
Comparison of the Correlations of Microbial Community and Volatile Compounds between Pit-Mud and Fermented Grains of Compound-Flavor Baijiu
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Determination of Physicochemical Indexes of FG and PM
2.3. Plate Counting of Culturable Microorganisms in FG and PM
2.4. High Throughput Sequencing
2.5. Analysis of Volatile Components by HS-SPME-GC-MS
2.5.1. Analysis of Volatile Components in FG
2.5.2. Analysis of Volatile Components in PM
2.6. Data Processing and Statistic Analysis
3. Results
3.1. Analysis of Physicochemical Indexes of FG and PM
3.2. Plate Counting of Culturable Microorganisms in FG and PM
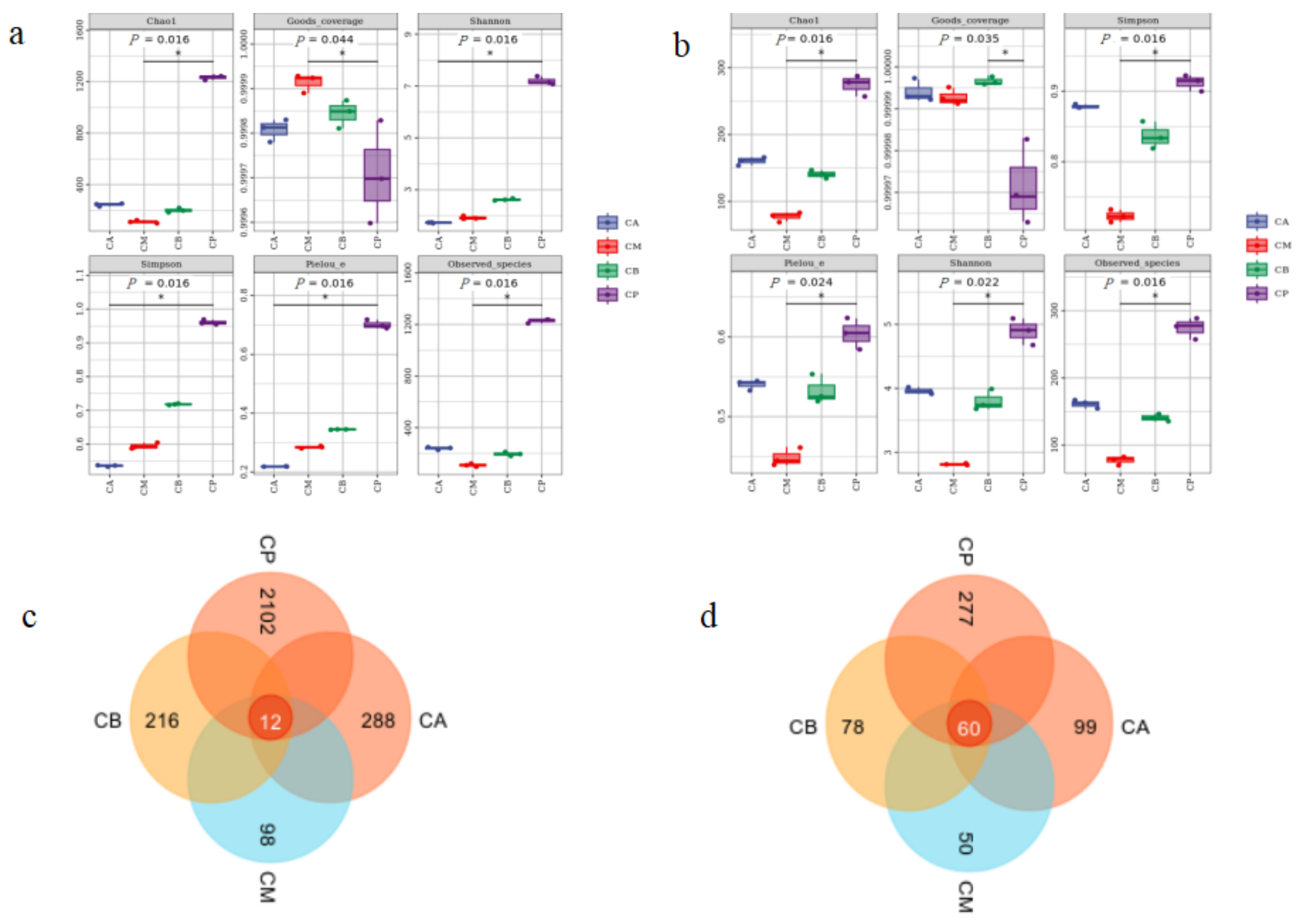
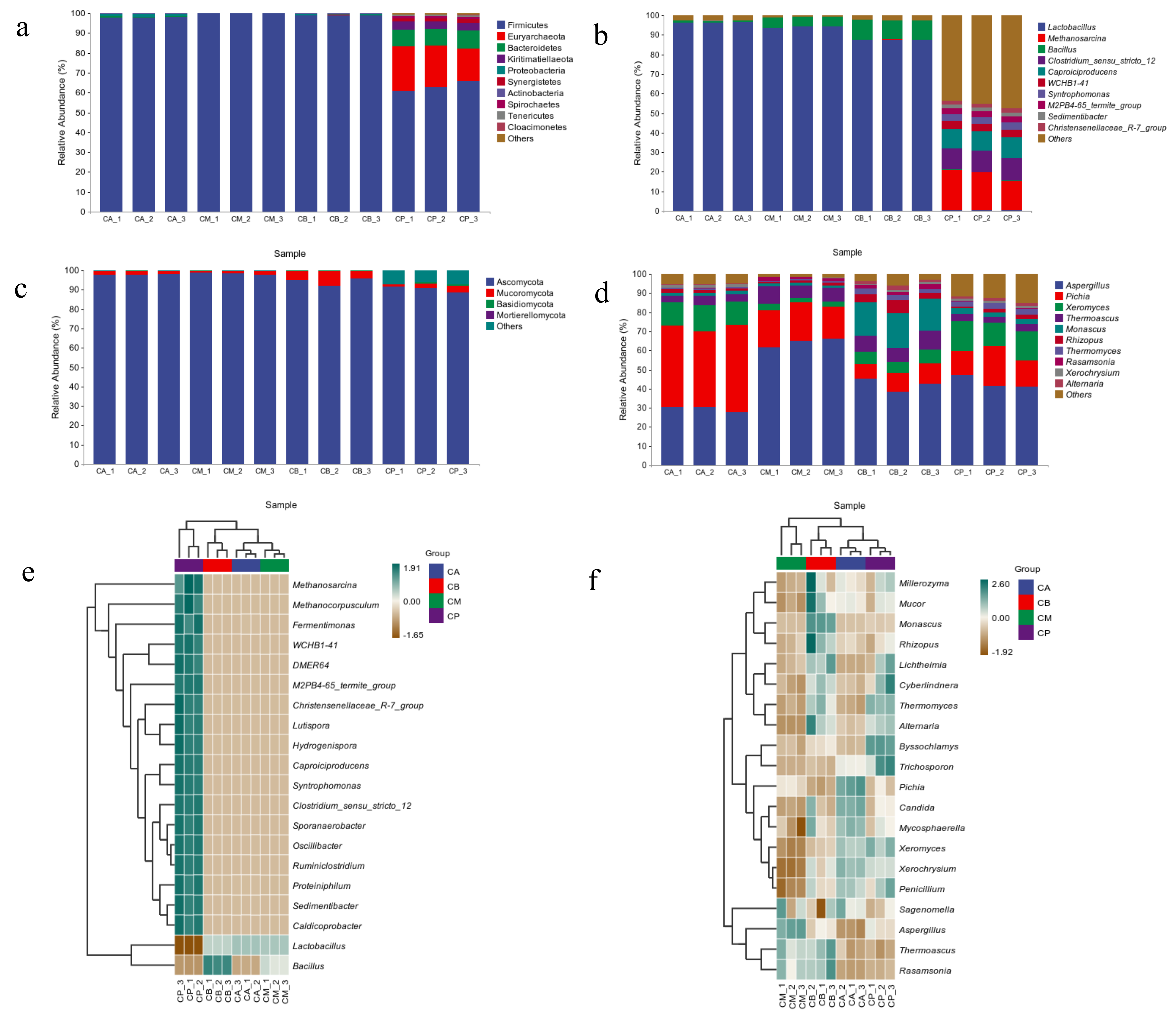
3.3. Analysis of Microbial Community Structure in FG and PM
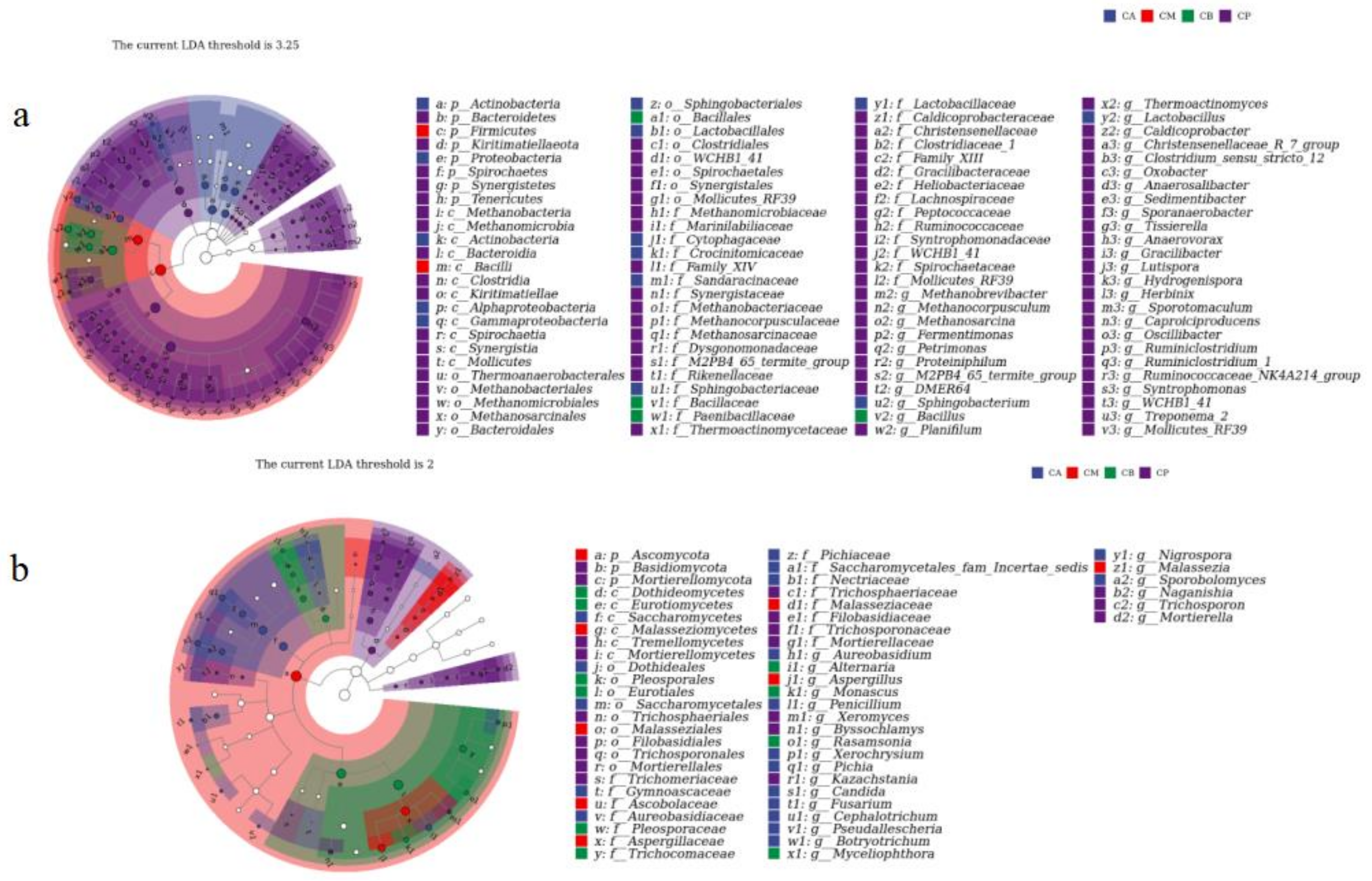
3.4. Microbial Community Structure, Heatmap, and LEfSe Analysis of FG and PM
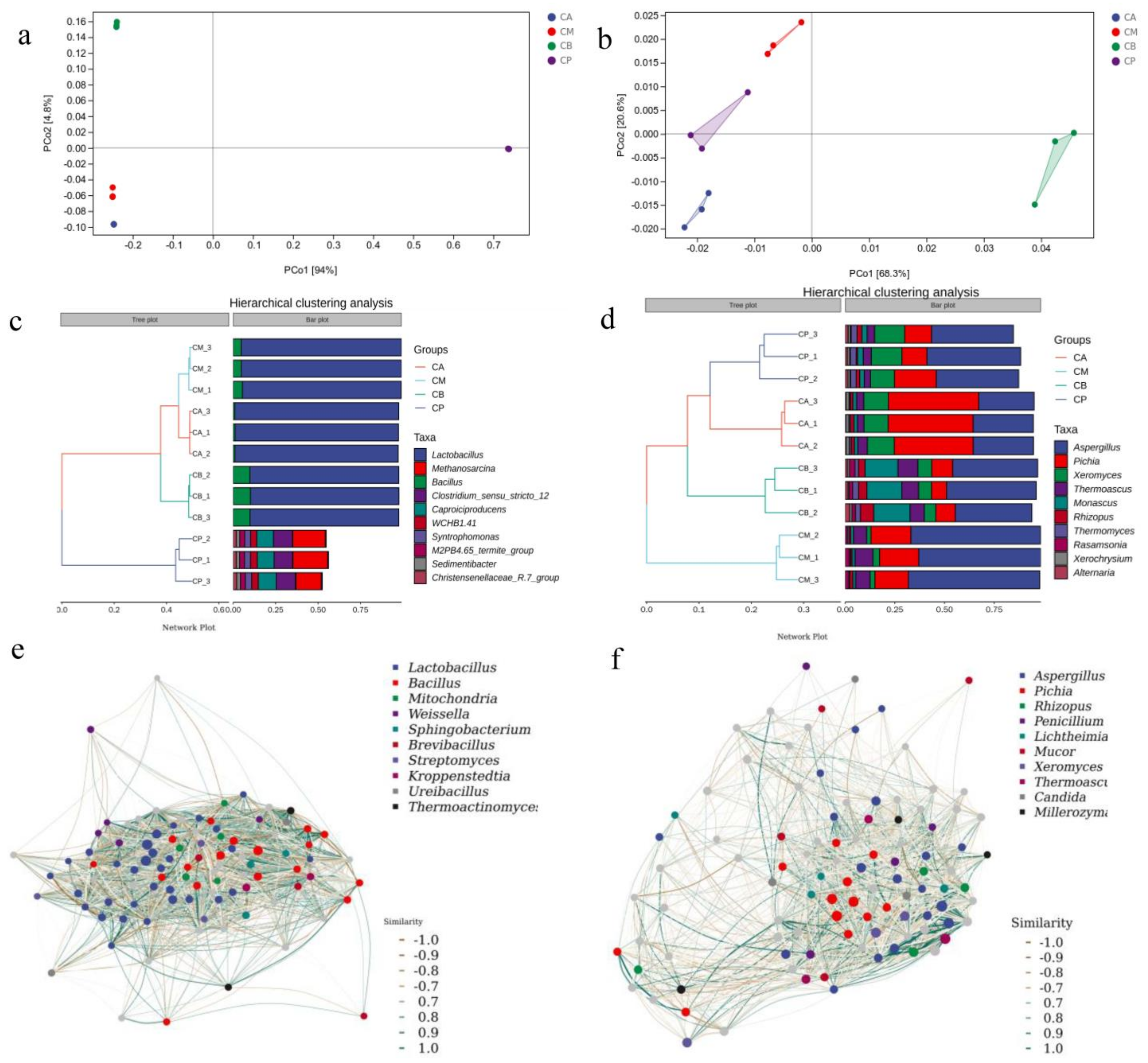
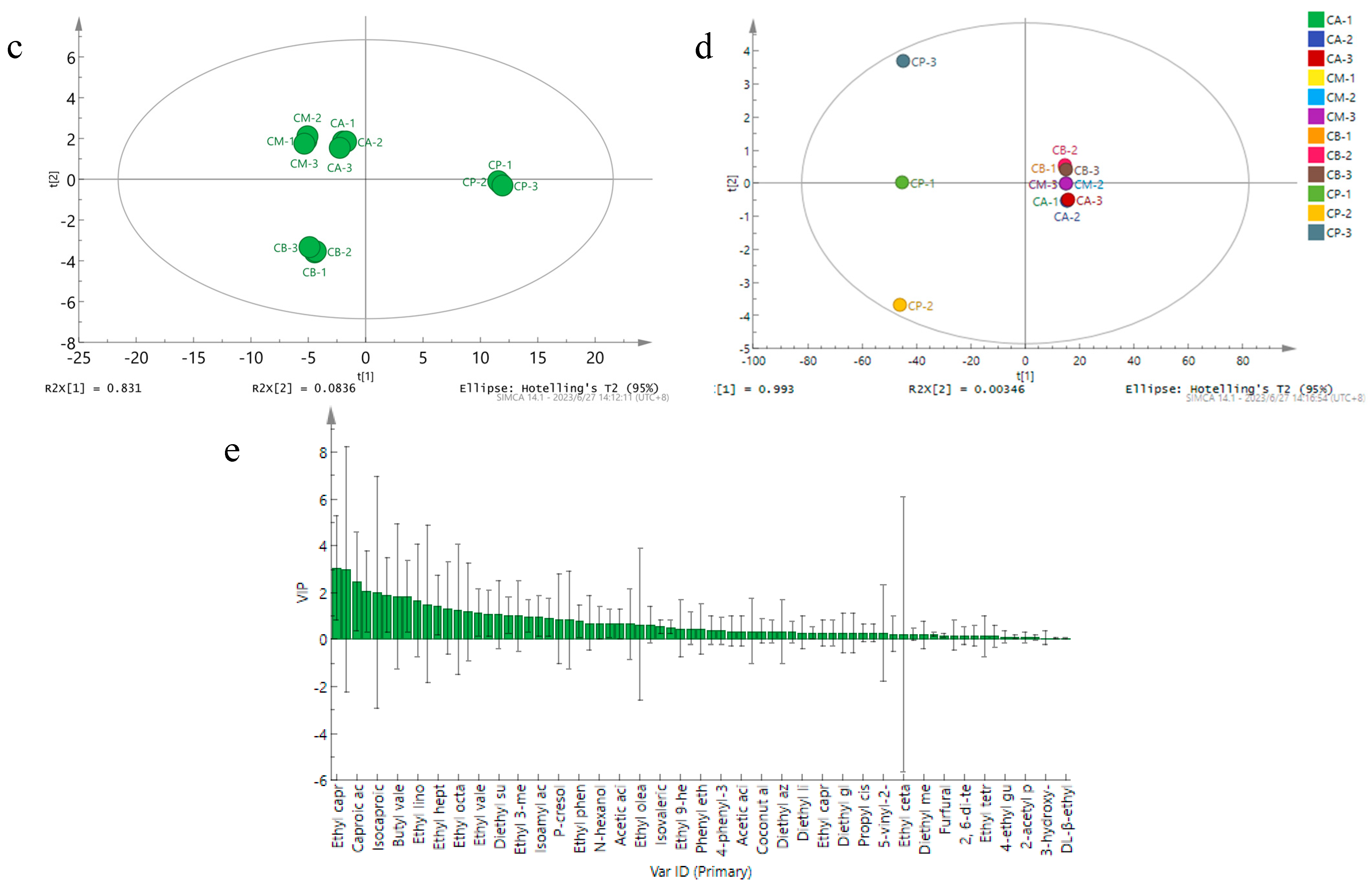
3.5. Principal Component Analysis (PCA), Hierarchical Clustering Analysis, and Network Correlation Analysis of Microbial Community in FG and PM
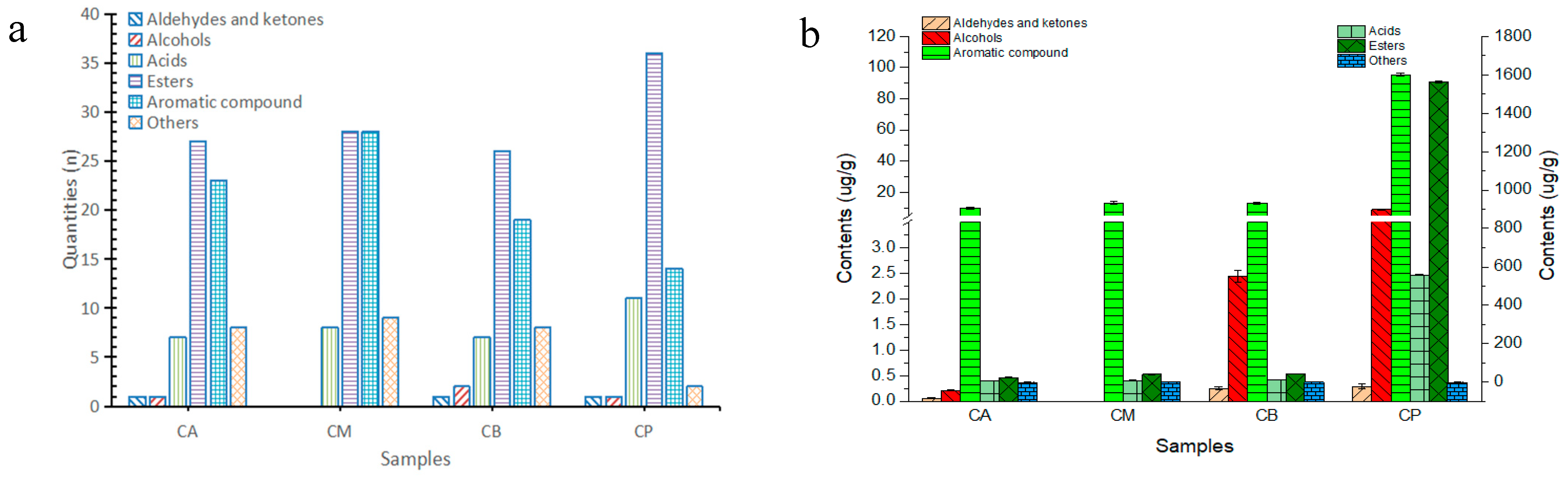
3.6. Analysis of Volatile Components in FG and PM
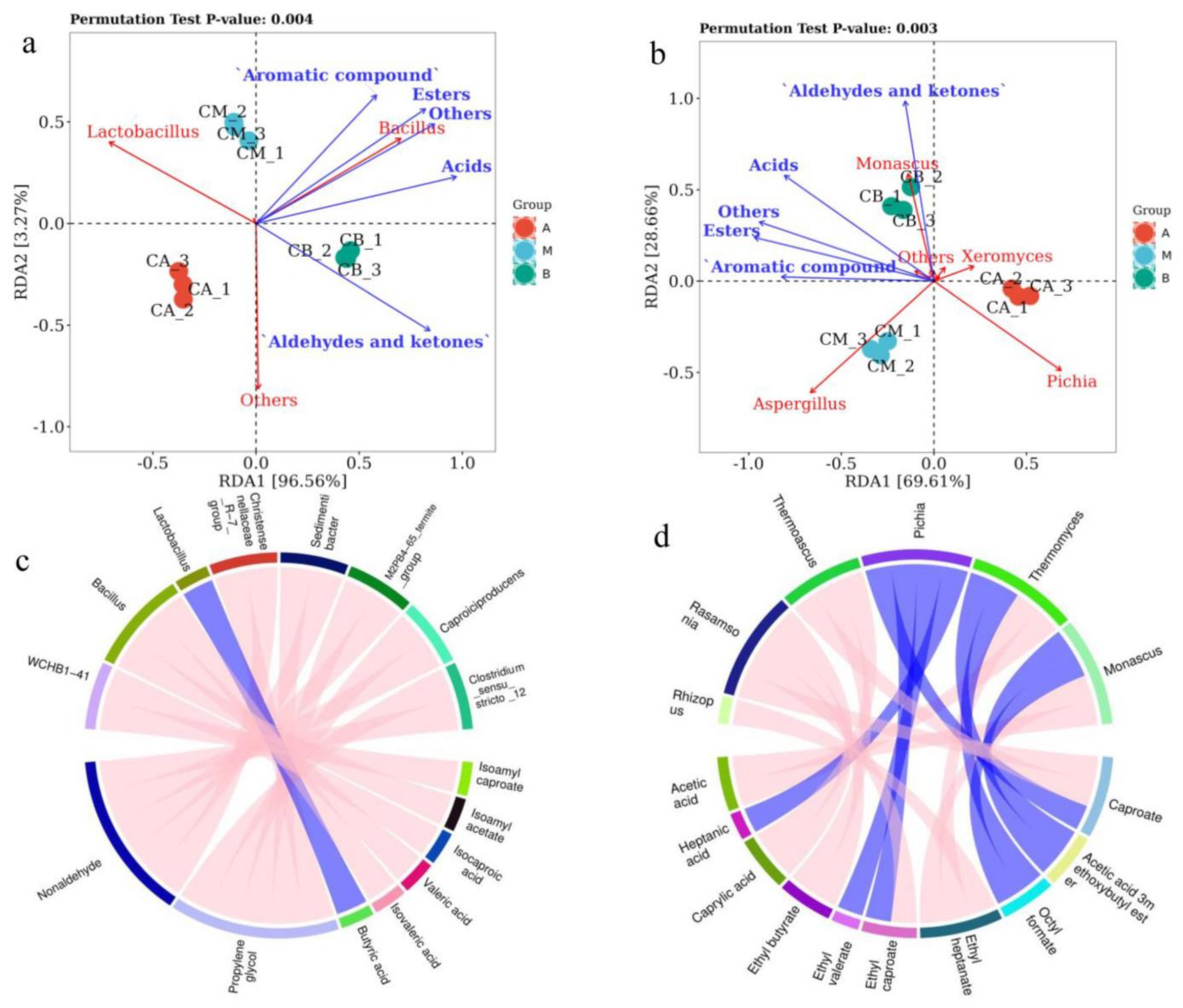
3.7. RDA and Correlation Perfume Circos of Microbial Community with Volatile Components of FG
4. Discussion
4.1. Physicochemical Indexes and Plate Counting of Culturable Microorganisms of FG and PM
4.2. Microbial Community Structure of FG and PM
4.3. Relationships between Microbial Communities and Volatile Compounds
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Duan, J.; Yang, S.; Li, H.; Qin, D.; Shen, Y.; Li, H.; Sun, J.; Zheng, F.; Sun, B. Why the key aroma compound of soy sauce aroma type baijiu has not been revealed yet? LWT-Food Sci. Technol. 2022, 154, 112735. [Google Scholar] [CrossRef]
