
Wild-Type Isocitrate Dehydrogenase-Dependent Oxidative Decarboxylation and Reductive Carboxylation in Cancer and Their Clinical Significance

Abstract
:Simple Summary
Abstract
1. Introduction
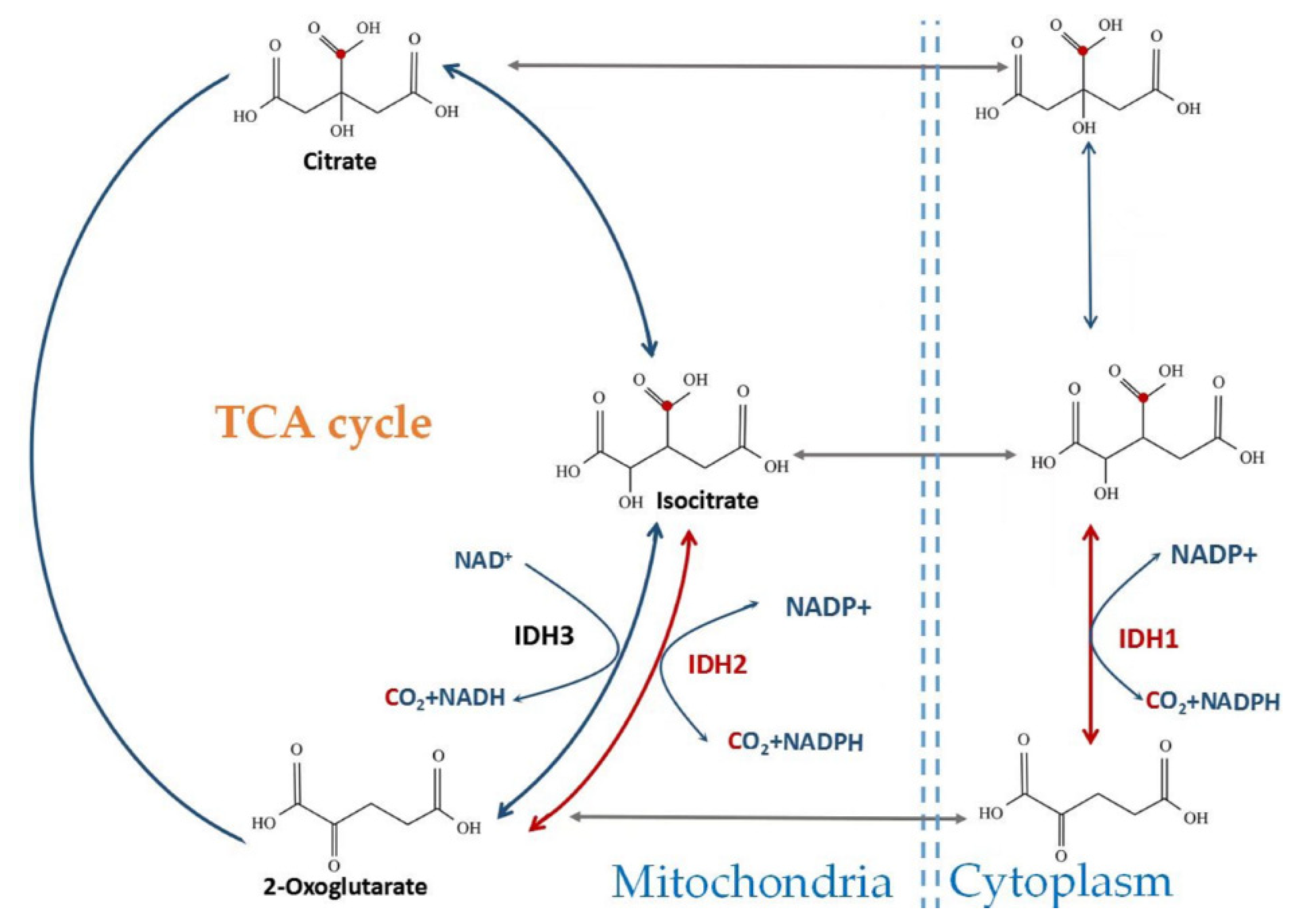
2. Catalytic Mechanism and Enzyme Structure of IDHs
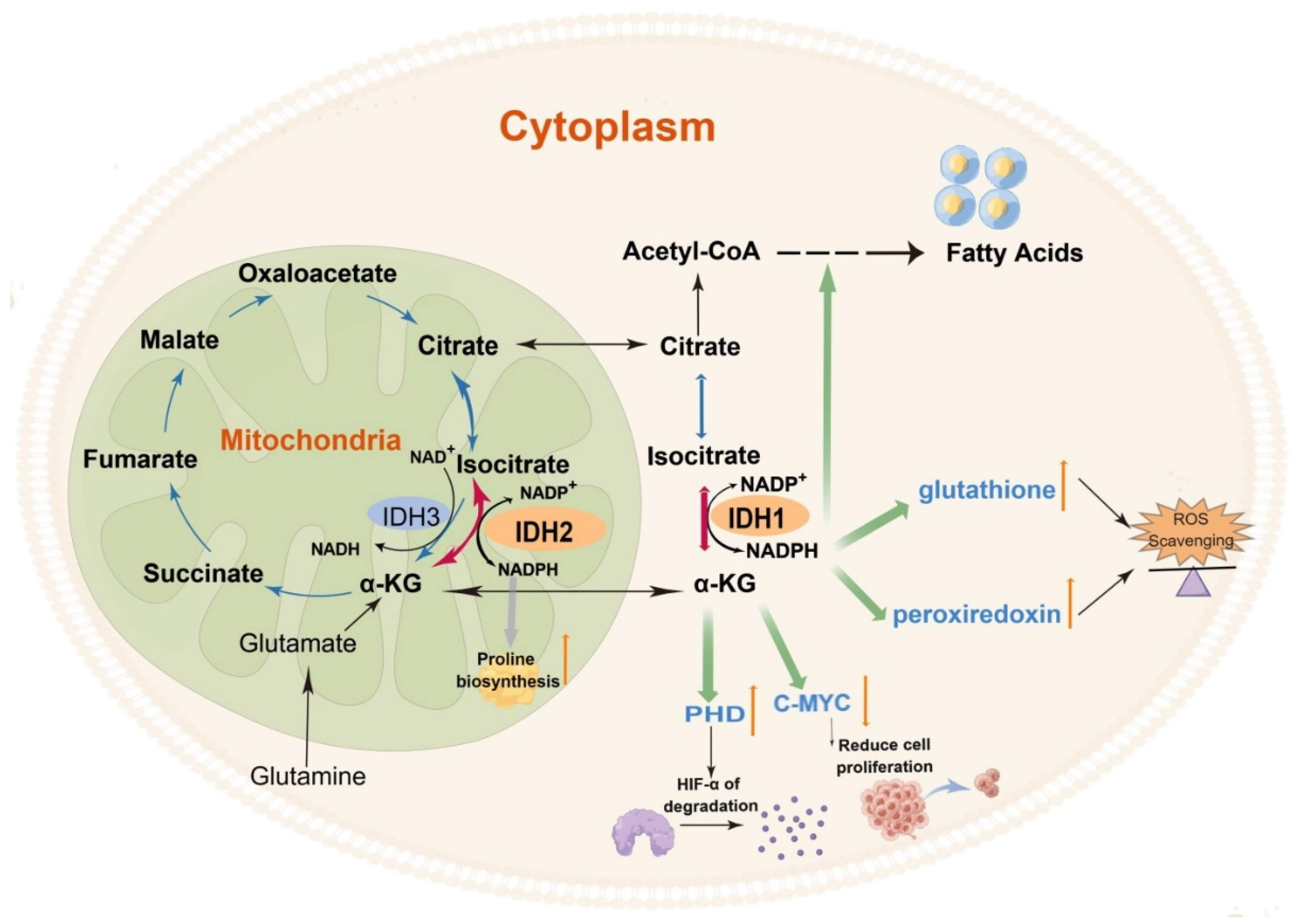
3. The Role of Wild-Type IDH1 and IDH2 in Cellular Metabolism
4. The Role of Wild-Type IDH3 in Cellular Metabolism
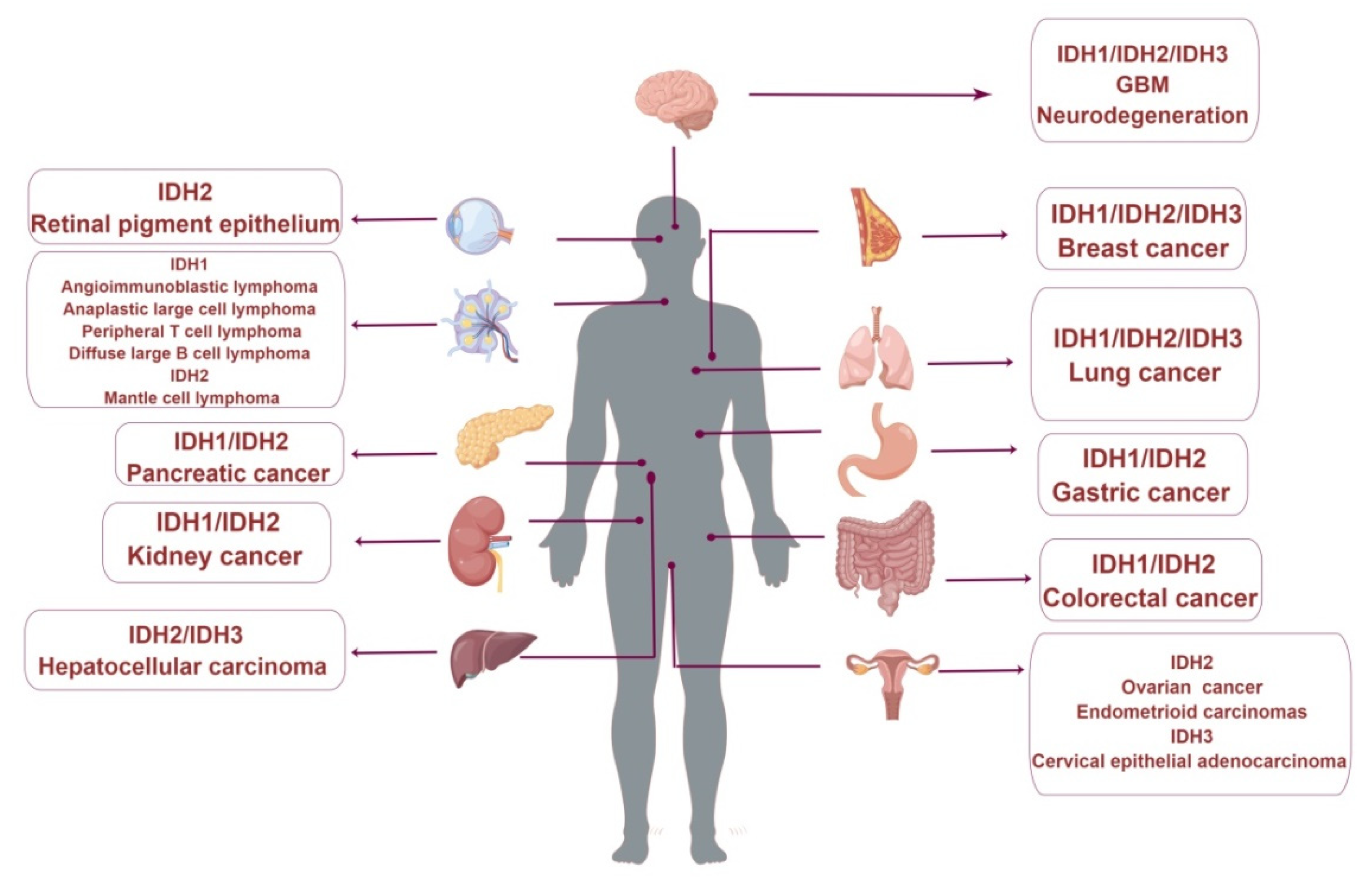
5. Research Progress of Wild-Type IDH1 in Cancers
6. Research Progress of Wild-Type IDH2 in Cancers
7. Research Progress of Wild-Type IDH3 in Cancers
8. Conclusions (Future Perspectives)
Author Contributions
Funding
Conflicts of Interest
References
- Cadoux-Hudson, T.; Schofield, C.J.; McCullagh, J.S.O. Isocitrate dehydrogenase gene variants in cancer and their clinical significance. Biochem. Soc. Trans. 2021, 49, 2561–2572. [Google Scholar] [CrossRef] [PubMed]
- Dang, L.; Jin, S.; Su, S.M. IDH mutations in glioma and acute myeloid leukemia. Trends Mol. Med. 2010, 16, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Cadoux-Hudson, T.; Schofield, C.J. Isocitrate dehydrogenase variants in cancer—Cellular consequences and therapeutic opportunities. Curr. Opin. Chem. Biol. 2020, 57, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Dang, L.; Su, S.-S.M. Isocitrate Dehydrogenase Mutation and (R)-2-Hydroxyglutarate: From Basic Discovery to Therapeutics Development. Annu. Rev. Biochem. 2017, 86, 305–331. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Weller, M.; Lee, E.Q.; Alexander, B.M.; Barnholtz-Sloan, J.S.; Barthel, F.P.; Batchelor, T.T.; Bindra, R.S.; Chang, S.M.; Chiocca, E.A.; et al. Glioblastoma in Adults: A Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) Consensus Review on Current Management and Future Directions. Neuro-Oncol. 2020, 22, 1073–1113. [Google Scholar] [CrossRef] [PubMed]
- Koppenol, W.H.; Bounds, P.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Mathupala, S.P.; Ko, Y.H.; Pedersen, P.L. Hexokinase-2 bound to mitochondria: Cancer‘s stygian link to the “Warburg effect” and a pivotal target for effective therapy. Semin. Cancer Biol. 2008, 19, 17–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peter, V.; Heinz, S.; Arnulf, M. The Warburg effect: Essential part of metabolic reprogramming and central contributor to cancer progression. Int. J. Radiat. Biol. 2019, 95, 912–919. [Google Scholar]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [Green Version]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2011, 481, 380–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaude, E.; Schmidt, C.; Gammage, P.A.; Dugourd, A.; Blacker, T.; Chew, S.P.; Saez-Rodriguez, J.; O’Neill, J.S.; Szabadkai, G.; Minczuk, M.; et al. NADH Shuttling Couples Cytosolic Reductive Carboxylation of Glutamine with Glycolysis in Cells with Mitochondrial Dysfunction. Mol. Cell 2018, 69, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Schwörer, S.; Berisa, M.; Kyung, Y.J.; Ryu, K.W.; Yi, J.; Jiang, X.; Cross, J.R.; Thompson, C.B. Mitochondrial NADP(H) generation is essential for proline biosynthesis. Science 2021, 372, 968–972. [Google Scholar] [CrossRef] [PubMed]
