
Near-Infrared Absorption Properties of Neutral Bis(1,2-dithiolene) Platinum(II) Complexes Using Density Functional Theory

 and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Methodology Section
2.2. Experimental Section
Synthesis of [Pt(iPr2timdt)2]
3. Results and Discussion
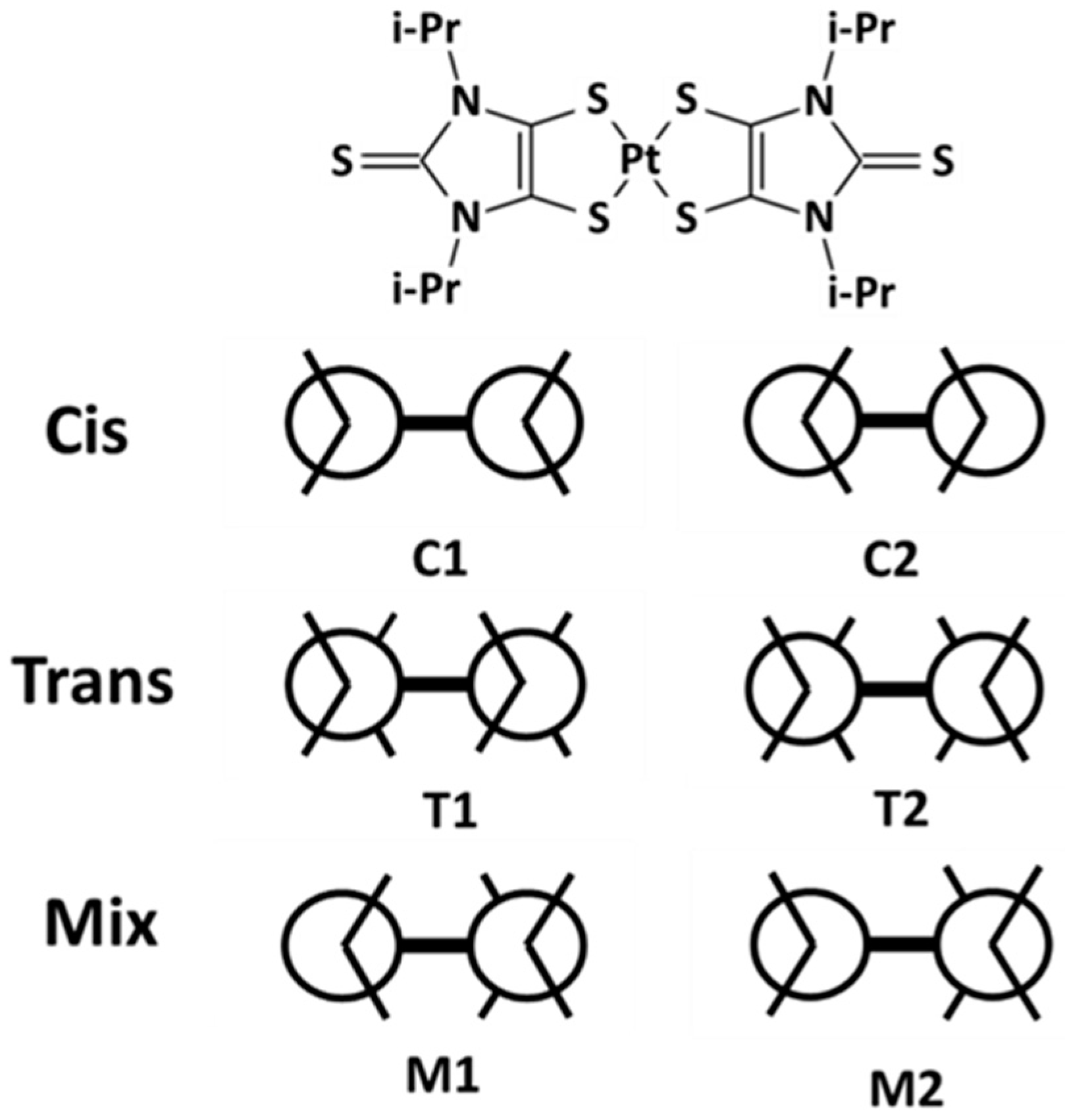
3.1. Equilibrated Structures
3.2. Optical Properties
3.3. Discussions
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pop, F.; Avarvari, N. Chiral metal-dithiolene complexes. Coord. Chem. Rev. 2017, 346, 20–31. [Google Scholar] [CrossRef]
- Espa, D.; Pilia, L.; Attar, S.; Serpe, A.; Deplano, P. Molecular engineering of heteroleptic metal-dithiolene complexes with optimized second-order NLO response. Inorg. Chim. Acta 2018, 470, 295–302. [Google Scholar] [CrossRef]
- Basu, P.; Colston, K.J.; Mogesa, B. Dithione, the antipodal redox partner of ene-1,2-dithiol ligands and their metal complexes. Coord. Chem. Rev. 2020, 409, 213211. [Google Scholar] [CrossRef]
- Jacob, K.; de Caro, D.; Faulmann, C.; Valade, L. Nanoparticles of Molecular Conductors and Superconductors: Progress Over the Last Ten Years. Eur. J. Inorg. Chem. 2020, 2020, 4237–4246. [Google Scholar] [CrossRef]
- Kobayashi, A.; Zhou, B.; Takagi, R.; Miyagawa, K.; Ishibashi, S.; Kobayashi, A.; Kawamura, T.; Nishibori, E.; Kanoda, K. Single-Component Molecular Conductors—Multi-Orbital Correlated π-d Electron Systems. Bull. Chem. Soc. Jpn. 2021, 94, 2540–2562. [Google Scholar] [CrossRef]
- Pintus, A.; Ambrosio, L.; Aragoni, M.C.; Binda, M.; Coles, S.J.; Hursthouse, M.B.; Isaia, F.; Lippolis, V.; Meloni, G.; Natali, D.; et al. Photoconducting Devices with Response in the Visible–Near-Infrared Region Based on Neutral Ni Complexes of Aryl-1,2-dithiolene Ligands. Inorg. Chem. 2020, 59, 6410–6421. [Google Scholar] [CrossRef]
- Yao, W.-W.; Xu, X.-Y.; Zhang, J.; Qian, Y.; Liu, J.-L.; Chen, X.-R.; Ren, X.-M. A Semiconductive Nature of Bis(dithiolato)nickelate Radical Salt Exhibiting Broadband Photoconduction. Inorg. Chem. 2021, 60, 15659–15666. [Google Scholar] [CrossRef] [PubMed]
- Naito, T.; Karasudani, T.; Mori, S.; Ohara, K.; Konishi, K.; Takano, T.; Takahashi, Y.; Inabe, T.; Nishihara, S.; Inoue, K. Molecular Photoconductor with Simultaneously Photocontrollable Localized Spins. J. Am. Chem. Soc. 2012, 134, 18656–18666. [Google Scholar] [CrossRef]
