
The Mitochondrial Citrate Carrier SLC25A1/CIC and the Fundamental Role of Citrate in Cancer, Inflammation and Beyond

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
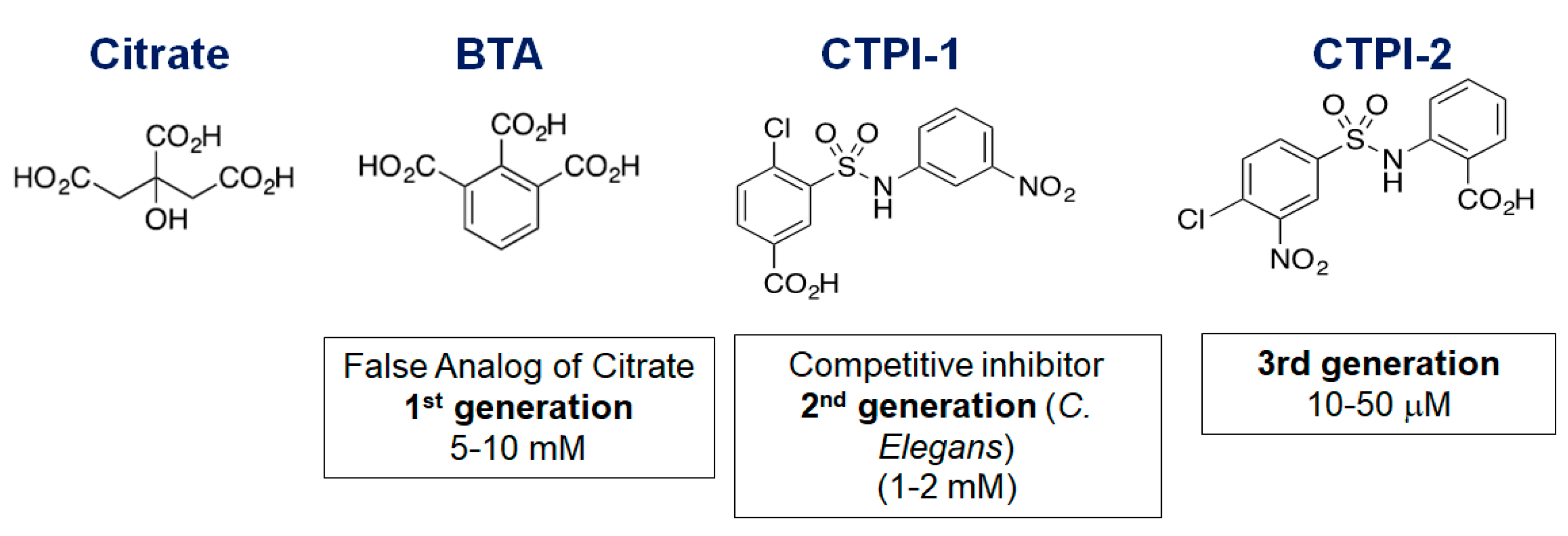
2. A Brief History of CIC Inhibitor Compounds
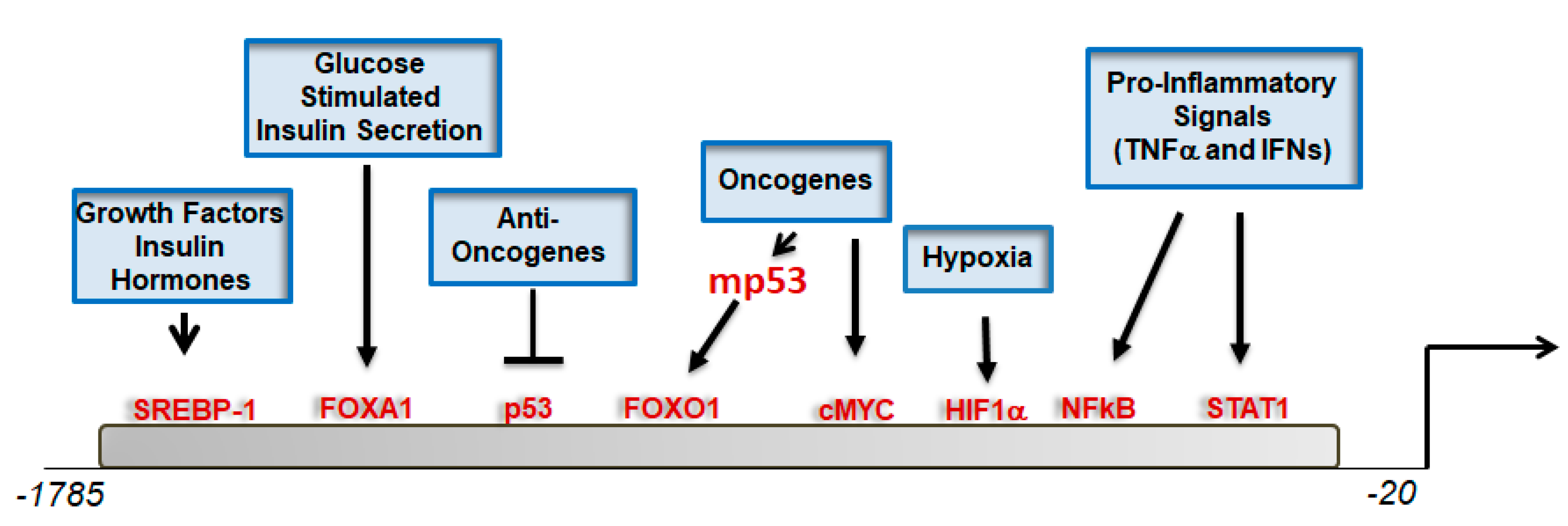
3. Regulation of CIC Expression Levels: Hints on Its Biological Functions
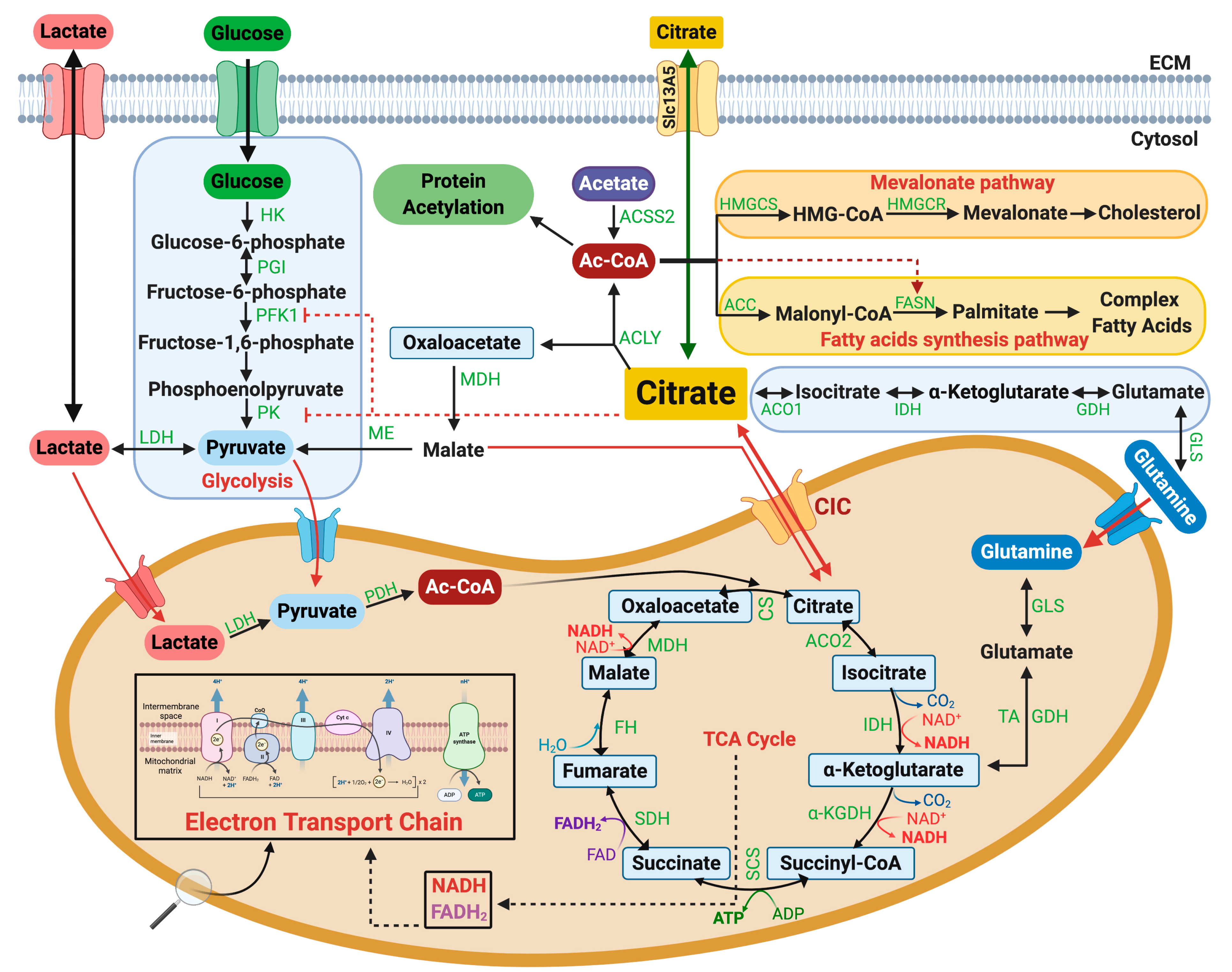
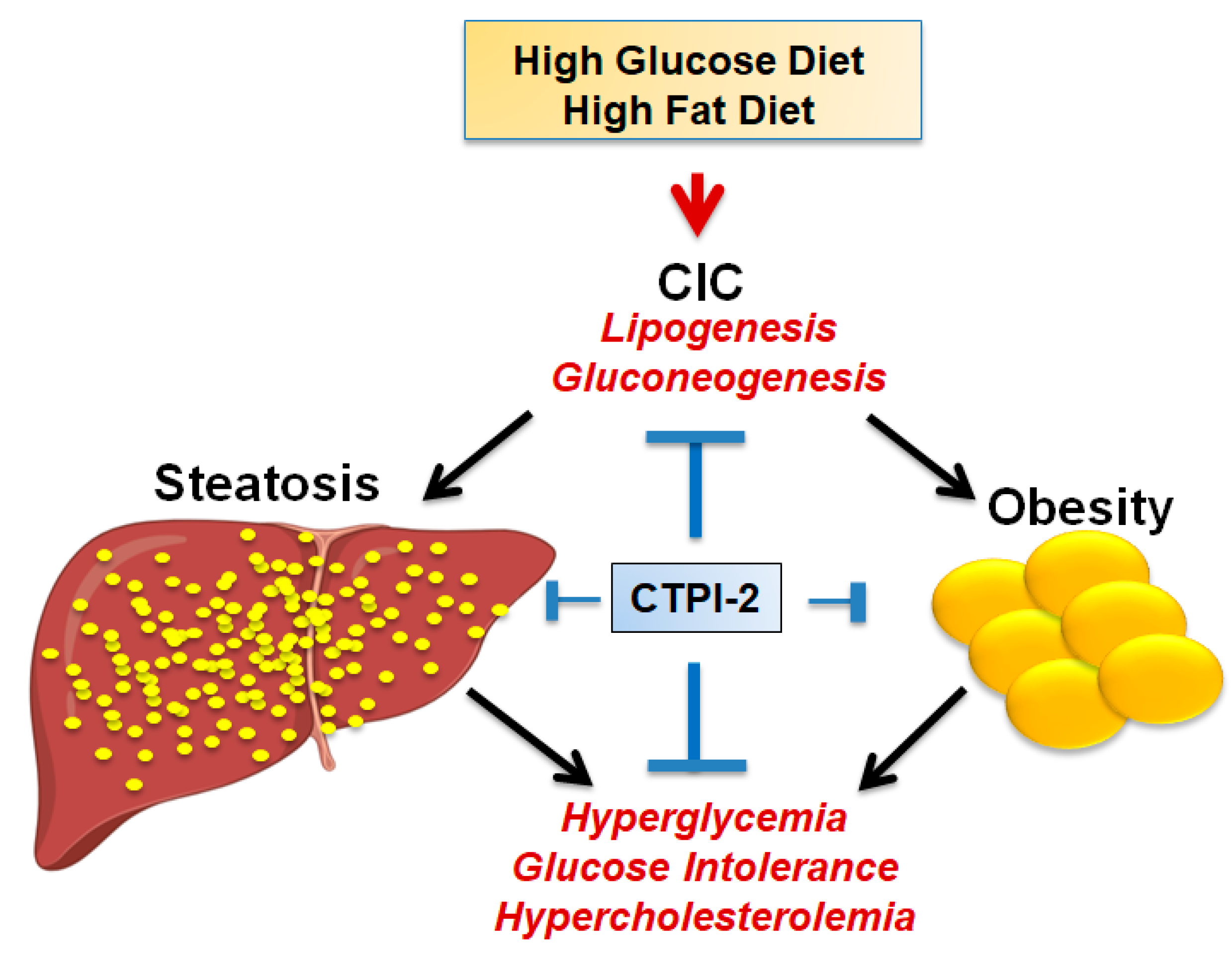