- Zeng, P.; Lu, W.; Tian, J.; Qiao, S.; Li, J.; Glorieux, C.; Wen, S.; Zhang, H.; Li, Y.; Huang, P. Reductive TCA cycle catalyzed by wild-type IDH2 promotes acute myeloid leukemia and is a metabolic vulnerability for potential targeted therapy. J. Hematol. Oncol. 2022, 15, 30. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.-F.; Jensen, M.V.; Gray, S.M.; El, K.; Wang, Y.; Lu, D.; Becker, T.C.; Campbell, J.E.; Newgard, C.B. Reductive TCA cycle metabolism fuels glutamine-and glucose-stimulated insulin secretion. Cell Metab. 2021, 33, 804–817. [Google Scholar] [CrossRef]
- Wise, D.R.; Ward, P.S.; Shay, J.E.S.; Cross, J.R.; Gruber, J.J.; Sachdeva, U.M.; Platt, J.M.; DeMatteo, R.G.; Simon, M.C.; Thompson, C.B. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of α-ketoglutarate to citrate to support cell growth and viability. Proc. Natl. Acad. Sci. USA 2011, 108, 19611–19616. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Shestov, A.A.; Swain, P.; Yang, C.; Parker, S.J.; Wang, Q.A.; Terada, L.S.; Adams, N.D.; McCabe, M.T.; Pietrak, B.; et al. Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature 2016, 532, 255–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claudiane, G.; Murthy, M.S.R.; Alexandre, A.; Erik, J.; Marc, P. A role for ATP-citrate lyase, malic enzyme, and pyruvate/citrate cycling in glucose-induced insulin secretion. J. Biol. Chem. 2007, 282, 35657–35665. [Google Scholar]
- Du, J.; Yanagida, A.; Knight, K.; Engel, A.L.; Vo, A.H.; Jankowski, C.; Sadilek, M.; Tran, V.T.B.; Manson, M.A.; Ramakrishnan, A.; et al. Reductive carboxylation is a major metabolic pathway in the retinal pigment epithelium. Proc. Natl. Acad. Sci. USA 2016, 113, 14710–14715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Ye, D.; Guan, K.-L.; Xiong, Y. IDH1 and IDH2 mutations in tumorigenesis: Mechanistic insights and clinical perspectives. Clin. Cancer Res. J. Am. Assoc. Cancer Res. 2012, 18, 5562–5571. [Google Scholar] [CrossRef] [Green Version]
- Leonardi, R.; Subramanian, C.; Jackowski, S.; Rock, C.O. Cancer-associated isocitrate dehydrogenase mutations inactivate NADPH-dependent reductive carboxylation. J. Biol. Chem. 2012, 287, 14615–14620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reitman, Z.J.; Yan, H. Isocitrate dehydrogenase 1 and 2 mutations in cancer: Alterations at a crossroads of cellular metabolism. J. Natl. Cancer Inst. 2010, 102, 932–941. [Google Scholar] [CrossRef] [Green Version]
- Rosiers, C.D.; Fernandez, C.A.; David, F.; Brunengraber, H. Reversibility of the mitochondrial isocitrate dehydrogenase reaction in the perfused rat liver. Evidence from isotopomer analysis of citric acid cycle intermediates. J. Biol. Chem. 1994, 269, 27179–27182. [Google Scholar] [CrossRef] [PubMed]
