
Site-Directed Mutations at Phosphorylation Sites in Zea mays PHO1 Reveal Modulation of Enzymatic Activity by Phosphorylation at S566 in the L80 Region
 , , ,
, , ,
Abstract
:1. Introduction
2. Results
2.1. Antibody Preparation and Specificity Detection
2.2. PHO1 Expression and Distribution Pattern
2.2.1. Transcripts of PHO1 Expression and Distribution Pattern in Developing Seeds
2.2.2. Protein (PHO1) Expression and Distribution Pattern in Developing Seeds
2.2.3. Phos-TagTM Enrichment of PHO1 Phosphorylated Proteins
2.2.4. Phosphorylation Sites Prediction
2.2.5. iTRAQ (Isobaric Tags for Relative and Absolute Quantitation) Identification of ZmPHO1 Phosphorylation Sites
2.2.6. Conservation of Identified Phosphorylation Sites in Monocots and Dicots
2.3. Site-Directed Point Mutations
2.4. Subcellular Localization and Stability of ZmPHO1
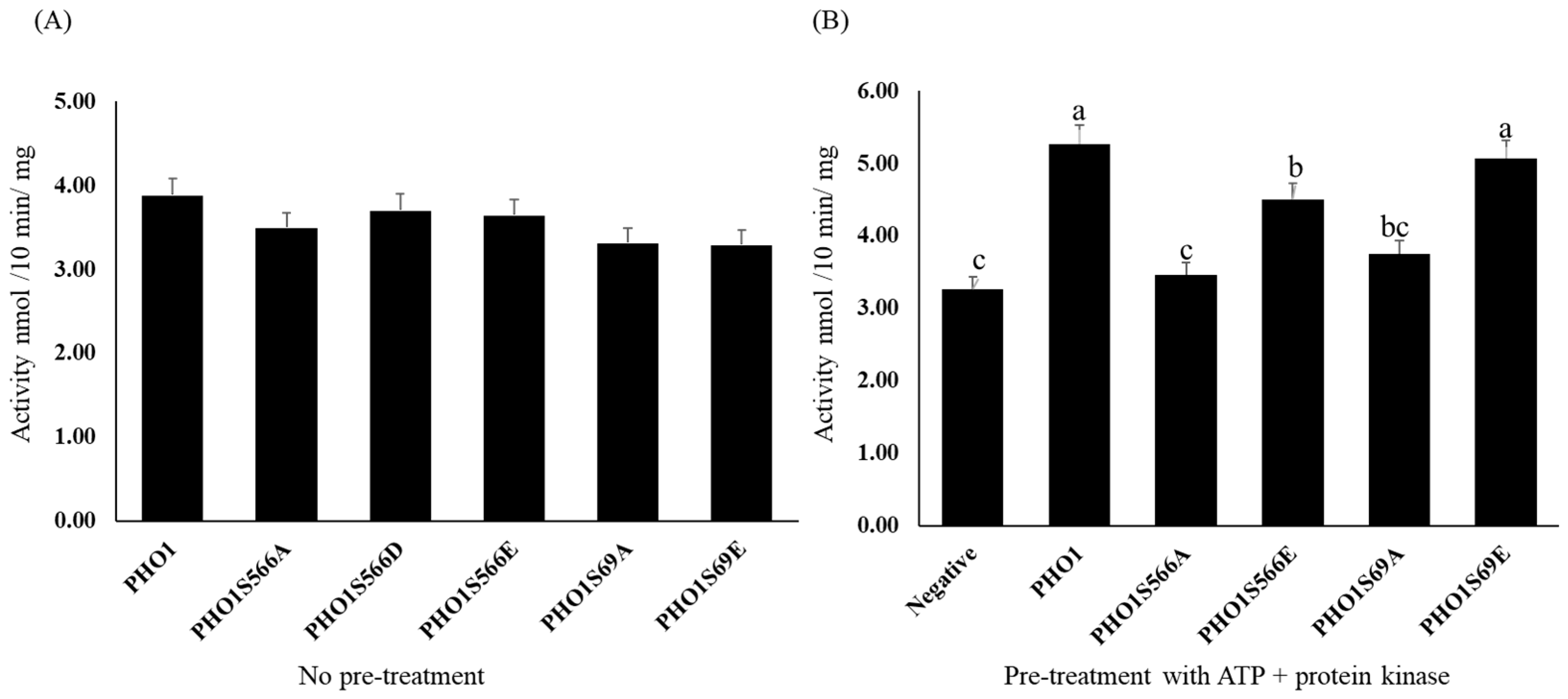
2.5. Activity of PHO1
2.5.1. Expression and Purification of Recombinant PHO1
2.5.2. Enzyme Assay
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Preparation of Plant Material
5.1.1. Transcripts of PHO1 Expression Pattern
5.1.2. PHO1 Protein Expression Pattern
5.1.3. Phos-tagTM Enrichment and Dephosphorylation
5.1.4. Phosphorylation Sites Prediction
5.1.5. iTRAQTM Labeling and Mass Spectrometry Analysis
5.2. PHO1 Gene Cloning and Recombination
5.3. Site-Directed Point Mutations
5.4. Expression and Purification of Recombinant PHO1
Enzyme Assay
5.5. Preparation of ZmPHO1 Antibody
5.6. Maize Leaf Protoplast Transformation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cori, G.T.; Cori, C.F.; Schmidt, G. The role of glucose-l-phosphate in the formation of blood sugar and synthesis of glycogen in the liver. J. Biol. Chem. 1939, 142, 355–366. [Google Scholar] [CrossRef]
- Shoaib, N.; Liu, L.; Ali, A.; Mughal, N.; Yu, G.; Huang, Y. Molecular Functions and Pathways of Plastidial Starch Phosphorylase (PHO1) in Starch Metabolism: Current and Future Perspectives. Int. J. Mol. Sci. 2021, 22, 10450. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.Y.; Nelson, O.E. Phosphorylases I and II of Maize Endosperm. Plant Physiol. 1968, 43, 103–112. [Google Scholar] [CrossRef]
- Subasinghe, R.M.; Liu, F.; Polack, U.C.; Lee, E.A.; Emes, M.J.; Tetlow, I.J. Multimeric states of starch phosphorylase determine protein-protein interactions with starch biosynthetic enzymes in amyloplasts. Plant Physiol. Biochem. 2014, 83, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Brisson, N.; Giroux, H.; Zollinger, M.; Camirand, A.; Simard, C. Maturation and subcellular compartmentation of potato starch phosphorylase. Plant Cell 1989, 1, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; Mori, H.; Fukui, T. Molecular cloning of cDNA encoding potato amyloplast alpha-glucan phosphorylase and the structure of its transit peptide. J. Biochem. 1989, 106, 691–695. [Google Scholar] [CrossRef]