4. CIC Activity in Glucose and Lipid Metabolism: Implications for Metabolic Diseases
5. Is CIC Rate Limiting for De Novo Lipid Synthesis?
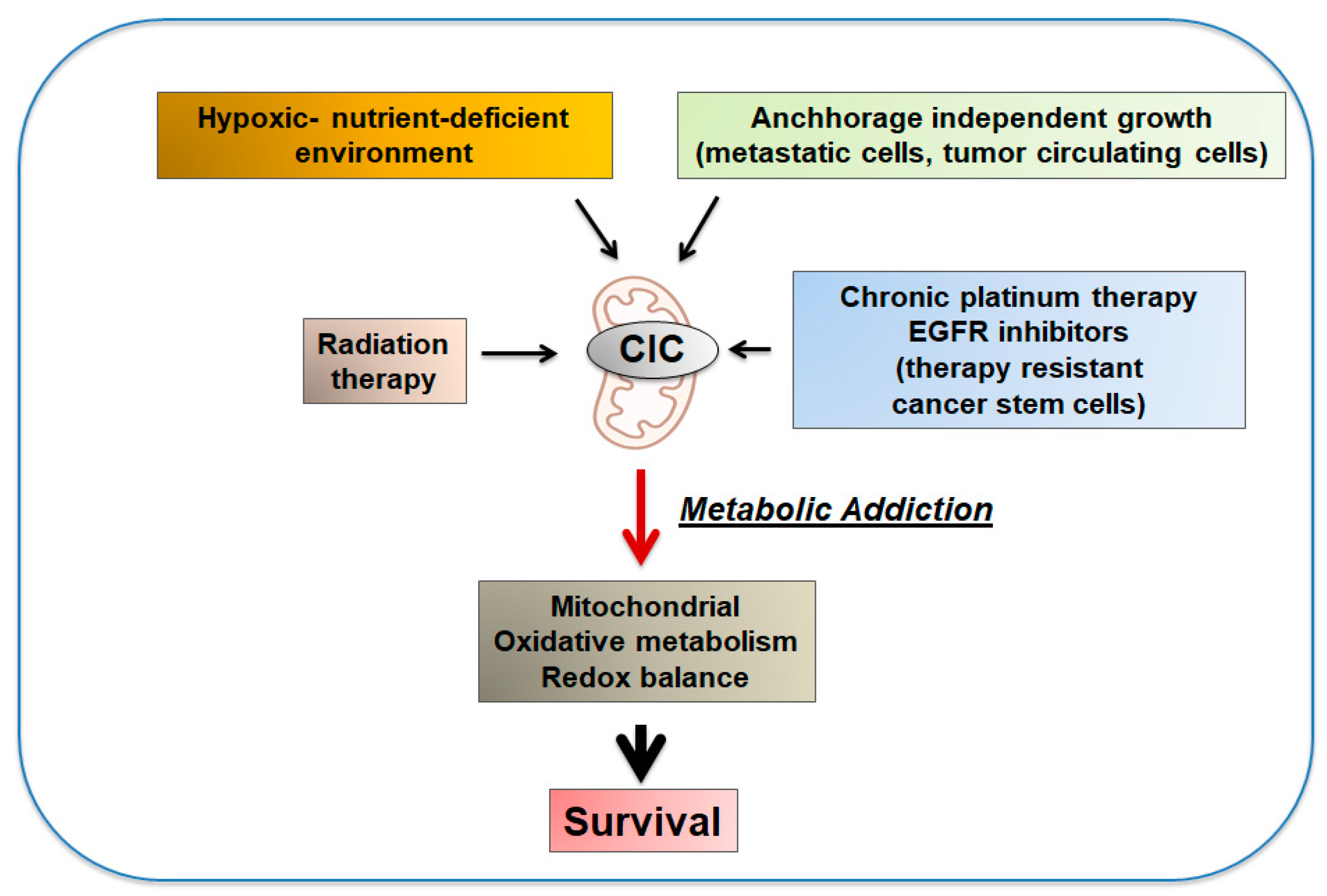
6. Pro-Oncogenic Activities of CIC, the Reversal of the Warburg Effect and the Phenomenon of Metabolic Addiction
7. CIC Inhibition Inhibits the Growth of Different Tumor Types
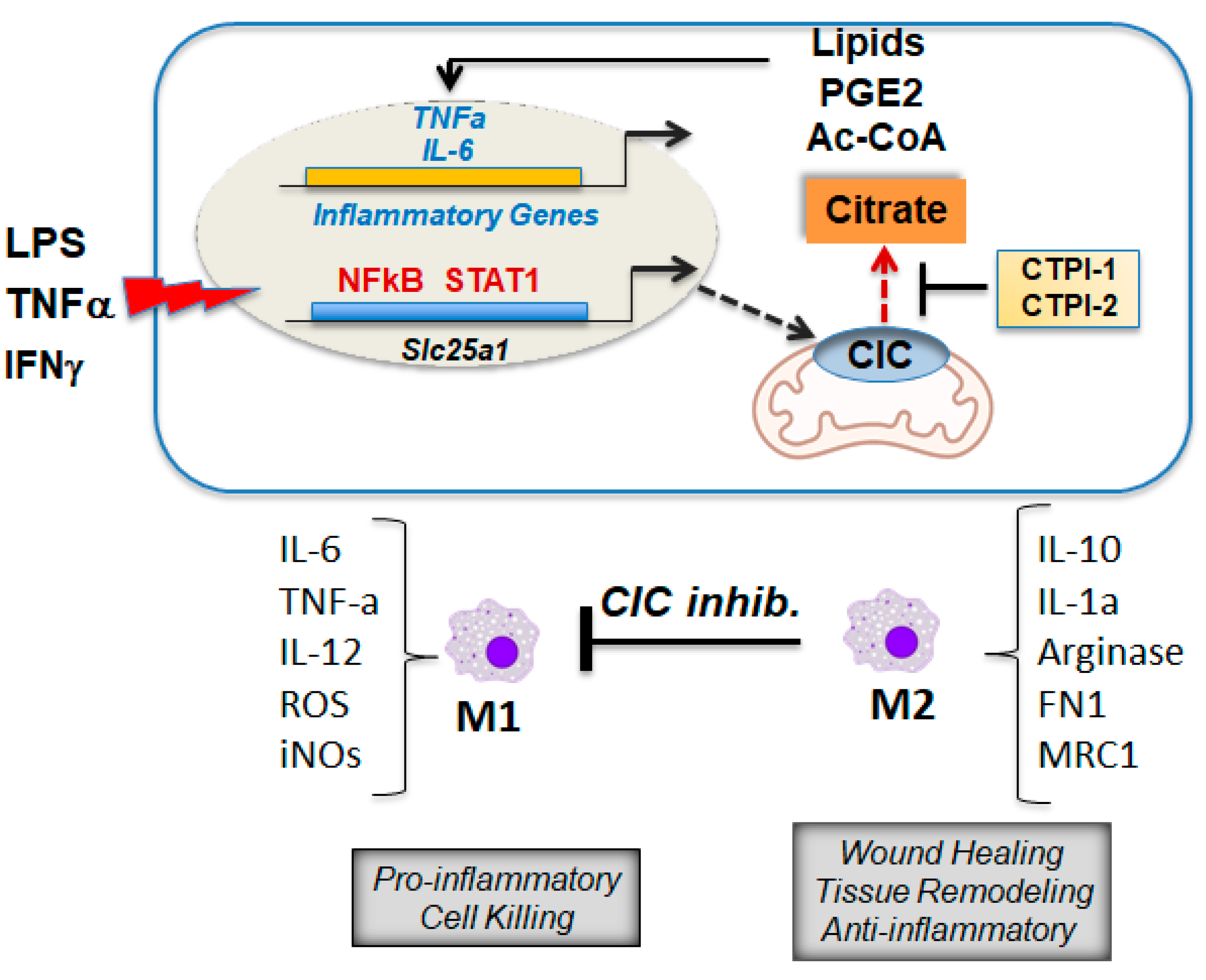
8. CIC and Citrate Are Important Mediators of Inflammation
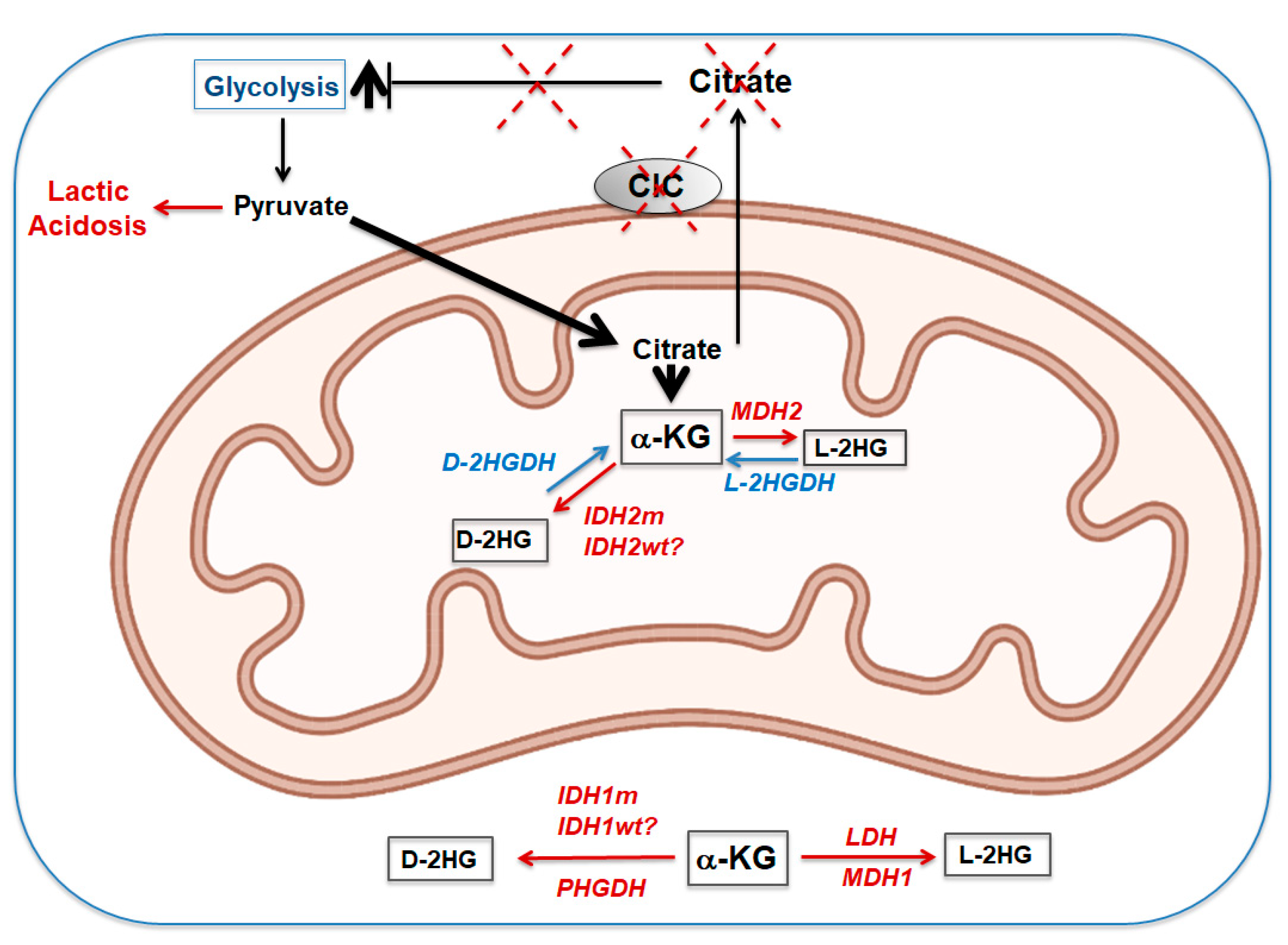
9. Loss of CIC during Embryogenesis and Development: The L-and D-2HGA “Affair”
- What levels of L-D-2HG in the context of SLC25A1 gene deficiency are to be considered pathogenic?
- Given that L- or D-HGs interfere with α-KG-dependent dioxygenases and affect methylation of histones and DNA, are alterations in L-D-2HG levels reflected in changes in the epigenetic landscape of CIC-deficient cells?
- What is the origin of L-D-2HG accumulation in CIC-deficient cells?
- Can correction of α-KG levels normalize any of the pathological manifestations seen in children with CIC deficiency?
- Are there differences in the lipid synthetic pathway in diseases sustained by CIC deficiency that aggravate the clinical manifestations?
- Is the multi-organ involvement seen in severe CIC deficiency syndromes due to systemic alterations (e.g., changes in circulating levels of TCA cycle intermediates, accumulation of toxic byproducts of the metabolism and elevated levels of 2-HGs) or to loss of tissue-specific activities of CIC?
