
Regulation of Cardiac Conduction and Arrhythmias by Ankyrin/Spectrin-Based Macromolecular Complexes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Cardiac Conduction System in Health and Disease
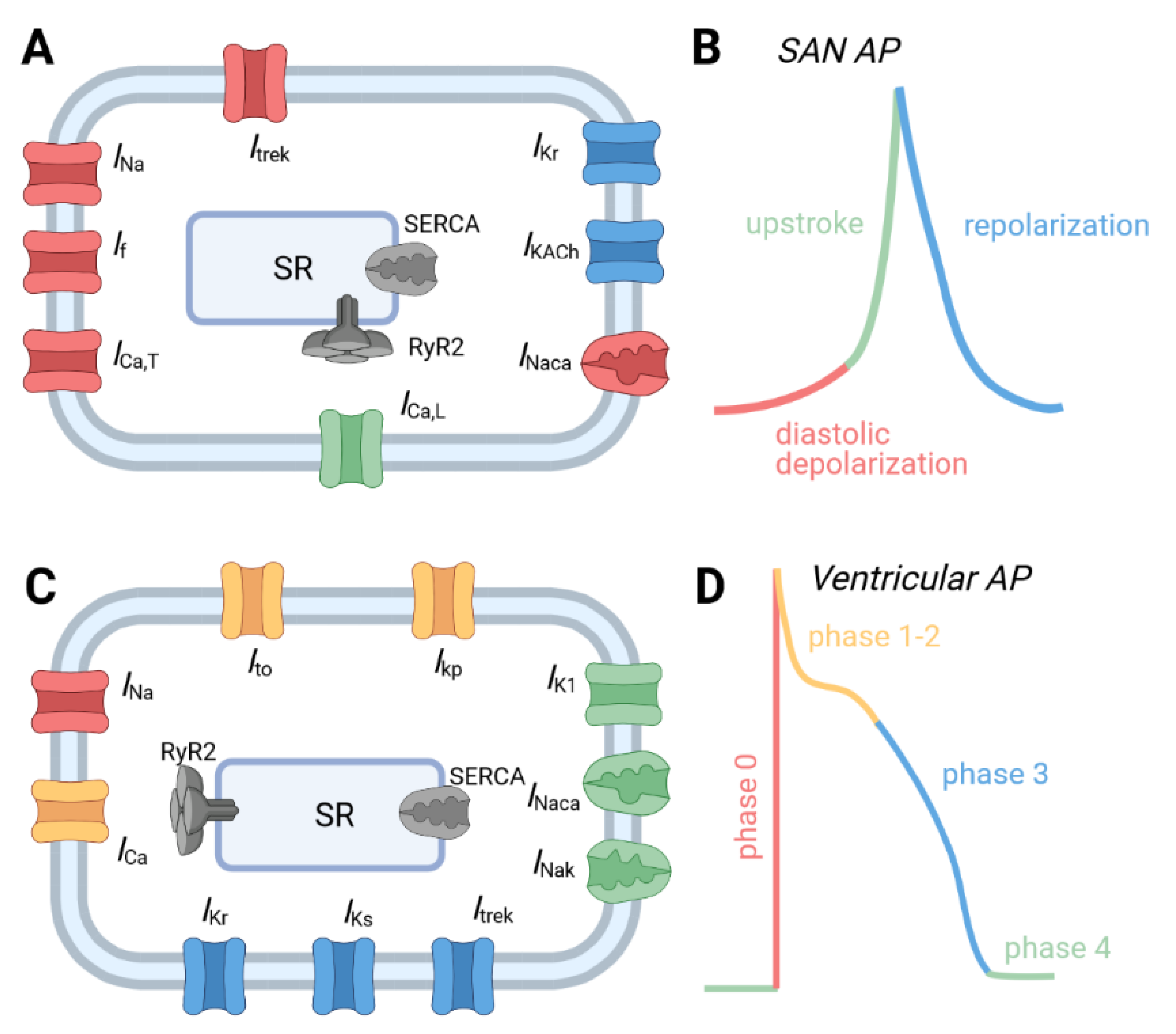
2.1. Normal Physiology of the Cardiac Conduction System
2.2. Cardiac Conduction System Dysfunction and Disease
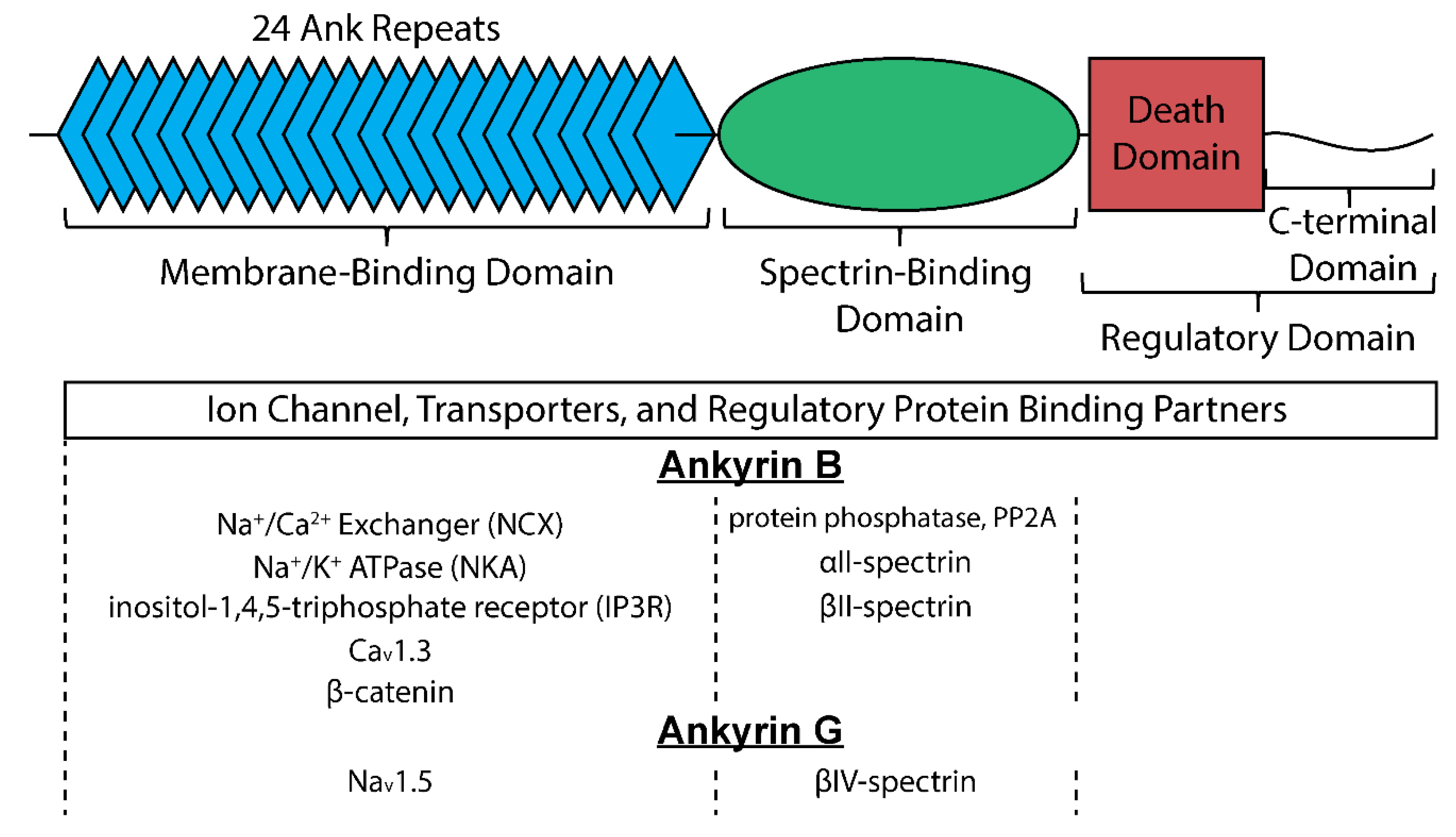
3. Ankyrins in the Cardiac Conduction System
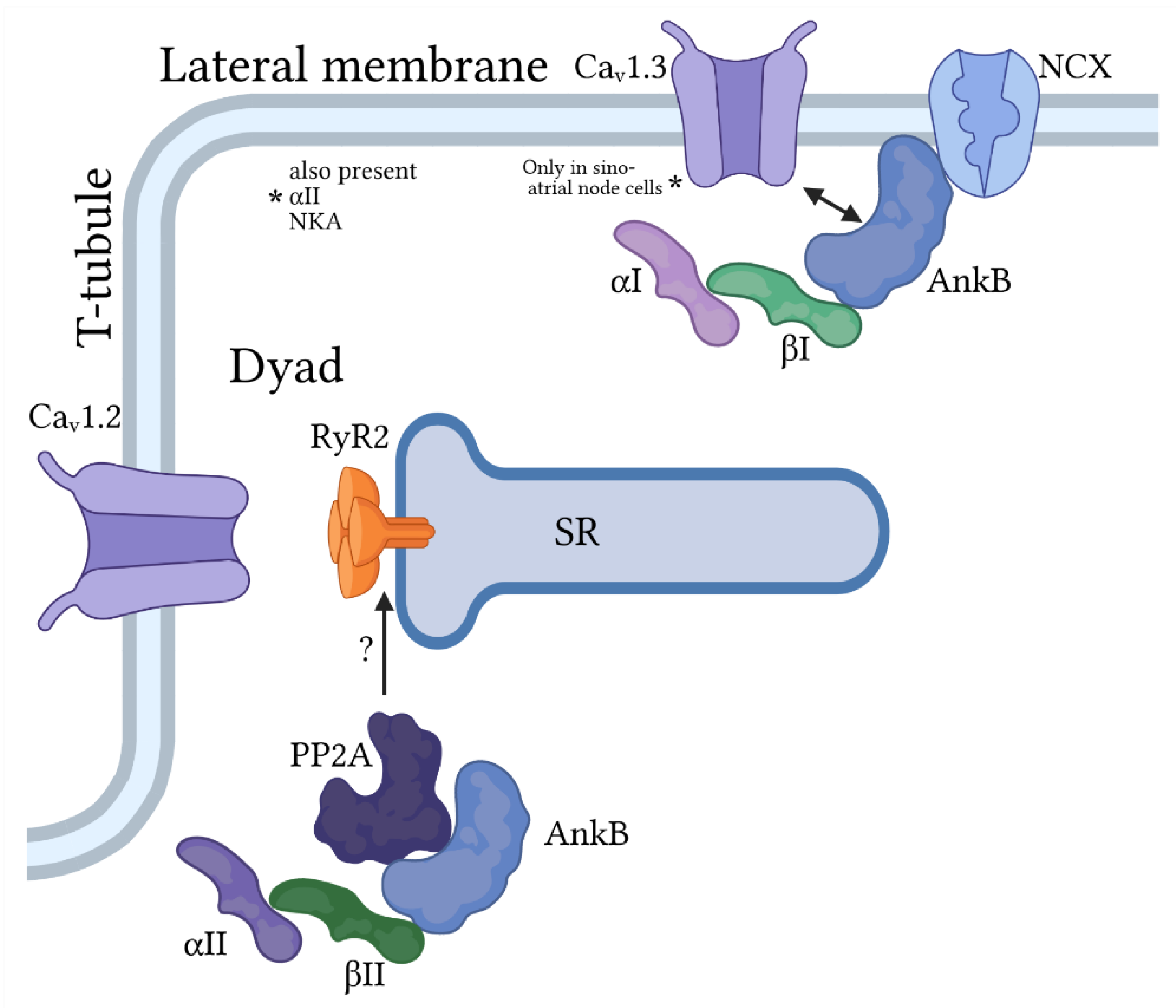
3.1. Ankyrin-B
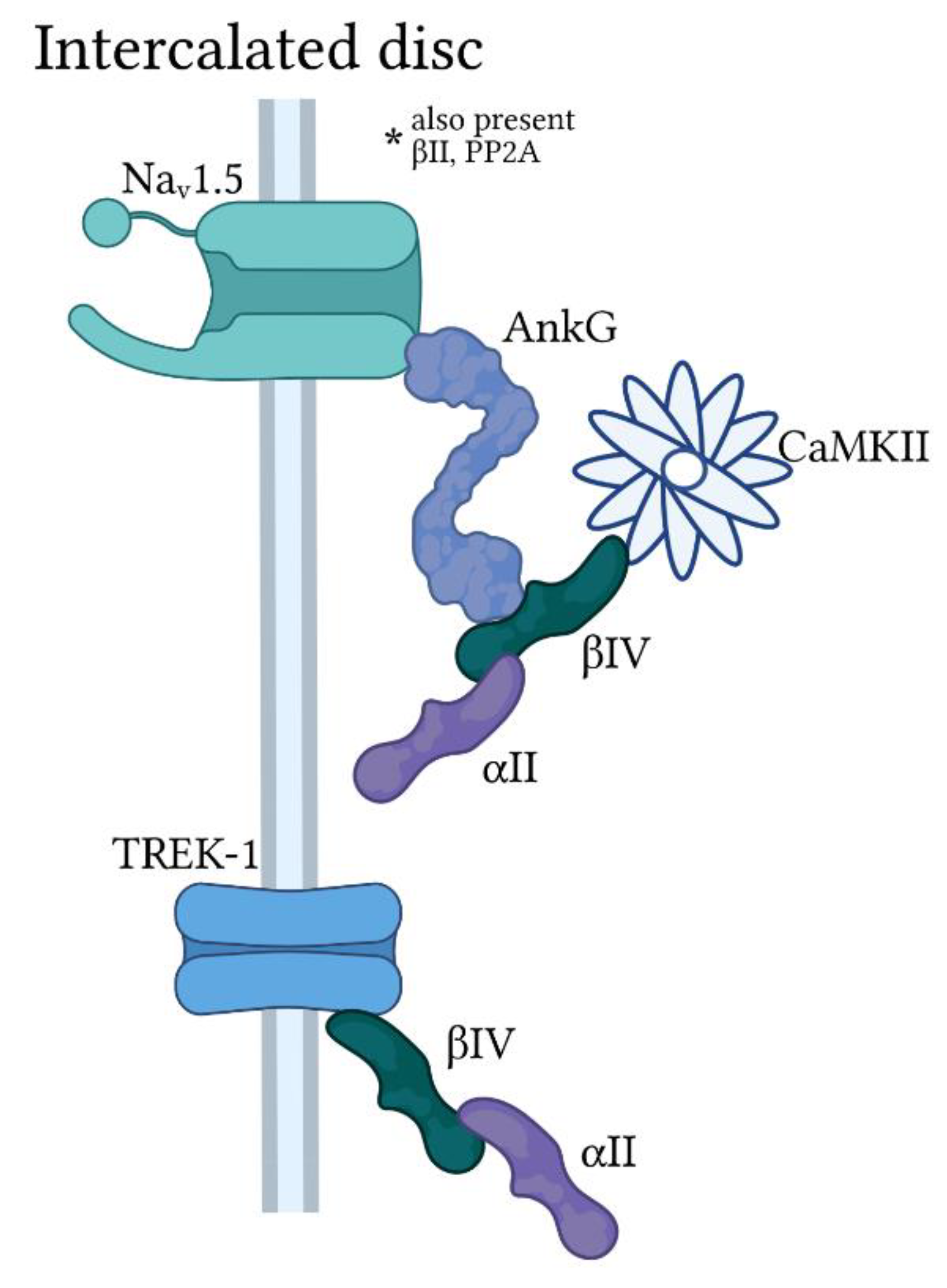
3.2. Ankyrin-G
4. Spectrins in the Cardiac Conduction System
4.1. αII-Spectrin
4.2. βII-Spectrin
4.3. βIV-Spectrin
5. Conclusions
Funding
Conflicts of Interest
References
- Raatikainen, M.J.P.; Arnar, D.O.; Merkely, B.; Nielsen, J.C.; Hindricks, G.; Heidbuchel, H.; Camm, J. A decade of information on the use of cardiac implantable electronic devices and interventional electrophysiological procedures in the European Society of Cardiology Countries: 2017 report from the European Heart Rhythm Association. Europace 2017, 19, ii1–ii90. [Google Scholar] [CrossRef]
- Dobrzynski, H.; Anderson, R.H.; Atkinson, A.; Borbas, Z.; D’Souza, A.; Fraser, J.F.; Inada, S.; Logantha, S.J.; Monfredi, O.; Morris, G.M.; et al. Structure, function and clinical relevance of the cardiac conduction system, including the atrioventricular ring and outflow tract tissues. Pharmacol. Ther. 2013, 139, 260–288. [Google Scholar] [CrossRef] [PubMed]
- Mesirca, P.; Fedorov, V.V.; Hund, T.J.; Torrente, A.G.; Bidaud, I.; Mohler, P.J.; Mangoni, M.E. Pharmacologic approach to sinoatrial node dysfunction. Annu. Rev. Pharmacol. Toxicol. 2021, 61, 757–778. [Google Scholar] [CrossRef]
- He, B.J.; Boyden, P.; Scheinman, M. Ventricular arrhythmias involving the His-Purkinje system in the structurally abnormal heart. Pacing Clin. Electrophysiol. 2018, 41, 1051–1059. [Google Scholar] [CrossRef]
- Park, D.S.; Fishman, G.I. The cardiac conduction system. Circulation 2011, 123, 904–915. [Google Scholar] [CrossRef] [Green Version]
- Mohler, P.J.; Wehrens, X.H. Mechanisms of human arrhythmia syndromes: Abnormal cardiac macromolecular interactions. Physiology 2007, 22, 342–350. [Google Scholar] [CrossRef] [Green Version]
- Bennett, V.; Baines, A.J. Spectrin and ankyrin-based pathways: Metazoan inventions for integrating cells into tissues. Physiol. Rev. 2001, 81, 1353–1392. [Google Scholar] [CrossRef] [Green Version]
- Unudurthi, S.D.; Greer-Short, A.; Patel, N.; Nassal, D.; Hund, T.J. Spectrin-based pathways underlying electrical and mechanical dysfunction in cardiac disease. Expert Rev. Cardiovasc. Ther. 2018, 16, 59–65. [Google Scholar] [CrossRef]
- Grimes, K.M.; Prasad, V.; McNamara, J.W. Supporting the heart: Functions of the cardiomyocyte’s non-sarcomeric cytoskeleton. J. Mol. Cell. Cardiol. 2019, 131, 187–196. [Google Scholar] [CrossRef]
