
In Silico Searching for Alternative Lead Compounds to Treat Type 2 Diabetes through a QSAR and Molecular Dynamics Study

 , ,
, ,
Abstract
:1. Introduction
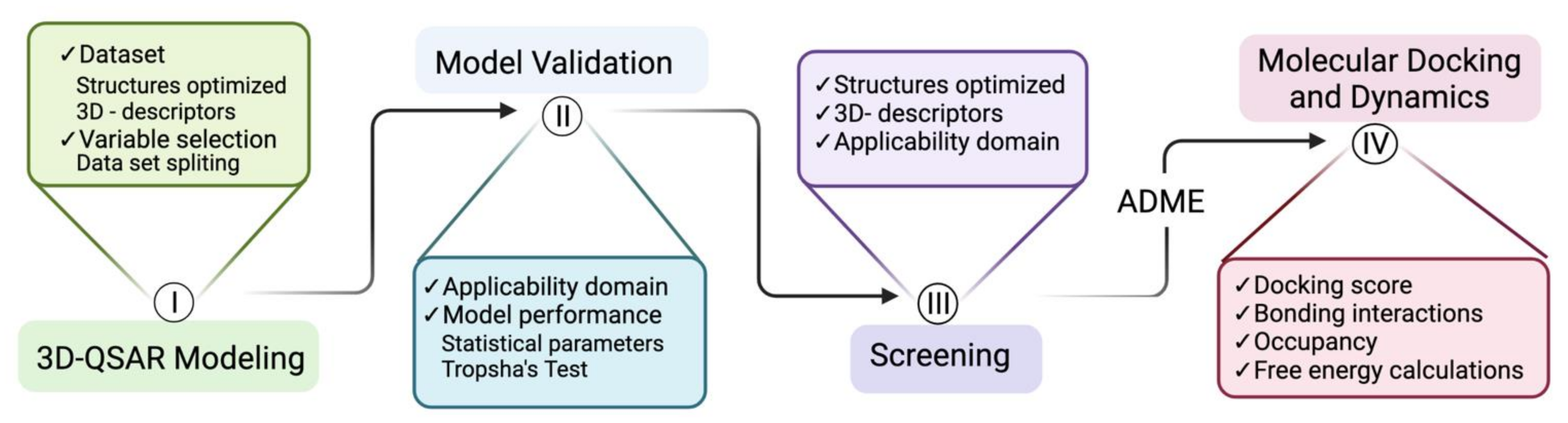
2. Materials and Methods
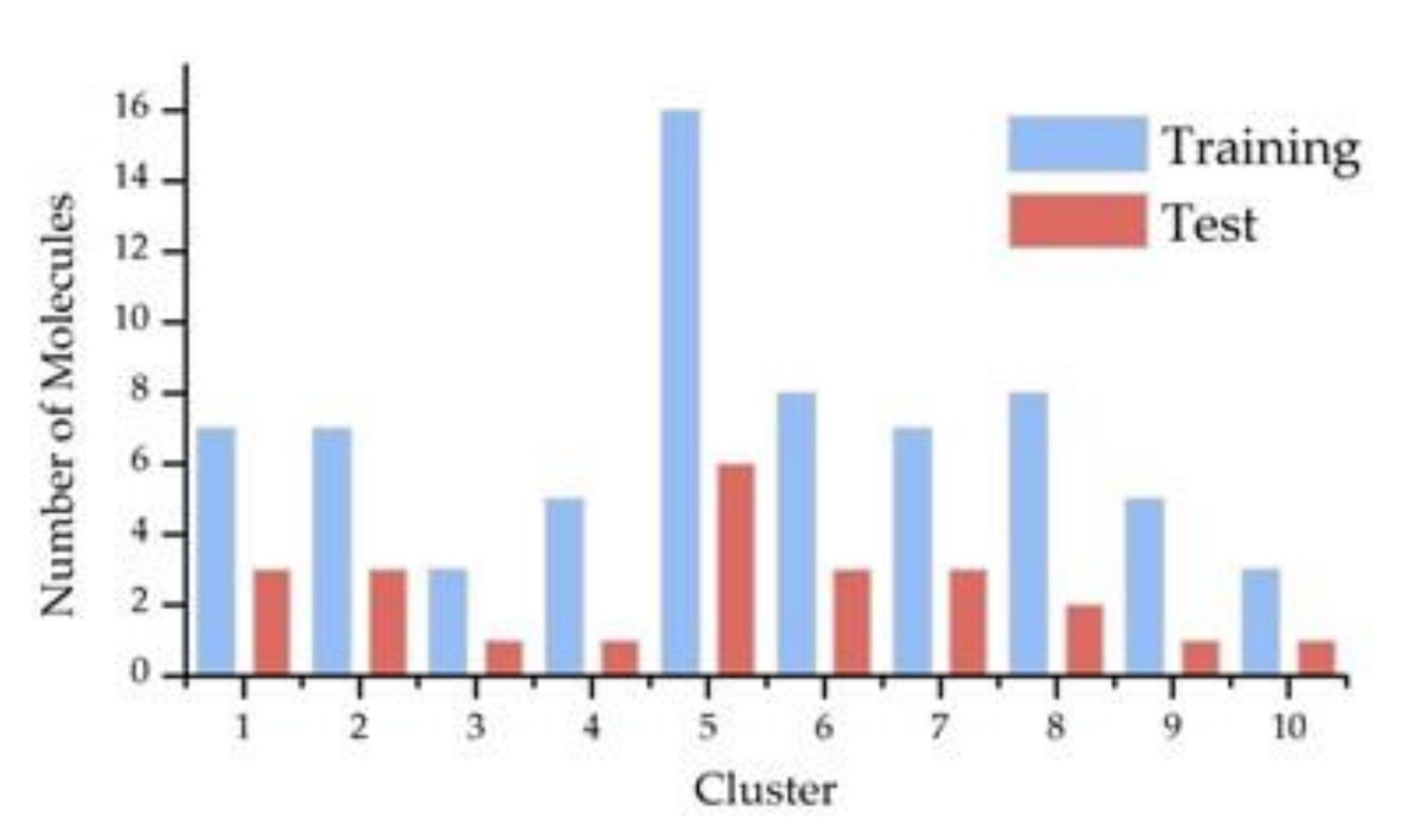
2.1. Dataset
2.2. Variable Selection
2.3. Applicability Domain
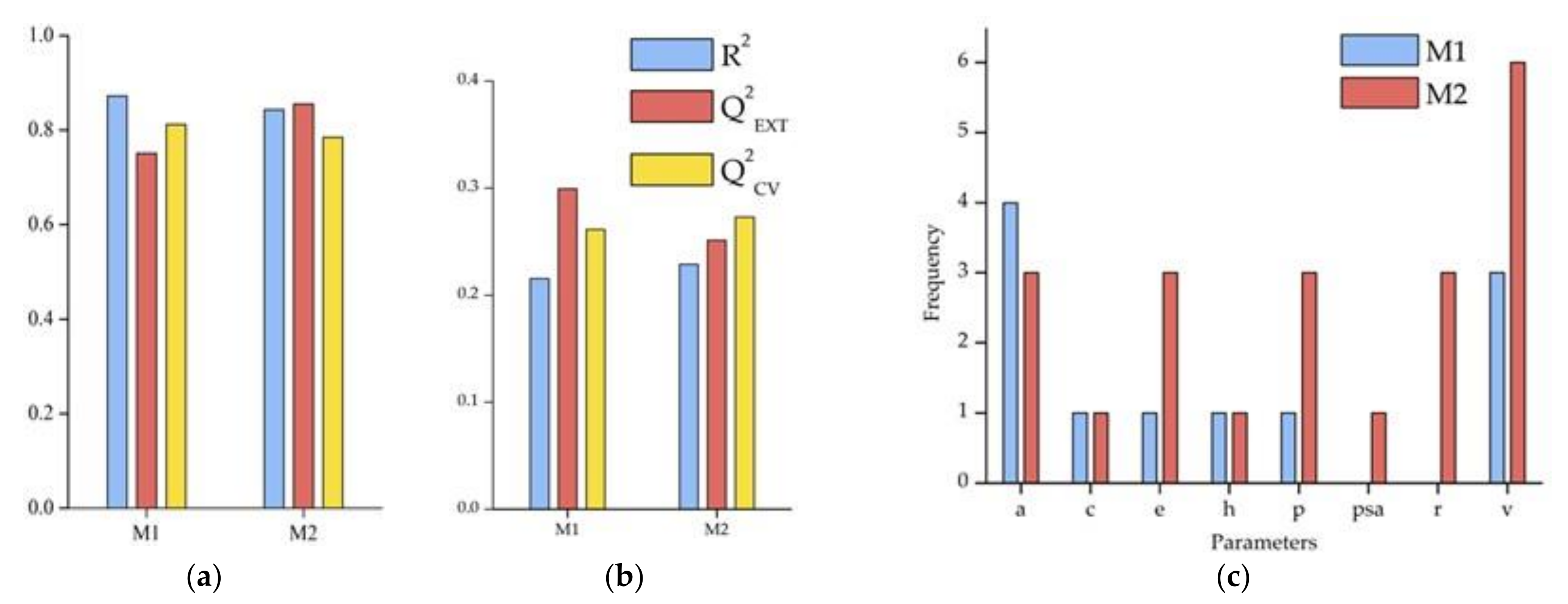
2.4. Model Performance
2.5. Screening of the DrugBank 5.1.7 and DiaNat Databases
2.6. Absorption, Distribution, Metabolism, and Excretion (ADME) Predictions
2.7. Molecular Docking
2.8. Molecular Dynamics
2.9. Free Energy Calculations
3. Results and Discussion
3.1. Dataset and Variable Selection
3.2. Applicability Domain
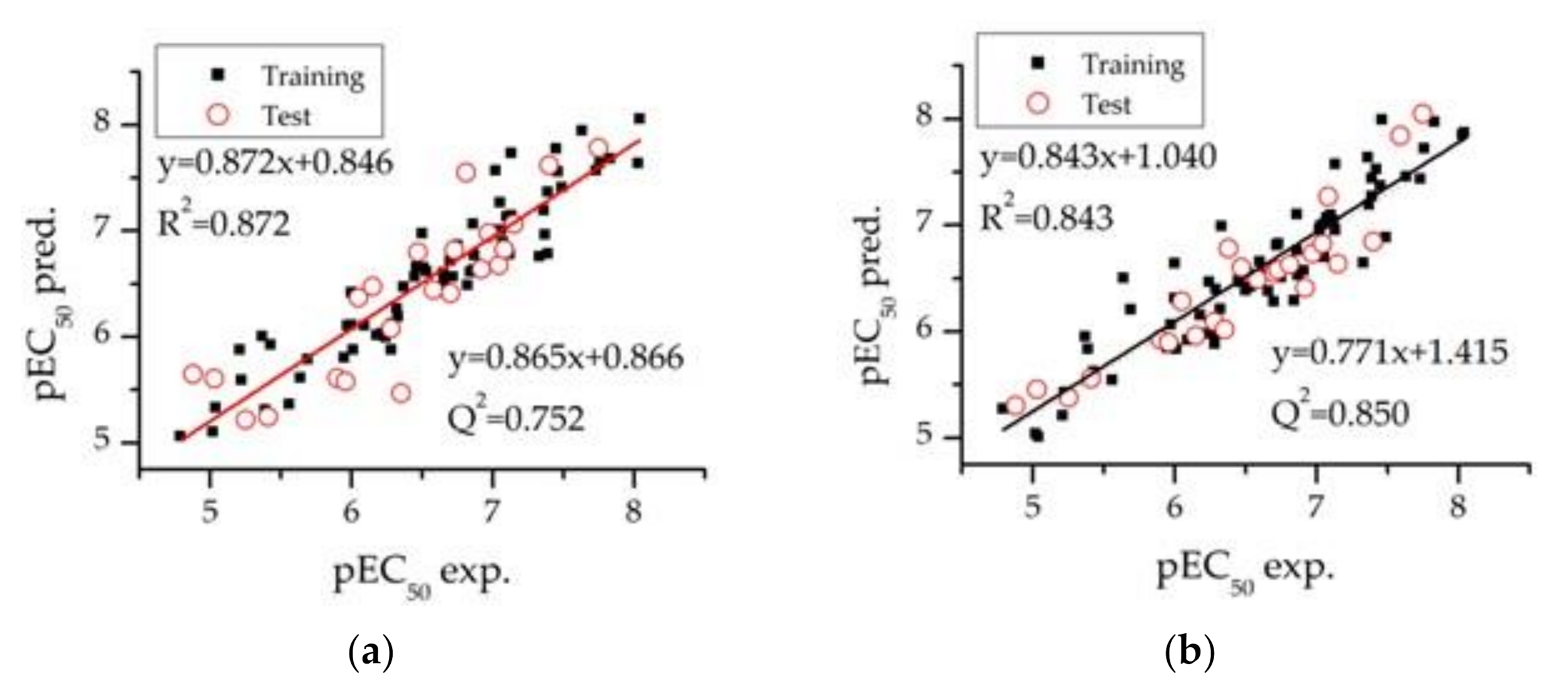
3.3. Model Validation
3.4. Screening of the DrugBank 5.1.7 and DiaNat Databases
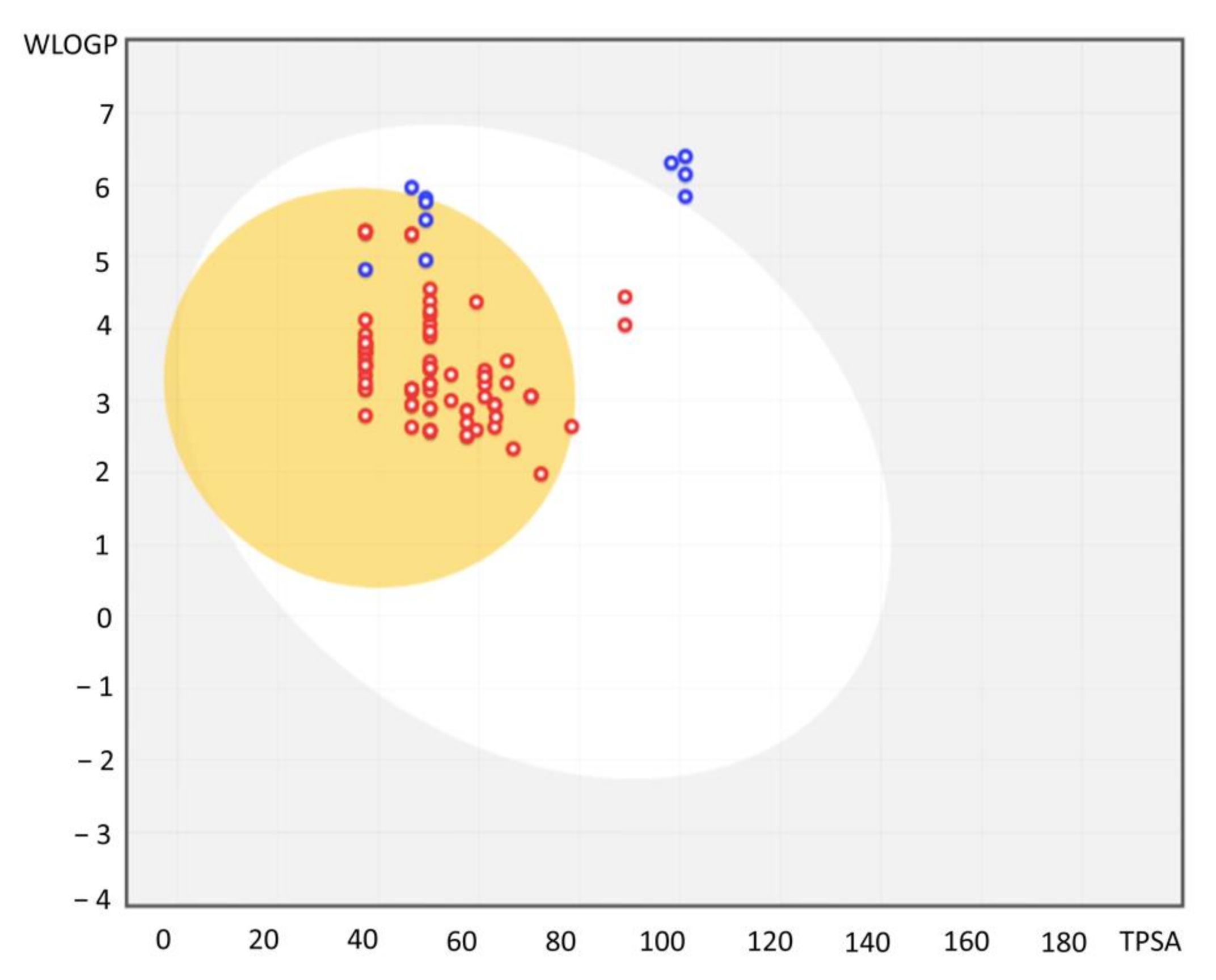
3.5. Absorption, Distribution, Metabolism, and Excretion (ADME) Predictions
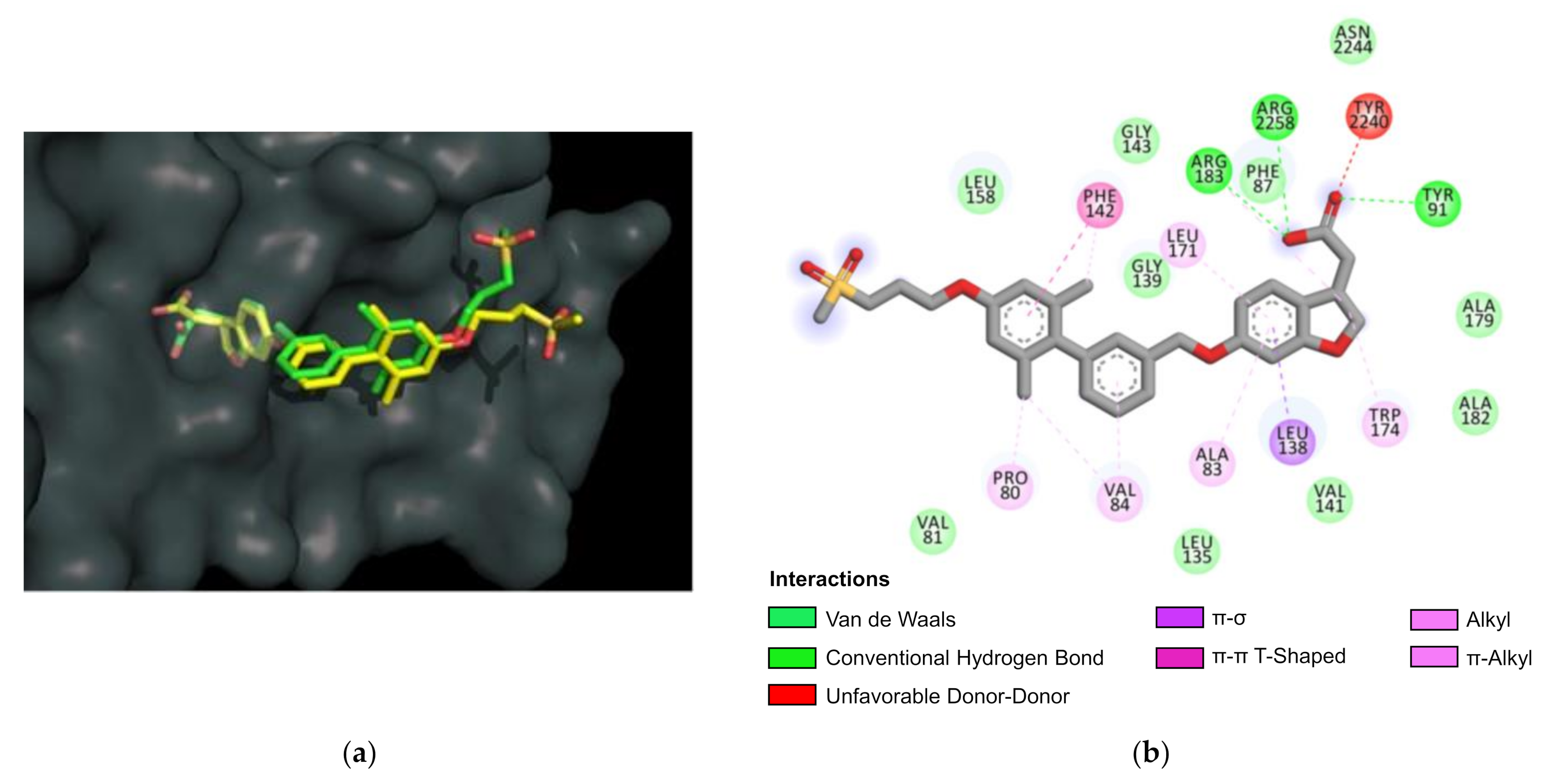
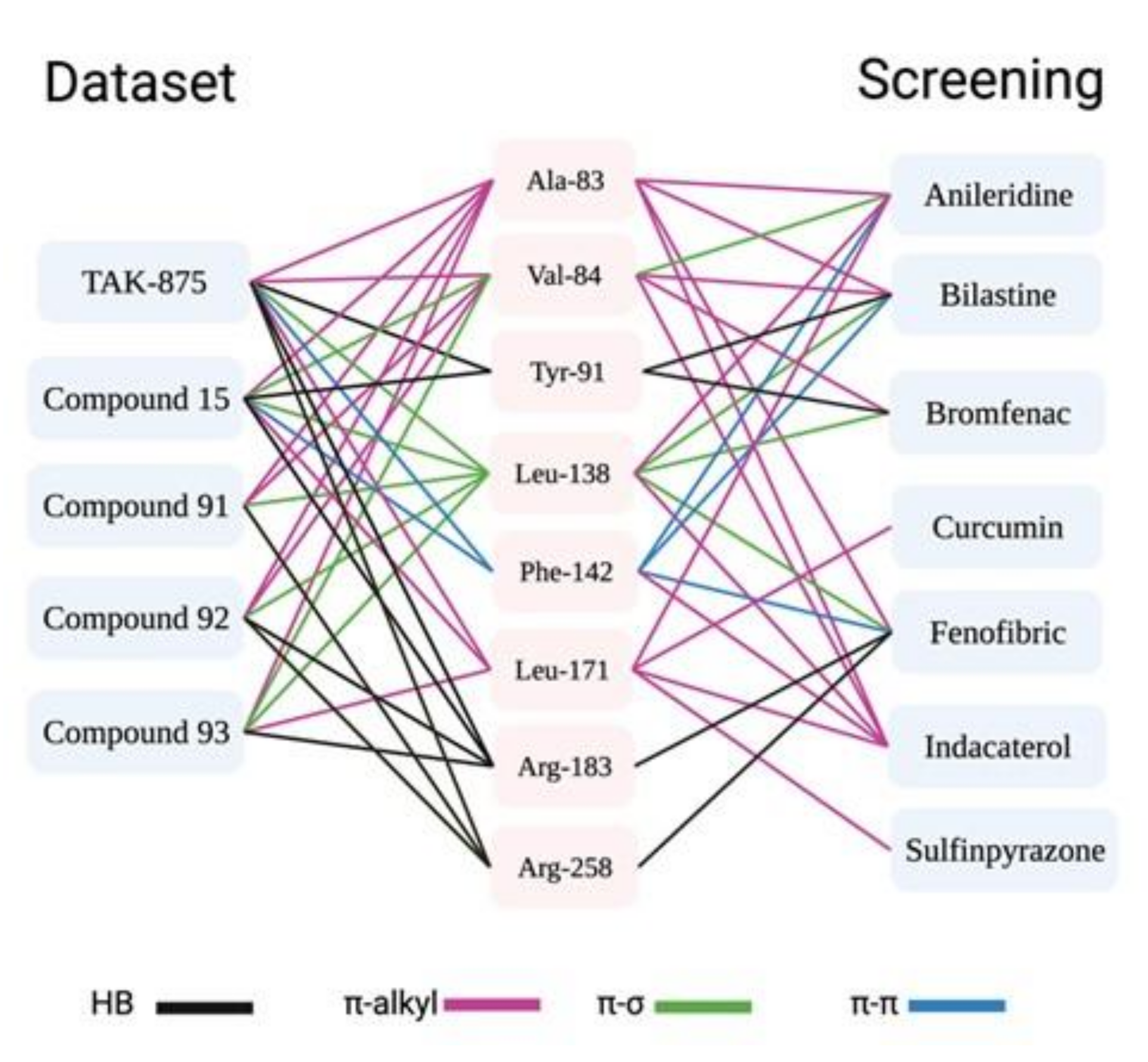
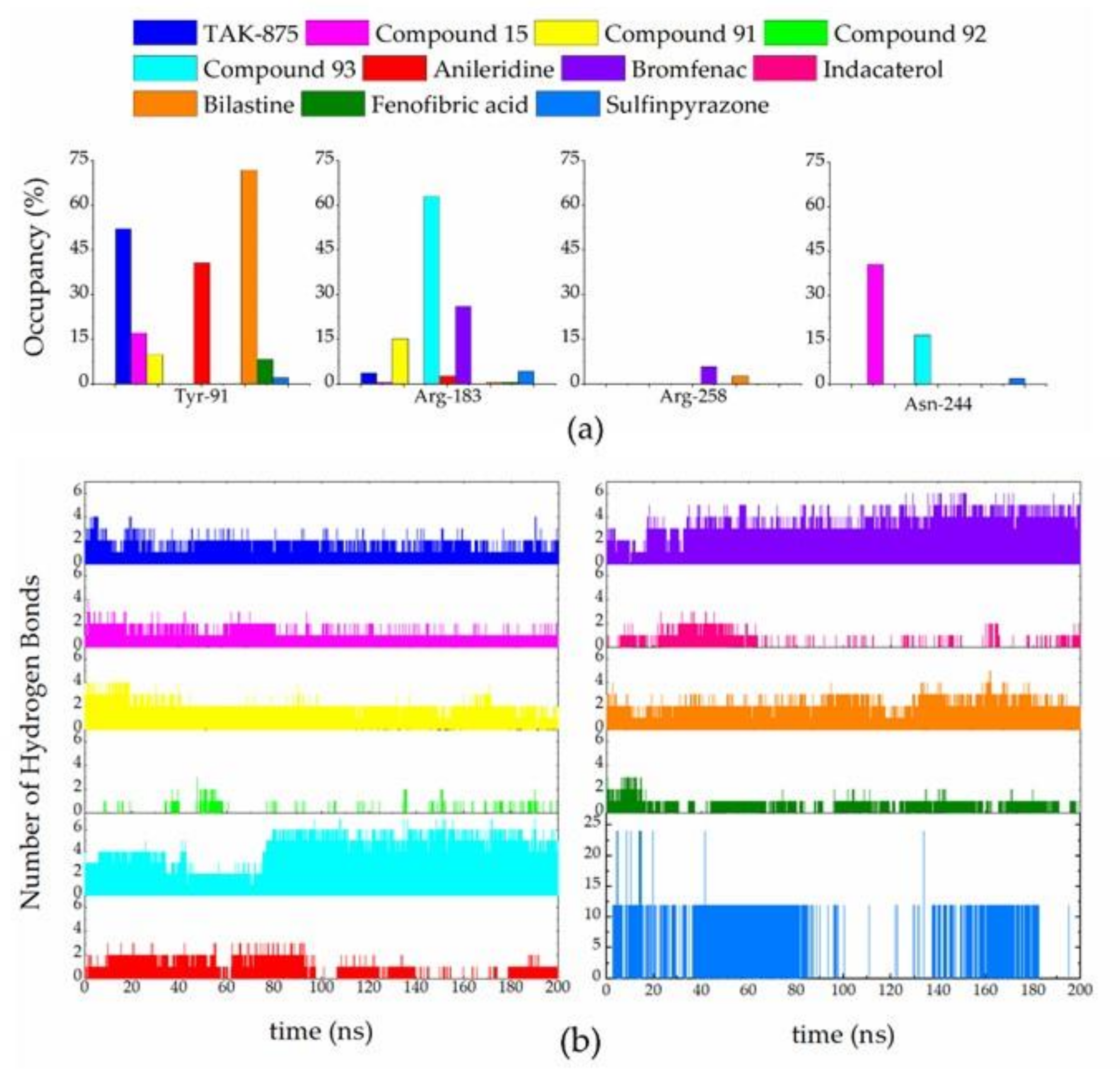
3.6. Molecular Docking
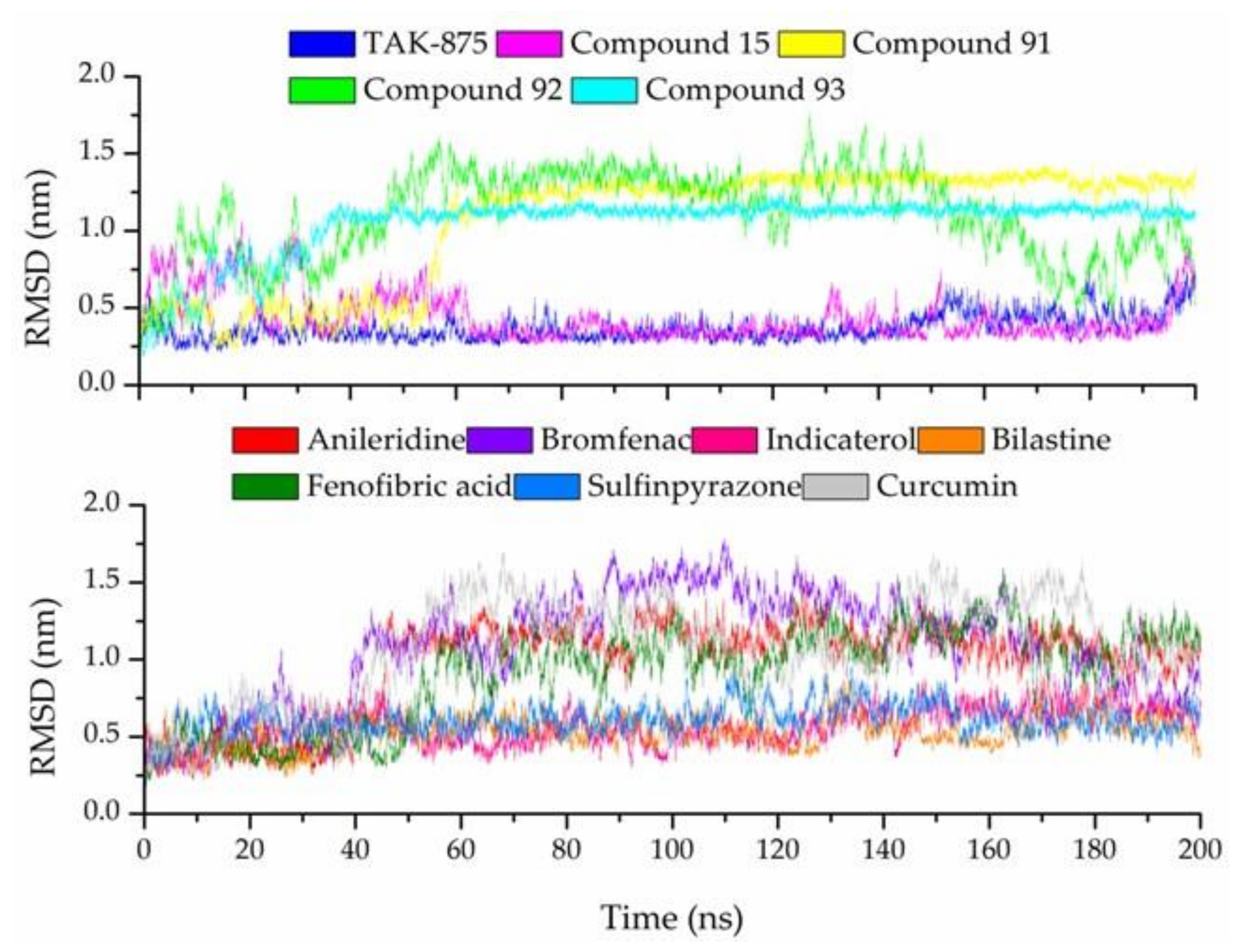
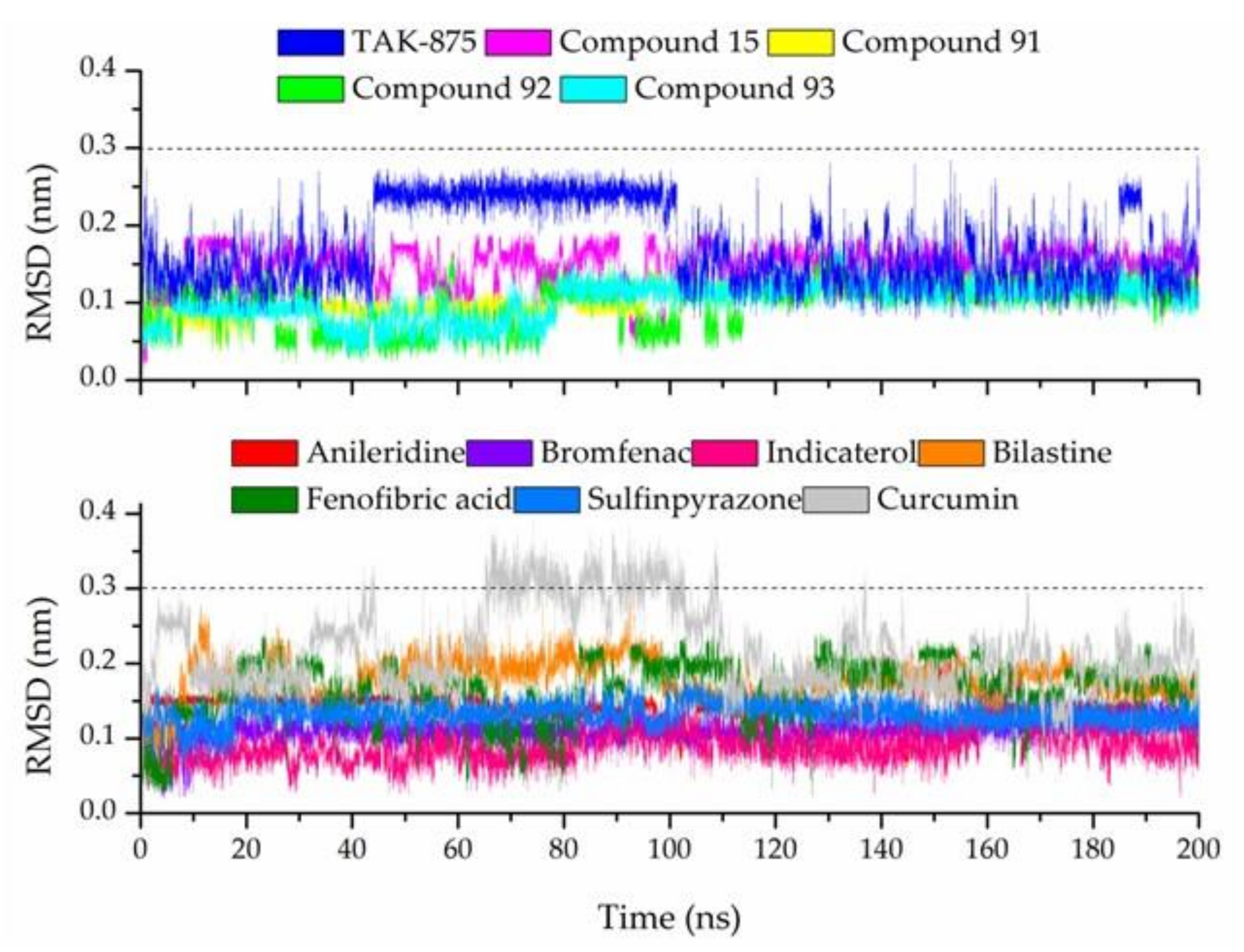
3.7. Molecular Dynamic Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Huang, Y.; Karuranga, S.; Malanda, B.; Williams, D.R.R. Call for data contribution to the IDF Diabetes Atlas 9th Edition 2019. Diabetes Res. Clin. Pract. 2018, 140, 351–352. [Google Scholar] [CrossRef] [PubMed]
- Eizirik, D.L.; Pasquali, L.; Cnop, M. Pancreatic beta-cells in type 1 and type 2 diabetes mellitus: Different pathways to failure. Nat. Rev. Endocrinol. 2020, 16, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Khunti, K.; Davies, M.J. Type 2 diabetes. Lancet 2017, 389, 2239–2251. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Ferrannini, E.; Groop, L.; Henry, R.R.; Herman, W.H.; Holst, J.J.; Hu, F.B.; Kahn, C.R.; Raz, I.; Shulman, G.I.; et al. Type 2 diabetes mellitus. Nat. Rev. Dis. Primers 2015, 1, 15019. [Google Scholar] [CrossRef] [PubMed]