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Stipani, I.; Kramer, R.; Palmieri, F.; Klingenberg, M. Citrate transport in liposomes reconstituted with Triton extracts from mitochondria. Biochem. Biophys. Res. Commun. 1980, 97, 1206–1214. [Google Scholar] [CrossRef]
- Stipani, I.; Palmieri, F. Purification of the active mitochondrial tricarboxylate carrier by hydroxylapatite chromatography. FEBS Lett. 1983, 161, 269–274. [Google Scholar] [CrossRef] [Green Version]
- Bisaccia, F.; de Palma, A.; Palmieri, F. Identification and purification of the tricarboxylate carrier from rat liver mitochondria. Biochim. Biophys. Acta Gen. Subj. 1989, 977, 171–176. [Google Scholar] [CrossRef]
- Kaplan, R.; Mayor, J.; Wood, D. The mitochondrial tricarboxylate transport protein. cDNA cloning, primary structure, and comparison with other mitochondrial transport proteins. J. Biol. Chem. 1993, 268, 13682–13690. [Google Scholar] [CrossRef]
- Palmieri, F.; Scarcia, P.; Monné, M. Diseases Caused by Mutations in Mitochondrial Carrier Genes SLC25: A Review. Biomolecules 2020, 10, 655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunji, E.R.S.; King, M.S.; Ruprecht, J.J.; Thangaratnarajah, C. The SLC25 Carrier Family: Important Transport Proteins in Mitochondrial Physiology and Pathology. Physiology 2020, 35, 302–327. [Google Scholar] [CrossRef]
- van Schaftingen, E.; Hers, H.G. Inhibition of fructose-1,6-bisphosphatase by fructose 2,6-biphosphate. Proc. Natl. Acad. Sci. USA 1981, 78, 2861–2863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Icard, P.; Fournel, L.; Coquerel, A.; Gligorov, J.; Alifano, M.; Lincet, H. Citrate targets FBPase and constitutes an emerging novel approach for cancer therapy. Cancer Cell Int. 2018, 18, 175. [Google Scholar] [CrossRef]
- Munday, M.R. Regulation of Mammalian Acetyl-CoA Carboxylase. Biochem. Soc. Trans. 2002, 30, 1059–1064. [Google Scholar] [CrossRef]
- Fernandez, H.R.; Gadre, S.M.; Tan, M.; Graham, G.T.; Mosaoa, R.; Ongkeko, M.S.; Kim, K.A.; Riggins, R.B.; Parasido, E.; Petrini, I.; et al. The mitochondrial citrate carrier, SLC25A1, drives stemness and therapy resistance in non-small cell lung cancer. Cell Death Differ. 2018, 25, 1239–1258. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Shestov, A.A.; Swain, P.; Yang, C.; Parker, S.J.; Wang, Q.A.; Terada, L.S.; Adams, N.D.; McCabe, M.T.; Pietrak, B.; et al. Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nat. Cell Biol. 2016, 532, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Morciano, P.; Carrisi, C.; Capobianco, L.; Mannini, L.; Burgio, G.; Cestra, G.; de Benedetto, G.E.; Corona, D.F.; Musio, A.; Cenci, G. A conserved role for the mitochondrial citrate transporter Sea/SLC25A1 in the maintenance of chromosome integrity. Hum. Mol. Genet. 2009, 18, 4180–4188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santarsiero, A.; Leccese, P.; Convertini, P.; Padula, A.; Abriola, P.; D’Angelo, S.; Bisaccia, F.; Infantino, V. New Insights into Behçet’s Syndrome Metabolic Reprogramming: Citrate Pathway Dysregulation. Mediat. Inflamm. 2018, 2018, 1419352. [Google Scholar] [CrossRef] [PubMed]
- Convertini, P.; Menga, A.; Andria, G.; Scala, I.; Santarsiero, A.; Morelli, M.A.C.; Iacobazzi, V.; Infantino, V. The contribution of the citrate pathway to oxidative stress in Down syndrome. Immunology 2016, 149, 423–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catalina-Rodriguez, O.; Kolukula, V.K.; Tomita, Y.; Preet, A.; Palmieri, F.; Wellstein, A.; Byers, S.; Giaccia, A.J.; Glasgow, E.; Albanese, C.; et al. The mitochondrial citrate transporter, CIC, is essential for mitochondrial homeostasis. Oncotarget 2012, 3, 1220–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, M.; Mosaoa, R.; Graham, G.T.; Kasprzyk-Pawelec, A.; Gadre, S.; Parasido, E.; Catalina-Rodriguez, O.; Foley, P.; Giaccone, G.; Cheema, A.; et al. Inhibition of the mitochondrial citrate carrier, Slc25a1, reverts steatosis, glucose intolerance, and inflammation in preclinical models of NAFLD/NASH. Cell Death Differ. 2020, 27, 2143–2157. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Aluvila, S.; Kotaria, R.; Mayor, J.A.; Walters, D.E.; Kaplan, R.S. Mitochondrial and Plasma Membrane Citrate Transporters: Discovery of Selective Inhibitors and Application to Structure/Function Analysis. Mol. Cell. Pharmacol. 2010, 2, 101–110. [Google Scholar]
- Infantino, V.; Iacobazzi, V.; de Santis, F.; Mastrapasqua, M.; Palmieri, F. Transcription of the mitochondrial citrate carrier gene: Role of SREBP-1, upregulation by insulin and downregulation by PUFA. Biochem. Biophys. Res. Commun. 2007, 356, 249–254. [Google Scholar] [CrossRef]
- Iacobazzi, V.; Infantino, V.; Bisaccia, F.; Castegna, A.; Palmieri, F. Role of FOXA in mitochondrial citrate carrier gene expression and insulin secretion. Biochem. Biophys. Res. Commun. 2009, 385, 220–224. [Google Scholar] [CrossRef]
- Kolukula, V.K.; Sahu, G.; Wellstein, A.; Rodriguez, O.C.; Preet, A.; Iacobazzi, V.; D’Orazi, G.; Albanese, C.; Palmieri, F.; Avantaggiati, M.L. SLC25A1, or CIC, is a novel transcriptional target of mutant p53 and a negative tumor prognostic marker. Oncotarget 2014, 5, 1212–1225. [Google Scholar] [CrossRef] [Green Version]
