
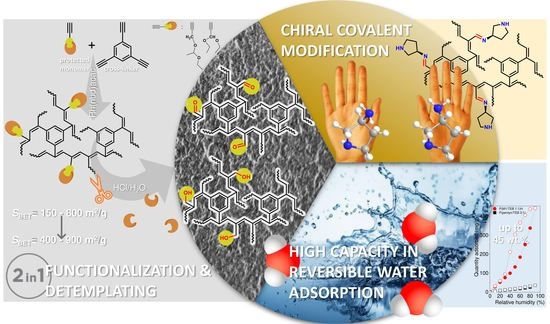
Combining Polymerization and Templating toward Hyper-Cross-Linked Poly(propargyl aldehyde)s and Poly(propargyl alcohol)s for Reversible H2O and CO2 Capture and Construction of Porous Chiral Networks
 , , , and
, , , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Copolymerization
2.3. Deprotection of Parent Networks
2.4. Techniques
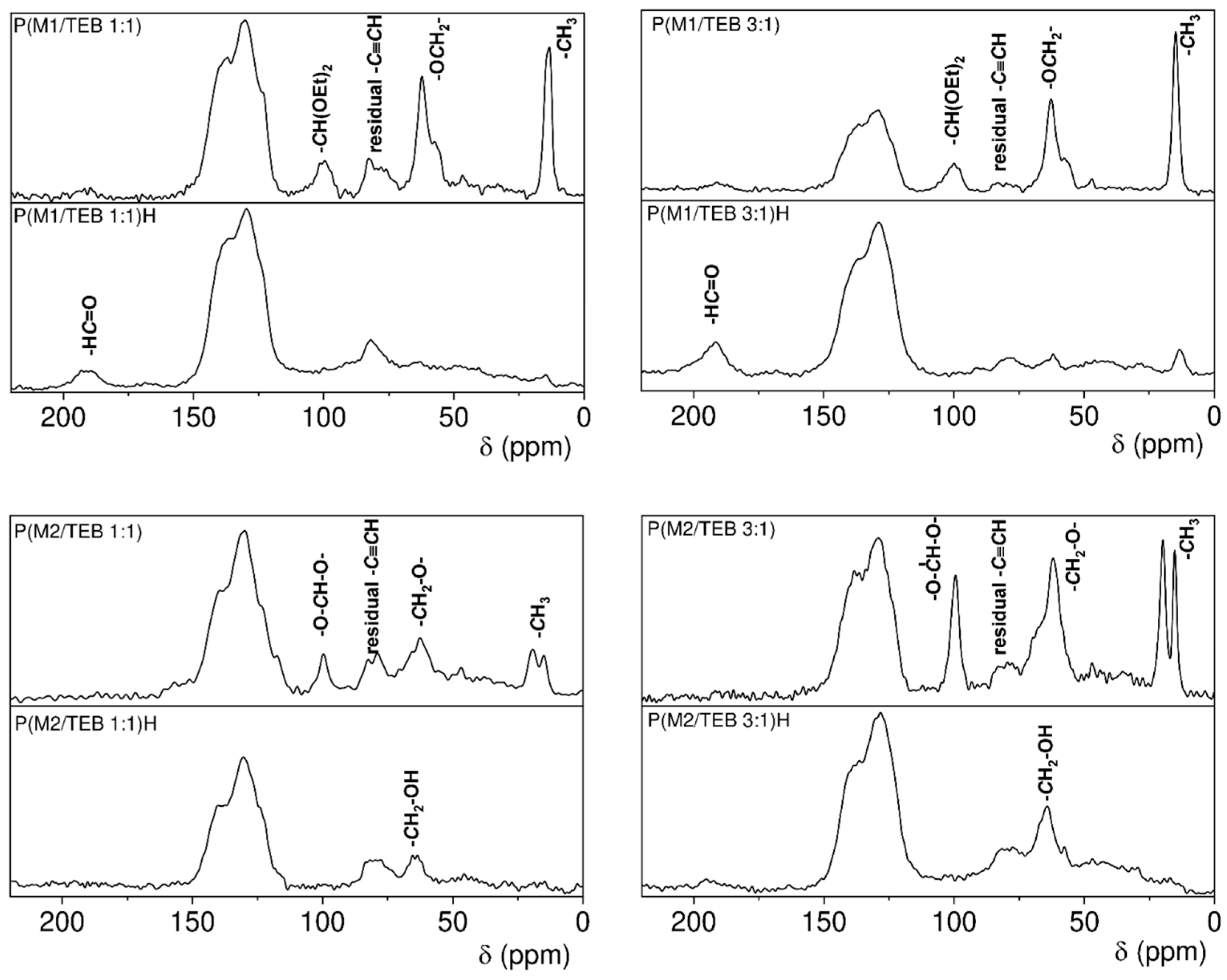
2.4.1. NMR
2.4.2. N2 and CO2 Adsorption
2.4.3. TGA
2.4.4. DVS
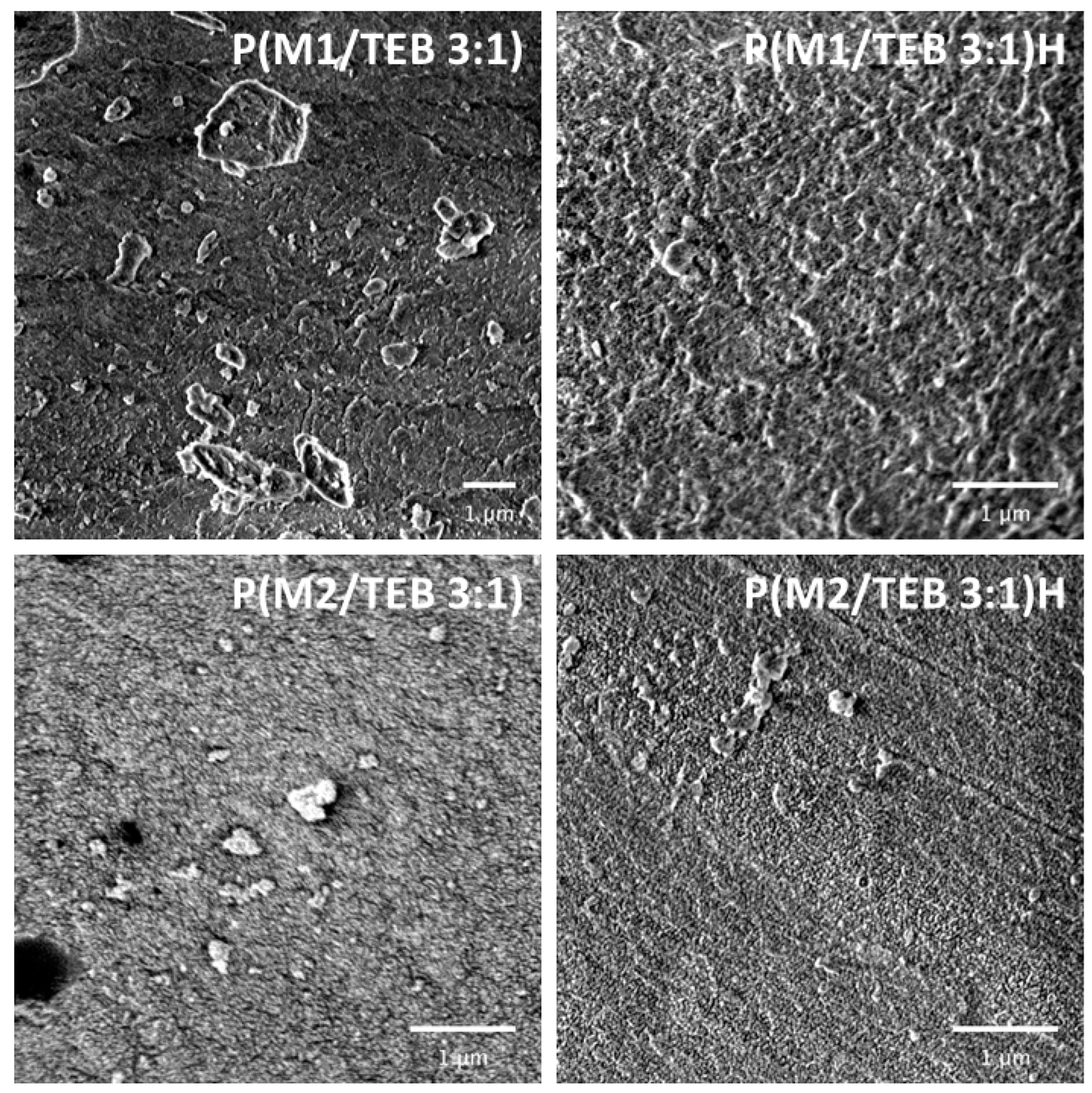
2.4.5. SEM
3. Results and Discussion
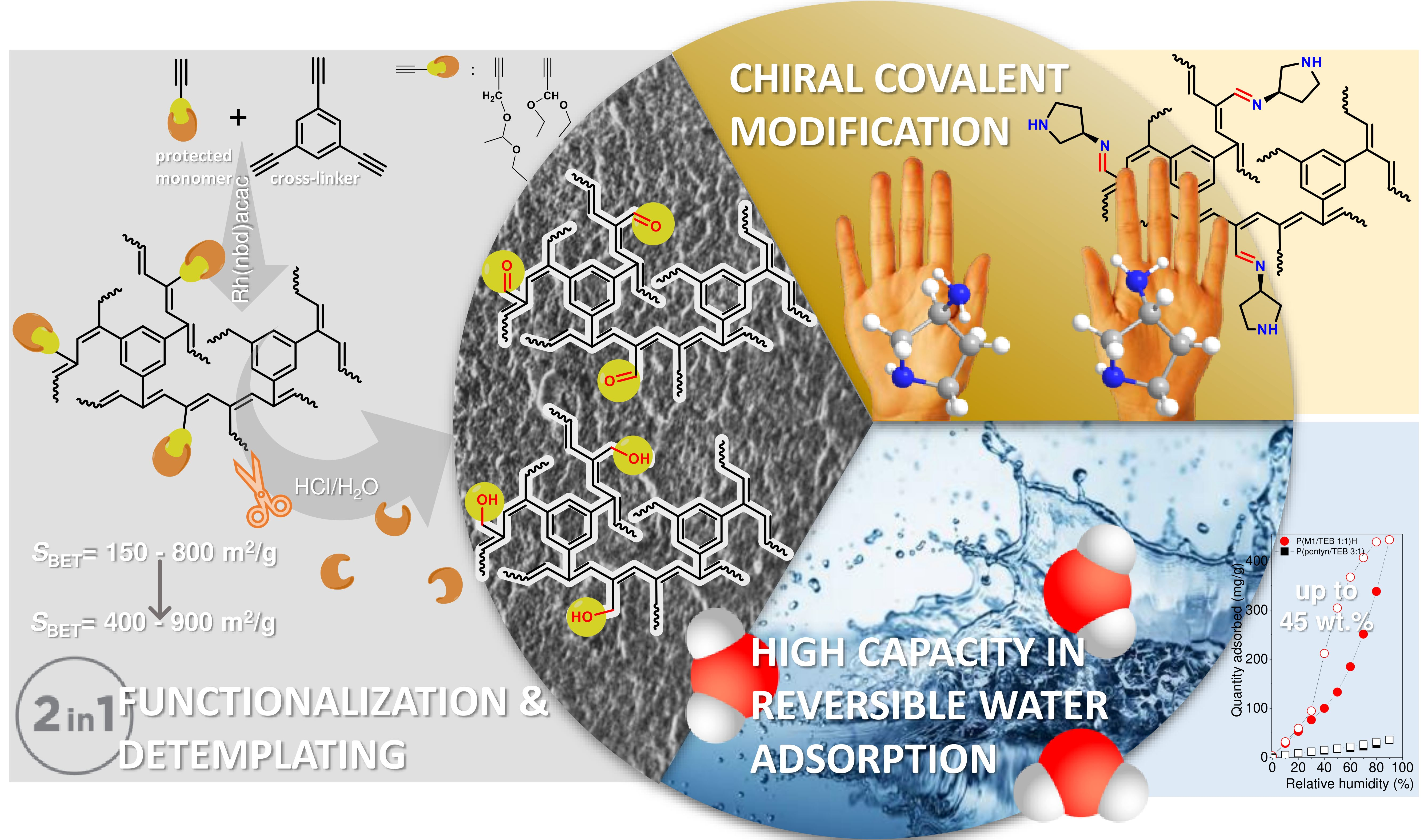
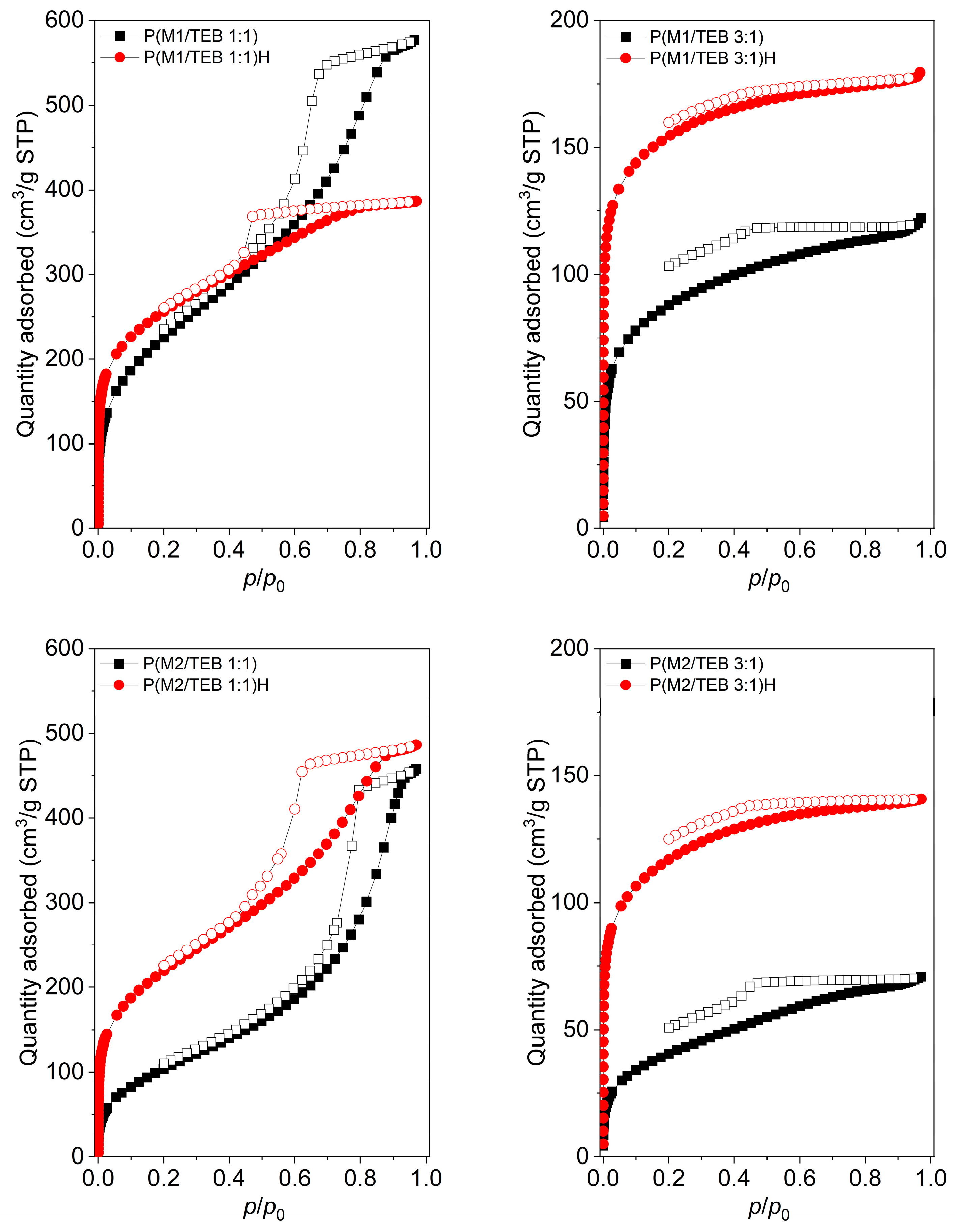
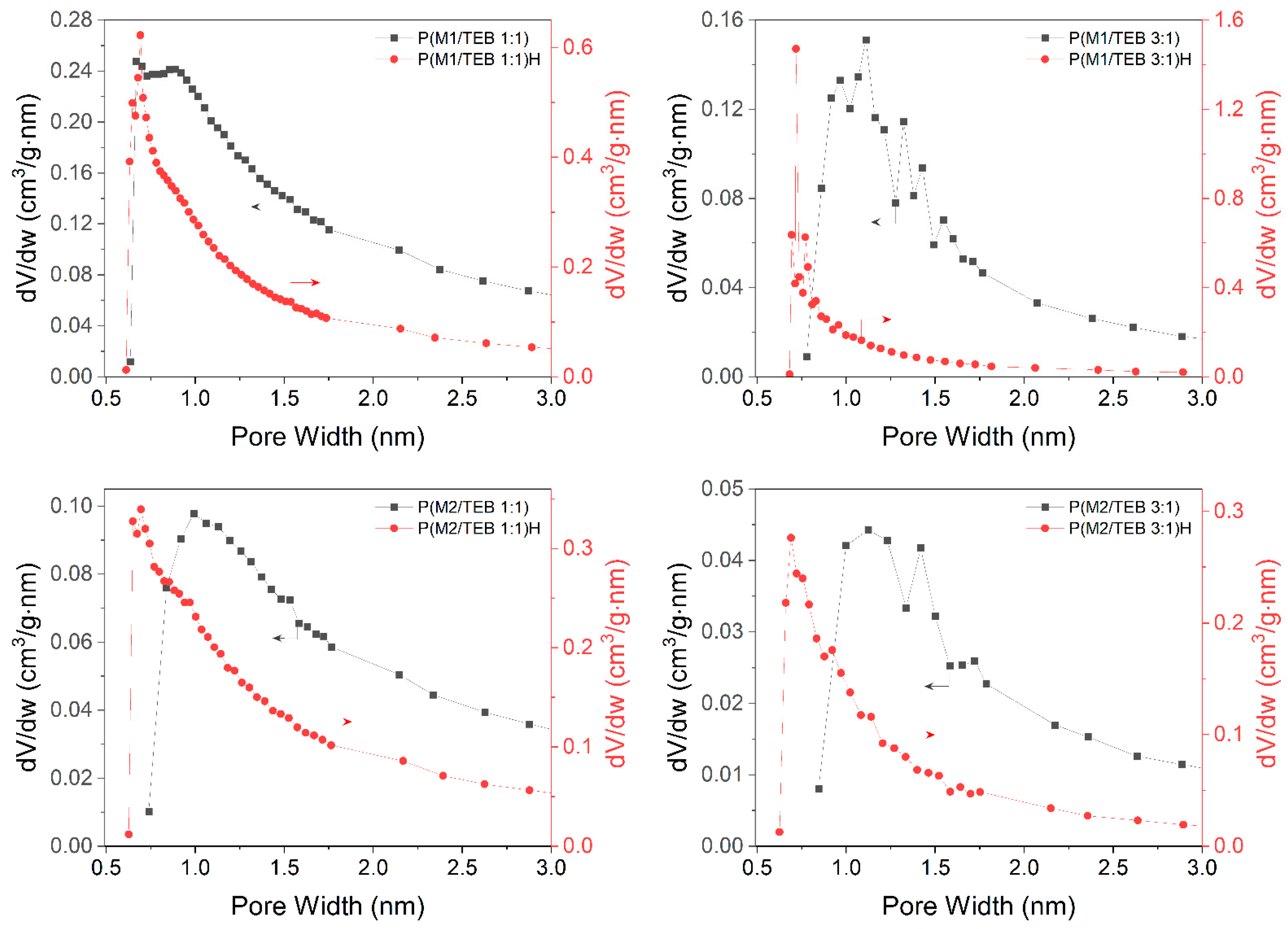
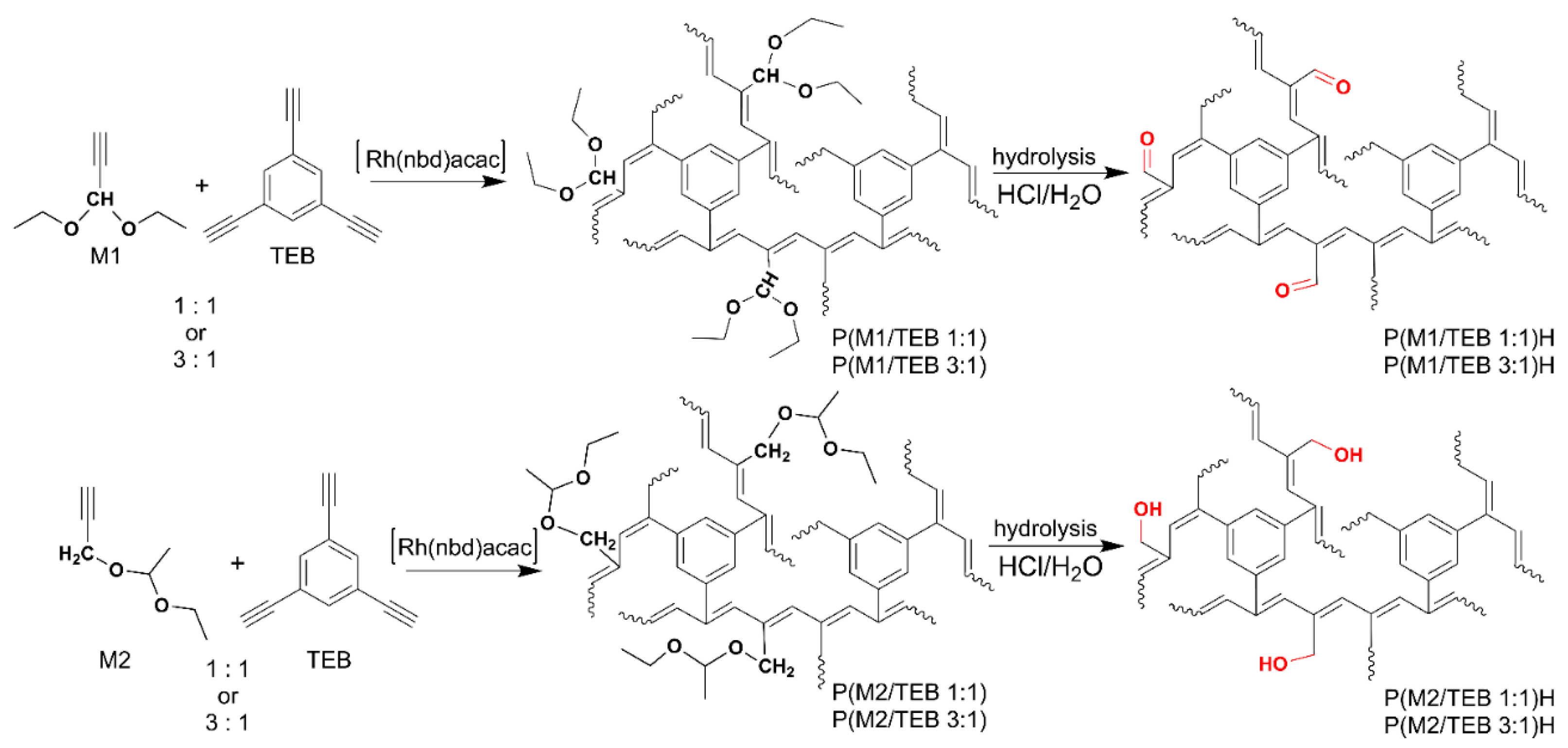
3.1. Synthesis and Characterization of the Porous Networks
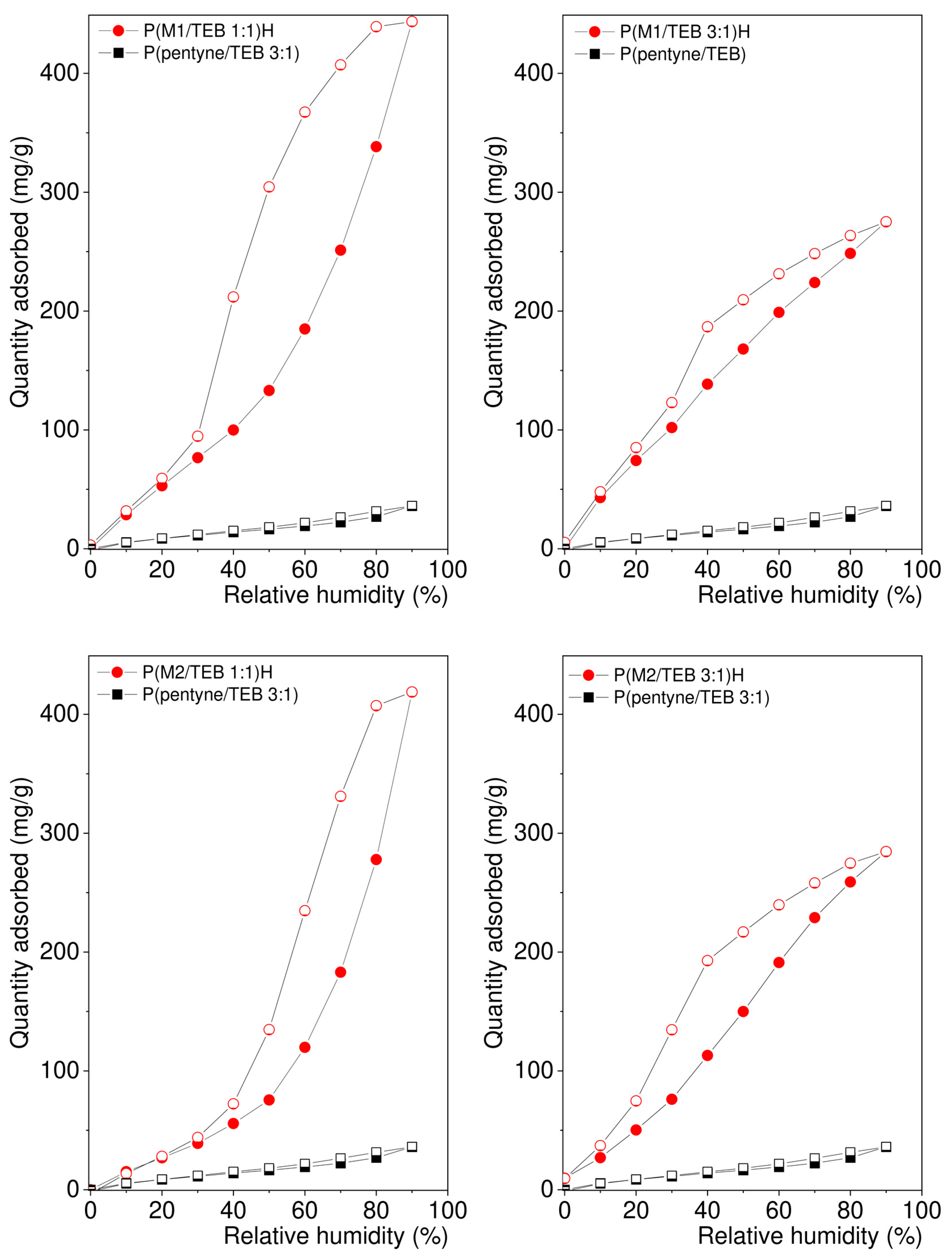
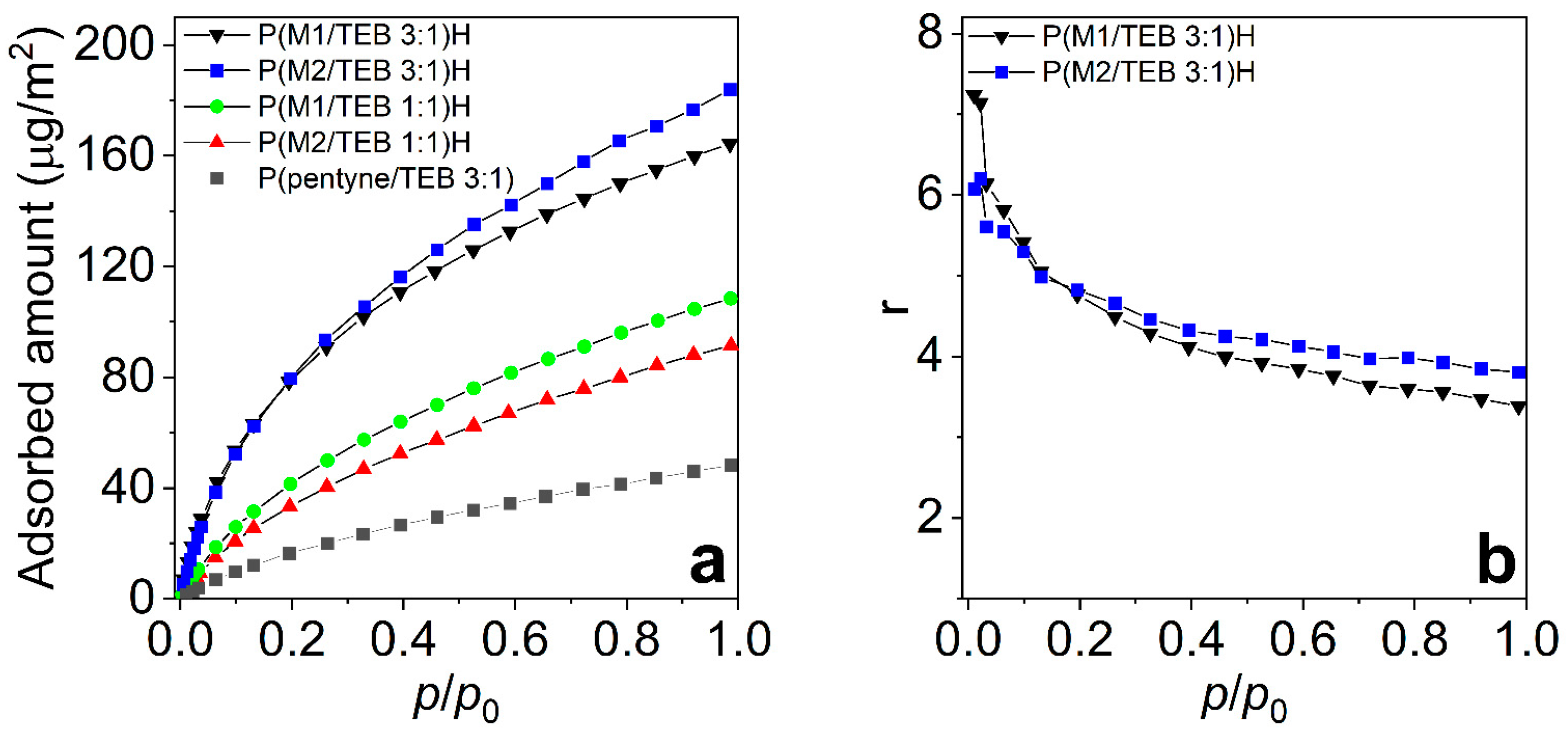
3.2. Water Vapour and Carbon Dioxide Capture
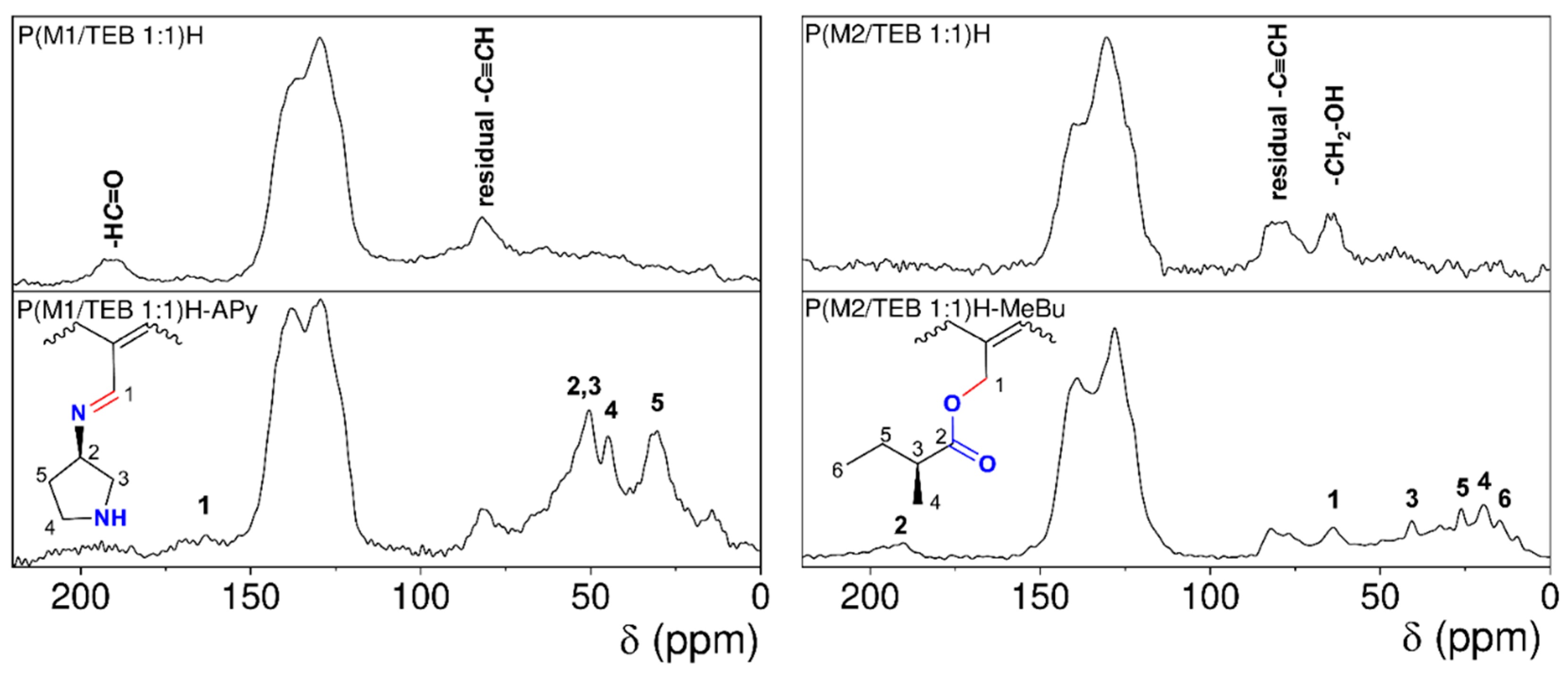
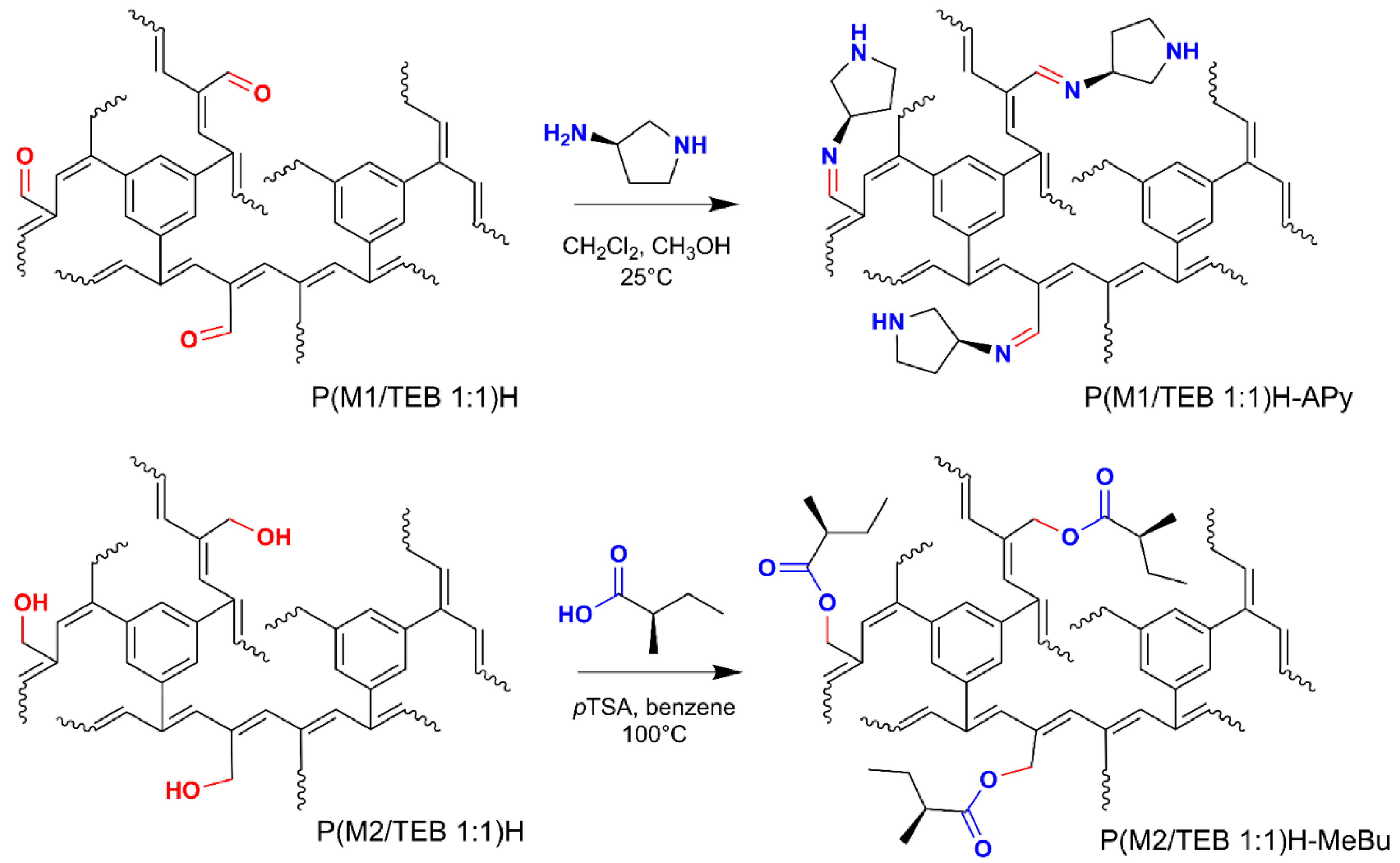
3.3. Covalent Modification of Hydrolyzed Networks
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Dawson, R.; Cooper, A.I.; Adams, D.J. Nanoporous organic polymer networks. Prog. Polym. Sci. 2011, 37, 530–563. [Google Scholar] [CrossRef]
- Taylor, D.; Dalgarno, S.J.; Xu, Z.; Vilela, F. Conjugated porous polymers: Incredibly versatile materials with far-reaching applications. Chem. Soc. Rev. 2020, 49, 3981–4042. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.-B.; Zhan, Z.-P. Conjugated Microporous Polymers for Heterogeneous Catalysis. Chem. Asian J. 2017, 13, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Bhanja, P.; Modak, A.; Bhaumik, A. Porous Organic Polymers for CO2 Storage and Conversion Reactions. Chemcatchem 2018, 11, 244–257. [Google Scholar] [CrossRef]
- Cousins, K.; Zhang, R. Highly Porous Organic Polymers for Hydrogen Fuel Storage. Polymers 2019, 11, 690. [Google Scholar] [CrossRef]
- Chowdhury, A.; Bhattacharjee, S.; Chatterjee, R.; Bhaumik, A. A new nitrogen rich porous organic polymer for ultra-high CO2 uptake and as an excellent organocatalyst for CO2 fixation reactions. J. CO2 Util. 2022, 65, 102236. [Google Scholar] [CrossRef]
- Song, K.S.; Fritz, P.W.; Coskun, A. Porous organic polymers for CO2 capture, separation and conversion. Chem. Soc. Rev. 2022, 51, 9831–9852. [Google Scholar] [CrossRef]