- Turner, R.C.; Cull, C.A.; Frighi, V.; Holman, R.R. Glycemic control with diet, sulfonylurea, metformin, or insulin in patients with type 2 diabetes mellitus: Progressive requirement for multiple therapies (UKPDS 49). UK Prospective Diabetes Study (UKPDS) Group. JAMA 1999, 281, 2005–2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, N.; Bhagat, S.; Chundawat, T.S. Recent Advances in Development of GPR40 Modulators (FFA1/FFAR1): An Emerging Target for Type 2 Diabetes. Mini Rev. Med. Chem. 2017, 17, 947–958. [Google Scholar] [CrossRef]
- Houthuijzen, J.M. For Better or Worse: FFAR1 and FFAR4 Signaling in Cancer and Diabetes. Mol. Pharmacol. 2016, 90, 738–743. [Google Scholar] [CrossRef] [Green Version]
- Teng, D.; Chen, J.; Li, D.; Wu, Z.; Li, W.; Tang, Y.; Liu, G. Computational Insights into Molecular Activation and Positive Cooperative Mechanisms of FFAR1 Modulators. J. Chem. Inf. Model. 2020, 60, 3214–3230. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Shin, D.; Lee, J.Y. G-protein coupled receptor 40 agonists as novel therapeutics for type 2 diabetes. Arch. Pharm. Res. 2014, 37, 435–439. [Google Scholar] [CrossRef]
- Kaku, K.; Enya, K.; Nakaya, R.; Ohira, T.; Matsuno, R. Long-term safety and efficacy of fasiglifam (TAK-875), a G-protein-coupled receptor 40 agonist, as monotherapy and combination therapy in Japanese patients with type 2 diabetes: A 52-week open-label phase III study. Diabetes Obes. Metab. 2016, 18, 925–929. [Google Scholar] [CrossRef] [PubMed]
- Marcinak, J.F.; Munsaka, M.S.; Watkins, P.B.; Ohira, T.; Smith, N. Correction to: Liver Safety of Fasiglifam (TAK-875) in Patients with Type 2 Diabetes: Review of the Global Clinical Trial Experience. Drug Saf. 2018, 41, 1431–1437. [Google Scholar] [CrossRef] [Green Version]
- Edfalk, S.; Steneberg, P.; Edlund, H. Gpr40 is expressed in enteroendocrine cells and mediates free fatty acid stimulation of incretin secretion. Diabetes 2008, 57, 2280–2287. [Google Scholar] [CrossRef] [Green Version]