- Nekrutenko, A.; Hillis, D.M.; Patton, J.C.; Bradley, R.D.; Baker, R.J. Cytosolic isocitrate dehydrogenase in humans, mice, and voles and phylogenetic analysis of the enzyme family. Mol. Biol. Evol. 1998, 15, 1674–1684. [Google Scholar] [CrossRef] [Green Version]
- Ma, T.; Peng, Y.; Huang, W.; Liu, Y.; Ding, J. The β and γ subunits play distinct functional roles in the α(2)βγ heterotetramer of human NAD-dependent isocitrate dehydrogenase. Sci Rep. 2017, 7, 41882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergaggio, E.; Piva, R. Wild-Type IDH Enzymes as Actionable Targets for Cancer Therapy. Cancers 2019, 11, 563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, P.F.; Colman, R.F. Diphosphopyridine nucleotide dependent isocitrate dehydrogenase from pig heart. Charactgerization of the active substrate and modes of regulation. Biochemistry 1972, 11, 1501–1508. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, R.S.; Colman, R.F. Binding of ligands to half of subunits of NAD-dependent isocitrate dehydrogenase from pig heart. Binding of manganous ion, isocitrate, ADP and NAD. J. Biol. Chem. 1981, 256, 1276–1282. [Google Scholar] [CrossRef]
- Ying, W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: Regulation and biological consequences. Antioxid. Redox Signal. 2008, 10, 1672. [Google Scholar] [CrossRef] [Green Version]
- Ogata, F.T.; Branco, V.; Vale, F.F.; Coppo, L. Glutaredoxin: Discovery, redox defense and much more. Redox Biol. 2021, 43, 101975. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.M.; Koh, H.-J.; Park, D.-C.; Song, B.J.; Huh, T.-L.; Park, J.-W. Cytosolic NADP + -dependent isocitrate dehydrogenase status modulates oxidative damage to cells. Free Radic. Biol. Med. 2002, 32, 1185–1196. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.A.; Parker, S.J.; Fiske, B.P.; McCloskey, D.; Gui, D.Y.; Green, C.R.; Vokes, N.I.; Feist, A.M.; Heiden, M.G.V.; Metallo, C.M. Tracing Compartmentalized NADPH Metabolism in the Cytosol and Mitochondria of Mammalian Cells. Mol. Cell 2014, 55, 253–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollak, N.; Niere, M.; Ziegler, M. NAD Kinase Levels Control the NADPH Concentration in Human Cells. J. Biol. Chem. 2007, 282, 33562–33571. [Google Scholar] [CrossRef]
- Nikiforov, A.; Dölle, C.; Niere, M.; Ziegler, M. Pathways and subcellular compartmentation of NAD biosynthesis in human cells: From entry of extracellular precursors to mitochondrial NAD generation. J. Biol. Chem. 2011, 286, 21767–21778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minard, K.I.; McAlister-Henn, L. Dependence of peroxisomal beta-oxidation on cytosolic sources of NADPH. J. Biol. Chem. 1999, 274, 3402–3406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itsumi, M.; Inoue, S.; Elia, A.J.; Murakami, K.; Sasaki, M.; Lind, E.F.; Brenner, D.; Harris, I.S.; Chio, I.I.C.; Afzal, S.; et al. Idh1 protects murine hepatocytes from endotoxin-induced oxidative stress by regulating the intracellular NADP(+)/NADPH ratio. Cell Death Differ. 2015, 22, 1837–1845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, J.; Ye, J.; Kamphorst, J.J.; Shlomi, T.; Thompson, C.B.; Rabinowitz, J.D. Quantitative flux analysis reveals folate-dependent NADPH production. Nature 2014, 510, 298–302. [Google Scholar] [CrossRef] [Green Version]
