
RNA-Binding Proteins in the Regulation of miRNA Activity: A Focus on Neuronal Functions

Abstract
:1. Introduction
1.1. MiRNA Biogenesis

1.2. RNA Modifications in miRNA Biogenesis
1.3. MiRNA Function
1.4. The miRNA Regulatory Network in the Neuronal Context and Its Alteration in Neurodegeneration
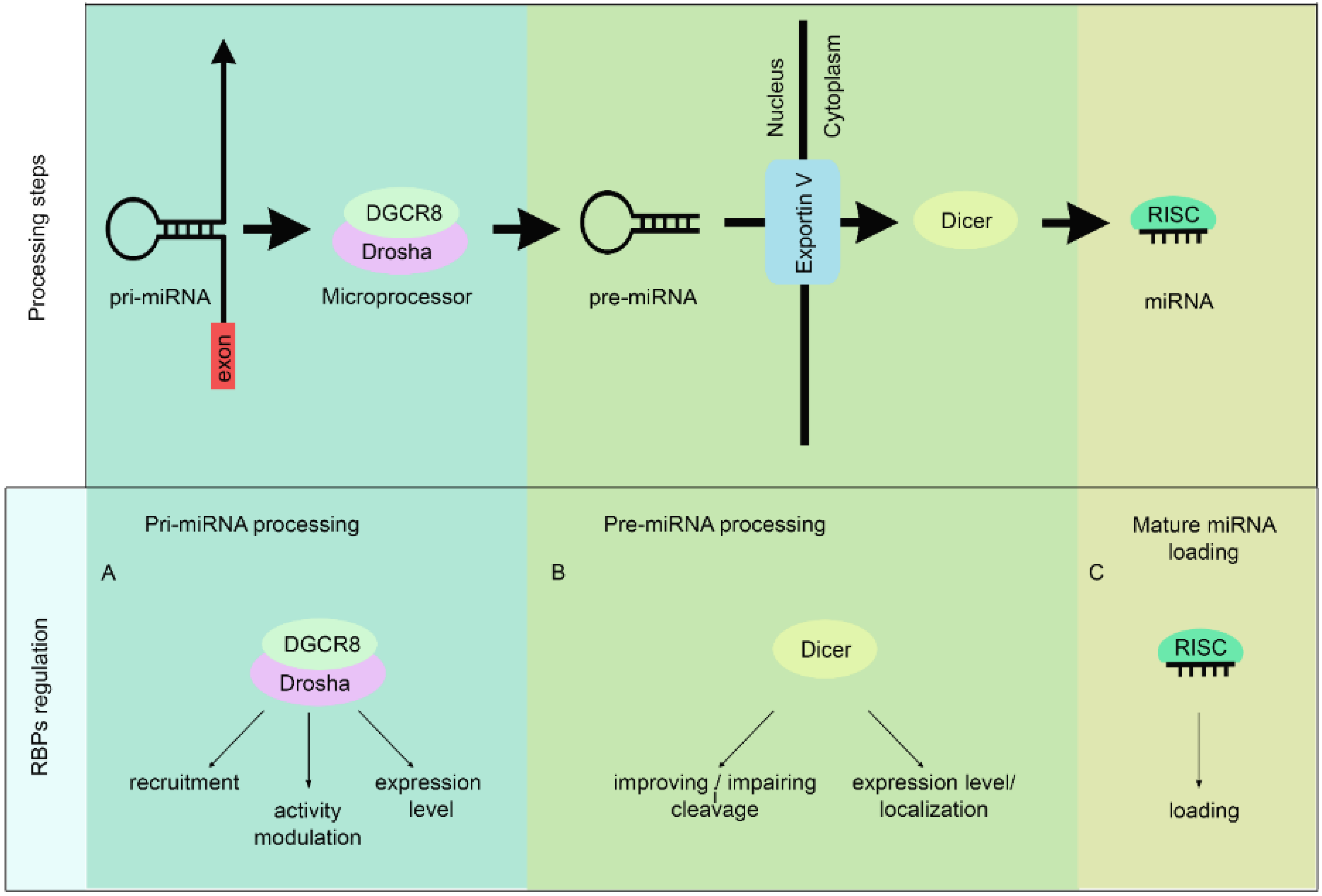
2. RBPs Regulate miRNAs Biogenesis

{kind=link}
{kind=link}
| RBP | Main RBPs Functions | miRNA Target | Affected Neuronal Functions | References * |
|---|---|---|---|---|
| TDP-43 ** | Transcriptional regulation, alternative splicing, mRNA transport and translation | miR-132 | Neuronal plasticity, synapse formation, neurite outgrowth [39,58,59] | [60] |
| FUS ** | miR-9; miR-125; miR-132; miR-200a; miR-141 | Synapse formation, neuronal plasticity, neurite outgrowth, neuronal differentiation and proliferation [39,53,58,59,61] | [62,63] | |
| TAF15 | Transcriptional regulation | miR-17 | Neuronal proliferation and survival [64] | [65] |
| Lin-28 | mRNA processing and translation | let-7 | Neuronal stem-cell commitment, neuronal proliferation, tissue-regeneration [66,67,68,69,70] | [67,71,72,73] |
| NF45/NF90 | mRNA transport/ stability | let-7 | See above for let-7 | [74] |
| DDX6 | mRNA translation and degradation | let-7 | See above for let-7 | [75] |
| hnRNP A1 | mRNA splicing and transport | miR 18a; let 7 | Neuronal survival and proliferation [64] See above for let-7 | [76,77] |
| MSI2/HuR | mRNA stability and localization | miR-7 | Synuclein levels regulation, neurite outgrowth [78,79] | [80] |
| DHX36 | mRNA stability | miR-134 | Neuronal plasticity [59,81,82] | [59] |
2.1. Pri-miRNA Processing
2.1.1. Drosha Recruitment to Pri-miRNA Transcription Sites
2.1.2. Modulation of Drosha RNAse III Activity
2.1.3. Regulation of Drosha Protein Expression
2.2. Pre-miRNA Processing
2.2.1. Pre-miRNA Export and Further Cleavage by Dicer
2.2.2. Regulation of Dicer Expression or Localization
2.3. RISC Loading
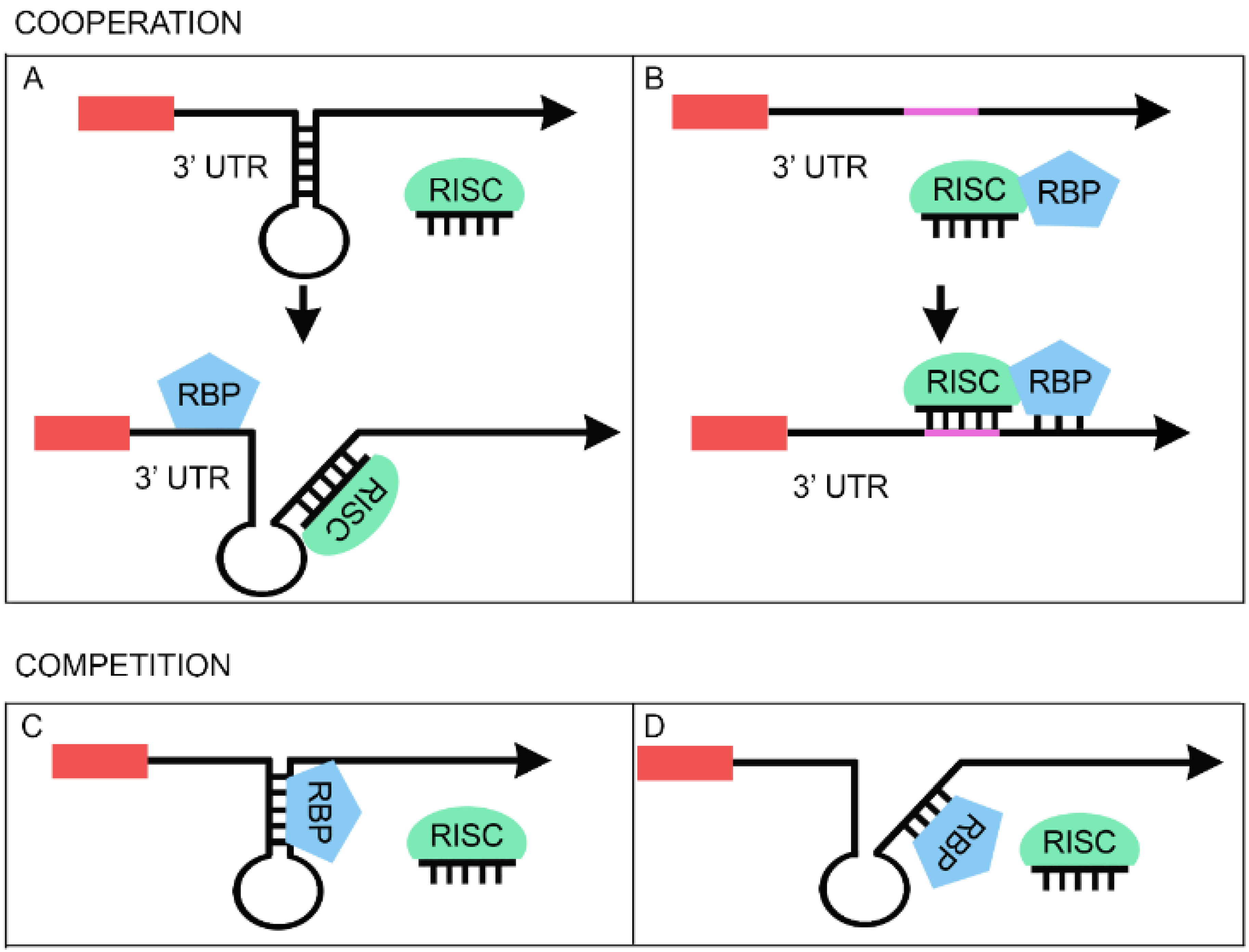
3. RBPs Regulate miRNA Function
| RBPs | miRNA | Common mRNA Target | Mechanism | References * |
|---|---|---|---|---|
| FMRP | miR-125b | NR2A | cooperation | [39] |
| FMRP | miR-125a | PSD-95 | cooperation | [124] |
| TDP-43 | miR-NID1 | NRXN1 | cooperation | [125] |
| HuR | miR-494 | NCL | competition | [126] |
| HuD | miR-129 | Kv1.1 | competition | [127] |
| hnRNP L | miR-297, miR-299 | VEGFA | competition | [128] |
3.1. Cooperative Regulation between miRNAs and RBPs

3.2. Competitive Regulation between miRNAs and RBPs
4. Perspectives and Conclusions
Acknowledgments
Conflicts of Interest
References
- Reinhart, B.J.; Slack, F.J.; Basson, M.; Pasquinelli, A.E.; Bettinger, J.C.; Rougvie, A.E.; Horvitz, H.R.; Ruvkun, G. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 2000, 403, 901–906. [Google Scholar] [PubMed]
- Kozomara, A.; Griffiths-Jones, S. Mirbase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yao, H.; Lin, S.; Zhu, X.; Shen, Z.; Lu, G.; Poon, W.S.; Xie, D.; Lin, M.C.; Kung, H.F. Transcriptional and epigenetic regulation of human microRNAs. Cancer Lett. 2013, 331, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Borchert, G.M.; Lanier, W.; Davidson, B.L. RNA polymerase III transcribes human microRNAs. Nat. Struct. Mol. Biol. 2006, 13, 1097–1101. [Google Scholar] [CrossRef] [PubMed]
- Parrott, A.M.; Mathews, M.B. Novel rapidly evolving hominid RNAs bind Nuclear Factor 90 and display tissue-restricted distribution. Nucleic Acids Res. 2007, 35, 6249–6258. [Google Scholar] [CrossRef] [PubMed]
- Landgraf, P.; Rusu, M.; Sheridan, R.; Sewer, A.; Iovino, N.; Aravin, A.; Pfeffer, S.; Rice, A.; Kamphorst, A.O.; Landthaler, M.; et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 2007, 129, 1401–1414. [Google Scholar] [CrossRef] [PubMed]
- Oler, A.J.; Alla, R.K.; Roberts, D.N.; Wong, A.; Hollenhorst, P.C.; Chandler, K.J.; Cassiday, P.A.; Nelson, C.A.; Hagedorn, C.H.; Graves, B.J.; et al. Human RNA polymerase III transcriptomes and relationships to Pol II promoter chromatin and enhancer-binding factors. Nat. Struct. Mol. Biol. 2010, 17, 620–628. [Google Scholar] [CrossRef] [PubMed]