- Kotarsky, K.; Nilsson, N.E.; Flodgren, E.; Owman, C.; Olde, B. A human cell surface receptor activated by free fatty acids and thiazolidinedione drugs. Biochem. Biophys. Res. Commun. 2003, 301, 406–410. [Google Scholar] [CrossRef]
- Stoddart, L.A.; Smith, N.J.; Milligan, G. International Union of Pharmacology. LXXI. Free fatty acid receptors FFA1, -2, and -3: Pharmacology and pathophysiological functions. Pharm. Rev. 2008, 60, 405–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, S.P.; Dransfield, P.J.; Vimolratana, M.; Jiao, X.; Zhu, L.; Pattaropong, V.; Sun, Y.; Liu, J.; Luo, J.; Zhang, J.; et al. Discovery of AM-1638: A Potent and Orally Bioavailable GPR40/FFA1 Full Agonist. ACS Med. Chem. Lett. 2012, 3, 726–730. [Google Scholar] [CrossRef] [Green Version]
- Holliday, N.; Watson, S.-J.; Brown, A. Drug Discovery Opportunities and Challenges at G Protein Coupled Receptors for Long Chain Free Fatty Acids. Front. Endocrinol. 2012, 2, 112. [Google Scholar] [CrossRef] [Green Version]
- Christiansen, E.; Urban, C.; Merten, N.; Liebscher, K.; Karlsen, K.K.; Hamacher, A.; Spinrath, A.; Bond, A.D.; Drewke, C.; Ullrich, S. Discovery of potent and selective agonists for the free fatty acid receptor 1 (FFA1/GPR40), a potential target for the treatment of type II diabetes. J. Med. Chem. 2008, 51, 7061–7064. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, E.; Urban, C.; Grundmann, M.; Due-Hansen, M.E.; Hagesaether, E.; Schmidt, J.; Pardo, L.; Ullrich, S.; Kostenis, E.; Kassack, M. Identification of a potent and selective free fatty acid receptor 1 (FFA1/GPR40) agonist with favorable physicochemical and in vitro ADME properties. J. Med. Chem. 2011, 54, 6691–6703. [Google Scholar] [CrossRef]
- Christiansen, E.; Due-Hansen, M.E.; Urban, C.; Grundmann, M.; Schröder, R.; Hudson, B.D.; Milligan, G.; Cawthorne, M.A.; Kostenis, E.; Kassack, M.U. Free fatty acid receptor 1 (FFA1/GPR40) agonists: Mesylpropoxy appendage lowers lipophilicity and improves ADME properties. J. Med. Chem. 2012, 55, 6624–6628. [Google Scholar] [CrossRef]