- Abriel, H.; Rougier, J.S.; Jalife, J. Ion channel macromolecular complexes in cardiomyocytes: Roles in sudden cardiac death. Circ. Res. 2015, 116, 1971–1988. [Google Scholar] [CrossRef] [Green Version]
- Benson, D.W.; Wang, D.W.; Dyment, M.; Knilans, T.K.; Fish, F.A.; Strieper, M.J.; Rhodes, T.H.; George, A.L., Jr. Congenital sick sinus syndrome caused by recessive mutations in the cardiac sodium channel gene (SCN5A). J. Clin. Invest. 2003, 112, 1019–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangrum, J.M.; DiMarco, J.P. The evaluation and management of bradycardia. N. Engl. J. Med. 2000, 342, 703–709. [Google Scholar] [CrossRef] [PubMed]
- Bennett, V.; Chen, L. Ankyrins and cellular targeting of diverse membrane proteins to physiological sites. Curr. Opin. Cell. Biol. 2001, 13, 61–67. [Google Scholar] [CrossRef]
- Cunha, S.R.; Mohler, P.J. Cardiac ankyrins: Essential components for development and maintenance of excitable membrane domains in heart. Cardiovasc. Res. 2006, 71, 22–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashemi, S.M.; Hund, T.J.; Mohler, P.J. Cardiac ankyrins in health and disease. J. Mol. Cell. Cardiol. 2009, 47, 203–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaely, P.; Tomchick, D.R.; Machius, M.; Anderson, R.G. Crystal structure of a 12 ANK repeat stack from human ankyrinR. EMBO J. 2002, 21, 6387–6396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curran, J.; Mohler, P.J. Coordinating electrical activity of the heart: Ankyrin polypeptides in human cardiac disease. Expert Opin. Ther. Targets. 2011, 15, 789–801. [Google Scholar] [CrossRef] [Green Version]
- Makara, M.A.; Curran, J.; Little, S.C.; Musa, H.; Polina, I.; Smith, S.A.; Wright, P.J.; Unudurthi, S.D.; Snyder, J.; Bennett, V.; et al. Ankyrin-G coordinates intercalated disc signaling platform to regulate cardiac excitability in vivo. Circ. Res. 2014, 115, 929–938. [Google Scholar] [CrossRef] [Green Version]
- Bennett, V.; Stenbuck, P.J. Identification and partial purification of ankyrin, the high affinity membrane attachment site for human erythrocyte spectrin. J. Biol. Chem. 1979, 254, 2533–2541. [Google Scholar] [CrossRef]
- Hall, T.G.; Bennett, V. Regulatory domains of erythrocyte ankyrin. J. Biol. Chem. 1987, 262, 10537–10545. [Google Scholar] [CrossRef]
- Davis, L.H.; Davis, J.Q.; Bennett, V. Ankyrin regulation: An alternatively spliced segment of the regulatory domain functions as an intramolecular modulator. J. Biol. Chem. 1992, 267, 18966–18972. [Google Scholar] [CrossRef]
- Mohler, P.J.; Yoon, W.; Bennett, V. Ankyrin-B targets beta2-spectrin to an intracellular compartment in neonatal cardiomyocytes. J. Biol. Chem. 2004, 279, 40185–40193. [Google Scholar] [CrossRef] [Green Version]
- Smith, S.; Curran, J.; Hund, T.J.; Mohler, P.J. Defects in cytoskeletal signaling pathways, arrhythmia, and sudden cardiac death. Front. Physiol. 2012, 3, 122. [Google Scholar] [CrossRef] [Green Version]