- Zhou, L.; Hu, Y.; Li, G. Conjugated Microporous Polymers with Built-In Magnetic Nanoparticles for Excellent Enrichment of Trace Hydroxylated Polycyclic Aromatic Hydrocarbons in Human Urine. Anal. Chem. 2016, 88, 6930–6938. [Google Scholar] [CrossRef]
- Havelková, L.; Hašková, A.; Bashta, B.; Brus, J.; Lhotka, M.; Vrbková, E.; Kindl, M.; Vyskočilová, E.; Sedláček, J. Synthesis of hyper-cross-linked microporous poly(phenylacetylene)s having aldehyde and other groups and their chemisorption and physisorption ability. Eur. Polym. J. 2019, 114, 279–286. [Google Scholar] [CrossRef]
- Zhang, Q.; Yu, S.; Wang, Q.; Xiao, Q.; Yue, Y.; Ren, S. Fluorene-Based Conjugated Microporous Polymers: Preparation and Chemical Sensing Application. Macromol. Rapid Commun. 2017, 38, 1700445. [Google Scholar] [CrossRef] [Green Version]
- Debruyne, M.; Van Speybroeck, V.; Van Der Voort, P.; Stevens, C.V. Porous organic polymers as metal free heterogeneous organocatalysts. Green Chem. 2021, 23, 7361–7434. [Google Scholar] [CrossRef]
- Sekerová, L.; Březinová, P.; Do, T.T.; Vyskočilová, E.; Krupka, J.; Červený, L.; Havelková, L.; Bashta, B.; Sedlacek, J. Sulfonated Hyper-cross-linked Porous Polyacetylene Networks as Versatile Heterogeneous Acid Catalysts. Chemcatchem 2019, 12, 1075–1084. [Google Scholar] [CrossRef]
- Zheng, Y.; Wang, X.; Liu, C.; Yu, B.; Li, W.; Wang, H.; Sun, T.; Jiang, J. Triptycene-supported bimetallic salen porous organic polymers for high efficiency CO2 fixation to cyclic carbonates. Inorg. Chem. Front. 2021, 8, 2880–2888. [Google Scholar] [CrossRef]
- Bonfant, G.; Balestri, D.; Perego, J.; Comotti, A.; Bracco, S.; Koepf, M.; Gennari, M.; Marchiò, L. Phosphine Oxide Porous Organic Polymers Incorporating Cobalt(II) Ions: Synthesis, Characterization, and Investigation of H2 Production. ACS Omega 2022, 7, 6104–6112. [Google Scholar] [CrossRef]
- Zhang, T.; Xing, G.; Chen, W.; Chen, L. Porous organic polymers: A promising platform for efficient photocatalysis. Mater. Chem. Front. 2019, 4, 332–353. [Google Scholar] [CrossRef]
- Yuan, D.; Lu, W.; Zhao, D.; Zhou, H.-C. Highly Stable Porous Polymer Networks with Exceptionally High Gas-Uptake Capacities. Adv. Mater. 2011, 23, 3723–3725. [Google Scholar] [CrossRef]