- Christiansen, E.; Due-Hansen, M.E.; Urban, C.; Grundmann, M.; Schmidt, J.; Hansen, S.V.; Hudson, B.D.; Zaibi, M.; Markussen, S.B.; Hagesaether, E. Discovery of a potent and selective free fatty acid receptor 1 agonist with low lipophilicity and high oral bioavailability. J. Med. Chem. 2013, 56, 982–992. [Google Scholar] [CrossRef]
- Mora, J.R.; Marrero-Ponce, Y.; García-Jacas, C.R.; Suarez Causado, A. Ensemble models based on QuBiLS-MAS features and shallow learning for the prediction of drug-induced liver toxicity: Improving deep learning and traditional approaches. Chem. Res. Toxicol. 2020. [Google Scholar] [CrossRef]
- Cabrera, N.; Mora, J.R.; Márquez, E.; Flores-Morales, V.; Calle, L.; Cortés, E. QSAR and molecular docking modelling of anti-leishmanial activities of organic selenium and tellurium compounds. SAR QSAR Environ. Res. 2020, 32, 29–50. [Google Scholar] [CrossRef] [PubMed]
- Gaba, V.; Rani, K.; Gupta, M.K. QSAR study on 4-alkynyldihydrocinnamic acid analogs as free fatty acid receptor 1 agonists and antidiabetic agents: Rationales to improve activity. Arab. J. Chem. 2019, 12, 1758–1764. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, A.; Yano, J.; Hirozane, Y.; Kefala, G.; Gruswitz, F.; Snell, G.; Lane, W.; Ivetac, A.; Aertgeerts, K.; Nguyen, J. High-resolution structure of the human GPR40 receptor bound to allosteric agonist TAK-875. Nature 2014, 513, 124–127. [Google Scholar] [CrossRef]
- Tikhonova, I.G.; Sum, C.S.; Neumann, S.; Thomas, C.J.; Raaka, B.M.; Costanzi, S.; Gershengorn, M.C. Bidirectional, iterative approach to the structural delineation of the functional "chemoprint" in GPR40 for agonist recognition. J. Med. Chem. 2007, 50, 2981–2989. [Google Scholar] [CrossRef] [Green Version]