- Cheng, W.; Chen, X.; Lan, W.; Liu, G.; Xue, X.; Li, R.; Pan, T.; Li, N.; Zhou, D.; Chen, X. Insights into the influence of physicochemical parameters on the microbial community and volatile compounds during the ultra-long fermentation of compound-flavor Baijiu. Front. Microbiol. 2023, 14, 1272559. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, S.; Sun, H.; Jiang, Z.; Xu, Y.; Mao, J.; Qian, B.; Wang, L.; Mao, J. Metagenomics-based insights into the microbial community profiling and flavor development potentiality of baijiu Daqu and huangjiu wheat Qu. Food Res. Int. 2022, 152, 110707. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhao, J.; Liu, X.; Zhang, C.; Zhao, Z.; Li, X.; Sun, B. Flavor mystery of Chinese traditional fermented baijiu: The great contribution of ester compounds. Food Chem. 2022, 369, 130920. [Google Scholar] [CrossRef]
- Cheng, W.; Chen, X.; Zhou, D.; Xiong, F. Applications and prospects of the automation of compound flavor baijiu production by solid-state fermentation. Int. J. Food Eng. 2022, 18, 737–749. [Google Scholar] [CrossRef]
- Jin, G.Y.; Zhu, Y.; Xu, Y. Mystery behind Chinese liquor fermentation. Trends Food Sci Technol. 2017, 63, 18–28. [Google Scholar] [CrossRef]
- Chai, L.J.; Qian, W.; Zhong, X.Z.; Zhang, X.J.; Lu, Z.M.; Zhang, S.Y.; Wang, S.T.; Shen, C.H.; Shi, J.S.; Xu, Z.H. Mining the factors driving the evolution of the pit mud microbiome under the impact of long-term production of strong-flavor baijiu. Appl. Environ. Microb. 2021, 87, e0088521. [Google Scholar] [CrossRef] [PubMed]
- Qian, W.; Lu, Z.M.; Chai, L.J.; Zhang, X.J.; Li, Q.; Wang, S.T.; Shen, C.H.; Shi, J.S.; Xu, Z.H. Cooperation within the microbial consortia of fermented grains and pit mud drives organic acid synthesis in strong-flavor baijiu production. Food Res. Int. 2021, 147, 110449. [Google Scholar] [CrossRef]
- Guan, T.; Wu, X.; Hou, R.; Tian, L.; Huang, Q.; Zhao, F.; Liu, Y.; Jiao, S.; Xiang, S.; Zhang, J.; et al. Application of Clostridium butyricum, Rummeliibacillus suwonensis, and Issatchenkia orientalis for Nongxiangxing baijiu fermentation: Improves the microbial communities and flavor of upper fermented grain. Food Res. Int. 2023, 169, 112885. [Google Scholar] [CrossRef]
- Wang, X.S.; Du, H.; Zhang, Y.; Xu, Y. Environmental microbiota drives microbial succession and metabolic profiles during Chinese liquor fermentation. Appl. Environ. Microb. 2018, 84, e02369-17. [Google Scholar] [CrossRef]