- Yu, Y.; Mu, H.H.; Wasserman, B.P.; Carman, G.M. Identification of the maize amyloplast stromal 112-kD protein as a plastidic starch phosphorylase. Plant Physiol. 2001, 125, 351–359. [Google Scholar] [CrossRef]
- Yu, G.; Shoaib, N.; Xie, Y.; Liu, L.; Mughal, N.; Li, Y.; Huang, H.; Zhang, N.; Zhang, J.; Liu, Y. Comparative Study of Starch Phosphorylase Genes and Encoded Proteins in Various Monocots and Dicots with Emphasis on Maize. Int. J. Mol. Sci. 2022, 23, 4518. [Google Scholar] [CrossRef]
- Cuesta-Seijo, J.A.; Nielsen, M.M.; Ruzanski, C.; Krucewicz, K.; Beeren, S.R.; Rydhal, M.G.; Yoshimura, Y.; Striebeck, A.; Motawia, M.S.; Willats, W.G.; et al. In vitro Biochemical Characterization of All Barley Endosperm Starch Synthases. Front. Plant Sci. 2015, 6, 1265. [Google Scholar] [CrossRef]
- Hwang, S.K.; Koper, K.; Satoh, H.; Okita, T.W. Rice Endosperm Starch Phosphorylase (Pho1) Assembles with Disproportionating Enzyme (Dpe1) to Form a Protein Complex That Enhances Synthesis of Malto-oligosaccharides. J. Biol. Chem. 2016, 291, 19994–20007. [Google Scholar] [CrossRef]
- Hwang, S.K.; Koper, K.; Okita, T.W. The plastid phosphorylase as a multiple-role player in plant metabolism. Plant Sci. 2020, 290, 110303. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, T.; Greve, B.; Pusch, K.; Kossmann, J.; Buchner, P.; Wobus, U.; Steup, M. Homodimers and heterodimers of Pho1-type phosphorylase isoforms in Solanum tuberosum L. as revealed by sequence-specific antibodies. Eur. J. Biochem. 1998, 251, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Chen, H.M.; Chou, I.M.; Chen, A.N.; Chen, C.P.; Young, G.H.; Lin, C.T.; Cheng, C.H.; Chang, S.C.; Juang, R.H. Plastidial starch phosphorylase in sweet potato roots is proteolytically modified by protein-protein interaction with the 20S proteasome. PLoS ONE 2012, 7, e35336. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Jiang, Q.T.; Zhang, X.W.; Lan, X.J.; Pu, Z.E.; Wei, Y.M.; Liu, C.; Lu, Z.X.; Zheng, Y.L. Structure and expression of barley starch phosphorylase genes. Planta 2013, 238, 1081–1093. [Google Scholar] [CrossRef]
- Goren, A.; Ashlock, D.; Tetlow, I.J. Starch formation inside plastids of higher plants. Protoplasma 2018, 255, 1855–1876. [Google Scholar] [CrossRef]
- Ozbun, J.L.; Hawker, J.S.; Greenberg, E.; Lammel, C.; Preiss, J. Starch Synthetase, Phosphorylase, ADPglucose Pyrophosphorylase, and UDPglucose Pyrophosphorylase in Developing Maize Kernels. Plant Physiol. 1973, 51, 1–5. [Google Scholar] [CrossRef]
- Dauvillee, D.; Chochois, V.; Steup, M.; Haebel, S.; Eckermann, N.; Ritte, G.; Ral, J.P.; Colleoni, C.; Hicks, G.; Wattebled, F.; et al. Plastidial phosphorylase is required for normal starch synthesis in Chlamydomonas reinhardtii. Plant J. 2006, 48, 274–285. [Google Scholar] [CrossRef]
