
A Fucan Sulfate with Pentasaccharide Repeating Units from the Sea Cucumber Holothuriafloridana and Its Anticoagulant Activity

Abstract
:1. Introduction
2. Results and Discussion
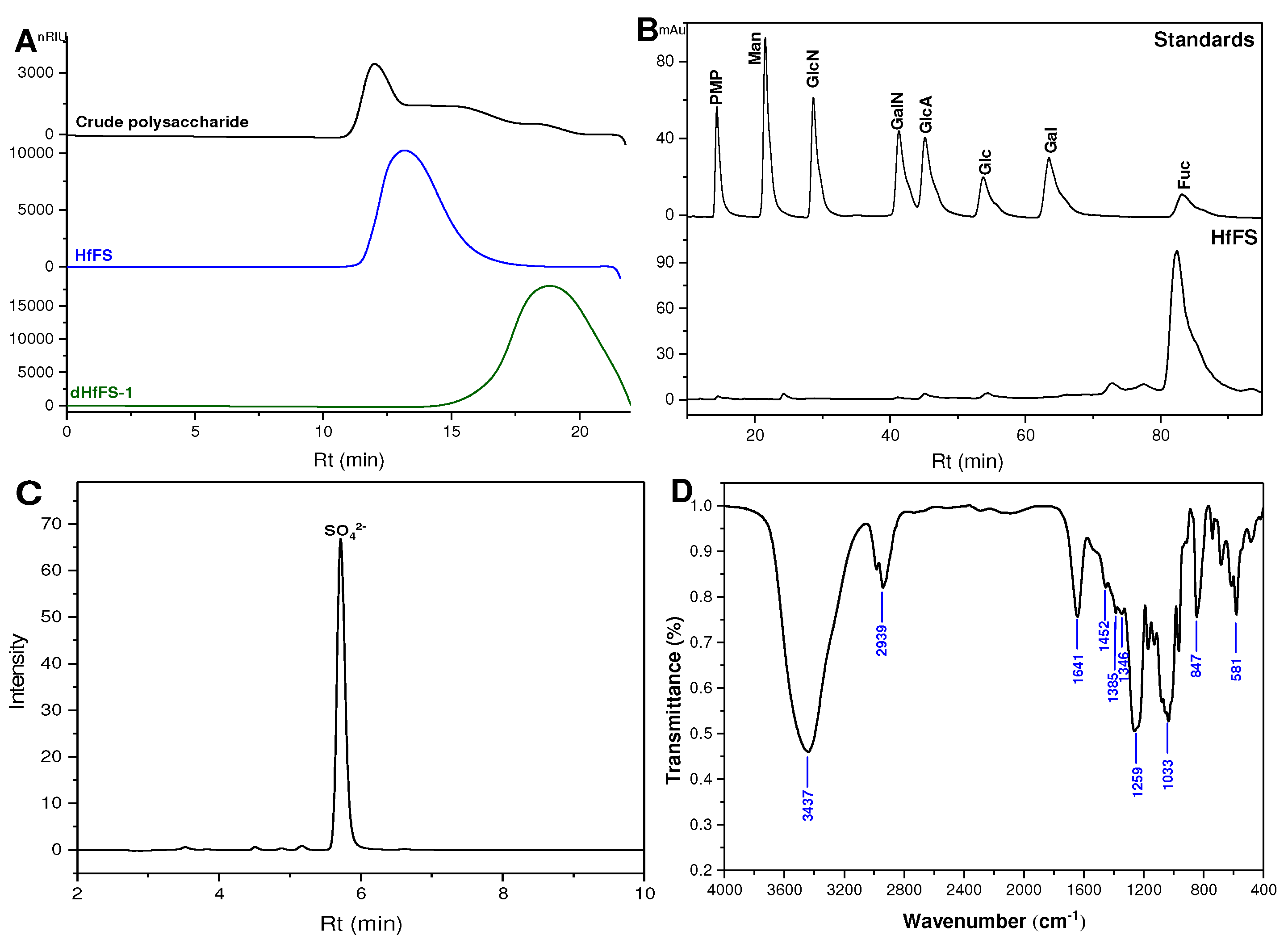
2.1. Purification and Physicochemical Properties of HfFS
2.2. Infrared (IR) Spectroscopy Analysis
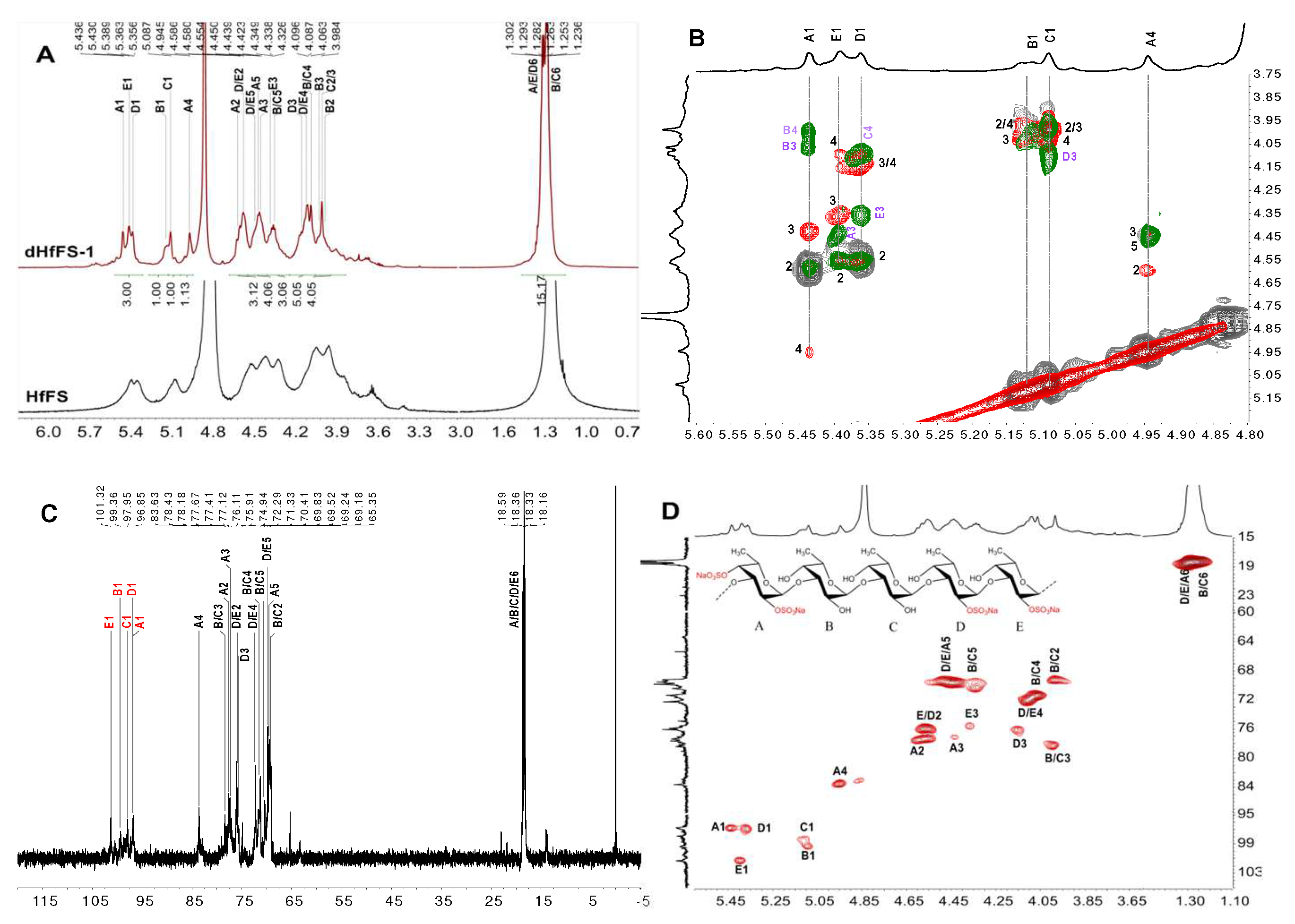
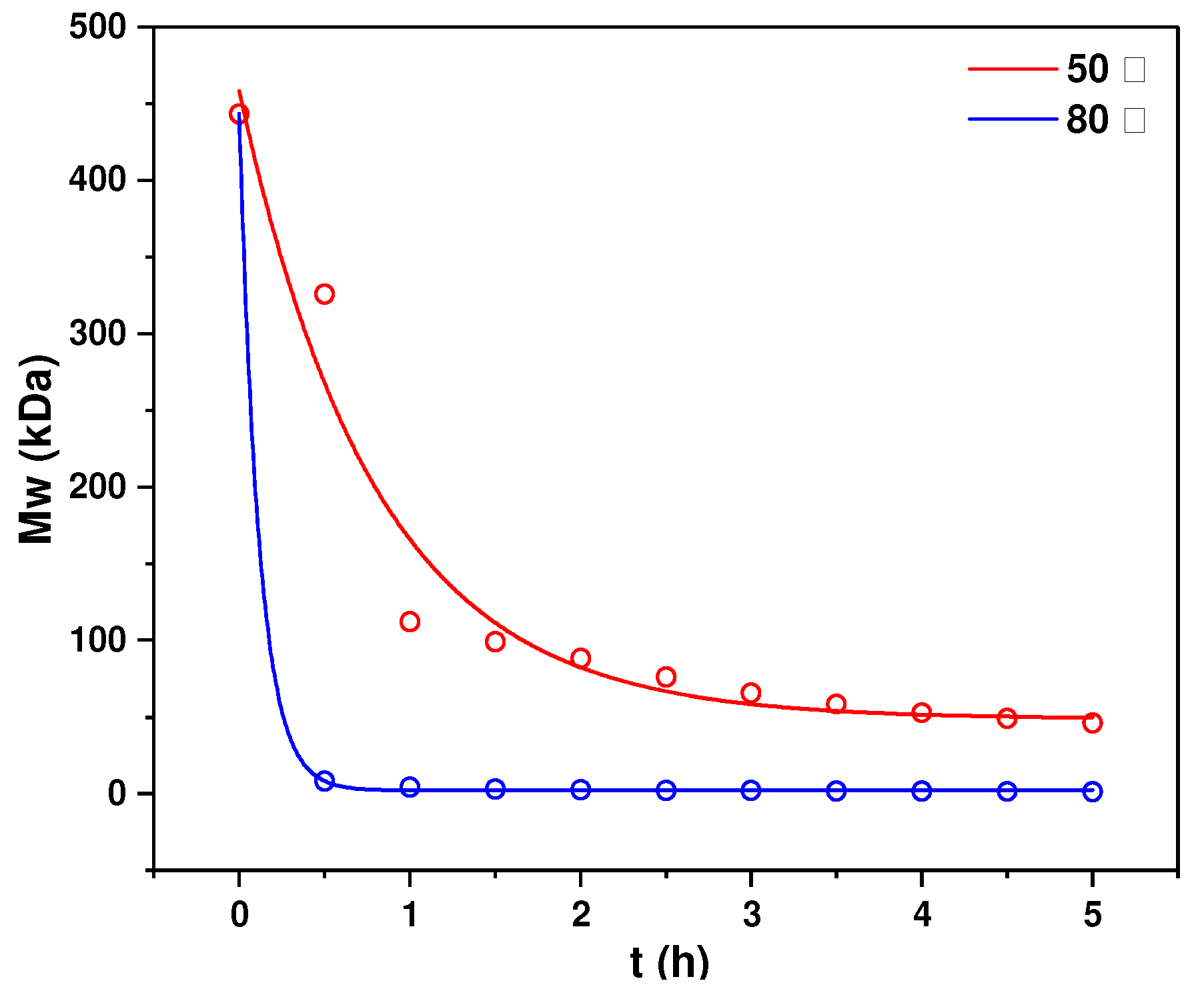
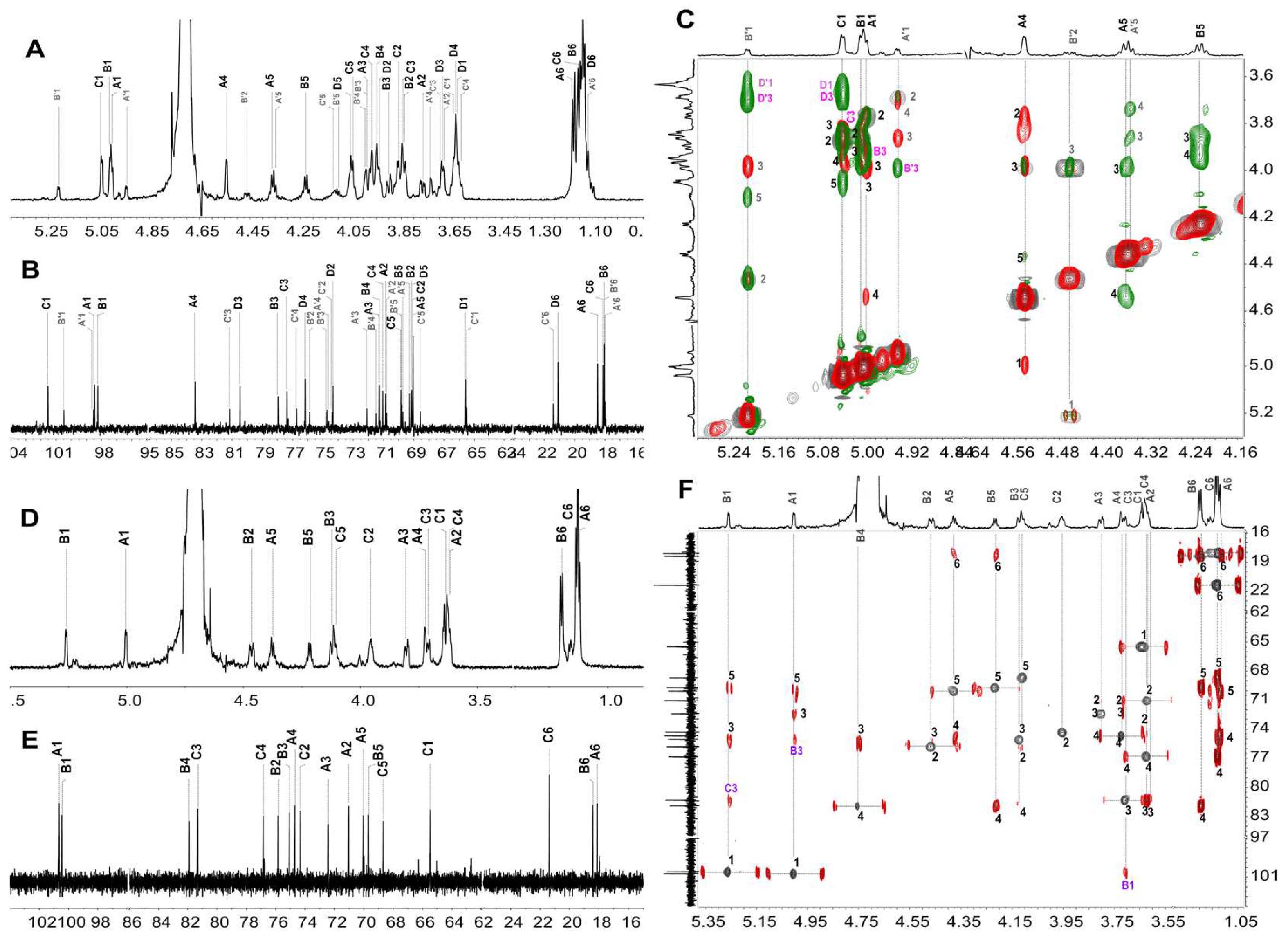
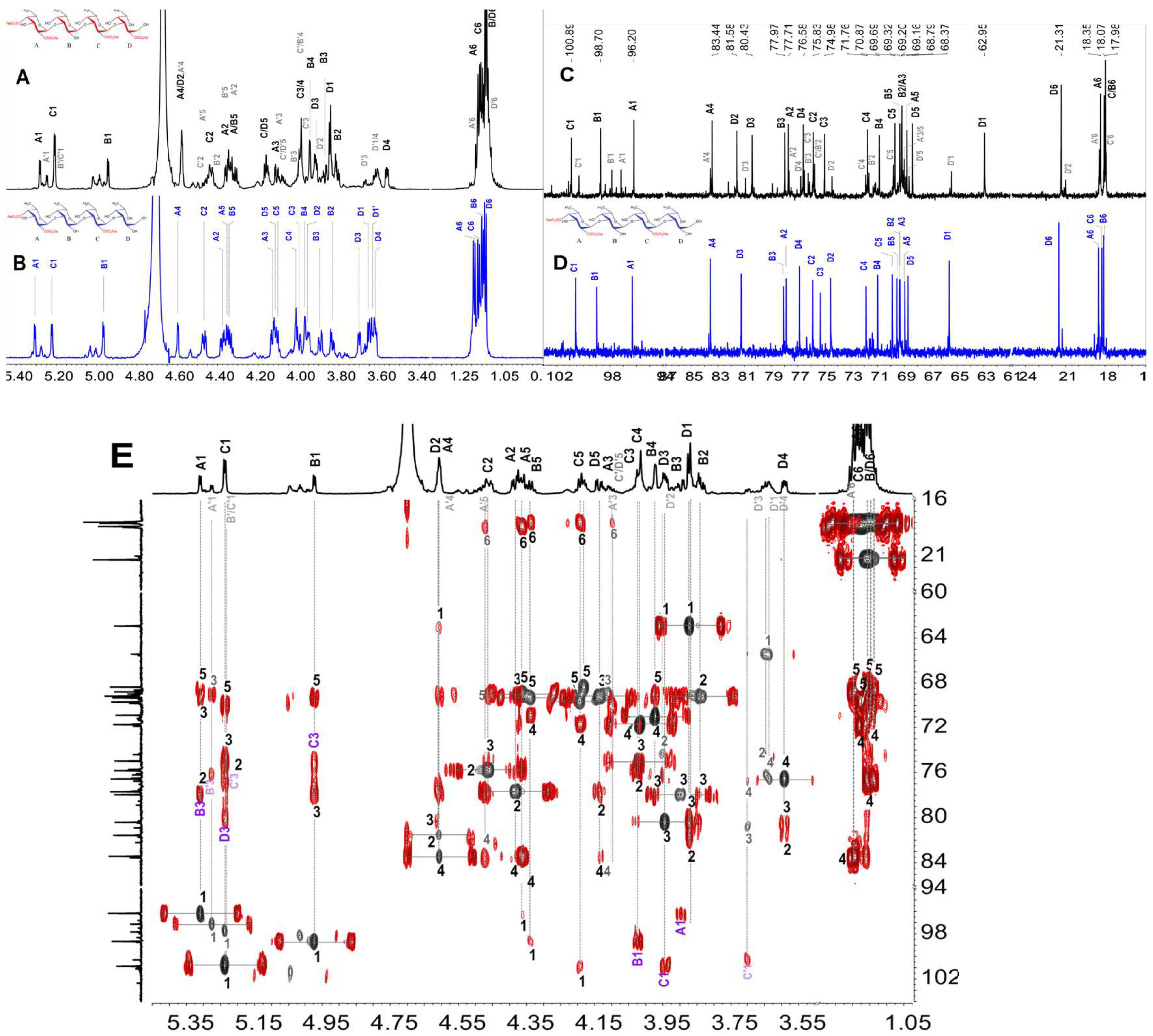
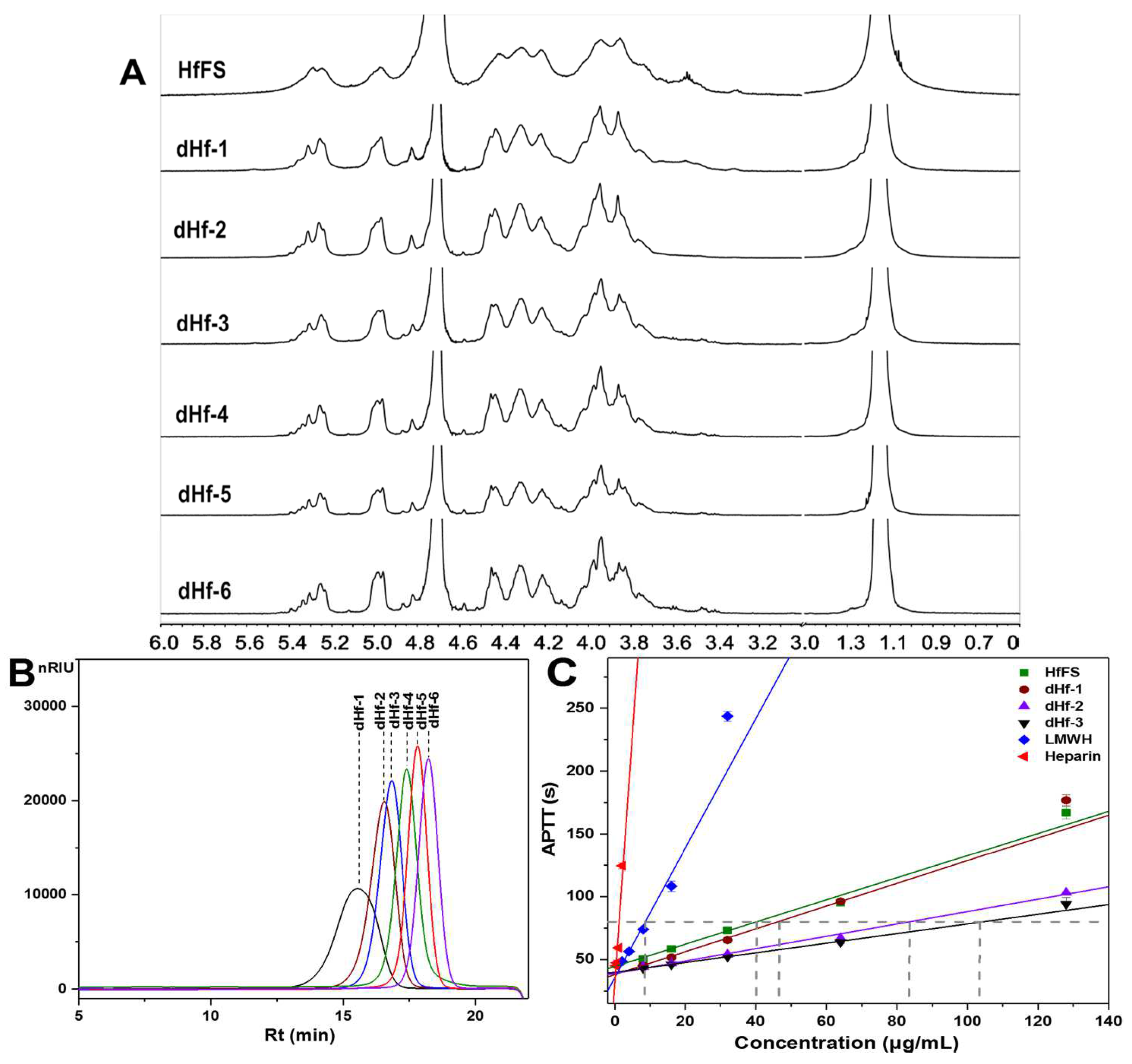
2.3. Structural Characterization of dHfFS-1 Prepared by Peroxidative Depolymerization
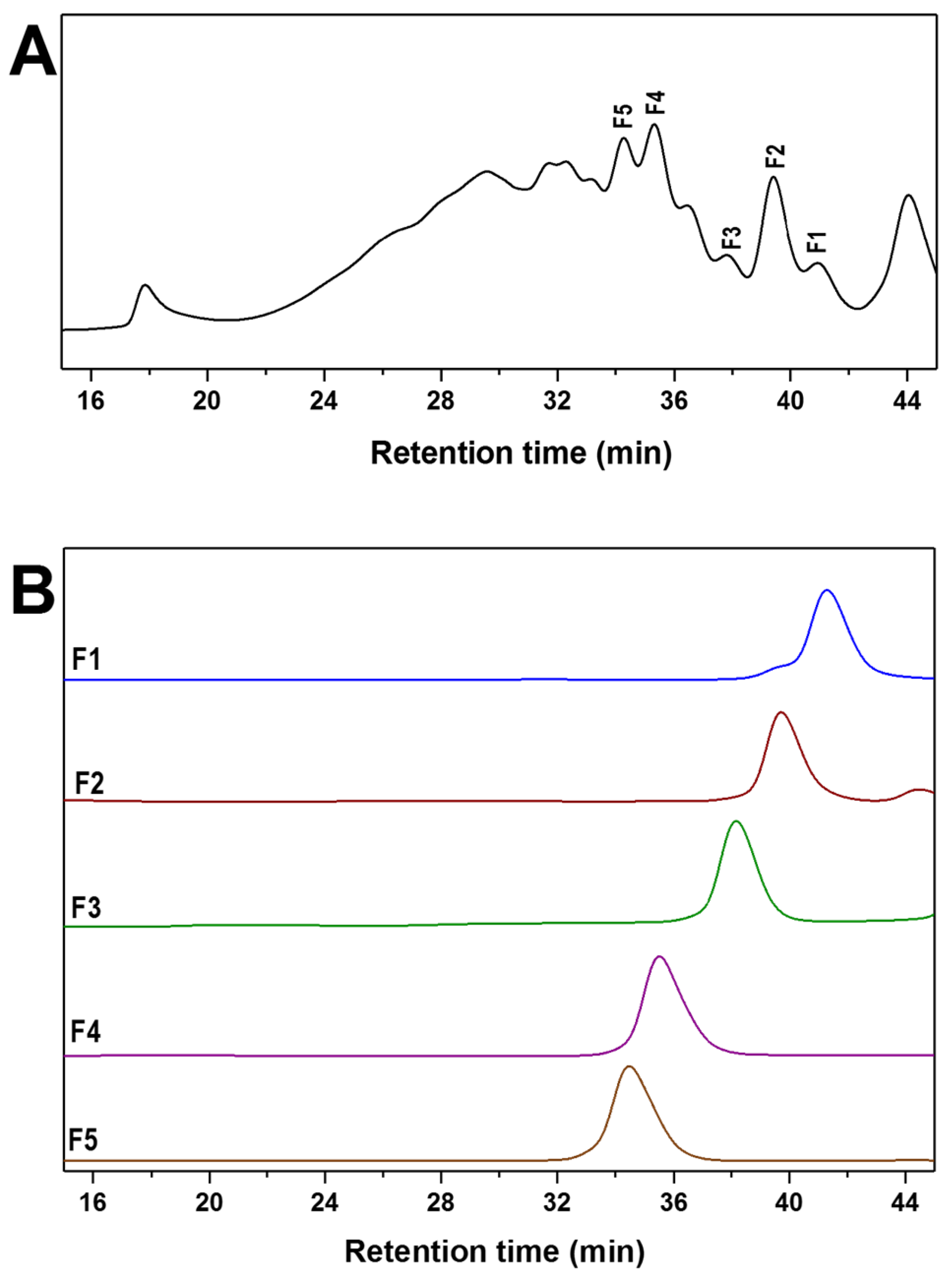
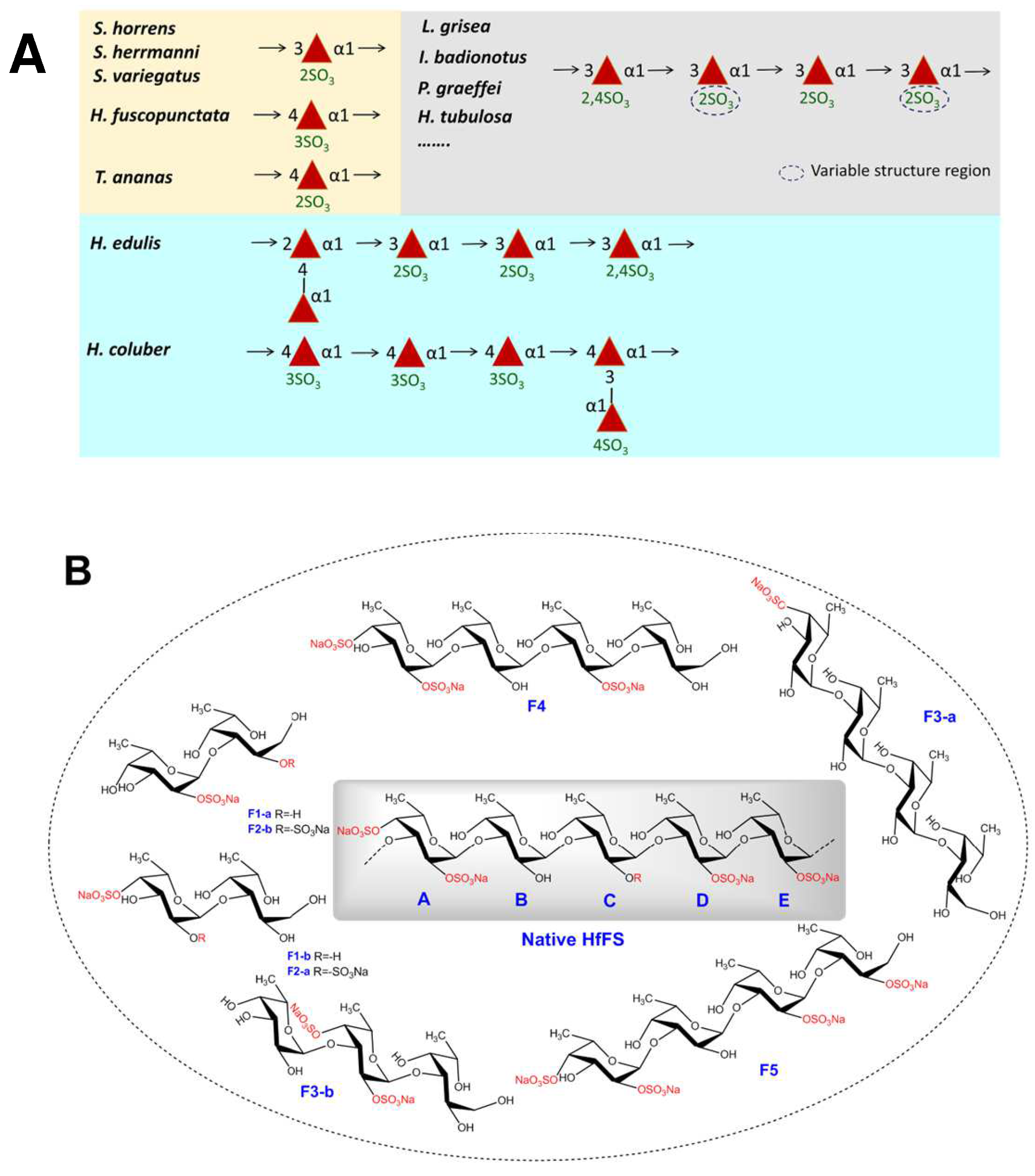
2.4. Mild Acid Hydrolysis of HfFS and Purification of Fucooligosaccharides
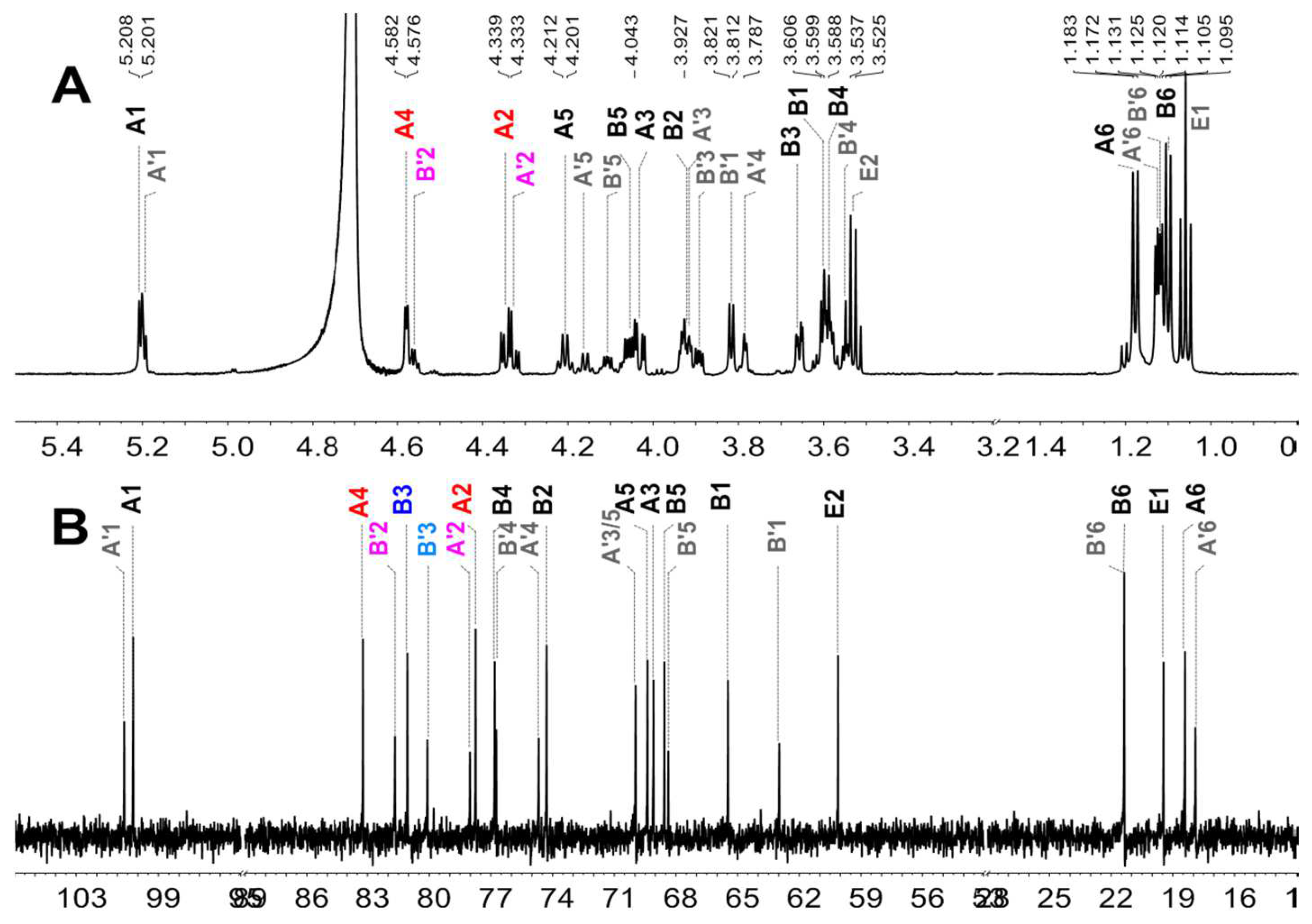
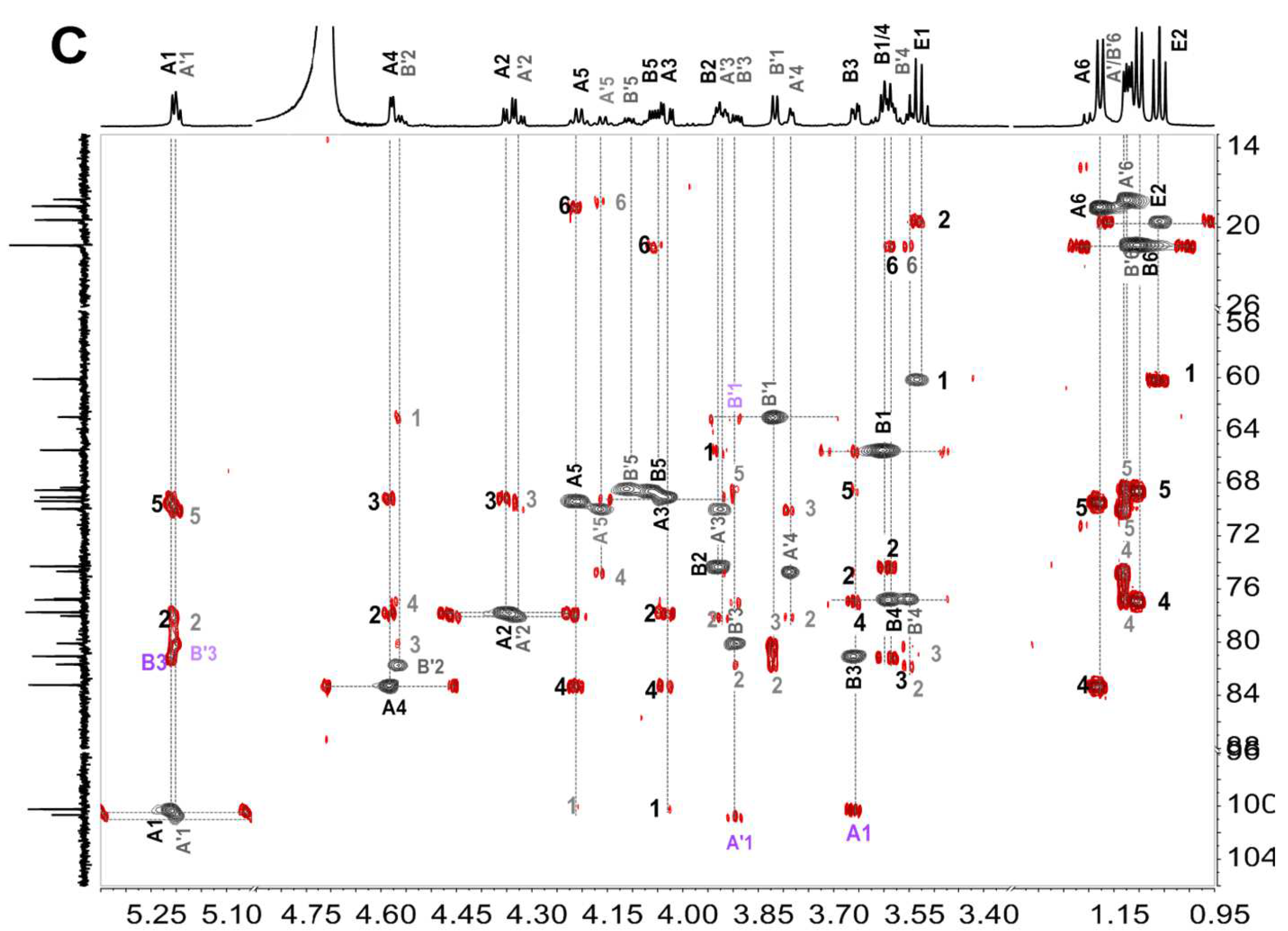
2.5. Structural Determination of Fucooligosaccharides by NMR

2.6. Anticoagulant Activity Analysis
3. Materials and Methods
3.1. Materials
3.2. Extraction and Purification of HfFS
3.3. Preparation of the Depolymerized Products dHfFS-1 by Peroxidative Depolymerization
3.4. Mild Acid Hydrolysis of HfFS
3.5. Purification of Fucooligosaccharides from dHfFS-2
3.6. Kinetics of Mild Acid Hydrolysis of HfFS at Different Temperatures
3.7. Chemical Composition and Physicochemical Properties of HfFS
3.8. IR and NMR Spectra Analysis
3.9. Mass Spectrometry Analysis
3.10. Anticoagulant Activities of HfFS and Its Low-Molecular-Weight Products In Vitro
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Adhikari, U.; Mateu, C.G.; Chattopadhyay, K.; Pujol, C.A.; Damonte, E.B.; Ray, B. Structure and antiviral activity of sulfated fucans from Stoechospermum marginatum. Phytochemistry 2006, 67, 2474–2482. [Google Scholar] [CrossRef] [PubMed]