- Ameres, S.L.; Zamore, P.D. Diversifying microRNA sequence and function. Nat. Rev. Mol. Cell Biol. 2013, 14, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 2010, 11, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Havens, M.A.; Reich, A.A.; Duelli, D.M.; Hastings, M.L. Biogenesis of mammalian microRNAs by a non-canonical processing pathway. Nucleic Acids Res. 2012, 40, 4626–4640. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Dai, Q.; Zheng, G.; He, C.; Parisien, M.; Pan, T. N6-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature 2015, 518, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Alarcon, C.R.; Lee, H.; Goodarzi, H.; Halberg, N.; Tavazoie, S.F. N6-methyladenosine marks primary microRNAs for processing. Nature 2015, 519, 482–485. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Chendrimada, T.P.; Wang, Q.; Higuchi, M.; Seeburg, P.H.; Shiekhattar, R.; Nishikura, K. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat. Struct. Mol. Biol. 2006, 13, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Kume, H.; Hino, K.; Galipon, J.; Ui-Tei, K. A-To-I editing in the miRNA seed region regulates target mRNA selection and silencing efficiency. Nucleic Acids Res. 2014, 42, 10050–10060. [Google Scholar] [CrossRef] [PubMed]
- Bahn, J.H.; Ahn, J.; Lin, X.; Zhang, Q.; Lee, J.H.; Civelek, M.; Xiao, X. Genomic analysis of ADAR1 binding and its involvement in multiple RNA processing pathways. Nat. Commun. 2015. [Google Scholar] [CrossRef] [PubMed]
- Huntzinger, E.; Kuzuoglu-Ozturk, D.; Braun, J.E.; Eulalio, A.; Wohlbold, L.; Izaurralde, E. The interactions of GW182 proteins with pabp and deadenylases are required for both translational repression and degradation of miRNA targets. Nucleic Acids Res. 2013, 41, 978–994. [Google Scholar] [CrossRef] [PubMed]
- Behm-Ansmant, I.; Rehwinkel, J.; Doerks, T.; Stark, A.; Bork, P.; Izaurralde, E. mRNA degradation by miRNAs and GW182 requires both CCR4:Not deadenylase and Dcp1:Dcp2 decapping complexes. Genes Dev. 2006, 20, 1885–1898. [Google Scholar] [CrossRef] [PubMed]
- Kuzuoglu-Ozturk, D.; Huntzinger, E.; Schmidt, S.; Izaurralde, E. The Caenorhabditis elegans GW182 protein AIN-1 interacts with PAB-1 and subunits of the PAN2-PAN3 and CCR4-not deadenylase complexes. Nucleic Acids Res. 2012, 40, 5651–5665. [Google Scholar] [CrossRef] [PubMed]
- Rouya, C.; Siddiqui, N.; Morita, M.; Duchaine, T.F.; Fabian, M.R.; Sonenberg, N. Human DDX6 effects miRNA-mediated gene silencing via direct binding to CNOT1. RNA 2014, 20, 1398–1409. [Google Scholar] [CrossRef] [PubMed]
- Mathys, H.; Basquin, J.; Ozgur, S.; Czarnocki-Cieciura, M.; Bonneau, F.; Aartse, A.; Dziembowski, A.; Nowotny, M.; Conti, E.; Filipowicz, W. Structural and biochemical insights to the role of the CCR4-NOT complex and DDX6 ATPase in microRNA repression. Mol. Cell 2014, 54, 751–765. [Google Scholar] [CrossRef]
- Chen, Y.; Boland, A.; Kuzuoglu-Ozturk, D.; Bawankar, P.; Loh, B.; Chang, C.T.; Weichenrieder, O.; Izaurralde, E. A DDX6-CNOT1 complex and w-binding pockets in CNOT9 reveal direct links between miRNA target recognition and silencing. Mol. Cell 2014, 54, 737–750. [Google Scholar] [CrossRef] [PubMed]
- Aitken, C.E.; Lorsch, J.R. A mechanistic overview of translation initiation in eukaryotes. Nat. Struct. Mol. Biol. 2012, 19, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Gingras, A.C.; Raught, B.; Sonenberg, N. EIF4 initiation factors: Effectors of mRNA recruitment to ribosomes and regulators of translation. Annu. Rev. Biochem. 1999, 68, 913–963. [Google Scholar] [CrossRef] [PubMed]
- Fukaya, T.; Iwakawa, H.O.; Tomari, Y. MicroRNAs block assembly of eIF4F translation initiation complex in Drosophila. Mol. Cell 2014, 56, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Meijer, H.A.; Kong, Y.W.; Lu, W.T.; Wilczynska, A.; Spriggs, R.V.; Robinson, S.W.; Godfrey, J.D.; Willis, A.E.; Bushell, M. Translational repression and eIF4A2 activity are critical for microRNA-mediated gene regulation. Science 2013, 340, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Fukao, A.; Mishima, Y.; Takizawa, N.; Oka, S.; Imataka, H.; Pelletier, J.; Sonenberg, N.; Thoma, C.; Fujiwara, T. MicroRNAs trigger dissociation of eIF4AI and eIF4AII from target mRNAs in humans. Mol. Cell 2014, 56, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Fukao, A.; Sasano, Y.; Imataka, H.; Inoue, K.; Sakamoto, H.; Sonenberg, N.; Thoma, C.; Fujiwara, T. The ELAV protein hud stimulates cap-dependent translation in a poly(A)- and eIF4A-dependent manner. Mol. Cell 2009, 36, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Fukaya, T.; Tomari, Y. PABP is not essential for microRNA-mediated translational repression and deadenylation in vitro. EMBO J. 2011, 30, 4998–5009. [Google Scholar] [CrossRef] [PubMed]
- Fukaya, T.; Tomari, Y. MicroRNAs mediate gene silencing via multiple different pathways in Drosophila. Mol. Cell 2012, 48, 825–836. [Google Scholar] [CrossRef] [PubMed]
