
MSC−sEV Treatment Polarizes Pro−Fibrotic M2 Macrophages without Exacerbating Liver Fibrosis in NASH
 , and
, and
Abstract
:1. Introduction
2. Results
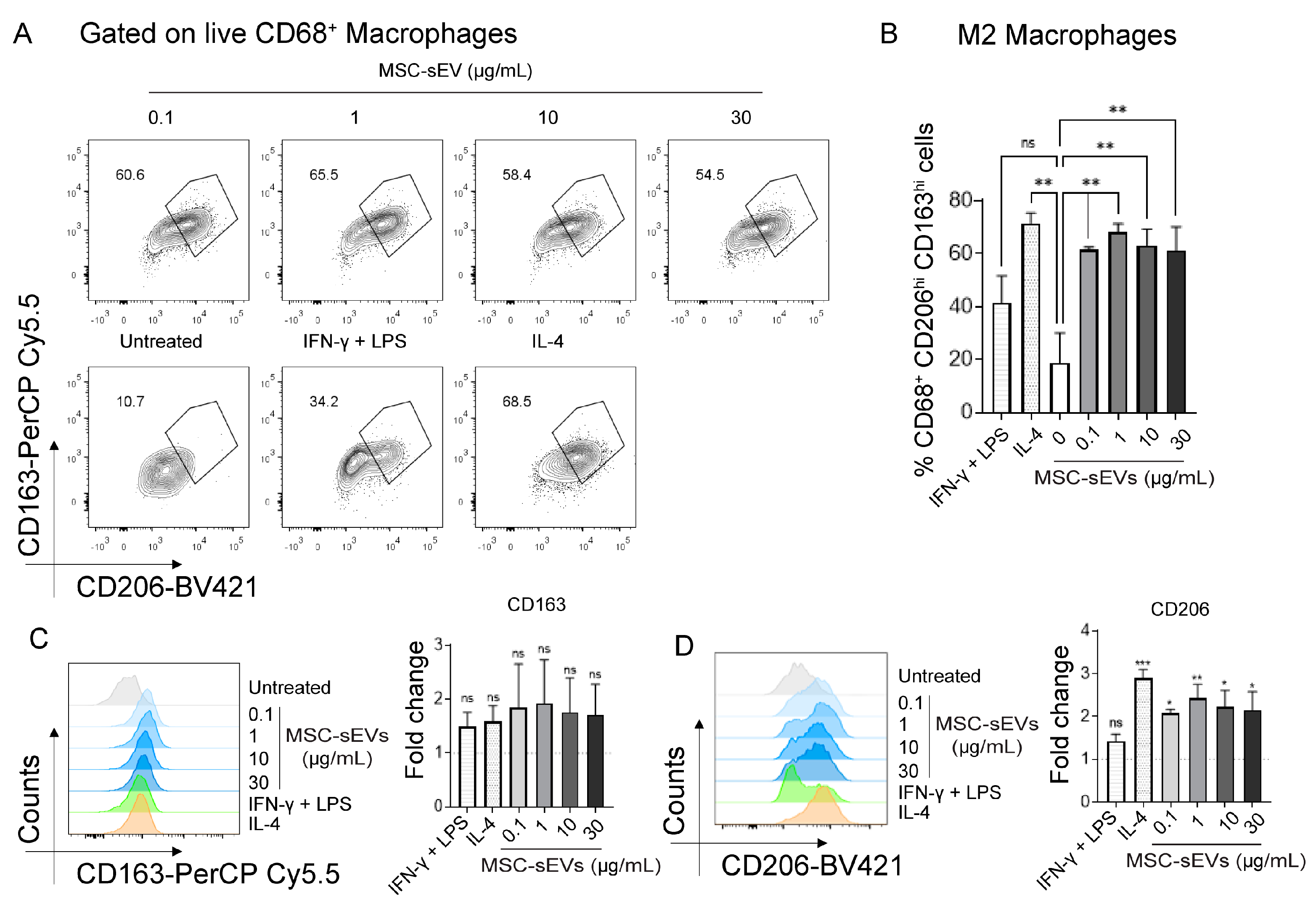
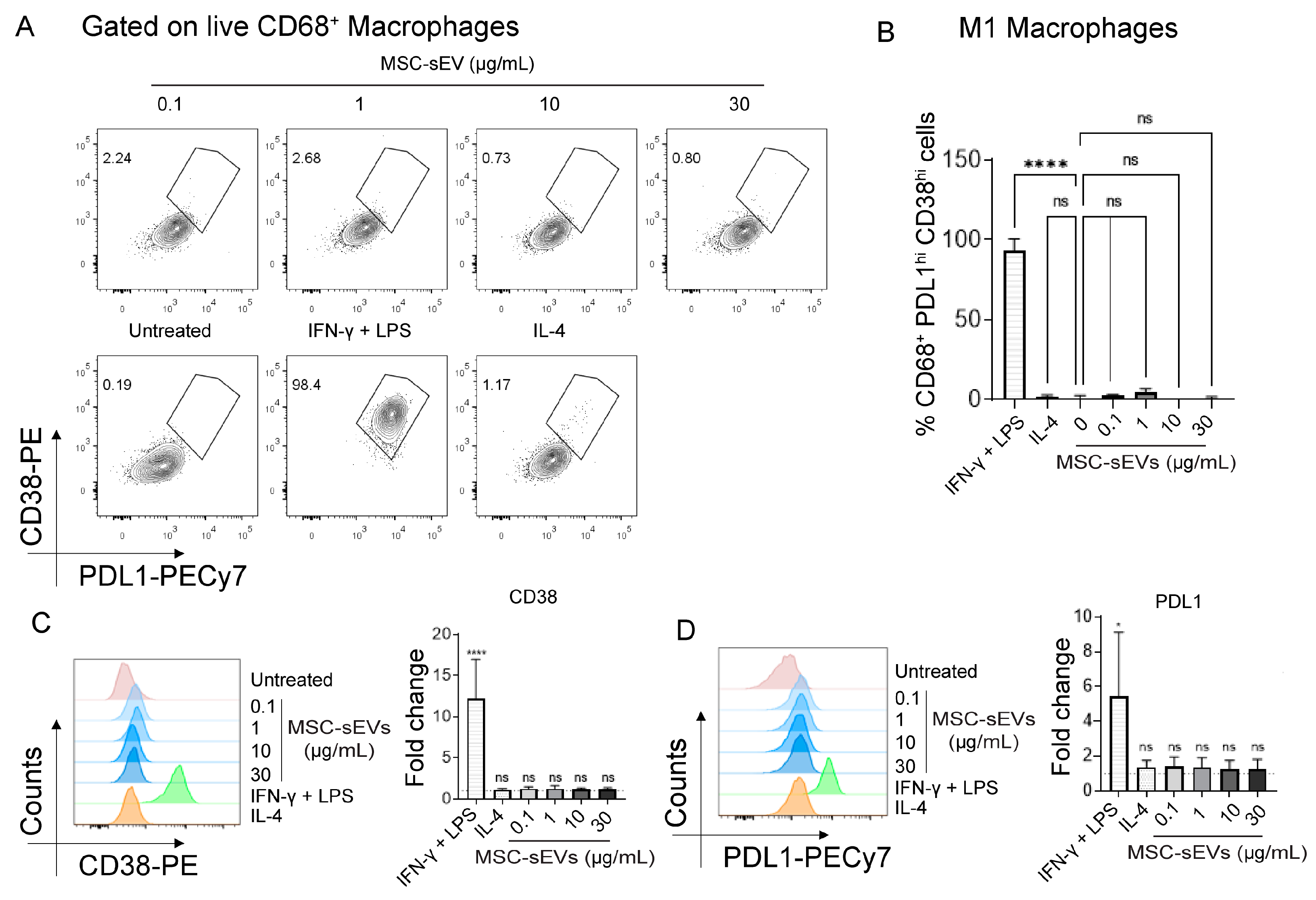
2.1. MSC−sEV Preparations Can Polarize M0 to M2 but Not M1 Macrophages
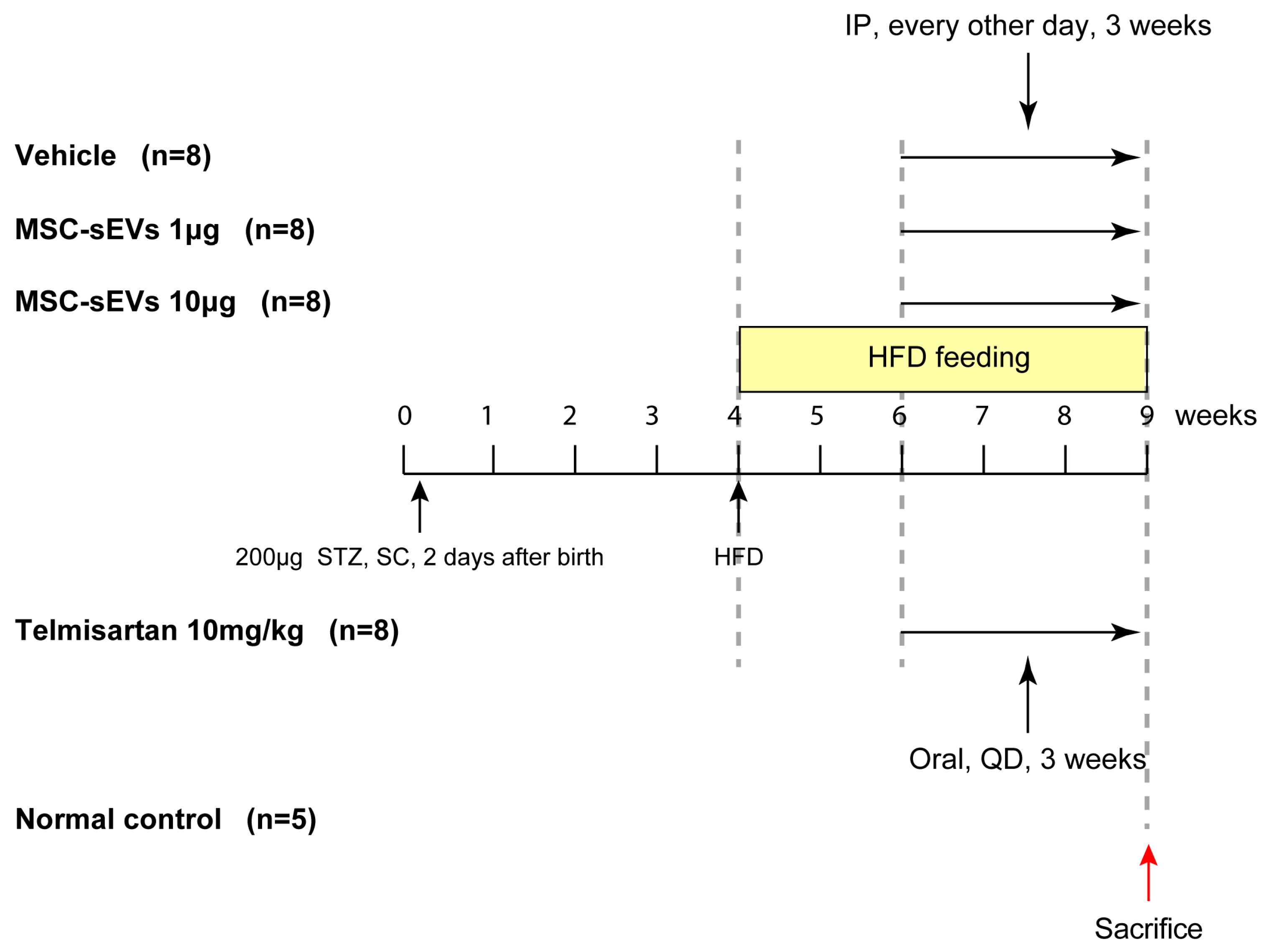
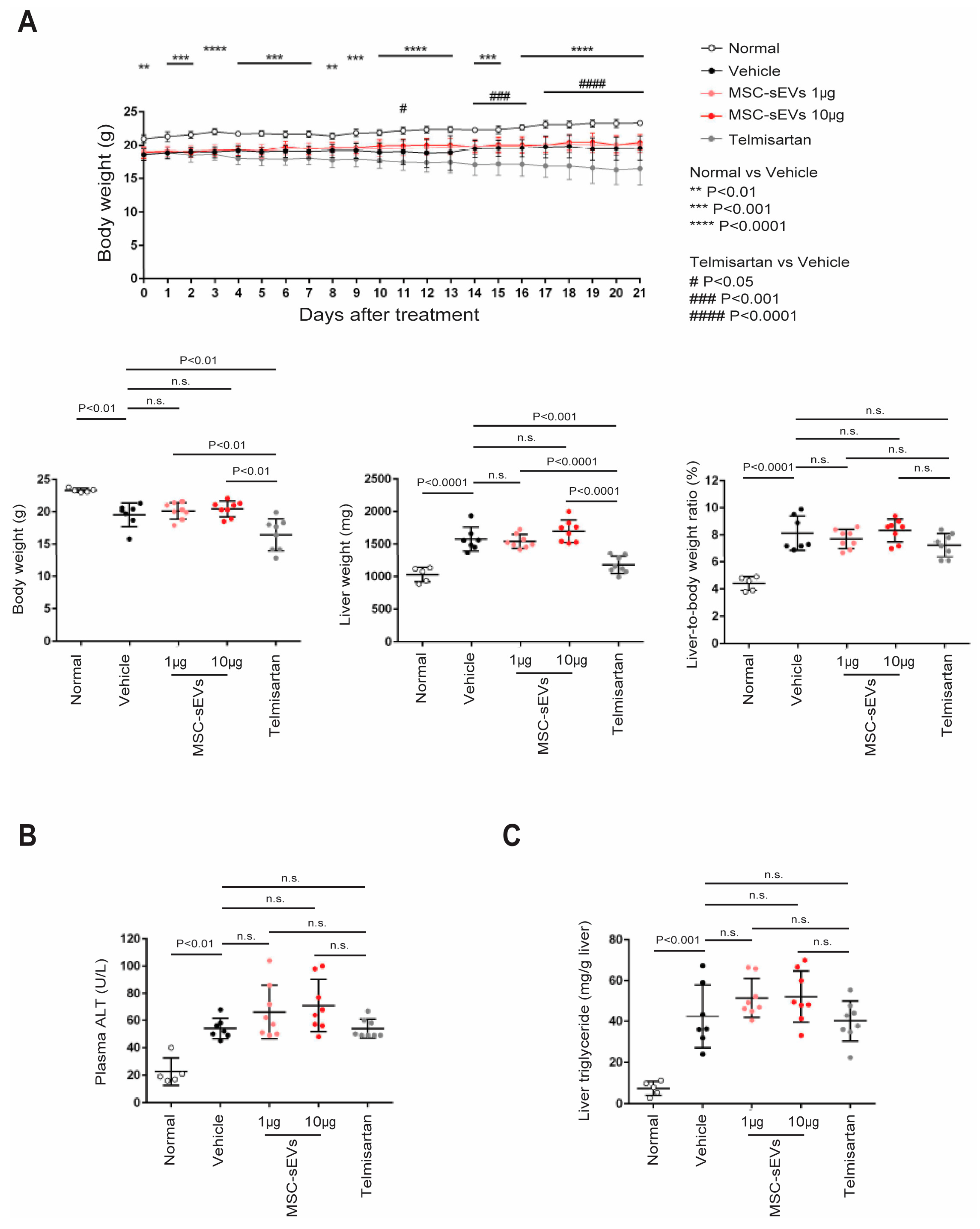