- Hund, T.J.; Wright, P.J.; Dun, W.; Snyder, J.S.; Boyden, P.A.; Mohler, P.J. Regulation of the ankyrin-B-based targeting pathway following myocardial infarction. Cardiovasc. Res. 2009, 81, 742–749. [Google Scholar] [CrossRef]
- Kashef, F.; Li, J.; Wright, P.; Snyder, J.; Suliman, F.; Kilic, A.; Higgins, R.S.; Anderson, M.E.; Binkley, P.F.; Hund, T.J.; et al. Ankyrin-B protein in heart failure: Identification of a new component of metazoan cardioprotection. J. Biol. Chem. 2012, 287, 30268–30281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dun, W.; Lowe, J.S.; Wright, P.; Hund, T.J.; Mohler, P.J.; Boyden, P.A. Ankyrin-G participates in INa remodeling in myocytes from the border zones of infarcted canine heart. PLoS ONE 2013, 8, e78087. [Google Scholar] [CrossRef] [PubMed]
- Makara, M.A.; Curran, J.; Lubbers, E.R.; Murphy, N.P.; Little, S.C.; Musa, H.; Smith, S.A.; Unudurthi, S.D.; Rajaram, M.V.S.; Janssen, P.M.L.; et al. Novel mechanistic roles for Ankyrin-G in cardiac remodeling and heart failure. JACC Basic Transl. Sci. 2018, 3, 675–689. [Google Scholar] [CrossRef]
- Sucharski, H.C.; Dudley, E.K.; Keith, C.B.R.; El Refaey, M.; Koenig, S.N.; Mohler, P.J. Mechanisms and alterations of cardiac ion channels leading to disease: Role of Ankyrin-B in cardiac function. Biomolecules 2020, 10, 211. [Google Scholar] [CrossRef] [Green Version]
- Cunha, S.R.; Mohler, P.J. Obscurin targets ankyrin-B and protein phosphatase 2A to the cardiac M-line. J. Biol. Chem. 2008, 283, 31968–31980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhasin, N.; Cunha, S.R.; Mduannayake, M.; Gigena, M.S.; Rogers, T.B.; Mohler, P.J. Molecular basis for PP2A regulatory subunit B56 alpha targeting in cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H109–H119. [Google Scholar] [CrossRef] [Green Version]
- Le Scouarnec, S.; Bhasin, N.; Vieyres, C.; Hund, T.J.; Cunha, S.R.; Koval, O.; Marionneau, C.; Chen, B.; Wu, Y.; Demolombe, S.; et al. Dysfunction in ankyrin-B-dependent ion channel and transporter targeting causes human sinus node disease. Proc. Natl. Acad. Sci. USA 2008, 105, 15617–15622. [Google Scholar] [CrossRef] [Green Version]
- Mohler, P.J.; Schott, J.J.; Gramolini, A.O.; Dilly, K.W.; Guatimosim, S.; duBell, W.H.; Song, L.S.; Haurogne, K.; Kyndt, F.; Ali, M.E.; et al. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature 2003, 421, 634–639. [Google Scholar] [CrossRef]
- Mohler, P.J.; Le Scouarnec, S.; Denjoy, I.; Lowe, J.S.; Guicheney, P.; Caron, L.; Driskell, I.M.; Schott, J.J.; Norris, K.; Leenhardt, A.; et al. Defining the cellular phenotype of “ankyrin-B syndrome” variants: Human ANK2 variants associated with clinical phenotypes display a spectrum of activities in cardiomyocytes. Circulation 2007, 115, 432–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunha, S.R.; Hund, T.J.; Hashemi, S.; Voigt, N.; Li, N.; Wright, P.; Koval, O.; Li, J.; Gudmundsson, H.; Gumina, R.J.; et al. Defects in ankyrin-based membrane protein targeting pathways underlie atrial fibrillation. Circulation 2011, 124, 1212–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, R.M.; Glynn, P.; Hashemi, S.; Zarei, K.; Mitchell, C.C.; Anderson, M.E.; Mohler, P.J.; Hund, T.J. Atrial fibrillation and sinus node dysfunction in human ankyrin-B syndrome: A computational analysis. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1253–H1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coban-Akdemir, Z.H.; Charng, W.L.; Azamian, M.; Paine, I.S.; Punetha, J.; Grochowski, C.M.; Gambin, T.; Valdes, S.O.; Cannon, B.; Zapata, G.; et al. Wolff-Parkinson-White syndrome: De novo variants and evidence for mutational burden in genes associated with atrial fibrillation. Am. J. Med. Genet. A 2020, 182, 1387–1399. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.D.; Murphy, N.P.; Hamilton, R.M.; Lubbers, E.R.; James, C.A.; Kline, C.F.; Gollob, M.H.; Krahn, A.D.; Sturm, A.C.; Musa, H.; et al. Ankyrin-B dysfunction predisposes to arrhythmogenic cardiomyopathy and is amenable to therapy. J. Clin. Invest. 2019, 129, 3171–3184. [Google Scholar] [CrossRef]
- Skogestad, J.; Aronsen, J.M.; Tovsrud, N.; Wanichawan, P.; Hougen, K.; Stokke, M.K.; Carlson, C.R.; Sjaastad, I.; Sejersted, O.M.; Swift, F. Coupling of the Na+/K+-ATPase to Ankyrin B controls Na+/Ca2+ exchanger activity in cardiomyocytes. Cardiovasc. Res. 2020, 116, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Camors, E.; Mohler, P.J.; Bers, D.M.; Despa, S. Ankyrin-B reduction enhances Ca spark-mediated SR Ca release promoting cardiac myocyte arrhythmic activity. J. Mol. Cell. Cardiol. 2012, 52, 1240–1248. [Google Scholar] [CrossRef] [Green Version]
- Chu, L.; Greenstein, J.L.; Winslow, R.L. Na(+) microdomains and sparks: Role in cardiac excitation-contraction coupling and arrhythmias in ankyrin-B deficiency. J. Mol. Cell. Cardiol. 2019, 128, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Lambert, S.; Malen, P.L.; Carpenter, S.; Boland, L.M.; Bennett, V. AnkyrinG is required for clustering of voltage-gated Na channels at axon initial segments and for normal action potential firing. J. Cell. Biol. 1998, 143, 1295–1304. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, S.M.; Bennett, V. Ankyrin-G coordinates assembly of the spectrin-based membrane skeleton, voltage-gated sodium channels, and L1 CAMs at Purkinje neuron initial segments. J. Cell. Biol. 2001, 155, 739–746. [Google Scholar] [CrossRef]
- Mohler, P.J.; Rivolta, I.; Napolitano, C.; LeMaillet, G.; Lambert, S.; Priori, S.G.; Bennett, V. Nav1.5 E1053K mutation causing Brugada syndrome blocks binding to ankyrin-G and expression of Nav1.5 on the surface of cardiomyocytes. Proc. Natl. Acad. Sci. USA 2004, 101, 17533–17538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowe, J.S.; Palygin, O.; Bhasin, N.; Hund, T.J.; Boyden, P.A.; Shibata, E.; Anderson, M.E.; Mohler, P.J. Voltage-gated Nav channel targeting in the heart requires an ankyrin-G dependent cellular pathway. J. Cell. Biol. 2008, 180, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Abriel, H. Roles and regulation of the cardiac sodium channel Na v 1.5: Recent insights from experimental studies. Cardiovasc. Res. 2007, 76, 381–389. [Google Scholar] [CrossRef] [Green Version]