- Hosios, A.M.; Heiden, M.G.V. The redox requirements of proliferating mammalian cells. J. Biol. Chem. 2018, 293, 7490–7498. [Google Scholar] [CrossRef] [Green Version]
- Katsyuba, E.; Romani, M.; Hofer, D.; Auwerx, J. NAD+ homeostasis in health and disease. Nat. Metab. 2020, 2, 9–31. [Google Scholar] [CrossRef]
- Xiao, W.; Loscalzo, J. Metabolic Responses to Reductive Stress. Antioxid. Redox Signal. 2020, 32, 1330–1347. [Google Scholar] [CrossRef] [Green Version]
- Ducker, G.S.; Rabinowitz, J.D. One-Carbon Metabolism in Health and Disease. Cell Metab. 2017, 25, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.; Deik, A.; Gonzalez, C.; Gonzalez, M.E.; Fu, F.; Ferrari, M.; Churchhouse, C.L.; Florez, J.C.; Jacobs, S.B.; Clish, C.B.; et al. Polyunsaturated Fatty Acid Desaturation Is a Mechanism for Glycolytic NAD+ Recycling. Cell Metab. 2019, 29, 856–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouleau, M.; Patel, A.; Hendzel, M.J.; Kaufmann, S.H.; Poirier, G.G. PARP inhibition: PARP1 and beyond. Nat. Rev. Cancer 2010, 10, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Borger, D.R.; Tanabe, K.K.; Fan, K.C.; Lopez, H.U.; Fantin, V.R.; Straley, K.S.; Schenkein, D.P.; Hezel, A.F.; Ancukiewicz, M.; Liebman, H.M.; et al. Frequent Mutation of Isocitrate Dehydrogenase (IDH)1 and IDH2 in Cholangiocarcinoma Identified Through Broad-Based Tumor Genotyping. Oncology 2012, 17, 72–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, M.R.; Kim, M.S.; Oh, J.E.; Kim, Y.R.; Song, S.Y.; Seo, S.I.; Lee, J.Y.; Yoo, N.J.; Lee, S.H. Mutational analysis of IDH1 codon 132 in glioblastomas and other common cancers. Int. J. Cancer 2009, 125, 353–355. [Google Scholar] [CrossRef]
- Paschka, P.; Schlenk, R.F.; Gaidzik, V.I.; Habdank, M.; Krönke, J.; Späth, L.B.D.; Kayser, S.; Zucknick, M.; Götze, K.; Horst, H.-A.; et al. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J. Clin. Oncol. J. Am. Soc. Clin. Oncol. 2010, 28, 3636–3643. [Google Scholar] [CrossRef]
- Mardis, E.R.; Ding, L.; Dooling, D.J.; Larson, D.E.; McLellan, M.D.; Chen, K.; Koboldt, D.C.; Fulton, R.S.; Delehaunty, K.D.; McGrath, S.D.; et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N. Engl. J. Med. 2009, 361, 1058–1066. [Google Scholar] [CrossRef] [Green Version]
- Jonsson, P.; Lin, A.L.; Young, R.J.; DiStefano, N.M.; Hyman, D.M.; Li, B.T.; Berger, M.F.; Zehir, A.; Ladanyi, M.; Solit, D.B.; et al. Genomic Correlates of Disease Progression and Treatment Response in Prospectively Characterized Gliomas. Clin. Cancer Res. 2019, 25, 5537–5547. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Lang, F.; Chou, F.-J.; Zaghloul, K.A.; Yang, C. Isocitrate Dehydrogenase Mutations in Glioma: Genetics, Biochemistry, and Clinical Indications. Biomedicines 2020, 8, 294. [Google Scholar] [CrossRef]