- Hašková, A.; Bashta, B.; Titlová, S.; Brus, J.; Vagenknechtová, A.; Vyskočilová, E.; Sedláček, J. Microporous Hyper-Cross-Linked Polymers with High and Tuneable Content of Pyridine Units: Synthesis and Application for Reversible Sorption of Water and Carbon Dioxide. Macromol. Rapid Commun. 2021, 42, e2100209. [Google Scholar] [CrossRef]
- Dawson, R.; Cooper, A.I.; Adams, D.J. Chemical functionalization strategies for carbon dioxide capture in microporous organic polymers. Polym. Int. 2013, 62, 345–352. [Google Scholar] [CrossRef]
- Byun, Y.; Je, S.H.; Talapaneni, S.N.; Coskun, A. Advances in Porous Organic Polymers for Efficient Water Capture. Chem. A Eur. J. 2019, 25, 10262–10283. [Google Scholar] [CrossRef] [PubMed]
- Byun, J.; Patel, H.; Thirion, D.; Yavuz, C. Reversible water capture by a charged metal-free porous polymer. Polymer 2017, 126, 308–313. [Google Scholar] [CrossRef]
- Mukherjee, S.; Zeng, Z.; Shirolkar, M.M.; Samanta, P.; Chaudhari, A.K.; Tan, J.; Ghosh, S.K. Self-Assembled, Fluorine-Rich Porous Organic Polymers: A Class of Mechanically Stiff and Hydrophobic Materials. Chem. A Eur. J. 2018, 24, 11771–11778. [Google Scholar] [CrossRef]
- Zhang, W.; Li, Y.; Wu, Y.; Fu, Y.; Chen, S.; Zhang, Z.; He, S.; Yan, T.; Ma, H. Fluorinated porous organic polymers for efficient recovery perfluorinated electronic specialty gas from exhaust gas of plasma etching. Sep. Purif. Technol. 2022, 287, 120561. [Google Scholar] [CrossRef]
- Ai, C.; Tang, J.; Zhang, Q.; Tang, X.; Wu, S.; Pan, C.; Yu, G.; Yuan, J. A knitting copolymerization Strategy to Build Porous Polytriazolium Salts for Removal of Anionic Dyes and MnO4−. Macromol. Rapid Commun. 2022. [Google Scholar] [CrossRef]
- Sedláček, J.; Sokol, J.; Zedník, J.; Faukner, T.; Kubů, M.; Brus, J.; Trhlíková, O. Homo- and Copolycyclotrimerization of Aromatic Internal Diynes Catalyzed with Co2(CO)8: A Facile Route to Microporous Photoluminescent Polyphenylenes with Hyperbranched or Crosslinked Architecture. Macromol. Rapid Commun. 2017, 39, 1700518. [Google Scholar] [CrossRef]
- Monterde, C.; Navarro, R.; Iglesias, M.; Sánchez, F. Adamantyl-BINOL as platform for chiral porous polymer aromatic frameworks. Multiple applications as recyclable catalysts. J. Catal. 2019, 377, 609–618. [Google Scholar] [CrossRef]