- Park, H.Y.; Han, M.H.; Park, C.; Jin, C.Y.; Kim, G.Y.; Choi, I.W.; Kim, N.D.; Nam, T.J.; Kwon, T.K.; Choi, Y.H. Anti-inflammatory effects of fucoidan through inhibition of NF-kappaB, MAPK and Akt activation in lipopolysaccharide-induced BV2 microglia cells. Food Chem. Toxicol. 2011, 49, 1745–1752. [Google Scholar] [CrossRef]
- Yao, Y.; Yim, E.K.F. Fucoidan for cardiovascular application and the factors mediating its activities. Carbohydr. Polym. 2021, 270, 118347. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Sun, X. A critical review of the abilities, determinants, and possible molecular mechanisms of seaweed polysaccharides antioxidants. Int. J. Mol. Sci. 2020, 21, 7774. [Google Scholar] [CrossRef] [PubMed]
- Deniaud-Bouët, E.; Hardouin, K.; Potin, P.; Kloareg, B.; Hervé, C. A review about brown algal cell walls and fucose-containing sulfated polysaccharides: Cell wall context, biomedical properties and key research challenges. Carbohydr. Polym. 2017, 175, 395–408. [Google Scholar] [CrossRef]
- Shang, F.; Mou, R.; Zhang, Z.; Gao, N.; Lin, L.; Li, Z.; Wu, M.; Zhao, J. Structural analysis and anticoagulant activities of three highly regular fucan sulfates as novel intrinsic factor Xase inhibitors. Carbohydr. Polym. 2018, 195, 257–266. [Google Scholar] [CrossRef]
- Mulloy, B.; Ribeiro, A.C.; Alves, A.P.; Vieira, R.P.; Mourão, P.A. Sulfated fucans from echinoderms have a regular tetrasaccharide repeating unit defined by specific patterns of sulfation at the O-2 and O-4 positions. J. Biol. Chem. 1994, 269, 22113–22123. [Google Scholar] [CrossRef]
- Yu, L.; Ge, L.; Xue, C.; Chang, Y.; Zhang, C.; Xu, X.; Wang, Y. Structural study of fucoidan from sea cucumber Acaudina molpadioides: A fucoidan containing novel tetrafucose repeating unit. Food Chem. 2014, 142, 197–200. [Google Scholar] [CrossRef]