- Naito, T.; Karasudani, T.; Ohara, K.; Takano, T.; Takahashi, Y.; Inabe, T.; Furukawa, K.; Nakamura, T. Simultaneous Control of Carriers and Localized Spins with Light in Organic Materials. Adv. Mater. 2012, 24, 6153–6157. [Google Scholar] [CrossRef]
- Noma, H.; Ohara, K.; Naito, T. Direct Control of Spin Distribution and Anisotropy in Cu-Dithiolene Complex Anions by Light. Inorganics 2016, 4, 7. [Google Scholar] [CrossRef] [Green Version]
- Crasto de Lima, F.; Ferreira, G.J.; Miwa, R.H. Quantum anomalous Hall effect in metal-bis(dithiolene), magnetic properties, doping and interfacing graphene. Phys. Chem. Chem. Phys. 2018, 20, 22652–22659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Zhang, Z.; Chen, X.; Xu, S.; Cao, S. Near-infrared absorbing dyes at 1064 nm: Soluble dithiolene nickel complexes with alkylated electron-donating groups as Peripheral substituents. Dye. Pigment. 2016, 128, 179–189. [Google Scholar] [CrossRef]
- Park, B.; Lee, K.M.; Park, S.; Yun, M.; Choi, H.-J.; Kim, J.; Lee, C.; Kim, H.; Kim, C. Deep tissue photoacoustic imaging of nickel(II) dithiolene-containing polymeric nanoparticles in the second near-infrared window. Theranostics 2020, 10, 2509–2521. [Google Scholar] [CrossRef] [PubMed]
- Artizzu, F.; Espa, D.; Marchiò, L.; Pilia, L.; Serpe, A.; Deplano, P. Progress and perspectives on strategies to control photochemical properties in Metallo-Dithiolene Donor-Acceptor systems. Inorg. Chim. Acta 2022, 531, 120731. [Google Scholar] [CrossRef]
- Artem’ev, A.V.; Petyuk, M.Y.; Berezin, A.S.; Gushchin, A.L.; Sokolov, M.N.; Bagryanskaya, I.Y. Synthesis and study of Re(I) tricarbonyl complexes based on octachloro-1,10-phenanthroline: Towards deep red-to-NIR emitters. Polyhedron 2021, 209, 115484. [Google Scholar] [CrossRef]
- Shekhovtsov, N.A.; Kokina, T.E.; Vinogradova, K.A.; Panarin, A.Y.; Rakhmanova, M.I.; Naumov, D.Y.; Pervukhina, N.V.; Nikolaenkova, E.B.; Krivopalov, V.P.; Czerwieniec, R.; et al. Near-infrared emitting copper(i) complexes with a pyrazolylpyrimidine ligand: Exploring relaxation pathways. Dalton Trans. 2022, 51, 2898–2911. [Google Scholar] [CrossRef]
- Zhao, Q.; Huang, C.; Li, F. Phosphorescent heavy-metal complexes for bioimaging. Chem. Soc. Rev. 2011, 40, 2508–2524. [Google Scholar] [CrossRef] [Green Version]
- Mebrouk, K.; Ciancone, M.; Vives, T.; Cammas-Marion, S.; Benvegnu, T.; Le Goff-Gaillard, C.; Arlot-Bonnemains, Y.; Fourmigué, M.; Camerel, F. Fine and Clean Photothermally Controlled NIR Drug Delivery from Biocompatible Nickel-bis(dithiolene)-Containing Liposomes. ChemMedChem 2017, 12, 1753–1758. [Google Scholar] [CrossRef]