- Kapplinger, J.D.; Tester, D.J.; Alders, M.; Benito, B.; Berthet, M.; Brugada, J.; Brugada, P.; Fressart, V.; Guerchicoff, A.; Harris-Kerr, C.; et al. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm 2010, 7, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Calloe, K.; Refaat, M.M.; Grubb, S.; Wojciak, J.; Campagna, J.; Thomsen, N.M.; Nussbaum, R.L.; Scheinman, M.M.; Schmitt, N. Characterization and mechanisms of action of novel NaV1.5 channel mutations associated with Brugada syndrome. Circ. Arrhythm. Electrophysiol. 2013, 6, 177–184. [Google Scholar] [CrossRef] [Green Version]
- Ellinor, P.T.; Nam, E.G.; Shea, M.A.; Milan, D.J.; Ruskin, J.N.; MacRae, C.A. Cardiac sodium channel mutation in atrial fibrillation. Heart Rhythm 2008, 5, 99–105. [Google Scholar] [CrossRef]
- Olesen, M.S.; Jespersen, T.; Nielsen, J.B.; Liang, B.; Moller, D.V.; Hedley, P.; Christiansen, M.; Varro, A.; Olesen, S.P.; Haunso, S.; et al. Mutations in sodium channel beta-subunit SCN3B are associated with early-onset lone atrial fibrillation. Cardiovasc. Res. 2011, 89, 786–793. [Google Scholar] [CrossRef]
- Watanabe, H.; Darbar, D.; Kaiser, D.W.; Jiramongkolchai, K.; Chopra, S.; Donahue, B.S.; Kannankeril, P.J.; Roden, D.M. Mutations in sodium channel beta1- and beta2-subunits associated with atrial fibrillation. Circ. Arrhythm. Electrophysiol. 2009, 2, 268–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivaud, M.R.; Delmar, M.; Remme, C.A. Heritable arrhythmia syndromes associated with abnormal cardiac sodium channel function: Ionic and non-ionic mechanisms. Cardiovasc. Res. 2020, 116, 1557–1570. [Google Scholar] [CrossRef] [Green Version]
- Cerrone, M.; Delmar, M. Desmosomes and the sodium channel complex: Implications for arrhythmogenic cardiomyopathy and Brugada syndrome. Trends Cardiovasc. Med. 2014, 24, 184–190. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Lacas-Gervais, S.; Morest, D.K.; Solimena, M.; Rasband, M.N. BetaIV spectrins are essential for membrane stability and the molecular organization of nodes of Ranvier. J. Neurosci. 2004, 24, 7230–7240. [Google Scholar] [CrossRef] [PubMed]
- Lacas-Gervais, S.; Guo, J.; Strenzke, N.; Scarfone, E.; Kolpe, M.; Jahkel, M.; De Camilli, P.; Moser, T.; Rasband, M.N.; Solimena, M. BetaIVSigma1 spectrin stabilizes the nodes of Ranvier and axon initial segments. J. Cell. Biol. 2004, 166, 983–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hund, T.J.; Koval, O.M.; Li, J.; Wright, P.J.; Qian, L.; Snyder, J.S.; Gudmundsson, H.; Kline, C.F.; Davidson, N.P.; Cardona, N.; et al. A betaIV spectrin/CaMKII signaling complex is essential for membrane excitability in mice. J. Clin. Invest. 2010, 120, 3508–3519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, V.; Stenbuck, P.J. The membrane attachment protein for spectrin is associated with band 3 in human erythrocyte membranes. Nature 1979, 280, 468–473. [Google Scholar] [CrossRef] [PubMed]