- Wu, Z.; Li, T.; Ding, Y.; Hu, A. Synthesis of Chiral Porous Organic Polymers Through Nucleophilic Substitution for Chiral Separation. ACS Appl. Polym. Mater. 2020, 2, 5414–5422. [Google Scholar] [CrossRef]
- Song, W.; Zhang, M.; Huang, X.; Chen, B.; Ding, Y.; Zhang, Y.; Yu, D.; Kim, I. Smart-borneol-loaded hierarchical hollow polymer nanospheres with antipollution and antibacterial capabilities. Mater. Today Chem. 2022, 26, 101252. [Google Scholar] [CrossRef]
- Tang, Y.; Varyambath, A.; Ding, Y.; Chen, B.; Huang, X.; Zhang, Y.; Yu, D.-G.; Kim, I.; Song, W. Porous organic polymers for drug delivery: Hierarchical pore structures, variable morphologies, and biological properties. Biomater. Sci. 2022, 10, 5369–5390. [Google Scholar] [CrossRef]
- Holst, J.R.; Stöckel, E.; Adams, D.J.; Cooper, A.I. High Surface Area Networks from Tetrahedral Monomers: Metal-Catalyzed Coupling, Thermal Polymerization, and “Click” Chemistry. Macromolecules 2010, 43, 8531–8538. [Google Scholar] [CrossRef]
- Xu, C.; Bacsik, Z.; Hedin, N. Adsorption of CO2 on a micro-/mesoporous polyimine modified with tris(2-aminoethyl)amine. J. Mater. Chem. A 2015, 3, 16229–16234. [Google Scholar] [CrossRef]
- Xiong, S.; Tang, X.; Pan, C.; Li, L.; Tang, J.; Yu, G. Carbazole-Bearing Porous Organic Polymers with a Mulberry-Like Morphology for Efficient Iodine Capture. ACS Appl. Mater. Interfaces 2019, 11, 27335–27342. [Google Scholar] [CrossRef]
- Bondarev, D.; Sivkova, R.; Šuly, P.; Polášková, M.; Krejčí, O.; Křikavová, R.; Trávníček, Z.; Zukal, A.; Kubů, M.; Sedláček, J. Microporous conjugated polymers via homopolymerization of 2,5-diethynylthiophene. Eur. Polym. J. 2017, 92, 213–219. [Google Scholar] [CrossRef]
- Zhang, Q.; Zeng, K.; Wang, C.; Wei, P.; Zhao, X.; Wu, F.; Liu, Z.-R. An imidazole functionalized porous organic polymer for the highly efficient extraction of uranium from aqueous solutions. New J. Chem. 2022, 46, 9238–9249. [Google Scholar] [CrossRef]
- Rodríguez-González, F.E.; Niebla, V.; Velázquez-Tundidor, M.; Tagle, L.H.; Martin-Trasanco, R.; Coll, D.; Ortiz, P.A.; Escalona, N.; Pérez, E.; Jessop, I.A.; et al. A new porous organic polymer containing Tröger’s base units: Evaluation of the catalytic activity in Knoevenagel condensation reaction. React. Funct. Polym. 2021, 167, 104998. [Google Scholar] [CrossRef]