- Chang, Y.; Hu, Y.; Yu, L.; McClements, D.J.; Xu, X.; Liu, G.; Xue, C. Primary structure and chain conformation of fucoidan extracted from sea cucumber Holothuria tubulosa. Carbohydr. Polym. 2016, 136, 1091–1097. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Ye, X.Q.; Sun, Y.J.; Wu, D.; Wu, N.A.; Hu, Y.Q.; Chen, S.G. Ultrasound effects on the degradation kinetics, structure, and antioxidant activity of sea cucumber fucoidan. J. Agric. Food Chem. 2014, 62, 1088–1095. [Google Scholar] [CrossRef]
- Hu, Y.; Li, S.; Li, J.; Ye, X.; Ding, T.; Liu, D.; Chen, J.; Ge, Z.; Chen, S. Identification of a highly sulfated fucoidan from sea cucumber Pearsonothuria graeffei with well-repeated tetrasaccharides units. Carbohydr. Polym. 2015, 134, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Xue, C.; Chang, Y.; Hu, Y.; Xu, X.; Ge, L.; Liu, G. Structure and rheological characteristics of fucoidan from sea cucumber Apostichopus japonicus. Food Chem. 2015, 180, 71–76. [Google Scholar] [CrossRef]
- Cai, Y.; Yang, W.; Yin, R.; Zhou, L.; Li, Z.; Wu, M.; Zhao, J. An anticoagulant fucan sulfate with hexasaccharide repeating units from the sea cucumber Holothuria albiventer. Carbohydr. Res. 2018, 464, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Mourão, P.A.S.; Vilanova, E.; Soares, P.A.G. Unveiling the structure of sulfated fucose-rich polysaccharides via nuclear magnetic resonance spectroscopy. Curr. Opin. Struct. Biol. 2018, 50, 33–41. [Google Scholar] [CrossRef]
- Pomin, V.H.; Pereira, M.S.; Valente, A.P.; Tollefsen, D.M.; Pavao, M.S.; Mourao, P.A. Selective cleavage and anticoagulant activity of a sulfated fucan: Stereospecific removal of a 2-sulfate ester from the polysaccharide by mild acid hydrolysis, preparation of oligosaccharides, and heparin cofactor II-dependent anticoagulant activity. Glycobiology 2005, 15, 369–381. [Google Scholar] [CrossRef] [Green Version]
- Pomin, V.H.; Valente, A.P.; Pereira, M.S.; Mourão, P.A.S. Mild acid hydrolysis of sulfated fucans: A selective 2-desulfation reaction and an alternative approach for preparing tailored sulfated oligosaccharides. Glycobiology 2005, 15, 1376–1385. [Google Scholar] [CrossRef] [PubMed]