- Bazzini, A.A.; Lee, M.T.; Giraldez, A.J. Ribosome profiling shows that miR-430 reduces translation before causing mRNA decay in zebrafish. Science 2012, 336, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Hendrickson, D.G.; Hogan, D.J.; McCullough, H.L.; Myers, J.W.; Herschlag, D.; Ferrell, J.E.; Brown, P.O. Concordant regulation of translation and mRNA abundance for hundreds of targets of a human microRNA. PLoS Biol. 2009, 7, e1000238. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Ingolia, N.T.; Weissman, J.S.; Bartel, D.P. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 2010, 466, 835–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subtelny, A.O.; Eichhorn, S.W.; Chen, G.R.; Sive, H.; Bartel, D.P. Poly(A)-tail profiling reveals an embryonic switch in translational control. Nature 2014, 508, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Eichhorn, S.W.; Guo, H.; McGeary, S.E.; Rodriguez-Mias, R.A.; Shin, C.; Baek, D.; Hsu, S.-H.; Ghoshal, K.; Villén, J.; Bartel, D.P. mRNA destabilization is the dominant effect of mammalian microRNAs by the time substantial repression ensues. Mol. Cell 2014, 56, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Schmiedel, J.M.; Klemm, S.L.; Zheng, Y.; Sahay, A.; Bluthgen, N.; Marks, D.S.; van Oudenaarden, A. Gene expression: MicroRNA control of protein expression noise. Science 2015, 348, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.K.; Young, A.G.; Bhutkar, A.; Zheng, G.X.; Bosson, A.D.; Nielsen, C.B.; Sharp, P.A. Genome-wide identification of AGO2 binding sites from mouse embryonic stem cells with and without mature microRNAs. Nat. Struct. Mol. Biol. 2011, 18, 237–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Jin, P. Roles of small regulatory RNAs in determining neuronal identity. Nat. Rev. Neurosci. 2010, 11, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Olde Loohuis, N.F.; Kos, A.; Martens, G.J.; van Bokhoven, H.; Nadif Kasri, N.; Aschrafi, A. MicroRNA networks direct neuronal development and plasticity. Cell. Mol. Life Sci. 2012, 69, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Edbauer, D.; Neilson, J.R.; Foster, K.A.; Wang, C.-F.; Seeburg, D.P.; Batterton, M.N.; Tada, T.; Dolan, B.M.; Sharp, P.A.; Sheng, M. Regulation of synaptic structure and function by FMRP-associated microRNAs miR-125b and miR-132. Neuron 2010, 65, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Coolen, M.; Katz, S.; Bally-Cuif, L. miR-9: A versatile regulator of neurogenesis. Front. Cell. Neurosci. 2013. [Google Scholar] [CrossRef] [PubMed]
- Yoo, A.S.; Sun, A.X.; Li, L.; Shcheglovitov, A.; Portmann, T.; Li, Y.; Lee-Messer, C.; Dolmetsch, R.E.; Tsien, R.W.; Crabtree, G.R. MicroRNA-mediated conversion of human fibroblasts to neurons. Nature 2011, 476, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Davis, T.H.; Cuellar, T.L.; Koch, S.M.; Barker, A.J.; Harfe, B.D.; McManus, M.T.; Ullian, E.M. Conditional loss of dicer disrupts cellular and tissue morphogenesis in the cortex and hippocampus. J. Neurosci. 2008, 28, 4322–4330. [Google Scholar] [CrossRef] [PubMed]
- Giraldez, A.J.; Cinalli, R.M.; Glasner, M.E.; Enright, A.J.; Thomson, J.M.; Baskerville, S.; Hammond, S.M.; Bartel, D.P.; Schier, A.F. MicroRNAs regulate brain morphogenesis in zebrafish. Science 2005, 308, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Wu, H.; Lin, Q.; Wei, W.; Lu, X.H.; Cantle, J.P.; Ao, Y.; Olsen, R.W.; Yang, X.W.; Mody, I.; et al. Deletion of astroglial dicer causes non-cell-autonomous neuronal dysfunction and degeneration. J. Neurosci. 2011, 31, 8306–8319. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; He, X.; Feng, J. Dicer is essential for neuronal polarity. Int. J. Dev. Neurosci. 2012, 30, 607–611. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Inoue, K.; Ishii, J.; Vanti, W.B.; Voronov, S.V.; Murchison, E.; Hannon, G.; Abeliovich, A. A microRNA feedback circuit in midbrain dopamine neurons. Science 2007, 317, 1220–1224. [Google Scholar] [CrossRef] [PubMed]
- Haramati, S.; Chapnik, E.; Sztainberg, Y.; Eilam, R.; Zwang, R.; Gershoni, N.; McGlinn, E.; Heiser, P.W.; Wills, A.M.; Wirguin, I.; et al. MiRNA malfunction causes spinal motor neuron disease. Proc. Natl. Acad. Sci. USA 2010, 107, 13111–13116. [Google Scholar] [CrossRef] [PubMed]
- Hebert, S.S.; de Strooper, B. Alterations of the microRNA network cause neurodegenerative disease. Trends Neurosci. 2009, 32, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Lau, P.; de Strooper, B. Dysregulated microRNAs in neurodegenerative disorders. Semin. Cell Dev. Biol. 2010, 21, 768–773. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, A.; O’Carroll, D.; Tan, C.L.; Hillman, D.; Sugimori, M.; Llinas, R.; Greengard, P. Cerebellar neurodegeneration in the absence of microRNAs. J. Exp. Med. 2007, 204, 1553–1558. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.T.; Chu, K.; Jung, K.H.; Kim, J.H.; Huh, J.Y.; Yoon, H.; Park, D.K.; Lim, J.Y.; Kim, J.M.; Jeon, D.; et al. miR-206 regulates brain-derived neurotrophic factor in Alzheimer Disease model. Ann. Neurol. 2012, 72, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Zhu, H.; Xu, Y.; Huang, L.; Ma, C.; Deng, W.; Liu, Y.; Qin, C. MicroRNA-153 negatively regulates the expression of amyloid precursor protein and amyloid precursor-like protein 2. Brain Res. 2012, 1455, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Packer, A.N.; Xing, Y.; Harper, S.Q.; Jones, L.; Davidson, B.L. The bifunctional microRNA miR-9/miR-9* regulates REST and COREST and is downregulated in Huntington’s disease. J. Neurosci. 2008, 28, 14341–14346. [Google Scholar] [CrossRef] [PubMed]