- Hund, T.J.; Snyder, J.S.; Wu, X.; Glynn, P.; Koval, O.M.; Onal, B.; Leymaster, N.D.; Unudurthi, S.D.; Curran, J.; Camardo, C.; et al. betaIV-Spectrin regulates TREK-1 membrane targeting in the heart. Cardiovasc. Res. 2014, 102, 166–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kline, C.F.; Wright, P.J.; Koval, O.M.; Zmuda, E.J.; Johnson, B.L.; Anderson, M.E.; Hai, T.; Hund, T.J.; Mohler, P.J. betaIV-Spectrin and CaMKII facilitate Kir6.2 regulation in pancreatic beta cells. Proc. Natl. Acad. Sci. USA 2013, 110, 17576–17581. [Google Scholar] [CrossRef] [Green Version]
- Komada, M.; Soriano, P. BetaIV-spectrin regulates sodium channel clustering through ankyrin-G at axon initial segments and nodes of Ranvier. J. Cell. Biol. 2002, 156, 337–348. [Google Scholar] [CrossRef] [Green Version]
- Sikorski, A.F.; Sangerman, J.; Goodman, S.R.; Critz, S.D. Spectrin (betaSpIIsigma1) is an essential component of synaptic transmission. Brain Res. 2000, 852, 161–166. [Google Scholar] [CrossRef]
- Patel, N.J.; Nassal, D.M.; Greer-Short, A.D.; Unudurthi, S.D.; Scandling, B.W.; Gratz, D.; Xu, X.; Kalyanasundaram, A.; Fedorov, V.V.; Accornero, F.; et al. betaIV-Spectrin/STAT3 complex regulates fibroblast phenotype, fibrosis, and cardiac function. JCI Insight 2019, 4, e131046. [Google Scholar] [CrossRef]
- Unudurthi, S.D.; Nassal, D.; Greer-Short, A.; Patel, N.; Howard, T.; Xu, X.; Onal, B.; Satroplus, T.; Hong, D.; Lane, C.; et al. betaIV-Spectrin regulates STAT3 targeting to tune cardiac response to pressure overload. J. Clin. Investig. 2018, 128, 5561–5572. [Google Scholar] [CrossRef] [PubMed]
- Lambert, M.W. Nuclear alpha spectrin: Critical roles in DNA interstrand cross-link repair and genomic stability. Exp. Biol. Med. 2016, 241, 1621–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syrbe, S.; Harms, F.L.; Parrini, E.; Montomoli, M.; Mutze, U.; Helbig, K.L.; Polster, T.; Albrecht, B.; Bernbeck, U.; van Binsbergen, E.; et al. Delineating SPTAN1 associated phenotypes: From isolated epilepsy to encephalopathy with progressive brain atrophy. Brain 2017, 140, 2322–2336. [Google Scholar] [CrossRef] [PubMed]
- Knierim, E.; Gill, E.; Seifert, F.; Morales-Gonzalez, S.; Unudurthi, S.D.; Hund, T.J.; Stenzel, W.; Schuelke, M. A recessive mutation in beta-IV-spectrin (SPTBN4) associates with congenital myopathy, neuropathy, and central deafness. Hum. Genet. 2017, 136, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.C.; Ortiz-Gonzalez, X.R.; Yum, S.W.; Gill, S.M.; White, A.; Kelter, E.; Seaver, L.H.; Lee, S.; Wiley, G.; Kelter, E.; et al. bIV spectrinopathies cause profound intellectual disability, congenital hypotonia, and motor axonal neuropathy. Am. J. Hum. Genet. 2018, 102, 1158–1168. [Google Scholar] [CrossRef] [Green Version]