- Yang, L.; Xian, C.; Li, P.; Wang, X.; Song, D.; Zhao, L.; Zhang, C. The spatio-temporal diversity and succession of microbial community and its environment driving factors during stacking fermentation of Maotai-flavor baijiu. Food Res. Int. 2023, 169, 112892. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, L.; Tan, Y.; Wang, H.; Yang, F.; Chen, L.; Hao, F.; Lv, X.; Du, H.; Xu, Y. Effect of Pichia on shaping the fermentation microbial community of sauce-flavor baijiu. Int. J. Food Microbiol. 2021, 336, 108898. [Google Scholar] [CrossRef] [PubMed]
- Pang, Z.; Li, W.; Hao, J.; Xu, Y.; Du, B.; Zhang, C.; Wang, K.; Zhu, H.; Wang, H.; Li, X.; et al. Correlational analysis of the physicochemical indexes, volatile flavor components, and microbial communities of high-temperature daqu in the northern region of China. Foods 2023, 12, 326. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Tian, R.; Wang, K.; Cao, Z.; Yan, P.; Li, F.; Li, X.; Li, S.; He, P. The prokaryotic community, physicochemical properties and flavors dynamics and their correlations in fermented grains for Chinese strong-flavor baijiu production. Food Res. Int. 2021, 148, 110626. [Google Scholar] [CrossRef]
- Zhang, M.; Wu, X.; Mu, D.; Xu, B.; Xu, X.; Chang, Q.; Li, X. Profiling the influence of physicochemical parameters on the microbial community and flavor substances of zaopei. J. Sci. Food Agr. 2021, 101, 6300–6310. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wu, X.; Mu, D.; Yang, W.; Jiang, S.; Sun, W.; Shen, Y.; Cai, J.; Zheng, Z.; Jiang, S.; et al. Profiling the effects of physicochemical indexes on the microbial diversity and its aroma substances in pit mud. Lett. Appl. Microbiol. 2020, 71, 667–678. [Google Scholar] [CrossRef]
- Hu, Y.; Wang, L.; Zhang, Z.; Yang, Q.; Chen, S.; Zhang, L.; Xia, X.; Tu, J.; Liang, Y.; Zhao, S. Microbial community changes during the mechanized production of light aroma Xiaoqu baijiu. Biotechnol. Biotec Eq. 2021, 35, 487–495. [Google Scholar] [CrossRef]