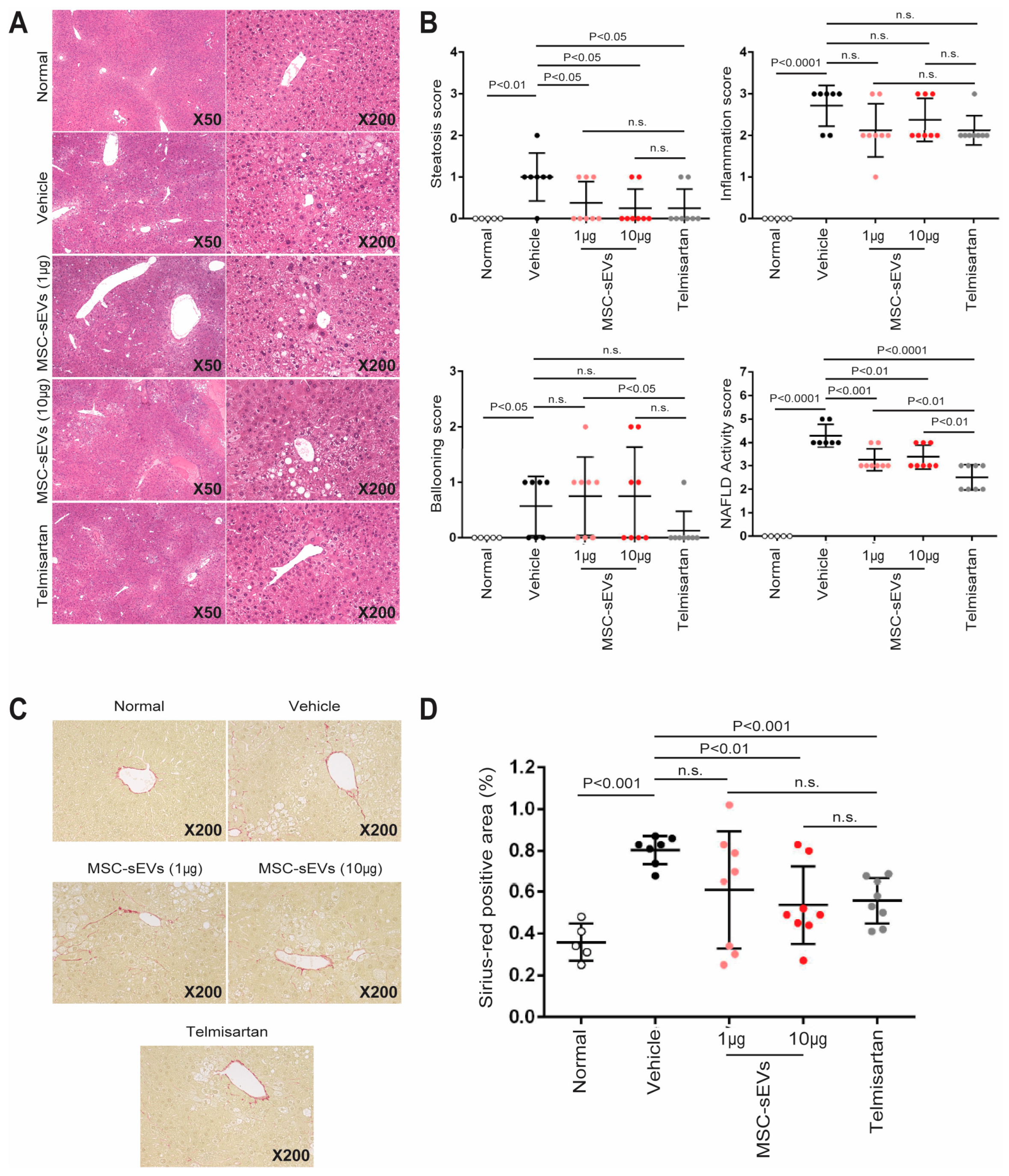
2.2. MSC−sEV Preparations Suppress Fibrosis in a Mouse Model of NASH
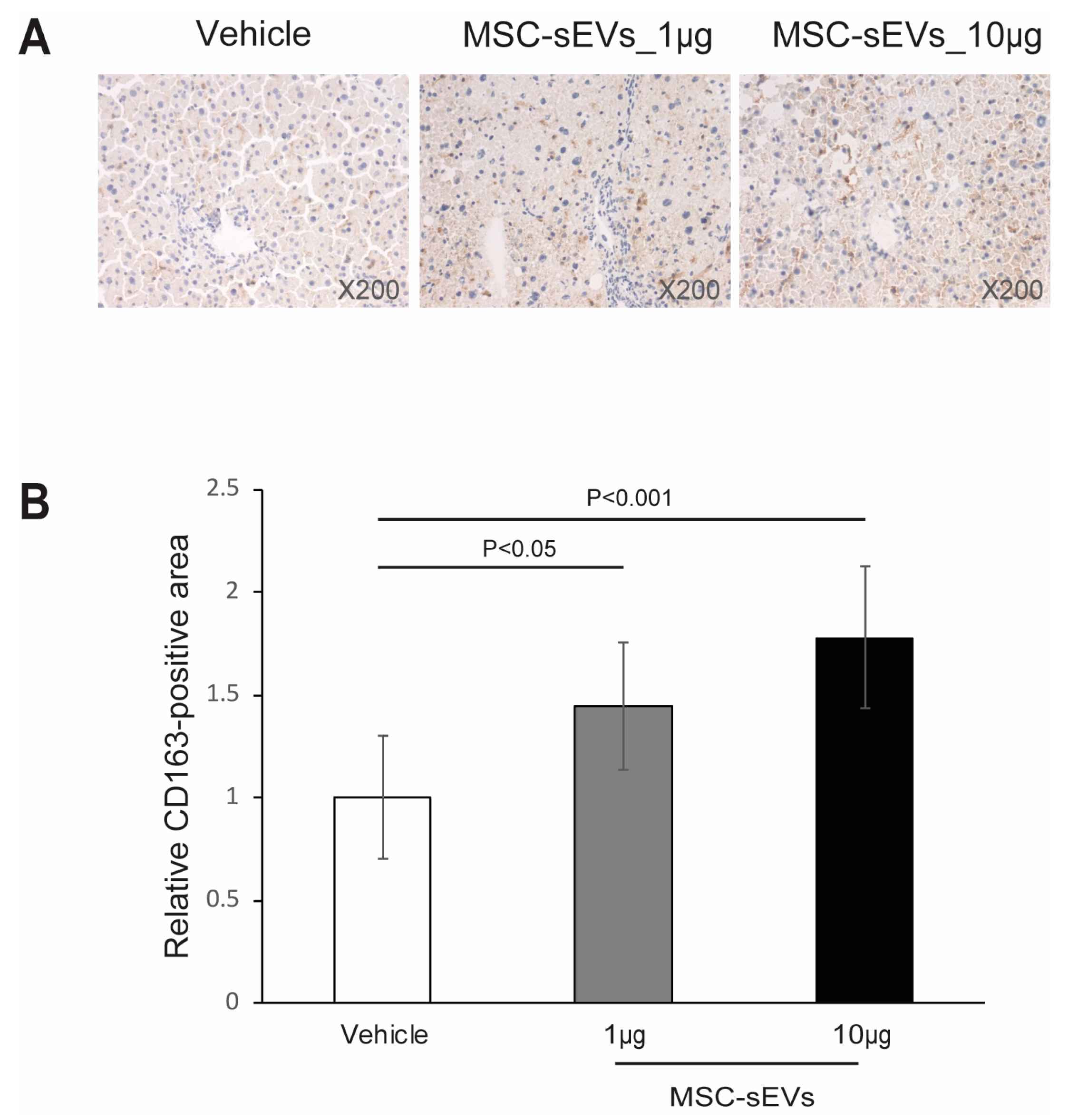
2.3. MSC−sEV Preparations Increase Anti−Inflammatory M2 Macrophages
3. Discussion
4. Materials and Methods
4.1. Culture of MSCs and Preparation of MSC−sEVs
4.2. Polarization of Macrophages by MSC−sEVs
4.3. HFD−Induced NASH Model
4.4. Liver Triglyceride Measurement