- Dawson, R.; Laybourn, A.; Clowes, R.; Khimyak, Y.Z.; Adams, D.J.; Cooper, A.I. Functionalized Conjugated Microporous Polymers. Macromolecules 2009, 42, 8809–8816. [Google Scholar] [CrossRef]
- Wang, T.; Zhao, Y.-C.; Zhang, L.-M.; Cui, Y.; Zhang, C.-S.; Han, B.-H. Novel approach to hydroxy-group-containing porous organic polymers from bisphenol A. Beilstein J. Org. Chem. 2017, 13, 2131–2137. [Google Scholar] [CrossRef]
- Wisser, F.M.; Eckhardt, K.; Wisser, D.; Böhlmann, W.; Grothe, J.; Brunner, E.; Kaskel, S. Tailoring Pore Structure and Properties of Functionalized Porous Polymers by Cyclotrimerization. Macromolecules 2014, 47, 4210–4216. [Google Scholar] [CrossRef]
- Li, H.; Han, X.; Yu, W.; Zhang, L.; Wei, M.; Wang, Z.; Kong, F.; Wang, W. Dimethoxypillar[5]arene knitted porous polymers for efficient removal of organic micropollutants from water. Chem. Eng. J. Adv. 2022, 12, 100384. [Google Scholar] [CrossRef]
- Dong, K.; Sun, Q.; Meng, X.; Xiao, F.-S. Strategies for the design of porous polymers as efficient heterogeneous catalysts: From co-polymerization to self-polymerization. Catal. Sci. Technol. 2017, 7, 1028–1039. [Google Scholar] [CrossRef]
- Stahlová, S.; Slováková, E.; Vaňkátová, P.; Zukal, A.; Kubů, M.; Brus, J.; Bondarev, D.; Moučka, R.; Sedláček, J. Chain-growth copolymerization of functionalized ethynylarenes with 1,4-diethynylbenzene and 4,4′-diethynylbiphenyl into conjugated porous networks. Eur. Polym. J. 2015, 67, 252–263. [Google Scholar] [CrossRef]
- Petrášová, S.; Zukal, A.; Brus, J.; Balcar, H.; Pastva, J.; Zedník, J.; Sedláček, J. New Hyper-Crosslinked Partly Conjugated Networks with Tunable Composition by Spontaneous Polymerization of Ethynylpyridines with Bis(bromomethyl)arenes: Synthesis, Spectral Properties, and Activity in CO2 Capture. Macromol. Chem. Phys. 2013, 214, 2856–2866. [Google Scholar] [CrossRef]
- Seo, M.; Kim, S.; Oh, J.; Kim, S.-J.; Hillmyer, M.A. Hierarchically Porous Polymers from Hyper-cross-linked Block Polymer Precursors. J. Am. Chem. Soc. 2015, 137, 600–603. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Seo, M. Downsizing of Block Polymer-Templated Nanopores to One Nanometer via Hyper-Cross-Linking of High χ–Low N Precursors. ACS Nano 2021, 15, 9154–9166. [Google Scholar] [CrossRef] [PubMed]