- Li, M.; Zhu, L.Z.; Lin, D.H. Toxicity of ZnO nanoparticles to Escherichia coli: Mechanism and the influence of medium components. Environ. Sci. Technol. 2011, 45, 1977–1983. [Google Scholar] [CrossRef]
- Martins, D.; English, A.M. Catalase activity is stimulated by H2O2 in rich culture medium and is required for H2O2 resistance and adaptation in yeast. Redox Biol. 2014, 2, 308–313. [Google Scholar] [CrossRef]
- Song, Z.; Du, H.; Zhang, Y.; Xu, Y. Unraveling core functional microbiota in traditional solid-state fermentation by high-throughput amplicons and metatranscriptomics sequencing. Front. Microbiol. 2017, 8, 1294. [Google Scholar] [CrossRef]
- Xu, Y.; Wu, M.; Niu, J.; Lin, M.; Zhu, H.; Wang, K.; Li, X.; Sun, B. Characteristics and correlation of the microbial communities and flavor compounds during the first three rounds of fermentation in Chinese sauce-flavor baijiu. Foods 2023, 12, 207. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sun, M.; Hou, P.; Wang, W.; Shen, X.; Zhang, L.; Han, S.; Pan, C. Analysis of microbial community structure and volatile compounds in pit mud used for manufacturing Taorong-type baijiu based on high-throughput sequencing. Sci. Rep. 2022, 12, 7347. [Google Scholar] [CrossRef]
- Sakandar, H.A.; Hussain, R.; Khan, Q.F.; Zhang, H.P. Functional microbiota in Chinese traditional baijiu and Mijiu Qu (starters): A review. Food Res. Int. 2020, 138, 109830. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.; Shen, H.; Yang, Q.; Chen, S.; Dun, Y.; Liang, Y.; Zheng, J.; Zhao, S. Deciphering succession and assembly patterns of microbial communities in a two-stage solid-state fermentation system. Microbiol. Spectr. 2021, 9, e00718–e00721. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.Y.; Yang, J.G.; Zhao, Q.S.; Zhang, K.Z.; Su, C. Research progress on flavor compounds and microorganisms of Maotai flavor baijiu. J. Food Sci. 2019, 84, 6–18. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Lian, B.; Ding, Y.; Nie, C.; Zhang, Q. Bacterial diversity in the central black component of Maotai Daqu and its flavor analysis. Ann. Microbiol. 2014, 64, 1659–1669. [Google Scholar] [CrossRef]
