C. et al. Final results from ILLUMINATE-204, a phase I/II trial of intratumoral tilsotolimod in combination with ipilimumab in PD-1 inhibitor refractory advanced melanoma. Ann. Oncol. 31, S672–S710 (2020).
Karapetyan, L., Luke, J. J. & Davar, D. Toll-like receptor 9 agonists in cancer. Onco Targets Ther. 13, 10039–10060 (2020).
Sabree, S. et al. Evaluation of the complexity of the immune response to CMP-001, a TLR9 agonist. Cancer Res. 80, Abstract nr 6666 (2020).
Lemke-Miltner, C. D. et al. Antibody opsonization of a TLR9 agonist-containing virus-like particle enhances in situ immunization. J. Immunol. 204, 1386–1394 (2020).
Milhem, M. et al. Intratumoral injection of CMP-001, a toll-like receptor 9 (TLR9) agonist, in combination with pembrolizumab reversed programmed death receptor 1 (PD-1) blockade resistance in advanced melanoma. J. Immunother. Cancer 8, A186–A187 (2020).
Davar, D. et al. Phase II trial of neoadjuvant nivolumab (Nivo) and intra-tumoral (IT) CMP-001 in high-risk resectable melanoma (Neo-C-Nivo): final results. J. Immunother. Cancer 8, A185–A186 (2020).
Negrao, M. et al. TLR9 agonist CMP-001 plus atezolizumab +/− radiation therapy in patients with PD-1 blockade resistant advanced NSCLC. J. Thoracic Onc. 16, S196 (2021).
O’Day, S. et al. Safety and preliminary efficacy of intratumoral cavrotolimod (AST-008), a spherical nucleic acid TLR9 agonist, in combination with pembrolizumab in patients with advanced solid tumors. J. Immunother. Cancer 8, A257–A258 (2020).
Milhem, M. M. et al. Phase 1b/2 study of an intratumoral TLR9 agonist spherical nucleic acid (AST-008) and pembrolizumab: evidence of immune activation. Cancer Res. 80, Abstract nr LB-140 (2020).
Dynavax. Dynavax announces strategic restructuring to focus on its vaccine business [press release]. 2019. Available at: https://investors.dynavax.com/news-releases/news-release-details/dynavax-announces-strategic-restructuring-focus-its-vaccine. Accessed 1 April 2022.
Milhem, M. M. et al. Phase 1b/2, open label, multicenter, study of the combination of SD-101 and pembrolizumab in patients with advanced melanoma who are naïve to anti-PD-1 therapy. J. Clin. Oncol. 37, 9534 (2019).
Dynavax. Overall response rate of 76% in advanced melanoma patients with Dynavax’s SD-101 in combination with KEYTRUDA® (pembrolizumab); data presented today at the 2019 ASCO annual meeting [press release]. Available at: https://investors.dynavax.com/news-releases/news-release-details/overall-response-rate-76-advanced-melanoma-patients-dynavaxs-sd. Accessed 1 April 2022 (2019).
Cohen, E. Intralesional SD-101 in combination with pembrolizumab in anti-PD-1 treatment-naïve head and neck squamous cell carcinoma: results from a multicenter, phase II trial. Clin. Cancer Res. 28, 1157–1166 (2022).
Dynavax. Dynavax presents phase 2 data on SD-101 in combination with KEYTRUDA® (pembrolizumab) for patients with head and neck squamous cell carcinoma at the 2019 ASCO annual meeting [press release]. Available at: https://investors.dynavax.com/news-releases/news-release-details/dynavax-presents-phase-2-data-sd-101-combination-keytrudar. Accessed 1 April 2022 (2019).
Idera Pharmaceuticals. Idera Pharmaceuticals provides update on IMO-2055 clinical development program [press release]. Available at: https://ir.iderapharma.com/news-releases/news-release-details/idera-pharmaceuticals-provides-update-imo-2055-clinical. Accessed 1 April 2022 (2011).
United States Food and Drug Administration. Adaptive designs for clinical trials of drugs and biologics. Guidance for industry. Available at: https://www.fda.gov/media/78495/download. Accessed 1 April 2022 (2019).
Acknowledgements
Medical writing support for this manuscript, under the direction of the authors, was provided by Tiffany DeSimone, PhD, of Ashfield MedComms, an Ashfield Health Company, and Paula Wun, of Sumitomo Pharma Oncology. Claudia Lebedinsky, MD, and Keisuke Matsubara, of Sumitomo Pharma Oncology, provided comments and medical review support. This support was funded by Sumitomo Pharma Oncology.
Author information
Authors and Affiliations
Contributions
C.R., E.G., P.M. and A.N. conceptualized the paper, provided oversight, contributed to data interpretation, and writing the manuscript. C.R., E.G., P.M., S.M., and A.N. contributed to literature search, data acquisition, data interpretation, and manuscript writing. All authors read, reviewed the manuscript, and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
C.R. has served as an advisory board member for Archer, Inivata, Merck Serono, Bristol-Myers Squibb, and Boston Pharmaceuticals; taken part in speaker events for Merck Sharpe & Dohme, Astra Zeneca, and Roche; received non-financial support from GuardantHealth and grants from the Lung Cancer Research Foundation and Pfizer Oncology collaboration, outside of the submitted work. E.G. reports no competing interests. P.M. was a salaried employee of Sumitomo Pharma Oncology at the time this manuscript was written. S.M. is a salaried employee of Sumitomo Pharma Oncology. A.N. received research funding from NCI, EMD Serono, MedImmune, Healios Onc. Nutrition, Atterocor/Millendo, Amplimmune, ARMO BioSciences, Karyopharm Therapeutics, Incyte, Novartis, Regeneron, Merck, Bristol-Myers Squibb, Pfizer, CytomX Therapeutics, Neon Therapeutics, Calithera Biosciences, TopAlliance Biosciences, Eli Lilly, Kymab, PsiOxus, Arcus Biosciences, NeoImmuneTech, ImmuneOncia, and Surface Oncology (paid to institution); was on the advisory boards of CytomX Therapeutics, Novartis, Genome & Company, OncoSec KEYNOTE-695, Kymab, and STCube Pharmaceuticals; and received travel and accommodation expense from ARMO BioSciences. Dr. Naing’s spouse received research funding from the Immune Deficiency Foundation, Jeffery Modell Foundation and chao physician-scientist, and Baxalta (paid to institution) and was on the advisory board of Takeda, CSL, Behring, Horizon and Pharming.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rolfo, C., Giovannetti, E., Martinez, P. et al. Applications and clinical trial landscape using Toll-like receptor agonists to reduce the toll of cancer. npj Precis. Onc. 7, 26 (2023). https://doi.org/10.1038/s41698-023-00364-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41698-023-00364-1