- Mizuno, S.; Kamiyoshihara, Y.; Shiba, H.; Shinmachi, F.; Watanabe, K.; Tateishi, A. Plastidial starch phosphorylase is highly associated with starch accumulation process in developing squash (Cucurbita sp.) fruit. Physiol. Plant. 2019, 167, 264–275. [Google Scholar] [CrossRef]
- Mu, H.H.; Yu, Y.; Wasserman, B.P.; Carman, G.M. Purification and characterization of the maize amyloplast stromal 112-kDa starch phosphorylase. Arch. Biochem. Biophys. 2001, 388, 155–164. [Google Scholar] [CrossRef]
- Fettke, J.; Albrecht, T.; Hejazi, M.; Mahlow, S.; Nakamura, Y.; Steup, M. Glucose 1-phosphate is efficiently taken up by potato (Solanum tuberosum) tuber parenchyma cells and converted to reserve starch granules. New Phytol. 2010, 185, 663–675. [Google Scholar] [CrossRef]
- Tetlow, I.J.; Bertoft, E. A Review of Starch Biosynthesis in Relation to the Building Block-Backbone Model. Int. J. Mol. Sci. 2020, 21, 7011. [Google Scholar] [CrossRef] [PubMed]
- Tetlow, I.J.; Wait, R.; Lu, Z.; Akkasaeng, R.; Bowsher, C.G.; Esposito, S.; Kosar-Hashemi, B.; Morell, M.K.; Emes, M.J. Protein phosphorylation in amyloplasts regulates starch branching enzyme activity and protein-protein interactions. Plant Cell 2004, 16, 694–708. [Google Scholar] [CrossRef] [PubMed]
- Tetlow, I.J.; Beisel, K.G.; Cameron, S.; Makhmoudova, A.; Liu, F.; Bresolin, N.S.; Wait, R.; Morell, M.K.; Emes, M.J. Analysis of protein complexes in wheat amyloplasts reveals functional interactions among starch biosynthetic enzymes. Plant Physiol. 2008, 146, 1878–1891. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Makhmoudova, A.; Lee, E.A.; Wait, R.; Emes, M.J.; Tetlow, I.J. The amylose extender mutant of maize conditions novel protein-protein interactions between starch biosynthetic enzymes in amyloplasts. J. Exp. Bot. 2009, 60, 4423–4440. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Ono, M.; Sawada, T.; Crofts, N.; Fujita, N.; Steup, M. Characterization of the functional interactions of plastidial starch phosphorylase and starch branching enzymes from rice endosperm during reserve starch biosynthesis. Plant Sci. 2017, 264, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, Z.; Tetlow, I.J.; Ahmed, R.; Morell, M.K.; Emes, M.J. Protein-protein interactions among enzymes of starch biosynthesis in high-amylose barley genotypes reveal differential roles of heteromeric enzyme complexes in the synthesis of A and B granules. Plant Sci. 2015, 233, 95–106. [Google Scholar] [CrossRef]
- Subasinghe, R.M. Role and Regulation of Starch Phosphorylase and Starch Synthase IV in Starch Biosynthesis in Maize Endosperm Amyloplast. Ph.D. Thesis, University of Guelph, Guelph, ON, Canada, 2014. [Google Scholar]
- Yu, G.; Lv, Y.; Shen, L.; Wang, Y.; Qing, Y.; Wu, N.; Li, Y.; Huang, H.; Zhang, N.; Liu, Y.; et al. The Proteomic Analysis of Maize Endosperm Protein Enriched by Phos-tag(tm) Reveals the Phosphorylation of Brittle-2 Subunit of ADP-Glc Pyrophosphorylase in Starch Biosynthesis Process. Int. J. Mol. Sci. 2019, 20, 986. [Google Scholar] [CrossRef]