- Yan, Q.; Zhang, K.Z.; Zou, W.; Hou, Y.C. Three main flavour types of Chinese baijiu: Characteristics, research, and perspectives. J. Inst. Brew. 2021, 127, 317–326. [Google Scholar] [CrossRef]
- Zhang, W.X.; Qiao, Z.W.; Tang, Y.Q.; Hu, C.; Sun, Q.; Morimura, S.; Kida, K. Analysis of the fungal community in Zaopei during the production of Chinese Luzhou-flavour liquor. J. Inst. Brew. 2007, 113, 21–27. [Google Scholar] [CrossRef]
- Li, H.; Liu, S.; Liu, Y.; Hui, M.; Pan, C. Functional microorganisms in baijiu Daqu: Research progress and fortification strategy for application. Front. Microbiol. 2023, 14, 1119675. [Google Scholar] [CrossRef]
- Tu, W.; Cao, X.; Cheng, J.; Li, L.; Zhang, T.; Wu, Q.; Xiang, P.; Shen, C.; Li, Q. Chinese baijiu: The perfect works of microorganisms. Front. Microbiol. 2022, 13, 919044. [Google Scholar] [CrossRef]
- Tong, W.; He, P.; Yang, Y.; Qiao, Z.; Huang, D.; Luo, H.; Feng, X. Occurrence, diversity, and character of bacillaceae in the solid fermentation process of strong aromatic liquors. Front. Microbiol. 2022, 12, 811788. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Huang, J.; Zhou, R.; Zhang, S.; Qin, H.; Dong, Y.; Wang, C.; Wang, X.; Pan, Q.; Tang, H. Comprehensive analysis for the bioturbation effect of space mutation and biofortification on strong-flavor Daqu by high-throughput sequencing, volatile analysis and metabolomics. Food Chem. 2023, 403, 134440. [Google Scholar] [CrossRef]
- Mu, Y.; Huang, J.; Zhou, R.; Zhang, S.; Qin, H.; Tang, H.; Pan, Q.; Tang, H. Characterization of the differences in aroma-active compounds in strong-flavor baijiu induced by bioaugmented Daqu using metabolomics and sensomics approaches. Food Chem. 2023, 424, 136429. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Wang, X.; Li, X.; Wei, N.; Jin, H.; Xu, Z.; Tang, Q.; Zhu, X. The functional potential and active populations of the pit mud microbiome for the production of Chinese strong-flavour liquor. Microb Biotechnol. 2017, 10, 1603–1615. [Google Scholar] [CrossRef]