4.5. Liver Histological Analysis
4.6. Immunohistochemistry Staining for CD163
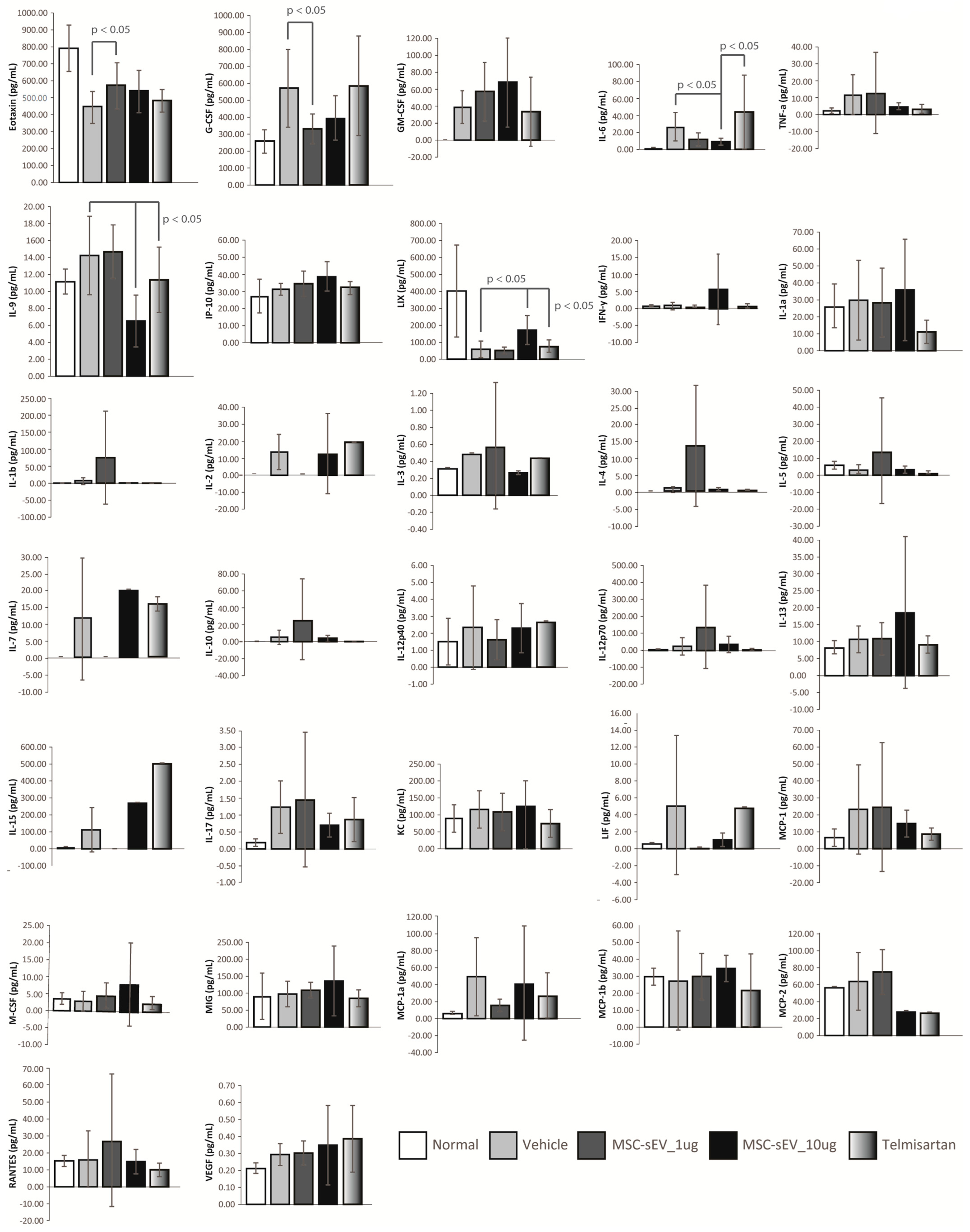
4.7. Mouse Plasmatic Cytokine/Chemokine Array
4.8. Statistical Tests
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper−Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Caplan, A.I. Mesenchymal Stem Cells: Time to Change the Name! Stem Cells Transl. Med. 2017, 6, 1445–1451. [Google Scholar] [CrossRef] [PubMed]