- Gaughwin, P.M.; Ciesla, M.; Lahiri, N.; Tabrizi, S.J.; Brundin, P.; Bjorkqvist, M. Hsa-miR-34b is a plasma-stable microRNA that is elevated in pre-manifest Huntington’s disease. Hum. Mol. Genet. 2011, 20, 2225–2237. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.H.; Valdez, G.; Moresi, V.; Qi, X.; McAnally, J.; Elliott, J.L.; Bassel-Duby, R.; Sanes, J.R.; Olson, E.N. MicroRNA-206 delays ALS progression and promotes regeneration of neuromuscular synapses in mice. Science 2009, 326, 1549–1554. [Google Scholar] [CrossRef] [PubMed]
- Butovsky, O.; Jedrychowski, M.P.; Cialic, R.; Krasemann, S.; Murugaiyan, G.; Fanek, Z.; Greco, D.J.; Wu, P.M.; Doykan, C.E.; Kiner, O.; et al. Targeting miR-155 restores abnormal microglia and attenuates disease in SOD1 mice. Ann. Neurol. 2015, 77, 75–99. [Google Scholar] [CrossRef] [PubMed]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Magill, S.T.; Cambronne, X.A.; Luikart, B.W.; Lioy, D.T.; Leighton, B.H.; Westbrook, G.L.; Mandel, G.; Goodman, R.H. MicroRNA-132 regulates dendritic growth and arborization of newborn neurons in the adult hippocampus. Proc. Natl. Acad. Sci. USA 2010, 107, 20382–20387. [Google Scholar] [CrossRef] [PubMed]
- Bicker, S.; Khudayberdiev, S.; Weiss, K.; Zocher, K.; Baumeister, S.; Schratt, G. The deah-box helicase DHX36 mediates dendritic localization of the neuronal precursor-microRNA-134. Genes Dev. 2013, 27, 991–996. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, Y.; Mieda-Sato, A. Tdp-43 promotes microRNA biogenesis as a component of the Drosha and Dicer complexes. Proc. Natl. Acad. Sci. USA 2012, 109, 3347–3352. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.; Singh, P.; Jauhari, A.; Singh, T.; Khan, F.; Pant, A.B.; Parmar, D.; Yadav, S. Critical role of the miR-200 family in regulating differentiation and proliferation of neurons. J. Neurochem. 2015, 133, 640–652. [Google Scholar] [CrossRef] [PubMed]
- Morlando, M.; Dini Modigliani, S.; Torrelli, G.; Rosa, A.; di Carlo, V.; Caffarelli, E.; Bozzoni, I. FUS stimulates microRNA biogenesis by facilitating co-transcriptional drosha recruitment. EMBO J. 2012, 31, 4502–4510. [Google Scholar] [CrossRef] [PubMed]
- Dini Modigliani, S.; Morlando, M.; Errichelli, L.; Sabatelli, M.; Bozzoni, I. An ALS-associated mutation in the FUS 3'-UTR disrupts a microRNA-FUS regulatory circuitry. Nat. Commun. 2014. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.S.; Chopp, M.; Wang, X.L.; Zhang, L.; Hozeska-Solgot, A.; Tang, T.; Kassis, H.; Zhang, R.L.; Chen, C.; Xu, J.; et al. MicroRNA-17-92 cluster mediates the proliferation and survival of neural progenitor cells after stroke. J. Biol. Chem. 2013, 288, 12478–12488. [Google Scholar] [CrossRef] [PubMed]
- Ballarino, M.; Jobert, L.; Dembélé, D.; de la Grange, P.; Auboeuf, D.; Tora, L. TAF15 is important for cellular proliferation and regulates the expression of a subset of cell cycle genes through miRNAs. Oncogene 2012, 32, 4646–4655. [Google Scholar] [CrossRef] [PubMed]
- Shyh-Chang, N.; Daley, G.Q. Lin28: Primal regulator of growth and metabolism in stem cells. Cell Stem. Cell 2013, 12, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Rybak, A.; Fuchs, H.; Smirnova, L.; Brandt, C.; Pohl, E.E.; Nitsch, R.; Wulczyn, F.G. A feedback loop comprising lin-28 and let-7 controls pre-let-7 maturation during neural stem-cell commitment. Nat. Cell Biol. 2008, 10, 987–993. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wang, X.; Gu, Y.; Chen, C.; Wang, Y.; Liu, J.; Hu, W.; Yu, B.; Wang, Y.; Ding, F.; et al. Let-7 microRNAs regenerate peripheral nerve regeneration by targeting nerve growth factor. Mol. Ther. 2014, 23, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Cimadamore, F.; Amador-Arjona, A.; Chen, C.; Huang, C.T.; Terskikh, A.V. Sox2-lin28/let-7 pathway regulates proliferation and neurogenesis in neural precursors. Proc. Natl. Acad. Sci. USA 2013, 110, E3017–E3026. [Google Scholar] [CrossRef] [PubMed]