- Tan, Y.; Du, H.; Zhang, H.; Fang, C.; Jin, G.; Chen, S.; Wu, Q.; Zhang, Y.; Zhang, M.; Xu, Y. Geographically associated fungus-bacterium interactions contribute to the formation of geography-dependent flavor during high-complexity spontaneous fermentation. Microbiol. Spectr. 2022, 10, e0184422. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Du, H.; Zhao, D.; Qiao, Z.; Zheng, J.; Yu, X.; Xu, Y. Stochastic processes drive the assembly and metabolite profiles of keystone taxa during Chinese strong-flavor baijiu fermentation. Microbiol. Spectr. 2023, 11, e0510322. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.J.; Song, L.; Han, Y.; Zhen, P.; Han, D.Y.; Zhao, X.; Zhou, X.; Wei, Y.H.; Yu, H.X.; Han, P.J.; et al. Microbial communities and their correlation with flavor compound formation during the mechanized production of light-flavor baijiu. Food Res. Int. 2023, 172, 113139. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Cheng, Y.; Shi, Q.; Ge, X.; Yang, Y.; Huang, Y. Metagenomic analyses reveal microbial communities and functional differences between Daqu from seven provinces. Food Res. Int. 2023, 172, 113076. [Google Scholar] [CrossRef]
- Kang, J.; Zheng, X.; Yang, X.; Li, H.; Cheng, J.; Fan, L.; Zhao, H.; Xue, Y.; Ding, Z.; Han, B. Contrasting summer versus winter dynamic microbial communities and their environmental driving factors in the solid-state saccharification process of Fuyu-flavor baijiu. Food Res. Int. 2022, 154, 111008. [Google Scholar] [CrossRef]
- Mu, Y.; Huang, J.; Zhou, R.; Mao, F.; Pan, Q.; Chen, S.; Lu, Z.; Du, L.; Xie, F. Exploring the response patterns of strong-flavor baijiu brewing microecosystem to fortified Daqu under different pit ages. Food Res. Int. 2022, 155, 111062. [Google Scholar] [CrossRef]
- Wang, X.; Wang, B.; Sun, Z.; Tan, W.; Zheng, J.; Zhu, W. Effects of modernized fermentation on the microbial community succession and ethyl lactate metabolism in Chinese baijiu fermentation. Food Res. Int. 2022, 159, 111566. [Google Scholar] [CrossRef]
- Liu, H.L.; Sun, B.G. Effect of fermentation processing on the flavor of Baijiu. J. Agr. Food Chem. 2018, 66, 5425–5432. [Google Scholar] [CrossRef]