- Slováková, E.; Ješelnik, M.; Žagar, E.; Zedník, J.; Sedláček, J.; Kovačič, S. Chain-Growth Insertion Polymerization of 1,3-Diethynylbenzene High Internal Phase Emulsions into Reactive π-Conjugated Foams. Macromolecules 2014, 47, 4864–4869. [Google Scholar] [CrossRef]
- Jurjevec, S.; Žerjav, G.; Pintar, A.; Žagar, E.; Kovačič, S. Tunable poly(aryleneethynylene) networks prepared by emulsion templating for visible-light-driven photocatalysis. Catal. Today 2020, 361, 146–151. [Google Scholar] [CrossRef]
- Huang, J.; Zhou, X.; Lamprou, A.; Maya, F.; Svec, F.; Turner, S.R. Nanoporous Polymers from Cross-Linked Polymer Precursors via tert-Butyl Group Deprotection and Their Carbon Dioxide Capture Properties. Chem. Mater. 2015, 27, 7388–7394. [Google Scholar] [CrossRef]
- Bashta, B.; Hašková, A.; Faukner, T.; Elsawy, M.A.; Šorm, D.; Brus, J.; Sedláček, J. Microporous hyper-cross-linked polyacetylene networks: Covalent structure and texture modification by reversible Schiff-base chemistry. Eur. Polym. J. 2020, 136, 109914. [Google Scholar] [CrossRef]
- Bashta, B.; Havelková, L.; Sokol, J.; Brus, J.; Sedláček, J. Microporous polymers prepared from non-porous hyper-cross-linked networks by removing covalently attached template molecules. Microporous Mesoporous Mater. 2021, 330, 111636. [Google Scholar] [CrossRef]
- Sedláček, J.; Balcar, H. Substituted Polyacetylenes Prepared with Rh Catalysts: From Linear to Network-Type Conjugated Polymers. Polym. Rev. 2016, 57, 31–51. [Google Scholar] [CrossRef]
- Masuda, T. Substituted Polyacetylenes: Synthesis, Properties, and Functions. Polym. Rev. 2016, 57, 1–14. [Google Scholar] [CrossRef]
- Inoue, Y.; Ishida, T.; Sano, N.; Yajima, T.; Sogawa, H.; Sanda, F. Platinum-Mediated Reversible Cross-linking/Decross-linking of Polyacetylenes Substituted with Phosphine Ligands: Catalytic Activity for Hydrosilylation. Macromolecules 2022, 55, 5711–5722. [Google Scholar] [CrossRef]
- Perlmutter, P. Propargyl Aldehyde. In Encyclopedia of Reagents for Organic Synthesis; John Wiley & Sons: Hoboken, NJ, USA, 2001; ISBN 9780470842898. [Google Scholar] [CrossRef]