- La Torre, A.; Georgi, S.; Reh, T.A. Conserved microRNA pathway regulates developmental timing of retinal neurogenesis. Proc. Natl. Acad. Sci. USA 2013, 110, E2362–E2370. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, S.R.; Daley, G.Q.; Gregory, R.I. Selective blockade of microRNA processing by lin28. Science 2008, 320, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Piskounova, E.; Polytarchou, C.; Thornton, J.E.; LaPierre, R.J.; Pothoulakis, C.; Hagan, J.P.; Iliopoulos, D.; Gregory, R.I. Lin28a and lin28b inhibit let-7 microRNA biogenesis by distinct mechanisms. Cell 2011, 147, 1066–1079. [Google Scholar] [CrossRef] [PubMed]
- Heo, I.; Joo, C.; Kim, Y.-K.; Ha, M.; Yoon, M.-J.; Cho, J.; Yeom, K.-H.; Han, J.; Kim, V.N. TUT4 in concert with lin28 suppresses microRNA biogenesis through pre-microRNA uridylation. Cell 2009, 138, 696–708. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, S.; Aoki, K.; Higuchi, T.; Todaka, H.; Morisawa, K.; Tamaki, N.; Hatano, E.; Fukushima, A.; Taniguchi, T.; Agata, Y. The NF90-NF45 complex functions as a negative regulator in the microRNA processing pathway. Mol. Cell. Biol. 2009, 29, 3754–3769. [Google Scholar] [CrossRef] [PubMed]
- Nicklas, S.; Okawa, S.; Hillje, A.L.; Gonzalez-Cano, L.; del Sol, A.; Schwamborn, J.C. The RNA helicase DDX6 regulates cell-fate specification in neural stem cells via miRNAs. Nucleic Acids Res. 2015, 43, 2638–2654. [Google Scholar] [CrossRef] [PubMed]
- Michlewski, G.; Cáceres, J.F. Antagonistic role of hnRNP A1 and KSRP in the regulation of let-7a biogenesis. Nat. Struct. Mol. Biol. 2010, 17, 1011–1018. [Google Scholar] [CrossRef] [PubMed]
- Guil, S.; Cáceres, J.F. The multifunctional RNA-binding protein hnRNP A1 is required for processing of miR-18a. Nat. Struct. Mol. Biol. 2007, 14, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Junn, E.; Lee, K.W.; Jeong, B.S.; Chan, T.W.; Im, J.Y.; Mouradian, M.M. Repression of -synuclein expression and toxicity by microRNA-7. Proc. Natl. Acad. Sci. USA 2009, 106, 13052–13057. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Shalom-Feuerstein, R.; Riley, J.; Zhang, S.D.; Tucci, P.; Agostini, M.; Aberdam, D.; Knight, R.A.; Genchi, G.; Nicotera, P.; et al. MiR-7 and miR-214 are specifically expressed during neuroblastoma differentiation, cortical development and embryonic stem cells differentiation, and control neurite outgrowth in vitro. Biochem. Biophys. Res. Commun. 2010, 394, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, N.R.; de Lima Alves, F.; de Andres-Aguayo, L.; Graf, T.; Caceres, J.F.; Rappsilber, J.; Michlewski, G. Tissue-specific control of brain-enriched miR-7 biogenesis. Genes Dev. 2013, 27, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Fiore, R.; Rajman, M.; Schwale, C.; Bicker, S.; Antoniou, A.; Bruehl, C.; Draguhn, A.; Schratt, G. MiR-134-dependent regulation of pumilio-2 is necessary for homeostatic synaptic depression. EMBO J. 2014, 33, 2231–2246. [Google Scholar] [CrossRef] [PubMed]
- Fiore, R.; Khudayberdiev, S.; Christensen, M.; Siegel, G.; Flavell, S.W.; Kim, T.K.; Greenberg, M.E.; Schratt, G. Mef2-mediated transcription of the miR379–410 cluster regulates activity-dependent dendritogenesis by fine-tuning pumilio2 protein levels. EMBO J. 2009, 28, 697–710. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.Y.; Pan, L.; Su, S.C.; Quinn, E.J.; Sasaki, M.; Jimenez, J.C.; Mackenzie, I.R.; Huang, E.J.; Tsai, L.H. Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat. Neurosci. 2013, 16, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Mastrocola, A.S.; Kim, S.H.; Trinh, A.T.; Rodenkirch, L.A.; Tibbetts, R.S. The RNA-binding protein fused in sarcoma (FUS) functions downstream of poly(ADP-ribose) polymerase (PARP) in response to DNA damage. J. Boil. Chem. 2013, 288, 24731–24741. [Google Scholar] [CrossRef] [PubMed]
- Vance, C.; Rogelj, B.; Hortobágyi, T.; de Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA processing protein, cause familial Amyotrophic Lateral Sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Rademakers, R.; Roeber, S.; Baker, M.; Kretzschmar, H.A.; Mackenzie, I.R.A. A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain 2009, 132, 2922–2931. [Google Scholar] [CrossRef] [PubMed]
- Merner, N.D.; Girard, S.L.; Catoire, H.; Bourassa, C.V.; Belzil, V.V.; Rivière, J.-B.; Hince, P.; Levert, A.; Dionne-Laporte, A.; Spiegelman, D.; et al. Exome sequencing identifies FUS mutations as a cause of essential tremor. Am. J. Hum. Genet. 2012, 91, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Robberecht, W.; Philips, T. The changing scene of amyotrophic lateral sclerosis. Nat. Rev. Neurosci. 2013, 14, 248–264. [Google Scholar] [CrossRef] [PubMed]