- Hou, Q.; Wang, Y.; Cai, W.; Ni, H.; Zhao, H.; Zhang, Z.; Liu, Z.; Liu, J.; Guo, Z. Metagenomic and physicochemical analyses reveal microbial community and functional differences between three types of low-temperature Daqu. Food Res. Int. 2022, 156, 111167. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Chai, L.J.; Fang, G.Y.; Mei, J.L.; Lu, Z.M.; Zhang, X.J.; Xiao, C.; Wang, S.T.; Shen, C.H.; Shi, J.S.; et al. Spatial heterogeneity of the microbiome and metabolome profiles of high-temperature Daqu in the same workshop. Food Res. Int. 2022, 156, 111298. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Chen, X.; Zeng, H.; Xue, X. Association between microbial community composition and quality indicators of strong-flavor Daqu of different producing regions in China. CyTA J. Food 2023, 21, 82–92. [Google Scholar] [CrossRef]
- Wang, H.Y.; Xu, Y. Effect of temperature on microbial composition of starter culture for Chinese light aroma style liquor fermentation. Lett. Appl. Microbiol. 2015, 60, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.Y.; Zhang, W.X.; Zhang, Q.S.; Hu, C.; Wang, R.; Liu, Z.H. Developing new sacchariferous starters for liquor production based on functional strains isolated from the pits of several famous Luzhou-flavor liquor brewers. J. Inst. Brew. 2009, 115, 111–115. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, X.; Liu, X.; Li, X.; Zhang, C.; Li, W.; Sun, X.; Wang, W.; Sun, B. Discovery and development of a novel short-chain fatty acid ester synthetic biocatalyst under aqueous phase from Monascus purpureus isolated from baijiu. Food Chem. 2021, 338, 128025. [Google Scholar] [CrossRef]
- Zhao, Q.S.; Yang, J.G.; Zhang, K.Z.; Wang, M.Y.; Zhao, X.X.; Su, C.; Cao, X.Z. Lactic acid bacteria in the brewing of traditional Daqu liquor. J. Inst. Brew. 2020, 126, 14–23. [Google Scholar] [CrossRef]
- Wang, S.; Xiong, W.; Wang, Y.; Nie, Y.; Wu, Q.; Xu, Y.; Geisen, S. Temperature-induced annual variation in microbial community changes and resulting metabolome shifts in a controlled fermentation system. Msystems 2020, 5, e00555-20. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Moisture Content (%) | Acidity (n mol/10 g) | Starch Content (g/100 g) | Reducing Sugars Content (g/100 g) |