- Zhou, L.; Kaiser, R.I.; Gao, L.G.; Chang, A.H.H.; Liang, M.; Yung, Y.L. Pathways to Oxygen-Bearing Molecules in the Interstellar Medium and in Planetary Atmospheres: Cyclopropenone (c-C3H2O) and Propynal (HCCCHO). Astrophys. J. 2008, 686, 1493–1502. [Google Scholar] [CrossRef]
- Sekerová, L.; Lhotka, M.; Vyskočilová, E.; Faukner, T.; Slováková, E.; Brus, J.; Červený, L.; Sedláček, J. Hyper-Cross-Linked Polyacetylene-Type Microporous Networks Decorated with Terminal Ethynyl Groups as Heterogeneous Acid Catalysts for Acetalization and Esterification Reactions. Chem. A Eur. J. 2018, 24, 14742–14749. [Google Scholar] [CrossRef]
- Metrane, A.; Delhali, A.; Ouikhalfan, M.; Assen, A.H.; Belmabkhout, Y. Water Vapor Adsorption by Porous Materials: From Chemistry to Practical Applications. J. Chem. Eng. Data 2022, 67, 1617–1653. [Google Scholar] [CrossRef]
- Chen, Q.; Liu, D.-P.; Zhu, J.-H.; Han, B.-H. Mesoporous Conjugated Polycarbazole with High Porosity via Structure Tuning. Macromolecules 2014, 47, 5926–5931. [Google Scholar] [CrossRef]
- Byun, Y.; Coskun, A. Epoxy-Functionalized Porous Organic Polymers via the Diels-Alder Cycloaddition Reaction for Atmospheric Water Capture. Angew. Chem. Int. Ed. 2018, 57, 3173–3177. [Google Scholar] [CrossRef]
- Karak, S.; Kandambeth, S.; Biswal, B.P.; Sasmal, H.S.; Kumar, S.; Pachfule, P.; Banerjee, R. Constructing Ultraporous Covalent Organic Frameworks in Seconds via an Organic Terracotta Process. J. Am. Chem. Soc. 2017, 139, 1856–1862. [Google Scholar] [CrossRef]
- Nguyen, H.L.; Hanikel, N.; Lyle, S.J.; Zhu, C.; Proserpio, D.M.; Yaghi, O.M. A Porous Covalent Organic Framework with Voided Square Grid Topology for Atmospheric Water Harvesting. J. Am. Chem. Soc. 2020, 142, 2218–2221. [Google Scholar] [CrossRef]
- Lu, H.; Shi, W.; Guo, Y.; Guan, W.; Lei, C.; Yu, G. Materials Engineering for Atmospheric Water Harvesting: Progress and Perspectives. Adv. Mater. 2022, 34, 2110079. [Google Scholar] [CrossRef]
- Singh, G.; Lee, J.; Karakoti, A.; Bahadur, R.; Yi, J.; Zhao, D.; AlBahily, K.; Vinu, A. Emerging trends in porous materials for CO2 capture and conversion. Chem. Soc. Rev. 2020, 49, 4360–4404. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Network Code | SBET (m2/g) | Vmi (cm3/g) | Vtot (cm3/g) | Dmi (nm) |
|---|---|---|---|---|
| P(M1/TEB 1:1) | 832 | 0.29 | 0.90 | 0.9 |
| P(M1/TEB 1:1)H | 911 | 0.35 | 0.60 | 0.7 |
| P(M1/TEB 3:1) | 313 | 0.12 | 0.19 | 1.1 |