- Sum, C.S.; Tikhonova, I.G.; Neumann, S.; Engel, S.; Raaka, B.M.; Costanzi, S.; Gershengorn, M.C. Identification of residues important for agonist recognition and activation in GPR40. J. Biol. Chem. 2007, 282, 29248–29255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madariaga-Mazón, A.; Naveja, J.J.; Medina-Franco, J.L.; Noriega-Colima, K.O.; Martinez-Mayorga, K. DiaNat-DB: A molecular database of antidiabetic compounds from medicinal plants. RSC Adv. 2021, 11, 5172–5178. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2017, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W., Jr. Computational methods in drug discovery. Pharm. Rev. 2013, 66, 334–395. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Hu, L.; Wang, X.; Zhou, Z.; Deng, L.; Xu, Y.; Zhang, L. Design, synthesis, and biological evaluation of novel dual FFA1 (GPR40)/PPARδ agonists as potential anti-diabetic agents. Bioorg. Chem. 2019, 92, 103254. [Google Scholar] [CrossRef]
- Li, Z.; Pan, M.; Su, X.; Dai, Y.; Fu, M.; Cai, X.; Shi, W.; Huang, W.; Qian, H. Discovery of novel pyrrole-based scaffold as potent and orally bioavailable free fatty acid receptor 1 agonists for the treatment of type 2 diabetes. Bioorg. Med. Chem. 2016, 24, 1981–1987. [Google Scholar] [CrossRef]
- Rappe, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A.; Skiff, W.M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Garcia-Jacas, C.R.; Marrero-Ponce, Y.; Acevedo-Martinez, L.; Barigye, S.J.; Valdes-Martini, J.R.; Contreras-Torres, E. QuBiLS-MIDAS: A parallel free-software for molecular descriptors computation based on multilinear algebraic maps. J. Comput. Chem. 2014, 35, 1395–1409. [Google Scholar] [CrossRef] [PubMed]
- García-Jacas, C.R.; Marrero-Ponce, Y.; Vivas-Reyes, R.; Suárez-Lezcano, J.; Martinez-Rios, F.; Terán, J.E.; Aguilera-Mendoza, L. Distributed and multicore QuBiLS-MIDAS software v2.0: Computing chiral, fuzzy, weighted and truncated geometrical molecular descriptors based on tensor algebra. J. Comput. Chem. 2020, 41, 1209–1227. [Google Scholar] [CrossRef]