- Johnson, L.N. Glycogen phosphorylase: Control by phosphorylation and allosteric effectors. FASEB J. 1992, 6, 2274–2282. [Google Scholar] [CrossRef]
- Sprang, S.R.; Acharya, K.R.; Goldsmith, E.J.; Stuart, D.I.; Varvill, K.; Fletterick, R.J.; Madsen, N.B.; Johnson, L.N. Structural changes in glycogen phosphorylase induced by phosphorylation. Nature 1988, 336, 215–221. [Google Scholar] [CrossRef]
- Yu, G.; Gaoyang, Y.; Liu, L.; Shoaib, N.; Deng, Y.; Zhang, N.; Li, Y.; Huang, Y. The Structure, Function, and Regulation of Starch Synthesis Enzymes SSIII with Emphasis on Maize. Agronomy 2022, 12, 1359. [Google Scholar] [CrossRef]
- Satoh, H.; Shibahara, K.; Tokunaga, T.; Nishi, A.; Tasaki, M.; Hwang, S.K.; Okita, T.W.; Kaneko, N.; Fujita, N.; Yoshida, M.; et al. Mutation of the plastidial alpha-glucan phosphorylase gene in rice affects the synthesis and structure of starch in the endosperm. Plant Cell 2008, 20, 1833–1849. [Google Scholar] [CrossRef] [PubMed]
- Grimaud, F.; Rogniaux, H.; James, M.G.; Myers, A.M.; Planchot, V. Proteome and phosphoproteome analysis of starch granule-associated proteins from normal maize and mutants affected in starch biosynthesis. J. Exp. Bot. 2008, 59, 3395–3406. [Google Scholar] [CrossRef] [PubMed]
- Leterrier, M.; Holappa, L.D.; Broglie, K.E.; Beckles, D.M. Cloning, characterisation and comparative analysis of a starch synthase IV gene in wheat: Functional and evolutionary implications. BMC Plant Biol. 2008, 8, 98. [Google Scholar] [CrossRef] [PubMed]
- Schupp, N.; Ziegler, P. The relation of starch phosphorylases to starch metabolism in wheat. Plant Cell Physiol. 2004, 45, 1471–1484. [Google Scholar] [CrossRef] [PubMed]
- Tickle, P.; Burrell, M.M.; Coates, S.A.; Emes, M.J.; Tetlow, I.J.; Bowsher, C.G. Characterization of plastidial starch phosphorylase in Triticum aestivum L. endosperm. J. Plant Physiol. 2009, 166, 1465–1478. [Google Scholar] [CrossRef]
- Tsai, C.Y.; Nelson, O.E. Two additional phosphorylases in developing maize seeds. Plant Physiol. 1969, 44, 159–167. [Google Scholar] [CrossRef]
- Cuesta-Seijo, J.A.; Ruzanski, C.; Krucewicz, K.; Meier, S.; Hagglund, P.; Svensson, B.; Palcic, M.M. Functional and structural characterization of plastidic starch phosphorylase during barley endosperm development. PLoS ONE 2017, 12, e0175488. [Google Scholar] [CrossRef]
- Lin, Y.-C.; Chang, S.-C.; Juang, R.-H. Plastidial α-glucan phosphorylase 1 complexes with disproportionating enzyme 1 in Ipomoea batatas storage roots for elevating malto-oligosaccharide metabolism. PLoS ONE 2017, 12, e0177115. [Google Scholar] [CrossRef]
- Nakamura, Y.; Ono, M.; Utsumi, C.; Steup, M. Functional interaction between plastidial starch phosphorylase and starch branching enzymes from rice during the synthesis of branched maltodextrins. Plant Cell Physiol. 2012, 53, 869–878. [Google Scholar] [CrossRef]