|---|---|---|---|---|
| CA | 62.05 ± 0.21 b | 4.05 ± 0.07 b | 8.58 ± 2.34 a | 0.43 ± 0.32 |
| CM | 63.68 ± 0.18 b | 4.40 ± 0.11 b | 7.63 ± 1.05 a | 0.35 ± 0.15 |
| CB | 67.67 ± 0.27 a | 5.39 ± 0.05 a | 5.75 ± 1.63 b | 0.29 ± 0.11 |
| Sample | Moisture (%) | pH | Ammonium (mg/100 g) | Available-P (mg/kg) | Available-K (mg/kg) | Calcium (g/kg) | Humus (g/kg) |
|---|---|---|---|---|---|---|---|
| CP | 46.58 ± 0.02 | 5.53 ± 0.31 | 31.48 ± 0.15 | 128.36 ± 0.01 | 5211.36 ± 30.01 | 0.86 ± 0.08 | 219.36 ± 0.01 |
| Samples | Total Colony Count (PCA, log CFU/g) | The Total Number of Bacterial Colonies (LB, log CFU/g) | The Total Number of Mold Colonies (CD, log CFU/g) | The Total Number of Yeast Colonies (YPD, log CFU/g) |
|---|---|---|---|---|
| CA | 7.55 ± 0.04 a | 5.74 ± 0.32 b | 7.54 ± 0.33 a | 6.37 ± 0.03 a |
| CM | 7.31 ± 0.64 a | 7.73 ± 0.16 a | 7.37 ± 3.33 a | 0.26 ± 1.58 c |
| CB | 7.21 ± 1.20 a | 7.64 ± 0.09 a | 6.38 ± 4.34 ab | 4.46 ± 1.69 ab |
| CP | 2.57 ± 0.13 b | 2.90 ± 0.00 c | 2.08 ± 1.90 b | 2.95 ± 0.05 b |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, W.; Chen, X.; Xue, X.; Lan, W.; Zeng, H.; Li, R.; Pan, T.; Li, N.; Gong, Z.; Yang, H. Comparison of the Correlations of Microbial Community and Volatile Compounds between Pit-Mud and Fermented Grains of Compound-Flavor Baijiu. Foods 2024, 13, 203. https://doi.org/10.3390/foods13020203
Cheng W, Chen X, Xue X, Lan W, Zeng H, Li R, Pan T, Li N, Gong Z, Yang H. Comparison of the Correlations of Microbial Community and Volatile Compounds between Pit-Mud and Fermented Grains of Compound-Flavor Baijiu. Foods. 2024; 13(2):203. https://doi.org/10.3390/foods13020203
Chicago/Turabian StyleCheng, Wei, Xuefeng Chen, Xijia Xue, Wei Lan, Huawei Zeng, Ruilong Li, Tianquan Pan, Na Li, Zilu Gong, and Hongwen Yang. 2024. "Comparison of the Correlations of Microbial Community and Volatile Compounds between Pit-Mud and Fermented Grains of Compound-Flavor Baijiu" Foods 13, no. 2: 203. https://doi.org/10.3390/foods13020203