- Turcan, S.; Rohle, D.; Goenka, A.; Walsh, L.A.; Fang, F.; Yilmaz, E.; Campos, C.; Fabius, A.W.M.; Lu, C.; Ward, P.S.; et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012, 483, 479–483. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef] [Green Version]
- Waitkus, M.S.; Diplas, B.H.; Yan, H. Isocitrate dehydrogenase mutations in gliomas. Neuro-Oncol. 2016, 18, 16–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golub, D.; Iyengar, N.; Dogra, S.; Wong, T.; Bready, D.; Tang, K.; Modrek, A.S.; Placantonakis, D.G. Mutant Isocitrate Dehydrogenase Inhibitors as Targeted Cancer Therapeutics. Front. Oncol. 2019, 9, 00417. [Google Scholar] [CrossRef] [PubMed]
- Ohka, F.; Ito, M.; Ranjit, M.; Senga, T.; Motomura, A.; Motomura, K.; Saito, K.; Kato, K.; Kato, Y.; Wakabayashi, T.; et al. Quantitative metabolome analysis profiles activation of glutaminolysis in glioma with IDH1 mutation. Tumor Biol. 2014, 35, 5911–5920. [Google Scholar] [CrossRef] [PubMed]
- Alzial, G.; Renoult, O.; Paris, F.; Gratas, C.; Clavreul, A.; Pecqueur, C. Wild-type isocitrate dehydrogenase under the spotlight in glioblastoma. Oncogene 2021, 41, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Al-Amodi, H.S.A.B.; Nabih, E.S.; Kamel, H.F.M.; El Sayed, M.A.; Dwedar, I.A.M. Wild-Type Isocitrate Dehydrogenase 1 Over-Expression is Related to Cancer Stem Cells Survival in Lung Adenocarcinoma. Cancer Investig. 2018, 36, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wang, Y.; Xiong, Y.; Peng, T.; Lu, M.; Zhang, L.; Guo, Z. Wild-type IDH1 inhibits the tumor growth through degrading HIF-α in renal cell carcinoma. Int. J. Biol. Sci. 2021, 17, 1250–1262. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.-Z.; Ye, J.C.; Pu, J.J.; Liu, H.; Ding, W.; Zheng, H.; Liu, D. Novel agents and regimens for hematological malignancies: Recent updates from 2020 ASH annual meeting. J. Hematol. Oncol. 2021, 14, 66. [Google Scholar] [CrossRef]
- Calvert, A.E.; Chalastanis, A.; Wu, Y.; Hurley, L.A.; Kouri, F.M.; Bi, Y.; Kachman, M.; May, J.L.; Bartom, E.; Hua, Y.; et al. Cancer-Associated IDH1 Promotes Growth and Resistance to Targeted Therapies in the Absence of Mutation. Cell Rep. 2017, 19, 1858–1873. [Google Scholar] [CrossRef] [Green Version]
- Zarei, M.; Lal, S.; Parker, S.J.; Nevler, A.; Vaziri-Gohar, A.; Dukleska, K.; Mambelli-Lisboa, N.C.; Moffat, C.; Blanco, F.F.; Chand, S.N.; et al. Posttranscriptional Upregulation of IDH1 by HuR Establishes a Powerful Survival Phenotype in Pancreatic Cancer Cells. Cancer Res. 2017, 77, 4460–4471. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.-F.; Wang, M.; Xu, J.-L.; Ning, Y.-J. Hypoxia promotes mitochondrial glutamine metabolism through HIF1α-GDH pathway in human lung cancer cells. Biochem. Biophys. Res. Commun. 2017, 483, 32–38. [Google Scholar] [CrossRef]
- Li, J.; He, Y.; Tan, Z.; Lu, J.; Li, L.; Song, X.; Shi, F.; Xie, L.; You, S.; Luo, X.; et al. Wild-type IDH2 promotes the Warburg effect and tumor growth through HIF1α in lung cancer. Theranostics 2018, 8, 4050–4061. [Google Scholar] [CrossRef]