- Roldan, I.; Wattebled, F.; Mercedes Lucas, M.; Delvalle, D.; Planchot, V.; Jimenez, S.; Perez, R.; Ball, S.; D’Hulst, C.; Merida, A. The phenotype of soluble starch synthase IV defective mutants of Arabidopsis thaliana suggests a novel function of elongation enzymes in the control of starch granule formation. Plant J. 2007, 49, 492–504. [Google Scholar] [CrossRef]
- Hwang, S.K.; Nishi, A.; Satoh, H.; Okita, T.W. Rice endosperm-specific plastidial alpha-glucan phosphorylase is important for synthesis of short-chain malto-oligosaccharides. Arch. Biochem. Biophys. 2010, 495, 82–92. [Google Scholar] [CrossRef]
- Tiessen, A.; Nerlich, A.; Faix, B.; Hummer, C.; Fox, S.; Trafford, K.; Weber, H.; Weschke, W.; Geigenberger, P. Subcellular analysis of starch metabolism in developing barley seeds using a non-aqueous fractionation method. J. Exp. Bot. 2012, 63, 2071–2087. [Google Scholar] [CrossRef]
- Fettke, J.; Eckermann, N.; Kotting, O.; Ritte, G.; Steup, M. Novel starch-related enzymes and carbohydrates. Cell Mol. Biol. 2007, 52, OL883–OL904. [Google Scholar] [PubMed]
- Liu, L.; Qing, Y.; Shoaib, N.; Di, R.; Liu, H.; Li, Y.; Hu, Y.; Huang, Y.; Yu, G. Preparation of Polyclonal Antibody against ZmBT1 Protein and Its Application in Hormone-Regulated Starch Synthesis. Agronomy 2023, 13, 1805. [Google Scholar] [CrossRef]
- Yu, G.; Shoaib, N.; Yang, Y.; Liu, L.; Mughal, N.; Mou, Y.; Huang, Y. Effect of Phosphorylation Sites Mutations on the Subcellular Localization and Activity of AGPase Bt2 Subunit: Implications for Improved Starch Biosynthesis in Maize. Agronomy 2023, 13, 2119. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Credibility | Peptide Sequence | Phosphorylation Site | ZmPHO1 Site |
|---|---|---|---|
| 99.35000038 | DRDVQGPVSPAEGLPSVLNS | Serine at number 9 | Serine 69 |
| 99.00000095 | LESEEVEAEEESSEDELDPFVK | Serine at number 13 | Serine 566 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shoaib, N.; Mughal, N.; Liu, L.; Raza, A.; Shen, L.; Yu, G. Site-Directed Mutations at Phosphorylation Sites in Zea mays PHO1 Reveal Modulation of Enzymatic Activity by Phosphorylation at S566 in the L80 Region. Plants 2023, 12, 3205. https://doi.org/10.3390/plants12183205
Shoaib N, Mughal N, Liu L, Raza A, Shen L, Yu G. Site-Directed Mutations at Phosphorylation Sites in Zea mays PHO1 Reveal Modulation of Enzymatic Activity by Phosphorylation at S566 in the L80 Region. Plants. 2023; 12(18):3205. https://doi.org/10.3390/plants12183205
Chicago/Turabian StyleShoaib, Noman, Nishbah Mughal, Lun Liu, Ali Raza, Leiyang Shen, and Guowu Yu. 2023. "Site-Directed Mutations at Phosphorylation Sites in Zea mays PHO1 Reveal Modulation of Enzymatic Activity by Phosphorylation at S566 in the L80 Region" Plants 12, no. 18: 3205. https://doi.org/10.3390/plants12183205