- Pereira, M.S.; Melo, F.R.; Mourão, P.A. Is there a correlation between structure and anticoagulant action of sulfated galactans and sulfated fucans? Glycobiology 2002, 12, 573–580. [Google Scholar] [CrossRef] [Green Version]
- An, Z.; Zhang, Z.; Zhang, X.; Yang, H.; Lu, H.; Liu, M.; Shao, Y.; Zhao, X.; Zhang, H. Oligosaccharide mapping analysis by HILIC-ESI-HCD-MS/MS for structural elucidation of fucoidan from sea cucumber Holothuria floridana. Carbohydr. Polym. 2022, 275, 118694. [Google Scholar] [CrossRef]
- Shi, X.; Guan, R.; Zhou, L.; Zuo, Z.; Tao, X.; Wang, P.; Zhou, Y.; Yin, R.; Zhao, L.; Gao, N.; et al. Structural characterization and heparanase inhibitory activity of fucosylated glycosaminoglycan from Holothuria floridana. Mar. Drugs 2021, 19, 162. [Google Scholar] [CrossRef]
- Grotjan, H.E.; Padrnos-Hicks, P.A.; Keel, B.A. Ion chromatographic method for the analysis of sulfate in complex carbohydrates. J. Chromatogr. A 1986, 367, 367–375. [Google Scholar] [CrossRef]
- Wu, M.; Xu, L.; Zhao, L.; Xiao, C.; Gao, N.; Luo, L.; Yang, L.; Li, Z.; Chen, L.; Zhao, J. Structural analysis and anticoagulant activities of the novel sulfated fucan possessing a regular well-defined repeating unit from sea cucumber. Mar. Drugs 2015, 13, 2063–2084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, N.; Chen, R.; Mou, R.; Xiang, J.; Zhou, K.; Li, Z.; Zhao, J. Purification, structural characterization and anticoagulant activities of four sulfated polysaccharides from sea cucumber Holothuria fuscopunctata. Int. J. Biol. Macromol. 2020, 164, 3421–3428. [Google Scholar] [CrossRef] [PubMed]
- Simha, R. Kinetics of degradation and size distribution of long chain polymers. J. Appl. Phys. 1941, 12, 569–578. [Google Scholar] [CrossRef]
- Pomin, V.H. Sulfated glycans in inflammation. Eur. J. Med. Chem. 2015, 92, 353–369. [Google Scholar] [CrossRef]