- Infantino, V.; Iacobazzi, V.; Menga, A.; Avantaggiati, M.L.; Palmieri, F. A key role of the mitochondrial citrate carrier (SLC25A1) in TNFα- and IFNγ-triggered inflammation. Biochim. Biophys. Acta Bioenerg. 2014, 1839, 1217–1225. [Google Scholar] [CrossRef] [Green Version]
- Damiano, F.; Gnoni, G.V.; Siculella, L. Citrate carrier promoter is target of peroxisome proliferator-activated receptor alpha and gamma in hepatocytes and adipocytes. Int. J. Biochem. Cell Biol. 2012, 44, 659–668. [Google Scholar] [CrossRef]
- Huang, Z.; Zhang, M.; Plec, A.A.; Estill, S.J.; Cai, L.; Repa, J.J.; McKnight, S.L.; Tu, B.P. ACSS2 promotes systemic fat storage and utilization through selective regulation of genes involved in lipid metabolism. Proc. Natl. Acad. Sci. USA 2018, 115, E9499–E9506. [Google Scholar] [CrossRef] [Green Version]
- Schug, Z.T.; Peck, B.; Jones, D.T.; Zhang, Q.; Grosskurth, S.; Alam, I.S.; Goodwin, L.M.; Smethurst, E.; Mason, S.; Blyth, K.; et al. Acetyl-CoA Synthetase 2 Promotes Acetate Utilization and Maintains Cancer Cell Growth under Metabolic Stress. Cancer Cell 2015, 27, 57–71. [Google Scholar] [CrossRef] [Green Version]
- Mazurek, M.P.; Prasad, P.D.; Gopal, E.; Fraser, S.P.; Bolt, L.; Rizaner, N.; Palmer, C.P.; Foster, C.S.; Palmieri, F.; Ganapathy, V.; et al. Molecular origin of plasma membrane citrate transporter in human prostate epithelial cells. EMBO Rep. 2010, 11, 431–437. [Google Scholar] [CrossRef]
- Mycielska, M.E.; Dettmer, K.; Rümmele, P.; Schmidt, K.M.; Prehn, C.; Milenkovic, V.M.; Jagla, W.; Madej, G.M.; Lantow, M.; Schladt, M.T.; et al. Extracellular Citrate Affects Critical Elements of Cancer Cell Metabolism and Supports Cancer Development In Vivo. Cancer Res. 2018, 78, 2513–2523. [Google Scholar] [CrossRef] [Green Version]
- Rogina, B. INDY—A New Link to Metabolic Regulation in Animals and Humans. Front. Genet. 2017, 8, 66. [Google Scholar] [CrossRef] [Green Version]
- Brachs, S.; Winkel, A.F.; Tang, H.; Birkenfeld, A.L.; Brunner, B.; Jahn-Hofmann, K.; Margerie, D.; Ruetten, H.; Schmoll, D.; Spranger, J. Inhibition of citrate cotransporter Slc13a5/mINDY by RNAi improves hepatic insulin sensitivity and prevents diet-induced non-alcoholic fatty liver disease in mice. Mol. Metab. 2016, 5, 1072–1082. [Google Scholar] [CrossRef]
- Mycielska, M.E.; Mohr, M.T.J.; Schmidt, K.; Drexler, K.; Rümmele, P.; Haferkamp, S.; Schlitt, H.J.; Gaumann, A.; Adamski, J.; Geissler, E.K. Potential Use of Gluconate in Cancer Therapy. Front. Oncol. 2019, 9, 522. [Google Scholar] [CrossRef]
- Mullen, A.R.; Wheaton, W.W.; Jin, E.S.; Chen, P.-H.; Sullivan, L.B.; Cheng, T.; Yang, Y.; Linehan, W.M.; Chandel, N.S.; de Berardinis, R.J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nat. Cell Biol. 2011, 481, 385–388. [Google Scholar] [CrossRef] [Green Version]
- Sun, R.C.; Denko, N.C. Hypoxic Regulation of Glutamine Metabolism through HIF1 and SIAH2 Supports Lipid Synthesis that Is Necessary for Tumor Growth. Cell Metab. 2014, 19, 285–292. [Google Scholar] [CrossRef] [Green Version]
- Fendt, S.-M.; Bell, E.L.; Keibler, M.A.; Olenchock, B.A.; Mayers, J.R.; Wasylenko, T.M.; Vokes, N.I.; Guarente, L.; Heiden, M.G.V.; Stephanopoulos, G. Reductive glutamine metabolism is a function of the α-ketoglutarate to citrate ratio in cells. Nat. Commun. 2013, 4, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Boufersaoui, A.; Yang, C.; Ko, B.; Rakheja, D.; Guevara, G.; Hu, Z.; de Berardinis, R.J. Quantitative metabolic flux analysis reveals an unconventional pathway of fatty acid synthesis in cancer cells deficient for the mitochondrial citrate transport protein. Metab. Eng. 2017, 43, 198–207. [Google Scholar] [CrossRef]
- Napoli, E.; Tassone, F.; Wong, S.; Angkustsiri, K.; Simon, T.J.; Song, G.; Giulivi, C. Mitochondrial Citrate Transporter-dependent Metabolic Signature in the 22q11.2 Deletion Syndrome. J. Biol. Chem. 2015, 290, 23240–23253. [Google Scholar] [CrossRef] [Green Version]
- Faubert, B.; Li, K.Y.; Cai, L.; Hensley, C.T.; Kim, J.; Zacharias, L.G.; Yang, C.; Brandon, F.; Doucette, S.; Burguete, D.; et al. Lactate Metabolism in Human Lung Tumors. Cell 2017, 171, 358–371.e9. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.-H.; Cai, L.; Huffman, K.; Yang, C.; Kim, J.; Faubert, B.; Boroughs, L.; Ko, B.; Sudderth, J.; McMillan, E.A.; et al. Metabolic Diversity in Human Non-Small Cell Lung Cancer Cells. Mol. Cell 2019, 76, 838–851.e5. [Google Scholar] [CrossRef]
- Sellers, K.; Fox, M.P.; Bousamra, M.; Slone, S.P.; Higashi, R.M.; Miller, D.M.; Wang, Y.; Yan, J.; Yuneva, M.O.; Deshpande, R.; et al. Pyruvate carboxylase is critical for non-small-cell lung cancer proliferation. J. Clin. Investig. 2015, 125, 687–698. [Google Scholar] [CrossRef] [Green Version]
- Vasan, K.; Werner, M.; Chandel, N.S. Mitochondrial Metabolism as a Target for Cancer Therapy. Cell Metab. 2020, 32, 341–352. [Google Scholar] [CrossRef]