- Hausler, M.G.; Begemann, M.; Lidov, H.G.; Kurth, I.; Darras, B.T.; Elbracht, M. A novel homozygous splice-site mutation in the SPTBN4 gene causes axonal neuropathy without intellectual disability. Eur. J. Med. Genet. 2020, 63, 103826. [Google Scholar] [CrossRef]
- Smith, S.A.; Sturm, A.C.; Curran, J.; Kline, C.F.; Little, S.C.; Bonilla, I.M.; Long, V.P.; Makara, M.; Polina, I.; Hughes, L.D.; et al. Dysfunction in the betaII spectrin-dependent cytoskeleton underlies human arrhythmia. Circulation 2015, 131, 695–708. [Google Scholar] [CrossRef] [Green Version]
- Baines, A.J.; Pinder, J.C. The spectrin-associated cytoskeleton in mammalian heart. Front. Biosci. 2005, 10, 3020–3033. [Google Scholar] [CrossRef] [Green Version]
- Stankewich, M.C.; Cianci, C.D.; Stabach, P.R.; Ji, L.; Nath, A.; Morrow, J.S. Cell organization, growth, and neural and cardiac development require alphaII-spectrin. J. Cell. Sci. 2011, 124, 3956–3966. [Google Scholar] [CrossRef] [Green Version]
- Lubbers, E.R.; Murphy, N.P.; Musa, H.; Huang, C.Y.; Gupta, R.; Price, M.V.; Han, M.; Daoud, E.; Gratz, D.; El Refaey, M.; et al. Defining new mechanistic roles for alphaII spectrin in cardiac function. J. Biol. Chem. 2019, 294, 9576–9591. [Google Scholar] [CrossRef]
- Benz, P.M.; Merkel, C.J.; Offner, K.; Abesser, M.; Ullrich, M.; Fischer, T.; Bayer, B.; Wagner, H.; Gambaryan, S.; Ursitti, J.A.; et al. Mena/VASP and alphaII-Spectrin complexes regulate cytoplasmic actin networks in cardiomyocytes and protect from conduction abnormalities and dilated cardiomyopathy. Cell. Commun. Signal. 2013, 11, 56. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Katuri, V.; Dillner, A.; Mishra, B.; Deng, C.X.; Mishra, L. Disruption of transforming growth factor-beta signaling in ELF beta-spectrin-deficient mice. Science 2003, 299, 574–577. [Google Scholar] [CrossRef] [Green Version]
- Berghs, S.; Aggujaro, D.; Dirkx, R., Jr.; Maksimova, E.; Stabach, P.; Hermel, J.M.; Zhang, J.P.; Philbrick, W.; Slepnev, V.; Ort, T.; et al. betaIV spectrin, a new spectrin localized at axon initial segments and nodes of ranvier in the central and peripheral nervous system. J. Cell. Biol. 2000, 151, 985–1002. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.H.; Stevens, S.R.; Teliska, L.H.; Stankewich, M.; Mohler, P.J.; Hund, T.J.; Rasband, M.N. Nodal beta spectrins are required to maintain Na(+) channel clustering and axon integrity. eLife 2020, 9, e52378. [Google Scholar] [CrossRef]
- Rasband, M.N. The axon initial segment and the maintenance of neuronal polarity. Nat. Rev. Neurosci. 2010, 11, 552–562. [Google Scholar] [CrossRef] [PubMed]
- Glynn, P.; Musa, H.; Wu, X.; Unudurthi, S.D.; Little, S.; Qian, L.; Wright, P.J.; Radwanski, P.B.; Gyorke, S.; Mohler, P.J.; et al. Voltage-gated sodium channel phosphorylation at Ser571 regulates late current, arrhythmia, and cardiac function in vivo. Circulation 2015, 132, 567–577. [Google Scholar] [CrossRef] [Green Version]