- Thinh, P.D.; Ly, B.M.; Usoltseva, R.V.; Shevchenko, N.M.; Rasin, A.B.; Anastyuk, S.D.; Malyarenko, O.S.; Zvyagintseva, T.N.; San, P.T.; Ermakova, S.P. A novel sulfated fucan from Vietnamese sea cucumber Stichopus variegatus: Isolation, structure and anticancer activity in vitro. Int. J. Biol. Macromol. 2018, 117, 1101–1109. [Google Scholar] [CrossRef]
- Li, X.; Li, S.; Liu, J.; Lin, L.; Sun, H.; Yang, W.; Cai, Y.; Gao, N.; Zhou, L.; Qin, H.; et al. A regular fucan sulfate from Stichopus herrmanni and its peroxide depolymerization: Structure and anticoagulant activity. Carbohydr. Polym. 2021, 256, 117513. [Google Scholar] [CrossRef]
- Ustyuzhanina, N.E.; Bilan, M.I.; Dmitrenok, A.S.; Borodina, E.Y.; Nifantiev, N.E.; Usov, A.I. A highly regular fucan sulfate from the sea cucumber Stichopus horrens. Carbohydr. Res. 2018, 456, 5–9. [Google Scholar] [CrossRef]
- Chen, S.; Xue, C.; Yin, L.A.; Tang, Q.; Yu, G.; Chai, W. Comparison of structures and anticoagulant activities of fucosylated chondroitin sulfates from different sea cucumbers. Carbohydr. Polym. 2011, 83, 688–696. [Google Scholar] [CrossRef]
- Yang, W.; Cai, Y.; Yin, R.; Lin, L.; Li, Z.; Wu, M.; Zhao, J. Structural analysis and anticoagulant activities of two sulfated polysaccharides from the sea cucumber Holothuria coluber. Int. J. Biol. Macromol. 2018, 115, 1055–1062. [Google Scholar] [CrossRef]
- Shi, D.; Qi, J.; Zhang, H.; Yang, H.; Yang, Y.; Zhao, X. Comparison of hydrothermal depolymerization and oligosaccharide profile of fucoidan and fucosylated chondroitin sulfate from Holothuria floridana. Int. J. Biol. Macromol. 2019, 132, 738–747. [Google Scholar] [CrossRef]
- Vismara, E.; Pierini, M.; Mascellani, G.; Liverani, L.; Lima, M.; Guerrini, M.; Torri, G. Low-molecular-weight heparin from Cu2+ and Fe2+ Fenton type depolymerisation processes. Thromb. Haemost. 2010, 103, 613–622. [Google Scholar] [PubMed] [Green Version]