- Hall, M.; Frank, E.; Holmes, G.; Pfahringer, B.; Reutemann, P.; Witten, I.H. The WEKA data mining software: An update. ACM SIGKDD Explor. Newsl. 2009, 11, 10–18. [Google Scholar] [CrossRef]
- Murtagh, F.; Legendre, P. Ward’s Hierarchical Agglomerative Clustering Method: Which Algorithms Implement Ward’s Criterion? J. Classif. 2014, 31, 274–295. [Google Scholar] [CrossRef] [Green Version]
- Leonard, J.T.; Roy, K. On selection of training and test sets for the development of predictive QSAR models. QSAR Comb. Sci. 2006, 25, 235–251. [Google Scholar] [CrossRef]
- Edraki, N.; Das, U.; Hemateenejad, B.; Dimmock, J.R.; Miri, R. Comparative QSAR analysis of 3, 5-bis (arylidene)-4-piperidone derivatives: The development of predictive cytotoxicity models. Iran. J. Pharm. Res. 2016, 15, 425. [Google Scholar]
- Cabrera, N.; Mora, J.R.; Marquez, E.A. Computational molecular modeling of Pin1 inhibition activity of quinazoline, benzophenone, and pyrimidine derivatives. J. Chem. 2019, 2019, 2954250. [Google Scholar] [CrossRef] [Green Version]
- Khan, P.M.; Roy, K. Current approaches for choosing feature selection and learning algorithms in quantitative structure–activity relationships (QSAR). Expert Opin. Drug Discov. 2018, 13, 1075–1089. [Google Scholar] [CrossRef]
- Hall, M.A. Correlation-Based Feature Selection for Machine Learning. Ph.D. Thesis, The University of Waikato, Hamilton, New Zealand, 1999. [Google Scholar]
- Hanser, T.; Barber, C.; Marchaland, J.F.; Werner, S. Applicability domain: Towards a more formal definition. SAR QSAR Environ. Res. 2016, 27, 865–881. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.; Kar, S.; Ambure, P. On a simple approach for determining applicability domain of QSAR models. Chemom. Intell. Lab. Syst. 2015, 145, 22–29. [Google Scholar] [CrossRef]