- Devillanova, F.A.; Du Mont, W.-W. Handbook of Chalcogen Chemistry: New Perspectives in Sulfur, Selenium and Tellurium; Royal Society of Chemistry: London, UK, 2013; Volume 1. [Google Scholar]
- Mogesa, B.; Perera, E.; Rhoda, H.M.; Gibson, J.K.; Oomens, J.; Berden, G.; van Stipdonk, M.J.; Nemykin, V.N.; Basu, P. Solution, Solid, and Gas Phase Studies on a Nickel Dithiolene System: Spectator Metal and Reactor Ligand. Inorg. Chem. 2015, 54, 7703–7716. [Google Scholar] [CrossRef] [Green Version]
- Mebrouk, K.; Camerel, F.; Jeannin, O.; Heinrich, B.; Donnio, B.; Fourmigué, M. High Photothermal Activity within Neutral Nickel Dithiolene Complexes Derived from Imidazolium-Based Ionic Liquids. Inorg. Chem. 2016, 55, 1296–1303. [Google Scholar] [CrossRef]
- Eisenberg, R.; Gray, H.B. Noninnocence in metal complexes: A dithiolene dawn. Inorg. Chem. 2011, 50, 9741–9751. [Google Scholar] [CrossRef] [PubMed]
- Periyasamy, G.; Burton, N.A.; Hillier, I.H.; Vincent, M.A.; Disley, H.; McMaster, J.; Garner, C.D. The dithiolene ligand—‘Innocent’or ‘non-innocent’? A theoretical and experimental study of some cobalt–dithiolene complexes. Faraday Discuss. 2007, 135, 469–488. [Google Scholar] [CrossRef] [PubMed]
- Masui, H. Metalloaromaticity. Coord. Chem. Rev. 2001, 219, 957–992. [Google Scholar] [CrossRef]
- Wan, H.C.; Zhang, J.-X.; Leung, C.S.; Sheong, F.K.; Lin, Z. Inter-ligand delocalisations in transition metal complexes containing multiple non-innocent ligands. Dalton Trans. 2019, 48, 14801–14807. [Google Scholar] [CrossRef]
- Avramopoulos, A.; Reis, H.; Mousdis, G.A.; Papadopoulos, M.G. Ni Dithiolenes—A Theoretical Study on Structure–Property Relationships. Eur. J. Inorg. Chem 2013, 2013, 4839–4850. [Google Scholar] [CrossRef]
- Avramopoulos, A.; Otero, N.; Reis, H.; Karamanis, P.; Papadopoulos, M.G. A computational study of photonic materials based on Ni bis(dithiolene) fused with benzene, possessing gigantic second hyperpolarizabilities. J. Mater. Chem. C 2018, 6, 91–110. [Google Scholar] [CrossRef]
- Deiana, C.; Aragoni, M.C.; Isaia, F.; Lippolis, V.; Pintus, A.; Slawin, A.M.; Woollins, J.D.; Arca, M. Structural tailoring of the NIR-absorption of bis (1,2-dichalcogenolene) Ni/Pt electrochromophores deriving from 1,3-dimethyl-2-chalcogenoxo-imidazoline-4, 5-dichalcogenolates. New J. Chem. 2016, 40, 8206–8210. [Google Scholar] [CrossRef] [Green Version]