| P(M1/TEB 3:1)H | 534 | 0.22 | 0.28 | 0.7 |
| P(M2/TEB 1:1) | 393 | 0.13 | 0.71 | 1 |
| P(M2/TEB 1:1)H | 794 | 0.29 | 0.76 | 0.7 |
| P(M2/TEB 3:1) | 149 | 0.05 | 0.11 | 1.1 |
| P(M2/TEB 3:1)H | 409 | 0.17 | 0.22 | 0.7 |
| Network Code | Polar Group Content (mmol/g) | Vtot (cm3/g) | aH2ORH40 (mg/g) | aH2ORH90 (mg/g) |
|---|---|---|---|---|
| P(M1/TEB 1:1)H | 4.90 | 0.60 | 100 | 445 |
| P(M1/TEB 3:1)H | 9.61 | 0.28 | 139 | 272 |
| P(M2/TEB 1:1)H | 4.85 | 0.76 | 56 | 422 |
| P(M2/TEB 3:1)H | 9.42 | 0.22 | 98 | 286 |
| P(pentyne/TEB 3:1) | 0 | 1.69 | 14 | 36 |
| Network Code | Polar Group Content (mmol/g) | SBET (m2/g) | 0.2 bar | 1 bar | Qst (kJ/mol) | ||
|---|---|---|---|---|---|---|---|
| aCO2 (mg/g) | aCO2/S (µg/m2) | aCO2 (mg/g) | aCO2/S (µg/m2) | ||||
| P(M1/TEB 1:1)H | 4.90 | 911 | 38.1 | 41.8 | 99.0 | 108.7 | 26 |
| P(M1/TEB 3:1)H | 9.61 | 534 | 42.2 | 79.0 | 87.6 | 164.0 | 29 |
| P(M2/TEB 1:1)H | 4.85 | 749 | 26.7 | 35.6 | 72.6 | 96.9 | 26 |
| P(M2/TEB 3:1)H | 9.42 | 409 | 32.9 | 80.4 | 75.2 | 184.0 | 27 |
| P(pentyne/TEB 3:1) | 0 | 810 | 13.5 | 16.7 | 39.2 | 48.3 | 23 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Havelková, L.; Bashta, B.; Hašková, A.; Vagenknechtová, A.; Vyskočilová, E.; Brus, J.; Sedláček, J. Combining Polymerization and Templating toward Hyper-Cross-Linked Poly(propargyl aldehyde)s and Poly(propargyl alcohol)s for Reversible H2O and CO2 Capture and Construction of Porous Chiral Networks. Polymers 2023, 15, 743. https://doi.org/10.3390/polym15030743
Havelková L, Bashta B, Hašková A, Vagenknechtová A, Vyskočilová E, Brus J, Sedláček J. Combining Polymerization and Templating toward Hyper-Cross-Linked Poly(propargyl aldehyde)s and Poly(propargyl alcohol)s for Reversible H2O and CO2 Capture and Construction of Porous Chiral Networks. Polymers. 2023; 15(3):743. https://doi.org/10.3390/polym15030743
Chicago/Turabian StyleHavelková, Lucie, Bogdana Bashta, Alena Hašková, Alice Vagenknechtová, Eliška Vyskočilová, Jiří Brus, and Jan Sedláček. 2023. "Combining Polymerization and Templating toward Hyper-Cross-Linked Poly(propargyl aldehyde)s and Poly(propargyl alcohol)s for Reversible H2O and CO2 Capture and Construction of Porous Chiral Networks" Polymers 15, no. 3: 743. https://doi.org/10.3390/polym15030743