- Hlouschek, J.; Hansel, C.; Jendrossek, V.; Matschke, J. The Mitochondrial Citrate Carrier (SLC25A1) Sustains Redox Homeostasis and Mitochondrial Metabolism Supporting Radioresistance of Cancer Cells with Tolerance to Cycling Severe Hypoxia. Front. Oncol. 2018, 8, 170. [Google Scholar] [CrossRef]
- Gandhi, N.; Das, G.M. Metabolic Reprogramming in Breast Cancer and Its Therapeutic Implications. Cells 2019, 8, 89. [Google Scholar] [CrossRef] [Green Version]
- Özkaya, A.B.; Ak, H.; Atay, S.; Aydin, H.H. Targeting Mitochondrial Citrate Transport in Breast Cancer Cell Lines. Anti-Cancer Agents Med. Chem. 2015, 15, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Bhalla, K.; Hwang, B.J.; Dewi, R.E.; Ou, L.; Twaddel, W.; Fang, H.-B.; Vafai, S.B.; Vazquez, F.; Puigserver, P.; Boros, L.; et al. PGC1 Promotes Tumor Growth by Inducing Gene Expression Programs Supporting Lipogenesis. Cancer Res. 2011, 71, 6888–6898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebadi, M.; Field, C.J.; Lehner, R.; Mazurak, V. Chemotherapy diminishes lipid storage capacity of adipose tissue in a preclinical model of colon cancer. Lipids Health Dis. 2017, 16, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, R.S.; Morris, H.P.; Coleman, P.S. Kinetic characteristics of citrate influx and efflux with mitochondria from Morris hepatomas 3924A and 16. Cancer Res. 1982, 42, 4399–4407. [Google Scholar]
- Zhu, P.; Wang, Y.; Wu, J.; Huang, G.; Liu, B.; Ye, B.; Du, Y.; Gao, G.; Tian, Y.; He, L.; et al. LncBRM initiates YAP1 signalling activation to drive self-renewal of liver cancer stem cells. Nat. Commun. 2016, 7, 13608. [Google Scholar] [CrossRef] [Green Version]
- Poolsri, W.-A.; Phokrai, P.; Suwankulanan, S.; Phakdeeto, N.; Phunsomboon, P.; Pekthong, D.; Richert, L.; Pongcharoen, S.; Srisawang, P. Combination of Mitochondrial and Plasma Membrane Citrate Transporter Inhibitors Inhibits De Novo Lipogenesis Pathway and Triggers Apoptosis in Hepatocellular Carcinoma Cells. BioMed Res. Int. 2018, 2018, 1–15. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, D.; Chen, L.; Gao, S.; Huang, X. Long Non-coding RNA BRM Promotes Proliferation and Invasion of Papillary Thyroid Carcinoma by Regulating the MicroRNA-331-3p/SLC25A1 Axis. Oncol. Lett. 2020, 19, 3071–3078. [Google Scholar] [CrossRef]
- Liu, Y.; Zuckier, L.S.; Ghesani, N.V. Dominant uptake of fatty acid over glucose by prostate cells: A potential new diagnostic and therapeutic approach. Anticancer Res. 2010, 30, 369–374. [Google Scholar]
- Mitra, R.; Le, T.T. Enhanced detection of metastatic prostate cancer cells in human plasma with lipid bodies staining. BMC Cancer 2014, 14, 91. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Ma, S.; Ruzzo, W.L. Spatial modeling of prostate cancer metabolic gene expression reveals extensive heterogeneity and selective vulnerabilities. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [Green Version]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef]
- Infantino, V.; Convertini, P.; Cucci, L.; Panaro, M.A.; di Noia, M.A.; Calvello, R.; Palmieri, F.; Iacobazzi, V. The mitochondrial citrate carrier: A new player in inflammation. Biochem. J. 2011, 438, 433–436. [Google Scholar] [CrossRef] [Green Version]
- Infantino, V.; Iacobazzi, V.; Palmieri, F.; Menga, A. ATP-citrate lyase is essential for macrophage inflammatory response. Biochem. Biophys. Res. Commun. 2013, 440, 105–111. [Google Scholar] [CrossRef]
- Assmann, N.; O’Brien, K.L.; Donnelly, R.P.; Dyck, L.; Zaiatz-Bittencourt, V.; Loftus, R.M.; Heinrich, P.; Oefner, P.J.; Lynch, L.; Gardiner, C.M.; et al. Srebp-controlled glucose metabolism is essential for NK cell functional responses. Nat. Immunol. 2017, 18, 1197–1206. [Google Scholar] [CrossRef]
- Mendez-Sanchez, N.; Valencia-Rodríguez, A.; Coronel-Castillo, C.; Vera-Barajas, A.; Contreras-Carmona, J.; Ponciano-Rodríguez, G.; Zamora-Valdés, D. The cellular pathways of liver fibrosis in non-alcoholic steatohepatitis. Ann. Transl. Med. 2020, 8, 400. [Google Scholar] [CrossRef]
- Li, H.; Zhou, Y.; Wang, H.; Zhang, M.; Qiu, P.; Zhang, R.; Zhao, Q.; Liu, J. Crosstalk Between Liver Macrophages and Surrounding Cells in Nonalcoholic Steatohepatitis. Front. Immunol. 2020, 11, 1169. [Google Scholar] [CrossRef]
- Wang, H.; Li, Z.; Niu, J.; Xu, Y.; Ma, L.; Lu, A.; Wang, X.; Qian, Z.; Huang, Z.; Jin, X.; et al. Antiviral effects of ferric ammonium citrate. Cell Discov. 2018, 4, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Stoffel, M.; Karayiorgou, M.; Espinosa, R.; Beau, M.M.L. The Human Mitochondrial Citrate Transporter Gene (SLC20A3) Maps to Chromosome Band 22q11 within a Region Implicated in DiGeorge Syndrome, Velo-Cardio-Facial Syndrome and Schizophrenia. Hum. Genet. 1996, 98, 113–115. [Google Scholar] [CrossRef]
- Maynard, T.M.; Meechan, D.W.; Dudevoir, M.L.; Gopalakrishna, D.; Peters, A.Z.; Heindel, C.C.; Sugimoto, T.J.; Wu, Y.; Lieberman, J.A.; la Mantia, A.-S. Mitochondrial localization and function of a subset of 22q11 deletion syndrome candidate genes. Mol. Cell. Neurosci. 2008, 39, 439–451. [Google Scholar] [CrossRef] [Green Version]