- Wu, M.; Xu, S.; Zhao, J.; Kang, H.; Ding, H. Physicochemical characteristics and anticoagulant activities of low molecular weight fractions by free-radical depolymerization of a fucosylated chondroitin sulphate from sea cucumber Thelenata ananas. Food Chem. 2010, 122, 716–723. [Google Scholar] [CrossRef]
- Gao, L.; Xu, C.; Tao, X.; Zuo, Z.; Ning, Z.; Wang, L.; Gao, N.; Zhao, J. Structure elucidation of fucan sulfate from sea cucumber Holothuria fuscopunctata through a bottom-up strategy and the antioxidant activity analysis. Int. J. Mol. Sci. 2022, 23, 4488. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.T.; Oneill, R.A. Monosaccharide composition analysis of oligosaccharides and glycoproteins by high-performance liquid chromatography. Anal. Biochem. 1995, 227, 377–384. [Google Scholar] [CrossRef]
- Pitt, J.J.; Gorman, J.J. Oligosaccharide characterization and quantitation using 1-phenyl-3-methyl-5-pyrazolone derivatization and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Anal. Biochem. 1997, 248, 63–75. [Google Scholar] [CrossRef]
- Zhou, L.; Gao, N.; Sun, H.; Xiao, C.; Yang, L.; Lin, L.; Yin, R.; Li, Z.; Zhang, H.; Ji, X.; et al. Effects of native fucosylated glycosaminoglycan, its depolymerized derivatives on intrinsic factor Xase, coagulation, thrombosis, and hemorrhagic risk. Thromb. Haemost. 2020, 120, 604–619. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Residue | Chemical Shift (ppm) | ||||||
|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | ||
| A -l-Fuc2S4S-α(1,3)- | H | 5.436 | 4.586 | 4.433 | 4.945 | 4.450 | 1.302 |
| C | 96.71 | 77.67 | 77.16 | 83.63 | 69.52 | 18.59 | |
| B -l-Fuc-α(1,3)- | H | 5.087 | 3.959 | 4.004 | 4.063 | 4.338 | 1.263 |
| C | 99.36 | 69.24 | 78.43 | 71.33 | 70.41 | 18.36 | |
| C -l-Fuc-α(1,3)- | H | 5.111 | 3.984 | 3.984 | 4.063 | 4.326 | 1.236 |
| C | 98.55 | 69.18 | 78.18 | 71.33 | 70.41 | 18.33 | |