- Wang, L.-N.; Tong, S.-W.; Hu, H.-D.; Ye, F.; Li, S.-L.; Ren, H.; Zhang, D.-Z.; Xiang, R.; Yang, Y.-X. Quantitative proteome analysis of ovarian cancer tissues using a iTRAQ approach. J. Cell Biochem. 2012, 113, 3762–3772. [Google Scholar] [CrossRef] [PubMed]
- Tian, G.-Y.; Zang, S.-F.; Wang, L.; Luo, Y.; Shi, J.-P.; Lou, G.-Q. Isocitrate Dehydrogenase 2 Suppresses the Invasion of Hepatocellular Carcinoma Cells via Matrix Metalloproteinase 9. Cell Physiol. Biochem. 2015, 37, 2405–2414. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Yang, D.; Hou, Y.; Liu, S.; Zhao, M.; Qin, Y.; Chen, R.; Teng, Y.; Liu, M. Intracellular citrate accumulation by oxidized ATM-mediated metabolism reprogramming via PFKP and CS enhances hypoxic breast cancer cell invasion and metastasis. Cell Death Dis. 2019, 10, 228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D. Isocitrate dehydrogenase 2 inhibits gastric cancer cell invasion via matrix metalloproteinase 7. Tumor Biol. 2015, 37, 5225–5230. [Google Scholar] [CrossRef]
- Guirguis, A.; Elishaev, E.; Oh, S.-H.; Tseng, G.C.; Zorn, K.; DeLoia, J.A. Use of gene expression profiles to stage concurrent endometrioid tumors of the endometrium and ovary. Gynecol. Oncol. 2008, 108, 370–376. [Google Scholar] [CrossRef]
- Xujun, L.; Yan, Q.; Xia, T.; Wenzhe, S. Isocitrate dehydrogenase 3A, a rate-limiting enzyme of the TCA cycle, promotes hepatocellular carcinoma migration and invasion through regulation of MTA1, a core component of the NuRD complex. Am. J. Cancer Res. 2020, 10, 3212–3229. [Google Scholar]
- May, J.L.; Kouri, F.M.; Hurley, L.A.; Liu, J.; Tommasini-Ghelfi, S.; Ji, Y.; Gao, P.; Calvert, A.E.; Lee, A.; Chandel, N.S.; et al. IDH3α regulates one-carbon metabolism in glioblastoma. Sci. Adv. 2019, 5, 0456. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Li, Z.-X.; Zhao, W.-C.; Huang, H.-B.; Wang, J.-Q.; Zhang, H.; Lu, J.-Y.; Wang, R.-N.; Li, W.; Cheng, Z.; et al. Identification and characterization of isocitrate dehydrogenase 1 (IDH1) as a functional target of marine natural product grincamycin B. Acta Pharmacol. Sin. 2021, 42, 801–813. [Google Scholar] [CrossRef]
- Mohammed, M.R.S.; Alzahrani, F.; Hosawi, S.; Choudhry, H.; Khan, M.I. Profiling the Effect of Targeting Wild Isocitrate Dehydrogenase 1 (IDH1) on the Cellular Metabolome of Leukemic Cells. Int. J. Mol. Sci. 2022, 23, 6653. [Google Scholar] [CrossRef]
- Al Tameemi, W.; Dale, T.P.; Al-Jumaily, R.M.K.; Forsyth, N.R. Hypoxia-Modified Cancer Cell Metabolism. Front. Cell Dev. Biol. 2019, 7, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chisari, A.; Golán, I.; Campisano, S.; Gélabert, C.; Moustakas, A.; Sancho, P.; Caja, L. Glucose and Amino Acid Metabolic Dependencies Linked to Stemness and Metastasis in Different Aggressive Cancer Types. Front. Pharmacol. 2021, 12, 723798. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [PubMed]
- Ying, M.; You, D.; Zhu, X.; Cai, L.; Zeng, S.; Hu, X. Lactate and glutamine support NADPH generation in cancer cells under glucose deprived conditions. Redox Biol. 2021, 46, 102065. [Google Scholar] [CrossRef] [PubMed]