- Sreedharan, J.; Brown, R.H., Jr. Amyotrophic Lateral Sclerosis: Problems and prospects. Ann. Neurol. 2013, 74, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Rademakers, R.; Neumann, M.; Mackenzie, I.R. Advances in understanding the molecular basis of frontotemporal dementia. Nat. Rev. Neurol. 2012, 8, 423–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, E.D. Understanding essential tremor: Progress on the biological front. Curr. Neurol. Neurosci. Rep. 2014. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Bentmann, E.; Dormann, D.; Jawaid, A.; DeJesus-Hernandez, M.; Ansorge, O.; Roeber, S.; Kretzschmar, H.A.; Munoz, D.G.; Kusaka, H.; et al. FET proteins TAF15 and EWS are selective markers that distinguish FTLD with FUS pathology from Amyotrophic Lateral Sclerosis with FUS mutations. Brain 2011, 134, 2595–2609. [Google Scholar] [CrossRef] [PubMed]
- Couthouis, J.; Hart, M.P.; Erion, R.; King, O.D.; Diaz, Z.; Nakaya, T.; Ibrahim, F.; Kim, H.J.; Mojsilovic-Petrovic, J.; Panossian, S.; et al. Evaluating the role of the FUS/TLS-related gene EWSR1 in Amyotrophic Lateral Sclerosis. Hum. Mol. Genet. 2012, 21, 2899–2911. [Google Scholar] [CrossRef] [PubMed]
- Cirulli, E.T.; Lasseigne, B.N.; Petrovski, S.; Sapp, P.C.; Dion, P.A.; Leblond, C.S.; Couthouis, J.; Lu, Y.F.; Wang, Q.; Krueger, B.J.; et al. Exome sequencing in Amyotrophic Lateral Sclerosis identifies risk genes and pathways. Science 2015, 347, 1436–1441. [Google Scholar] [CrossRef] [PubMed]
- Sohn, E.J.; Park, J.; Kang, S.-I.; Wu, Y.-P. Accumulation of pre-let-7g and downregulation of mature let-7g with the depletion of EWS. Biochem. Biophys. Res. Commun. 2012, 426, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Gregory, R.I.; Yan, K.-P.; Amuthan, G.; Chendrimada, T.; Doratotaj, B.; Cooch, N.; Shiekhattar, R. The microprocessor complex mediates the genesis of microRNAs. Nature 2004, 432, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Ballarino, M.; Pagano, F.; Girardi, E.; Morlando, M.; Cacchiarelli, D.; Marchioni, M.; Proudfoot, N.J.; Bozzoni, I. Coupled RNA processing and transcription of intergenic primary microRNAs. Mol. Cell. Biol. 2009, 29, 5632–5638. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.Y.; Hwang, Y.J.; Jung, M.K.; Choe, J.; Kim, Y.; Kim, S.; Lee, C.J.; Ahn, H.; Lee, J.; Kowall, N.W.; et al. A multifunctional protein EWS regulates the expression of Drosha and microRNAs. Cell Death Differ. 2013, 21, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Polymenidou, M.; Lagier-Tourenne, C.; Hutt, K.R.; Bennett, C.F.; Cleveland, D.W.; Yeo, G.W. Misregulated RNA processing in Amyotrophic Lateral Sclerosis. Brain Res. 2012, 1462, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Walsh, M.J.; Cooper-Knock, J.; Dodd, J.E.; Stopford, M.J.; Mihaylov, S.R.; Kirby, J.; Shaw, P.J.; Hautbergue, G.M. Invited review: Decoding the pathophysiological mechanisms that underlie RNA dysregulation in neurodegenerative disorders: A review of the current state of the art. Neuropathol. Appl. Neurobiol. 2015, 41, 109–134. [Google Scholar] [CrossRef] [PubMed]
- Scotter, E.L.; Chen, H.J.; Shaw, C.E. TDP-43 proteinopathy and ALS: Insights into disease mechanisms and therapeutic targets. Neurotherapeutics 2015, 12, 352–363. [Google Scholar] [CrossRef] [PubMed]
- Di Carlo, V.; Grossi, E.; Laneve, P.; Morlando, M.; Dini Modigliani, S.; Ballarino, M.; Bozzoni, I.; Caffarelli, E. TDP-43 regulates the microprocessor complex activity during in vitro neuronal differentiation. Mol. Neurobiol. 2013, 48, 952–963. [Google Scholar] [CrossRef] [PubMed]
- Thornton, J.E.; Gregory, R.I. How does lin28 let-7 control development and disease? Trends Cell Biol. 2012, 22, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Newman, M.A.; Thomson, J.M.; Hammond, S.M. Lin-28 interaction with the let-7 precursor loop mediates regulated microRNA processing. RNA 2008, 14, 1539–1549. [Google Scholar] [CrossRef] [PubMed]
- Schwamborn, J.C.; Berezikov, E.; Knoblich, J.A. The TRIM-NHL protein TRIM32 activates microRNAs and prevents self-renewal in mouse neural progenitors. Cell 2009, 136, 913–925. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.H.; Moss, E.G. Temporally regulated expression of lin-28 in diverse tissues of the developing mouse. Gene Expression Patterns 2003, 3, 719–726. [Google Scholar] [CrossRef]
- Moss, E.G.; Lee, R.C.; Ambros, V. The cold shock domain protein lin-28 controls developmental timing in C. elegans and is regulated by the lin-4 RNA. Cell 1997, 88, 637–646. [Google Scholar] [CrossRef]