- Jaworska, J.; Nikolova-Jeliazkova, N. How can structural similarity analysis help in category formation? SAR QSAR Environ. Res. 2007, 18, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Golbraikh, A.; Tropsha, A. Predictive QSAR modeling based on diversity sampling of experimental datasets for the training and test set selection. Mol. Divers. 2002, 5, 231–243. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. iLOGP: A simple, robust, and efficient description of n-octanol/water partition coefficient for drug design using the GB/SA approach. J. Chem. Inf. Model. 2014, 54, 3284–3301. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.; Zhao, Y.; Li, X.; Lin, F.; Xu, Y.; Zhang, X.; Li, Y.; Wang, R.; Lai, L. Computation of octanol-water partition coefficients by guiding an additive model with knowledge. J. Chem. Inf. Model. 2007, 47, 2140–2148. [Google Scholar] [CrossRef]
- Wildman, S.A.; Crippen, G.M. Prediction of physicochemical parameters by atomic contributions. J. Chem. Inf. Comput. Sci. 1999, 39, 868–873. [Google Scholar] [CrossRef]
- Moriguchi, I.; HIRONO, S.; LIU, Q.; NAKAGOME, I.; MATSUSHITA, Y. Simple method of calculating octanol/water partition coefficient. Chem. Pharm. Bull. 1992, 40, 127–130. [Google Scholar] [CrossRef] [Green Version]
- Sanders, M.P.; Barbosa, A.J.; Zarzycka, B.; Nicolaes, G.A.; Klomp, J.P.; de Vlieg, J.; Del Rio, A. Comparative analysis of pharmacophore screening tools. J. Chem. Inf. Model. 2012, 52, 1607–1620. [Google Scholar] [CrossRef]
- Daina, A.; Zoete, V. A BOILED-Egg to predict gastrointestinal absorption and brain penetration of small molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [Green Version]
- PyMOL, version 1.8; Schrödinger LLC.: New York, NY, USA, 2015.
- Sanner, M.; Huey, R.; Dallakyan, S.; Karnati, S.; Lindstrom, W.; Morris, G.; Norledge, B.; Omelchenko, A.; Stoffler, D.; Vareille, G. AutoDockTools, version 1.4.5.; The Scripps Research Institute: La Jolla, CA, USA, 2007. [Google Scholar]
- Discovery Studio Visualizer; Dassault Systemes BIOVIA: San Diego, CA, USA, 2017; p. 936.