- Aragoni, M.C.; Caltagirone, C.; Lippolis, V.; Podda, E.; Slawin, A.M.Z.; Woollins, J.D.; Pintus, A.; Arca, M. Diradical Character of Neutral Heteroleptic Bis(1,2-dithiolene) Metal Complexes: Case Study of [Pd(Me2timdt)(mnt)] (Me2timdt = 1,3-Dimethyl-2,4,5-trithioxoimidazolidine; mnt2– = 1,2-Dicyano-1,2-ethylenedithiolate). Inorg. Chem. 2020, 59, 17385–17401. [Google Scholar] [CrossRef]
- Bigoli, F.; Deplano, P.; Mercuri, M.L.; Pellinghelli, M.A.; Pintus, G.; Trogu, E.F.; Zonnedda, G.; Wang, H.H.; Williams, J.M. Novel oxidation and reduction products of the neutral nickel-dithiolene Ni(Pr2itimdt)2 (Pr2itimdt is the monoanion of 1,3-diisopropylimidazolidine-2,4,5-trithione). Inorg. Chim. Acta 1998, 273, 175–183. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Hedin, L. New method for calculating the one-particle Green’s function with application to the electron-gas problem. Phys. Rev. 1965, 139, A796. [Google Scholar] [CrossRef]
- Albrecht, S.; Reining, L.; Del Sole, R.; Onida, G. Ab initio calculation of excitonic effects in the optical spectra of semiconductors. Phys. Rev. Lett. 1998, 80, 4510. [Google Scholar] [CrossRef] [Green Version]
- Rohlfing, M.; Louie, S.G. Electron-hole excitations and optical spectra from first principles. Phys. Rev. B 2000, 62, 4927. [Google Scholar] [CrossRef]
- Pham, N.N.T.; Han, S.H.; Park, J.S.; Lee, S.G. Optical and Electronic Properties of Organic NIR-II Fluorophores by Time-Dependent Density Functional Theory and Many-Body Perturbation Theory: GW-BSE Approaches. Nanomaterials 2021, 11, 2293. [Google Scholar] [CrossRef] [PubMed]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758. [Google Scholar] [CrossRef]
- Sander, T.; Maggio, E.; Kresse, G. Beyond the Tamm-Dancoff approximation for extended systems using exact diagonalization. Phys. Rev. B 2015, 92, 045209. [Google Scholar] [CrossRef]
- Dresselhaus, M. Solid state physics part ii optical properties of solids. Lect. Notes 2001, 17, 15–16. [Google Scholar]
- Stoffel, P. The preparation of parabanic acids from 1,1,3-trisubstituted ureas via a Hofmann elimination reaction. J. Org. Chem. 1964, 29, 2794–2796. [Google Scholar] [CrossRef]
- Ulrich, H.; Sayigh, A. The reaction of oxalyl chloride with substituted ureas and thioureas. J. Org. Chem. 1965, 30, 2781–2783. [Google Scholar] [CrossRef]
- LauraáMercuri, M.; AngelaáPellinghelli, M. New neutral nickel dithiolene complexes derived from 1, 3-dialkylimidazolidine-2,4,5-trithione, showing remarkable near-IR absorption. J. Chem. Soc. Chem. Commun. 1995, 3, 371–372. [Google Scholar]