- Lehrbach, N.J.; Armisen, J.; Lightfoot, H.L.; Murfitt, K.J.; Bugaut, A.; Balasubramanian, S.; Miska, E.A. Lin-28 and the poly(U) polymerase PUP-2 regulate let-7 microRNA processing in Caenorhabditis elegans. Nat. Struct. Mol. Biol. 2009, 16, 1016–1020. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Chen, Y.; Ito, H.; Watanabe, A.; Ge, X.; Kodama, T.; Aburatani, H. Identification and characterization of lin-28 homolog B (LIN28B) in human hepatocellular carcinoma. Gene 2006, 384, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Charlesworth, A.; Meijer, H.A.; de Moor, C.H. Specificity factors in cytoplasmic polyadenylation. Wiley Interdiscip. Rev. RNA 2013, 4, 437–461. [Google Scholar] [CrossRef] [PubMed]
- Chaudhury, A.; Chander, P.; Howe, P.H. Heterogeneous nuclear ribonucleoproteins (hnRNPs) in cellular processes: Focus on hnRNP E1’s multifunctional regulatory roles. RNA 2010, 16, 1449–1462. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-Synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Giasson, B.I.; Duda, J.E.; Murray, I.V.; Chen, Q.; Souza, J.M.; Hurtig, H.I.; Ischiropoulos, H.; Trojanowski, J.Q.; Lee, V.M. Oxidative damage linked to neurodegeneration by selective α-synuclein nitration in synucleinopathy lesions. Science 2000, 290, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Berson, A.; Barbash, S.; Shaltiel, G.; Goll, Y.; Hanin, G.; Greenberg, D.S.; Ketzef, M.; Becker, A.J.; Friedman, A.; Soreq, H. Cholinergic-associated loss of hnRNP-A/B in Alzheimer’s disease impairs cortical splicing and cognitive function in mice. EBMO Mol. Med. 2012, 4, 730–742. [Google Scholar] [CrossRef] [PubMed]
- Bekenstein, U.; Soreq, H. Heterogeneous nuclear ribonucleoprotein A1 in health and neurodegenerative disease: From structural insights to post-transcriptional regulatory roles. Mol. Cell. Neurosci. 2013, 56, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Trabucchi, M.; Briata, P.; Garcia-Mayoral, M.; Haase, A.D.; Filipowicz, W.; Ramos, A.; Gherzi, R.; Rosenfeld, M.G. The RNA-binding protein KSRP promotes the biogenesis of a subset of microRNAs. Nature 2009, 459, 1010–1014. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Pedersen, J.S.; Kwon, S.C.; Belair, C.D.; Kim, Y.-K.; Yeom, K.-H.; Yang, W.-Y.; Haussler, D.; Blelloch, R.; Kim, V.N. Posttranscriptional crossregulation between Drosha and DGCR8. Cell 2009, 136, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Schratt, G.M.; Tuebing, F.; Nigh, E.A.; Kane, C.G.; Sabatini, M.E.; Kiebler, M.; Greenberg, M.E. A brain-specific microRNA regulates dendritic spine development. Nature 2006, 439, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wang, W.-Y.; Mao, Y.-W.; Gräff, J.; Guan, J.-S.; Pan, L.; Mak, G.; Kim, D.; Su, S.C.; Tsai, L.-H. A novel pathway regulates memory and plasticity via SIRT1 and miR-134. Nature 2010, 466, 1105–1109. [Google Scholar] [CrossRef] [PubMed]
- Hagan, J.P.; Piskounova, E.; Gregory, R.I. Lin28 recruits the TUTase Zcchc11 to inhibit let-7 maturation in mouse embryonic stem cells. Nat. Struct. Mol. Biol. 2009, 16, 1021–1025. [Google Scholar] [CrossRef] [PubMed]
- Abdelmohsen, K.; Tominaga-Yamanaka, K.; Srikantan, S.; Yoon, J.H.; Kang, M.J.; Gorospe, M. RNA-binding protein AUF1 represses dicer expression. Nucleic Acids Res. 2012, 40, 11531–11544. [Google Scholar] [CrossRef] [PubMed]
- O’Carroll, D.; Schaefer, A. General principals of miRNA biogenesis and regulation in the brain. Neuropsychopharmacology 2013, 38, 39–54. [Google Scholar] [CrossRef] [PubMed]
- King, I.N.; Yartseva, V.; Salas, D.; Kumar, A.; Heidersbach, A.; Ando, D.M.; Stallings, N.R.; Elliott, J.L.; Srivastava, D.; Ivey, K.N. The RNA-binding protein TDP-43 selectively disrupts microRNA-1/206 incorporation into the RNA-induced silencing complex. J. Biol. Chem. 2014, 289, 14263–14271. [Google Scholar] [CrossRef] [PubMed]
- Muddashetty, R.S.; Nalavadi, V.C.; Gross, C.; Yao, X.; Xing, L.; Laur, O.; Warren, S.T.; Bassell, G.J. Reversible inhibition of PSD-95 mRNA translation by miR-125a, FMRP phosphorylation, and mGLUR signaling. Mol. Cell 2011, 42, 673–688. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Chen, X.; Chen, R. Transcriptome-wide analysis of TDP-43 binding small RNAs identifies miR-NID1 (miR-8485), a novel miRNA that represses NRXN1 expression. Genomics 2014, 103, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, K.; Srikantan, S.; Lee, E.K.; Subaran, S.S.; Martindale, J.L.; Abdelmohsen, K.; Gorospe, M. Competitive regulation of nucleolin expression by HuR and miR-494. Mol. Cell. Biol. 2011, 31, 4219–4231. [Google Scholar] [CrossRef] [PubMed]
- Sosanya, N.M.; Huang, P.P.C.; Cacheaux, L.P.; Chen, C.J.; Nguyen, K.; Perrone-Bizzozero, N.I.; Raab-Graham, K.F. Degradation of high affinity HuD targets releases kv1.1 mRNA from miR-129 repression by mTORC1. J. Cell Biol. 2013, 202, 53–69. [Google Scholar] [CrossRef] [PubMed]