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [Green Version]
- Sousa da Silva, A.W.; Vranken, W.F. ACPYPE—AnteChamber PYthon Parser interfacE. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Murugan, N.A.; Kumar, S.; Jeyakanthan, J.; Srivastava, V. Searching for target-specific and multi-targeting organics for Covid-19 in the Drugbank database with a double scoring approach. Sci. Rep. 2020, 10, 19125. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa--a GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- García-Jacas, C.s.R.; Marrero-Ponce, Y.; Cortés-Guzmán, F.; Suárez-Lezcano, J.; Martinez-Rios, F.O.; García-González, L.A.; Pupo-Meriño, M.; Martinez-Mayorga, K. Enhancing acute oral toxicity predictions by using consensus modeling and algebraic form-based 0D-to-2D molecular encodes. Chem. Res. Toxicol. 2019, 32, 1178–1192. [Google Scholar] [CrossRef]
- Roy, K.; Das, R.N.; Ambure, P.; Aher, R.B. Be aware of error measures. Further studies on validation of predictive QSAR models. Chemom. Intell. Lab. Syst. 2016, 152, 18–33. [Google Scholar] [CrossRef]
- Martin, Y.C. A bioavailability score. J. Med. Chem. 2005, 48, 3164–3170. [Google Scholar] [CrossRef]
- Carrano, A.; Snkhchyan, H.; Kooij, G.; van der Pol, S.; van Horssen, J.; Veerhuis, R.; Hoozemans, J.; Rozemuller, A.; de Vries, H.E. ATP-binding cassette transporters P-glycoprotein and breast cancer related protein are reduced in capillary cerebral amyloid angiopathy. Neurobiol. Aging 2014, 35, 565–575. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Delaney, J.S. ESOL: Estimating aqueous solubility directly from molecular structure. J. Chem. Inf. Comput. Sci. 2004, 44, 1000–1005. [Google Scholar] [CrossRef]
- Ali, J.; Camilleri, P.; Brown, M.B.; Hutt, A.J.; Kirton, S.B. Revisiting the general solubility equation: In silico prediction of aqueous solubility incorporating the effect of topographical polar surface area. J. Chem. Inf. Model. 2012, 52, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.P.H.; Keating, G.M. Fenofibric Acid. Am. J. Cardiovasc. Drugs 2009, 9, 401–409. [Google Scholar] [CrossRef]
- Moen, M.D. Indacaterol. Drugs 2010, 70, 2269–2280. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.C.; Patchva, S.; Aggarwal, B.B. Therapeutic Roles of Curcumin: Lessons Learned from Clinical Trials. AAPS J. 2013, 15, 195–218. [Google Scholar] [CrossRef] [Green Version]
- Verta, R.; Grange, C.; Gurrieri, M.; Borga, S.; Nardini, P.; Argenziano, M.; Ghe, C.; Cavalli, R.; Benetti, E.; Miglio, G.; et al. Effect of Bilastine on Diabetic Nephropathy in DBA2/J Mice. Int. J. Mol. Sci. 2019, 20, 2554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinna, A.; Blasetti, F.; Ricci, G.D.; Boscia, F. Bromfenac eyedrops in the treatment of diabetic macular edema: A pilot study. Eur. J. Ophthalmol. 2017, 27, 326–330. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Function |
|---|---|
| R2 | Global evaluator of the model. |
| Q2CV, and Q2ext | Evaluators of the predictability of the model. |
| (Q2boot) | Evaluate the randomly generating training sets with 5000 sample repetition, and the predicted response of each sample is obtained. |
| Y-scrambling analysis | A random perturbation of the response variable. |
| M1 | Cross-Validation | External Validation | ||
| Criterion | Result | Assessment | Result | Assessment |
| R2 > 0.6 | 0.872 | PASS | 0.872 | PASS |
| Q2Val > 0.5 | 0.816 | PASS | 0.752 | PASS |
| (Q2Val − R02)/Q2Val < 0.1 | 0.001 | PASS | 0.023 | PASS |
| (Q2Val − R0′2)/Q2Val < 0.1 | 0.037 | PASS | 0.024 | PASS |
| abs(R02 − R0′2) < 0.1 | 0.030 | PASS | 0.001 | PASS |
| 0.85 < k < 1.15 | 0.999 | PASS | 0.998 | PASS |
| 0.85 < k′ < 1.15 | 0.998 | PASS | 0.998 | PASS |
| M2 | Cross-Validation | External Validation | ||
| Criterion | Result | Assessment | Result | Assessment |
| R2 > 0.6 | 0.843 | PASS | 0.843 | PASS |
| Q2Val> 0.5 | 0.778 | PASS | 0.850 | PASS |
| (Q2Val − R02)/Q2Val < 0.1 | 0.002 | PASS | 0.037 | PASS |
| (Q2Val − R0′2)/Q2Val < 0.1 | 0.054 | PASS | 0.001 | PASS |
| abs(R02 − R0′2) < 0.1 | 0.041 | PASS | 0.032 | PASS |
| 0.85 < k < 1.15 | 0.998 | PASS | 1.005 | PASS |
| 0.85 < k′ < 1.15 | 0.999 | PASS | 0.993 | PASS |
| Molecule | pEC50 | ESOL Class | Ali Class | Silicos-IT Class | Consensus Log Po/w |
|---|---|---|---|---|---|
| 52 | 8.03 | Ms | Ps | Ps | 4.93 |
| 49 | 7.75 | Ms | Ps | Ps | 5.24 |
| 48 | 7.73 | Ms | Ms | Ps | 4.59 |
| 47 | 7.63 | Ms | Ps | Ps | 5.22 |
| 15 | 7.49 | Ms | Ms | Ms | 3.83 |
| 93 | 7.45 | S | Ms | Ms | 3.37 |
| 92 | 7.42 | Ms | Ms | Ms | 3.51 |
| 91 | 7.4 | Ms | Ms | Ms | 3.57 |
| Compound | Van der Waals Energy | Electrostatic Energy | SASA Energy | Binding Energy | pEC50 Pred. |
|---|---|---|---|---|---|
| TAK-875 | −55.80 | −9.78 | −5.28 | −30.88 | 8.45 |
| 15 | −38.75 | −5.40 | −3.97 | −25.15 | 6.88 |
| 91 | −42.97 | −8.39 | −4.07 | −26.96 | 6.84 |
| 92 | −40.11 | −2.14 | −4.00 | −28.70 | 7.53 |
| 93 | −43.84 | −18.10 | −4.29 | −28.61 | 7.37 |
| Anileridine | −50.91 | −7.76 | −4.92 | −32.69 | 7.82 |
| Bromfenac | −38.08 | −32.20 | −3.80 | −15.22 | 8.47 |
| Sulfinpyrazone | −53.32 | −5.75 | −5.14 | −24.49 | 7.55 |
| Indacaterol | −43.90 | −0.91 | −4.43 | −28.63 | 7.41 |
| Bilastine | −59.13 | −16.58 | −5.83 | −36.97 | 7.57 |
| Fenofibric acid | −35.25 | −4.67 | −3.70 | −20.70 | 7.92 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cabrera, N.; Cuesta, S.A.; Mora, J.R.; Calle, L.; Márquez, E.A.; Kaunas, R.; Paz, J.L. In Silico Searching for Alternative Lead Compounds to Treat Type 2 Diabetes through a QSAR and Molecular Dynamics Study. Pharmaceutics 2022, 14, 232. https://doi.org/10.3390/pharmaceutics14020232
Cabrera N, Cuesta SA, Mora JR, Calle L, Márquez EA, Kaunas R, Paz JL. In Silico Searching for Alternative Lead Compounds to Treat Type 2 Diabetes through a QSAR and Molecular Dynamics Study. Pharmaceutics. 2022; 14(2):232. https://doi.org/10.3390/pharmaceutics14020232
Chicago/Turabian StyleCabrera, Nicolás, Sebastián A. Cuesta, José R. Mora, Luis Calle, Edgar A. Márquez, Roland Kaunas, and José Luis Paz. 2022. "In Silico Searching for Alternative Lead Compounds to Treat Type 2 Diabetes through a QSAR and Molecular Dynamics Study" Pharmaceutics 14, no. 2: 232. https://doi.org/10.3390/pharmaceutics14020232