| D -l-Fuc2S-α(1,3)- | H | 5.363 | 4.554 | 4.150 | 4.096 | 4.472 | 1.282 |
| C | 96.96 | 76.11 | 76.11 | 72.29 | 69.83 | 18.23 | |
| E -l-Fuc2S-α(1,3)- | H | 5.389 | 4.554 | 4.368 | 4.087 | 4.472 | 1.293 |
| C | 101.32 | 75.91 | 75.62 | 72.29 | 69.83 | 18.16 | |
| Fucooligosaccharide | Residue | Chemical Shift (ppm) | ||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | |||
| F2-a | A: l-Fuc2S4S-α(1) | H | 5.208 (J1,2 = 3.96) | 4.345 | 4.032 | 4.578 | 4.206 | 1.178 |
| C | 100.25 | 77.76 | 69.10 | 83.23 | 69.40 | 18.41 | ||
| B: -3)-l-Fuc-ol | H | 3.612 (J1,1’ = 11.34) | 3.926 | 3.657 | 3.585 | 4.058 | 1.100 | |
| 3.583 (J1,2 = 5.04) | ||||||||
| C | 65.48 | 74.30 | 81.07 | 76.82 | 68.57 | 21.37 | ||
| F2-b | A’: l-Fuc2S-α(1) | H | 5.195 (J1,2 = 3.96) | 4.327 | 3.921 | 3.784 | 4.159 | 1.125 |
| C | 100.68 | 78.03 | 69.97 | 74.68 | 69.97 | 17.91 | ||
| B’: -3)-l-Fuc2S-ol | H | 3.816 (J1,2 = 5.34) | 4.558 | 3.892 | 3.546 | 4.107 | 1.119 | |
| C | 62.98 | 81.68 | 80.10 | 76.74 | 68.38 | 21.37 | ||
| Fucooligosaccharide | Residue | Chemical Shift (ppm) | ||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | |||
| F4 | A: l-Fuc2S4S-α(1)- | H | 5.309 (J1,2 = 3.84) | 4.383 | 4.138 | 4.600 | 4.355 | 1.191 |
| C | 93.49 | 75.02 | 66.46 | 80.72 | 66.10 | 15.66 | ||
| B: -3)-l-Fuc-α(1)- | H | 4.971 (J1,2 = 4.00) | 3.834 | 3.895 | 3.971 | 4.339 | 1.143 | |
| C | 96.13 | 66.51 | 75.23 | 68.15 | 66.69 | 15.28 | ||
| C: -3)-l-Fuc2S-α(1)- | H | 5.225 (J1,2 = 4.08) | 4.470 | 4.012 | 3.993 | 4.107 | 1.169 | |
| C | 97.73 | 73.03 | 72.44 | 69.01 | 67.05 | 15.40 | ||
| D: -3)-l-Fuc-ol | H | 3.672 (J1, 1’ = 8.38) 3.656 (J1,2 = 4.80) | 3.954 | 3.702 | 3.622 | 4.125 | 1.136 | |
| C | 62.76 | 71.68 | 78.40 | 74.01 | 65.86 | 18.64 | ||
| F5 | A: l-Fuc2S4S-α(1)- | H | 5.281 (J1,2 = 3.92) | 4.105 | 4.109 | 4.576 | 4.335 | 1.165 |
| C | 96.20 | 77.71 | 69.16 | 83.44 | 68.79 | 18.35 | ||