- Mehta, M.; Basalingappa, K.; Griffith, J.N.; Andrade, D.; Babu, A.; Amreddy, N.; Muralidharan, R.; Gorospe, M.; Herman, T.; Ding, W.-Q.; et al. HuR silencing elicits oxidative stress and DNA damage and sensitizes human triple-negative breast cancer cells to radiotherapy. Oncotarget 2016, 7, 64820–64835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, W.-J.; Cheng, S.; Campbell, C.; Wright, A.; Furneaux, H. Cloning and Characterization of HuR, a Ubiquitously Expressed Elav-like Protein. J. Biol. Chem. 1996, 271, 8144–8151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.; Ooi, W.F.; Qamra, A.; Tan, J.; Chua, B.Y.-J.; Ho, S.W.T.; Das, K.; Isa, Z.F.A.; Li, Z.; Yao, X.; et al. HNF4α pathway mapping identifies wild-type IDH1 as a targetable metabolic node in gastric cancer. Gut 2020, 69, 231–242. [Google Scholar] [CrossRef]
- Nagtegaal, I.D.; Knijn, N.; Hugen, N.; Marshall, H.; Sugihara, K.; Tot, T.; Ueno, H.; Quirke, P. Tumor Deposits in Colorectal Cancer: Improving the Value of Modern Staging—A Systematic Review and Meta-Analysis. J. Clin. Oncol. 2017, 35, 1119–1127. [Google Scholar] [CrossRef]
- Wu, J.; Wang, F.; Liu, X.; Zhang, T.; Liu, F.; Ge, X.; Mao, Y.; Hua, D. Correlation of IDH1 and B7 H3 expression with prognosis of CRC patients. Eur. J. Surg. Oncol. (EJSO) 2018, 44, 1254–1260. [Google Scholar] [CrossRef]
- Sun, R.C.; Denko, N.C. Hypoxic Regulation of Glutamine Metabolism through HIF1 and SIAH2 Supports Lipid Synthesis that Is Necessary for Tumor Growth. Cell Metab. 2014, 19, 285–292. [Google Scholar] [CrossRef] [Green Version]
- Herr, C.Q.; Hausinger, R.P. Amazing Diversity in Biochemical Roles of Fe(II)/2-Oxoglutarate Oxygenases. Trends Biochem. Sci. 2018, 43, 517–532. [Google Scholar] [CrossRef]
- Islam, S.; Leissing, T.M.; Chowdhury, R.; Hopkinson, R.J.; Schofield, C.J. 2-Oxoglutarate-Dependent Oxygenases. Annu. Rev. Biochem. 2018, 87, 585–620. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhang, Y. TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 2017, 18, 517–534. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, T.; Han, Y.; Ren, Z.; Zou, J.; Liu, J.; Xi, S. lncRNA OTUD6B-AS1 Exacerbates As2O3-Induced Oxidative Damage in Bladder Cancer via miR-6734-5p-Mediated Functional Inhibition of IDH2. Oxidative Med. Cell Longev. 2020, 2020, 3035624. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Xu, W.; Jiang, W.; Yu, W.; Lin, Y.; Zhang, T.; Yao, J.; Zhou, L.; Zeng, Y.; Li, H.; et al. Regulation of Cellular Metabolism by Protein Lysine Acetylation. Science 2010, 327, 1000–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, W.; Dittenhafer-Reed, K.E.; Denu, J.M. SIRT3 Protein Deacetylates Isocitrate Dehydrogenase 2 (IDH2) and Regulates Mitochondrial Redox Status. J. Biol. Chem. 2012, 287, 14078–14086. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Denu, R.A.; Krautkramer, K.A.; Grindle, K.M.; Yang, D.T.; Asimakopoulos, F.; Hematti, P.; Denu, J.M. Loss of SIRT3 Provides Growth Advantage for B Cell Malignancies. J. Biol. Chem. 2016, 291, 3268–3279. [Google Scholar] [CrossRef] [Green Version]