- McCleverty, J.A. Metal 1, 2-dithiolene and related complexes. Prog. Inorg. Chem. 1968, 10, 49–221. [Google Scholar]
- Ray, K.; Weyhermüller, T.; Neese, F.; Wieghardt, K. Electronic structure of square planar bis (benzene-1,2-dithiolato) metal complexes [M(L)2]z (z = 2−, 1−, 0; M = Ni, Pd, Pt, Cu, Au): An experimental, density functional, and correlated ab initio study. Inorg. Chem. 2005, 44, 5345–5360. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | Bond Length (Å) (Deviation) | Bond Angle (°) (Deviation) | ||||||
|---|---|---|---|---|---|---|---|---|
| C–C | C–S | Pt–S | C–N | S–Pt–S | C–S–Pt | C–N–C | N–C–S | |
| a | b | c | d | θ1 | θ2 | θ3 | θ4 | |
| C1 | 1.406 (0.000) | 1.700 (0.000) | 2.271 (0.000) | 1.470 (0.000) | 87.8 (0.0) | 100.5 (0.0) | 127.2 (0.0) | 127.3 (0.0) |
| C2 | 1.404 (0.000) | 1.703 (0.000) | 2.267 (0.000) | 1.464 (0.001) | 88.3 (0.0) | 101.3 (0.0) | 123.2 (0.0) | 127.1 (0.0) |
| T1 | 1.404 (0.000) | 1.702 (0.002) | 2.269 (0.000) | 1.466 (0.004) | 88.0 (0.0) | 100.9 (0.2) | 125.1 (1.9) | 127.2 (1.4) |
| T2 | 1.404 (0.000) | 1.702 (0.002) | 2.269 (0.000) | 1.466 (0.004) | 88.0 (0.2) | 100.9 (0.2) | 125.1 (1.9) | 127.2 (1.4) |
| M1 | 1.403 (0.001) | 1.701 (0.001) | 2.270 (0.002) | 1.468 (0.003) | 87.9 (0.1) | 100.7 (0.2) | 126.1 (1.5) | 127.2 (1.0) |
| M2 | 1.405 (0.001) | 1.702 (0.001) | 2.268 (0.002) | 1.465 (0.002) | 88.1 (0.1) | 101.1 (0.2) | 124.1 (1.4) | 127.1 (1.0) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luong, X.-H.; Pham, N.N.T.; An, K.-L.; Lee, S.U.; Kim, S.S.; Park, J.S.; Lee, S.G. Near-Infrared Absorption Properties of Neutral Bis(1,2-dithiolene) Platinum(II) Complexes Using Density Functional Theory. Nanomaterials 2022, 12, 1704. https://doi.org/10.3390/nano12101704
Luong X-H, Pham NNT, An K-L, Lee SU, Kim SS, Park JS, Lee SG. Near-Infrared Absorption Properties of Neutral Bis(1,2-dithiolene) Platinum(II) Complexes Using Density Functional Theory. Nanomaterials. 2022; 12(10):1704. https://doi.org/10.3390/nano12101704
Chicago/Turabian StyleLuong, Xuan-Hoang, Nguyet N. T. Pham, Kyoung-Lyong An, Seong Uk Lee, Shi Surk Kim, Jong S. Park, and Seung Geol Lee. 2022. "Near-Infrared Absorption Properties of Neutral Bis(1,2-dithiolene) Platinum(II) Complexes Using Density Functional Theory" Nanomaterials 12, no. 10: 1704. https://doi.org/10.3390/nano12101704