- Jafarifar, F.; Yao, P.; Eswarappa, S.M.; Fox, P.L. Repression of VEGFA by Ca-rich element-binding microRNAs is modulated by hnRNP L. EMBO J. 2011, 30, 1324–1334. [Google Scholar] [CrossRef] [PubMed]
- Ciafrè, S.A.; Galardi, S. MicroRNAs and RNA-binding proteins. RNA Biol. 2014, 10, 934–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, P.; Coller, H. Functional interactions between microRNAs and RNA binding proteins. MicroRNA 2012, 1, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Caudy, A.A.; Myers, M.; Hannon, G.J.; Hammond, S.M. Fragile X-related protein and VIG associate with the RNA interference machinery. Genes Dev. 2002, 16, 2491–2496. [Google Scholar] [CrossRef] [PubMed]
- Santoro, M.R.; Bray, S.M.; Warren, S.T. Molecular mechanisms of Fragile X syndrome: A twenty-year perspective. Ann. Rev. Pathol. 2012, 7, 219–245. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Biou, V.; Xu, W.; Schlüter, O.; Malenka, R.C. A critical role for PSD-95/AKAP interactions in endocytosis of synaptic AMPA receptors. Nat. Neurosci. 2009, 12, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Schutt, J.; Falley, K.; Richter, D.; Kreienkamp, H.J.; Kindler, S. Fragile X mental retardation protein regulates the levels of scaffold proteins and glutamate receptors in postsynaptic densities. J. Biol. Chem. 2009, 284, 25479–25487. [Google Scholar] [CrossRef] [PubMed]
- Polymenidou, M.; Lagier-Tourenne, C.; Hutt, K.R.; Huelga, S.C.; Moran, J.; Liang, T.Y.; Ling, S.C.; Sun, E.; Wancewicz, E.; Mazur, C.; et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci. 2011, 14, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Caudle, W.M.; Kitsou, E.; Li, J.; Bradner, J.; Zhang, J. A role for a novel protein, nucleolin, in Parkinson’s disease. Neurosci. Lett. 2009, 459, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Tsoi, H.; Chan, H.Y. Expression of expanded CAG transcripts triggers nucleolar stress in Huntington’s disease. Cerebellum 2013, 12, 310–312. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.H.; Kuwano, Y.; Srikantan, S.; Lee, E.K.; Martindale, J.L.; Gorospe, M. HuR recruits let-7/RISC to repress C-mMc expression. Genes Dev. 2009, 23, 1743–1748. [Google Scholar] [CrossRef] [PubMed]
- Filipowicz, W.; Bhattacharyya, S.N.; Sonenberg, N. Mechanisms of post-transcriptional regulation by microRNAs: Are the answers in sight? Nat. Rev. Genet. 2008, 2008, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Ouyang, K.; Huang, J.; Zhou, Y.; Ouyang, H.; Li, H.; Wang, G.; Wu, Q.; Wei, C.; Bi, Y.; et al. Direct conversion of fibroblasts to neurons by reprogramming PTB-regulated microRNA circuits. Cell 2013, 152, 82–96. [Google Scholar] [CrossRef] [PubMed]
- Kundu, P.; Fabian, M.R.; Sonenberg, N.; Bhattacharyya, S.N.; Filipowicz, W. Hur protein attenuates miRNA-mediated repression by promoting miRISC dissociation from the target RNA. Nucleic Acids Res. 2012, 40, 5088–5100. [Google Scholar] [CrossRef] [PubMed]
- Shih, S.C.; Claffey, K.P. Regulation of human vascular endothelial growth factor mRNA stability in hypoxia by heterogeneous nuclear ribonucleoprotein l. J. Biol. Chem. 1999, 274, 1359–1365. [Google Scholar] [CrossRef] [PubMed]
- Moreau, C.; Gosset, P.; Kluza, J.; Brunaud-Danel, V.; Lassalle, P.; Marchetti, P.; Defebvre, L.; Destée, A.; Devos, D. Deregulation of the hypoxia inducible factor-1α pathway in monocytes from sporadic Amyotrophic Lateral Sclerosis patients. Neuroscience 2011, 172, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Wan, W.; Cao, L.; Kalionis, B.; Xia, S.; Tai, X. Applications of induced pluripotent stem cells in studying the neurodegenerative diseases. Stem Cells Int. 2015. [Google Scholar] [CrossRef] [PubMed]
- Ruggieri, M.; Riboldi, G.; Brajkovic, S.; Bucchia, M.; Bresolin, N.; Comi, G.P.; Corti, S. Induced neural stem cells: Methods of reprogramming and potential therapeutic applications. Prog. Neurobiol. 2014, 114, 15–24. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loffreda, A.; Rigamonti, A.; Barabino, S.M.L.; Lenzken, S.C. RNA-Binding Proteins in the Regulation of miRNA Activity: A Focus on Neuronal Functions. Biomolecules 2015, 5, 2363-2387. https://doi.org/10.3390/biom5042363
Loffreda A, Rigamonti A, Barabino SML, Lenzken SC. RNA-Binding Proteins in the Regulation of miRNA Activity: A Focus on Neuronal Functions. Biomolecules. 2015; 5(4):2363-2387. https://doi.org/10.3390/biom5042363
Chicago/Turabian StyleLoffreda, Alessia, Aurora Rigamonti, Silvia M. L. Barabino, and Silvia C. Lenzken. 2015. "RNA-Binding Proteins in the Regulation of miRNA Activity: A Focus on Neuronal Functions" Biomolecules 5, no. 4: 2363-2387. https://doi.org/10.3390/biom5042363