| B: -3)-l-Fuc–α(1)- | H | 4.944 (J1,2 = 4.00) | 3.807 | 3.869 | 3.943 | 4.310 | 1.122 | |
| C | 98.70 | 69.20 | 77.97 | 70.87 | 69.32 | 17.98 | ||
| C: -3)-l-Fuc2S-α(1)- | H | 5.209 (J1,2 = 3.92) | 4.432 | 3.996 | 3.990 | 4.166 | 1.150 | |
| C | 100.89 | 75.83 | 74.98 | 71.76 | 69.69 | 18.07 | ||
| D: -3)-l-Fuc2S-ol | H | 3.844 (J1,2 = 5.52) | 4.582 | 3.914 | 3.563 | 4.153 | 1.130 | |
| C | 62.95 | 81.58 | 80.43 | 76.58 | 68.37 | 21.31 | ||
| Sample | Mw a (kDa) | Mn (kDa) | PDI | APTT b (μg/mL) | PT (μg/mL) | TT (μg/mL) |
|---|---|---|---|---|---|---|
| HfFS | 443.41 | 136.88 | 3.24 | 46.17 | >128 | >128 |
| dHf-1 | 74.74 | 49.75 | 1.50 | 42.49 | >128 | 74.85 |
| dHf-2 | 27.68 | 21.76 | 1.27 | 82.61 | >128 | >128 |
| dHf-3 | 20.32 | 15.75 | 1.30 | 95.43 | >128 | >128 |
| dHf-4 | 11.50 | 8.88 | 1.30 | >128 | >128 | >128 |
| dHf-5 | 8.26 | 6.86 | 1.20 | >128 | >128 | >128 |
| dHf-6 | 5.99 | 4.80 | 1.25 | >128 | >128 | >128 |
| LMWH | ~4.40 | / | / | 9.60 | / | 1.98 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ning, Z.; Wang, P.; Zuo, Z.; Tao, X.; Gao, L.; Xu, C.; Wang, Z.; Wu, B.; Gao, N.; Zhao, J. A Fucan Sulfate with Pentasaccharide Repeating Units from the Sea Cucumber Holothuriafloridana and Its Anticoagulant Activity. Mar. Drugs 2022, 20, 377. https://doi.org/10.3390/md20060377
Ning Z, Wang P, Zuo Z, Tao X, Gao L, Xu C, Wang Z, Wu B, Gao N, Zhao J. A Fucan Sulfate with Pentasaccharide Repeating Units from the Sea Cucumber Holothuriafloridana and Its Anticoagulant Activity. Marine Drugs. 2022; 20(6):377. https://doi.org/10.3390/md20060377
Chicago/Turabian StyleNing, Zimo, Pin Wang, Zhichuang Zuo, Xuelin Tao, Li Gao, Chen Xu, Zhiyue Wang, Bin Wu, Na Gao, and Jinhua Zhao. 2022. "A Fucan Sulfate with Pentasaccharide Repeating Units from the Sea Cucumber Holothuriafloridana and Its Anticoagulant Activity" Marine Drugs 20, no. 6: 377. https://doi.org/10.3390/md20060377