- Lv, Q.; Xing, S.; Li, Z.; Li, J.; Gong, P.; Xu, X.; Chang, L.; Jin, X.; Gao, F.; Li, W.; et al. Altered expression levels of IDH2 are involved in the development of colon cancer. Exp. Ther. Med. 2012, 4, 801–806. [Google Scholar] [CrossRef] [Green Version]
- Ting, L.; Song, C.; Tao, G.; Hong, Z.X.; Liu, T.; Dandan, L.; Michael, C.; Yuan, Z.Y.; Chen, F.Y.; Nicole, C.; et al. Visualization of endogenous p27 and Ki67 reveals the importance of a c-Myc-driven metabolic switch in promoting survival of quiescent cancer cells. Theranostics 2021, 11, 9605–9622. [Google Scholar]
- Zeng, L.; Morinibu, A.; Kobayashi, M.; Zhu, Y.; Wang, X.; Goto, Y.; Yeom, C.J.; Zhao, T.; Hirota, K.; Shinomiya, K.; et al. Aberrant IDH3α expression promotes malignant tumor growth by inducing HIF-1-mediated metabolic reprogramming and angiogenesis. Oncogene 2015, 34, 4758–4766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Wang, Y.; Shi, Z.; Liu, J.; Sun, P.; Hou, X.; Zhang, J.; Zhao, S.; Zhou, B.P.; Mi, J. Metabolic Reprogramming of Cancer-Associated Fibroblasts by IDH3α Downregulation. Cell Rep. 2015, 10, 1335–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.-H.; Yan, Z.-Z.; Luo, S.-D.; Hu, J.-J.; Wu, M.; Zhao, J.; Liu, W.-F.; Li, C.; Liu, K.-X. Gut microbiota-derived succinate aggravates acute lung injury after intestinal ischemia/reperfusion in mice. Eur. Respir. J. 2022, 60, 00840. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| IDH Type | Overexpressed | Downregulated |
|---|---|---|
| IDH1 | Lung adenocarcinoma [55] | Kidney cancer [56] |
| Angioimmunoblastic lymphoma [57] | ||
| Peripheral T cell lymphoma [57] | ||
| Diffuse large B cell lymphoma [57] | ||
| Glioblastoma [58] | ||
| Pancreatic ductal adenocarcinoma [59] | ||
| IDH2 | Lung cancer [60] | Kidney cancer [61] |
| Ovarian cancer [62] | Hepatocellular carcinoma [63] | |
| Breast cancer [64] | Gastric cancer [65] | |
| Endometroid carcinomas [66] | Glioblastoma [15,16] | |
| Acute lymphocytic leukemia [13] | ||
| IDH3 | Hepatocellular carcinoma [67] | |
| Glioblastoma [68] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, Q.; Chen, J.; Xie, Z.; Chen, Z. Wild-Type Isocitrate Dehydrogenase-Dependent Oxidative Decarboxylation and Reductive Carboxylation in Cancer and Their Clinical Significance. Cancers 2022, 14, 5779. https://doi.org/10.3390/cancers14235779
He Q, Chen J, Xie Z, Chen Z. Wild-Type Isocitrate Dehydrogenase-Dependent Oxidative Decarboxylation and Reductive Carboxylation in Cancer and Their Clinical Significance. Cancers. 2022; 14(23):5779. https://doi.org/10.3390/cancers14235779
Chicago/Turabian StyleHe, Qiwei, Junxiong Chen, Zijing Xie, and Zhenzhou Chen. 2022. "Wild-Type Isocitrate Dehydrogenase-Dependent Oxidative Decarboxylation and Reductive Carboxylation in Cancer and Their Clinical Significance" Cancers 14, no. 23: 5779. https://doi.org/10.3390/cancers14235779