- Greer-Short, A.; Musa, H.; Alsina, K.M.; Ni, L.; Word, T.A.; Reynolds, J.O.; Gratz, D.; Lane, C.; El-Refaey, M.; Unudurthi, S.; et al. Calmodulin kinase II regulates atrial myocyte late sodium current, calcium handling, and atrial arrhythmia. Heart Rhythm 2020, 17, 503–511. [Google Scholar] [CrossRef]
- Honore, E. The neuronal background K2P channels: Focus on TREK1. Nat. Rev. Neurosci. 2007, 8, 251–261. [Google Scholar] [CrossRef]
- Unudurthi, S.D.; Wu, X.; Qian, L.; Amari, F.; Onal, B.; Li, N.; Makara, M.A.; Smith, S.A.; Snyder, J.; Fedorov, V.V.; et al. Two-pore K+ channel TREK-1 regulates sinoatrial node membrane excitability. J. Am. Heart Assoc. 2016, 5, e002865. [Google Scholar] [CrossRef] [Green Version]
- Lugenbiel, P.; Wenz, F.; Syren, P.; Geschwill, P.; Govorov, K.; Seyler, C.; Frank, D.; Schweizer, P.A.; Franke, J.; Weis, T.; et al. TREK-1 (K2P2.1) K+ channels are suppressed in patients with atrial fibrillation and heart failure and provide therapeutic targets for rhythm control. Basic Res. Cardiol. 2017, 112, 8. [Google Scholar] [CrossRef]
- Decher, N.; Ortiz-Bonnin, B.; Friedrich, C.; Schewe, M.; Kiper, A.K.; Rinne, S.; Seemann, G.; Peyronnet, R.; Zumhagen, S.; Bustos, D.; et al. Sodium permeable and "hypersensitive" TREK-1 channels cause ventricular tachycardia. EMBO Mol. Med. 2017, 9, 403–414. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, C.; Wiedmann, F.; Kallenberger, S.M.; Ratte, A.; Schulte, J.S.; Scholz, B.; Muller, F.U.; Voigt, N.; Zafeiriou, M.P.; Ehrlich, J.R.; et al. Stretch-activated two-pore-domain (K2P) potassium channels in the heart: Focus on atrial fibrillation and heart failure. Prog. Biophys. Mol. Biol. 2017, 130, 233–243. [Google Scholar] [CrossRef]
- Rinne, S.; Ortiz-Bonnin, B.; Stallmeyer, B.; Kiper, A.K.; Fortmuller, L.; Schindler, R.F.R.; Herbort-Brand, U.; Kabir, N.S.; Dittmann, S.; Friedrich, C.; et al. POPDC2 a novel susceptibility gene for conduction disorders. J. Mol. Cell. Cardiol. 2020, 145, 74–83. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nassal, D.; Yu, J.; Min, D.; Lane, C.; Shaheen, R.; Gratz, D.; Hund, T.J. Regulation of Cardiac Conduction and Arrhythmias by Ankyrin/Spectrin-Based Macromolecular Complexes. J. Cardiovasc. Dev. Dis. 2021, 8, 48. https://doi.org/10.3390/jcdd8050048
Nassal D, Yu J, Min D, Lane C, Shaheen R, Gratz D, Hund TJ. Regulation of Cardiac Conduction and Arrhythmias by Ankyrin/Spectrin-Based Macromolecular Complexes. Journal of Cardiovascular Development and Disease. 2021; 8(5):48. https://doi.org/10.3390/jcdd8050048
Chicago/Turabian StyleNassal, Drew, Jane Yu, Dennison Min, Cemantha Lane, Rebecca Shaheen, Daniel Gratz, and Thomas J. Hund. 2021. "Regulation of Cardiac Conduction and Arrhythmias by Ankyrin/Spectrin-Based Macromolecular Complexes" Journal of Cardiovascular Development and Disease 8, no. 5: 48. https://doi.org/10.3390/jcdd8050048