- Chaouch, A.; Porcelli, V.; Cox, D.J.; Edvardson, S.; Scarcia, P.; de Grassi, A.; Pierri, C.L.; Cossins, J.; Laval, S.H.; Griffin, H.; et al. Mutations in the Mitochondrial Citrate Carrier SLC25A1 are Associated with Impaired Neuromuscular Transmission. J. Neuromuscul. Dis. 2014, 1, 75–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Futaisi, A.; Ahmad, F.; Al-Kasbi, G.; Al-Thihli, K.; Koul, R.; Al-Maawali, A. Missense mutations in SLC25A1 are associated with congenital myasthenic syndrome type 23. Clin. Genet. 2019, 97, 666–667. [Google Scholar] [CrossRef]
- Edvardson, S.; Porcelli, V.; Jalas, C.; Soiferman, D.; Kellner, Y.; Shaag, A.; Korman, S.H.; Pierri, C.L.; Scarcia, P.; Fraenkel, N.D.; et al. Agenesis of corpus callosum and optic nerve hypoplasia due to mutations inSLC25A1encoding the mitochondrial citrate transporter. J. Med. Genet. 2013, 50, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Nota, B.; Struys, E.A.; Pop, A.; Jansen, E.E.; Ojeda, M.R.F.; Kanhai, W.A.; Kranendijk, M.; van Dooren, S.J.; Bevova, M.R.; Sistermans, E.A.; et al. Deficiency in SLC25A1, Encoding the Mitochondrial Citrate Carrier, Causes Combined D-2- and L-2-Hydroxyglutaric Aciduria. Am. J. Hum. Genet. 2013, 92, 627–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasun, P.; Young, S.; Salomons, G.; Werneke, A.; Jiang, Y.-H.; Struys, E.; Paige, M.; Avantaggiati, M.L.; McDonald, M. Expanding the Clinical Spectrum of Mitochondrial Citrate Carrier (SLC25A1) Deficiency: Facial Dysmorphism in Siblings with Epileptic Encephalopathy and Combined D,L-2-Hydroxyglutaric Aciduria. JIMD Rep. 2014, 19, 111–115. [Google Scholar] [CrossRef]
- Smith, A.; McBride, S.; Marcadier, J.L.; Michaud, J.; Al-Dirbashi, O.Y.; Schwartzentruber, J.; Beaulieu, C.L.; Katz, S.L.; Majewski, J. Severe Neonatal Presentation of Mitochondrial Citrate Carrier (SLC25A1) Deficiency. JIMD Rep. 2016, 30, 73–79. [Google Scholar] [CrossRef] [Green Version]
- Cohen, I.; Staretz-Chacham, O.; Wormser, O.; Perez, Y.; Golubitzky, A.; Kadir, R.; Birk, O.S. A novel homozygous SLC25A1 mutation with impaired mitochondrial complex V: Possible phenotypic expansion. Am. J. Med. Genet. Part A 2017, 176, 330–336. [Google Scholar] [CrossRef]
- Eguchi, M.; Ozaki, E.; Yamauchi, T.; Ohta, M.; Higaki, T.; Masuda, K.; Imoto, I.; Ishii, E.; Eguchi-Ishimae, M. Manifestation of Recessive Combined D-2-, L-2-Hydroxyglutaric Aciduria in Combination with 22q11.2 Deletion Syndrome. Am. J. Med. Genet. A 2018, 176, 351–358. [Google Scholar] [CrossRef]
- Ye, D.; Guan, K.-L.; Xiong, Y. Metabolism, Activity, and Targeting of D- and L-2-Hydroxyglutarates. Trends Cancer 2018, 4, 151–165. [Google Scholar] [CrossRef] [Green Version]
- Rzem, R.; Vincent, M.-F.; van Schaftingen, E.; da Cunha, M.V. l-2-Hydroxyglutaric aciduria, a defect of metabolite repair. J. Inherit. Metab. Dis. 2007, 30, 681–689. [Google Scholar] [CrossRef]
- Kranendijk, M.; Struys, E.A.; Salomons, G.S.; van der Knaap, M.S.; Jakobs, C. Progress in understanding 2-hydroxyglutaric acidurias. J. Inherit. Metab. Dis. 2012, 35, 571–587. [Google Scholar] [CrossRef] [Green Version]
- Pop, A.; Williams, M.; Struys, E.A.; Monné, M.; Jansen, E.E.W.; de Grassi, A.; Kanhai, W.A.; Scarcia, P.; Ojeda, M.R.F.; Porcelli, V.; et al. An overview of combined D-2- and L-2-hydroxyglutaric aciduria: Functional analysis of CIC variants. J. Inherit. Metab. Dis. 2018, 41, 169–180. [Google Scholar] [CrossRef] [Green Version]
- Matsunaga, H.; Futakuchi-Tsuchida, A.; Takahashi, M.; Ishikawa, T.; Tsuji, M.; Ando, O. IDH1 and IDH2 Have Critical Roles in 2-Hydroxyglutarate Production in D-2-Hydroxyglutarate Dehydrogenase Depleted Cells. Biochem. Biophys. Res. Commun. 2012, 423, 553–556. [Google Scholar] [CrossRef]
- Ježek, P. 2-Hydroxyglutarate in Cancer Cells. Antioxid. Redox Signal. 2020, 33, 903–926. [Google Scholar] [CrossRef] [Green Version]
- Intlekofer, A.M.; Wang, B.; Liu, H.; Shah, H.; Carmona-Fontaine, C.; Rustenburg, A.S.; Salah, S.; Gunner, S.S.M.R.; Chodera, C.C. L-2-Hydroxyglutarate production arises from noncanonical enzyme function at acidic pH. Nat. Chem. Biol. 2017, 13, 494–500. [Google Scholar] [CrossRef]
- Fan, J.; Teng, X.; Liu, L.; Mattaini, K.R.; Looper, R.E.; Heiden, M.G.V.; Rabinowitz, J.D. Human Phosphoglycerate Dehydrogenase Produces the Oncometabolite d-2-Hydroxyglutarate. ACS Chem. Biol. 2014, 10, 510–516. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mosaoa, R.; Kasprzyk-Pawelec, A.; Fernandez, H.R.; Avantaggiati, M.L. The Mitochondrial Citrate Carrier SLC25A1/CIC and the Fundamental Role of Citrate in Cancer, Inflammation and Beyond. Biomolecules 2021, 11, 141. https://doi.org/10.3390/biom11020141
Mosaoa R, Kasprzyk-Pawelec A, Fernandez HR, Avantaggiati ML. The Mitochondrial Citrate Carrier SLC25A1/CIC and the Fundamental Role of Citrate in Cancer, Inflammation and Beyond. Biomolecules. 2021; 11(2):141. https://doi.org/10.3390/biom11020141
Chicago/Turabian StyleMosaoa, Rami, Anna Kasprzyk-Pawelec, Harvey R. Fernandez, and Maria Laura Avantaggiati. 2021. "The Mitochondrial Citrate Carrier SLC25A1/CIC and the Fundamental Role of Citrate in Cancer, Inflammation and Beyond" Biomolecules 11, no. 2: 141. https://doi.org/10.3390/biom11020141