- Caplan, A.I.; Dennis, J.E. Mesenchymal stem cells as trophic mediators. J. Cell Biochem. 2006, 98, 1076–1084. [Google Scholar] [CrossRef] [PubMed]
- Timmers, L.; Lim, S.-K.; Arslan, F.; Armstrong, J.S.; Hoefler, I.E.; Doevendans, P.A.; Piek, J.J.; El Oakley, R.M.; Choo, A.; Lee, C.N.; et al. Reduction of myocardial infarct size by human mesenchymal stem cell conditioned medium. Stem Cell Res. 2008, 1, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Bruno, S.; Grange, C.; Deregibus, M.C.; Calogero, R.A.; Saviozzi, S.; Collino, F.; Morando, L.; Busca, A.; Falda, M.; Bussolati, B.; et al. Mesenchymal stem cell−derived microvesicles protect against acute tubular injury. J. Am. Soc. Nephrol. 2009, 20, 1053–1067. [Google Scholar] [CrossRef]
- Lai, R.C.; Arslan, F.; Lee, M.M.; Sze, N.S.; Choo, A.; Chen, T.S.; Salto−Tellez, M.; Timmers, L.; Lee, C.N.; El Oakley, R.M.; et al. Exosome secreted by MSC reduces myocardial ischemia/reperfusion injury. Stem Cell Res. 2010, 4, 214–222. [Google Scholar] [CrossRef]
- Bruno, S.; Tapparo, M.; Collino, F.; Chiabotto, G.; Deregibus, M.C.; Soares Lindoso, R.; Neri, F.; Kholia, S.; Giunti, S.; Wen, S. Renal regenerative potential of different extracellular vesicle populations derived from bone marrow mesenchymal stromal cells. Tissue Eng. Part A 2017, 23, 1262–1273. [Google Scholar] [CrossRef] [PubMed]
- Gimona, M.; Brizzi, M.F.; Choo, A.B.H.; Dominici, M.; Davidson, S.M.; Grillari, J.; Hermann, D.M.; Hill, A.F.; de Kleijn, D.; Lai, R.C.; et al. Critical considerations for the development of potency tests for therapeutic applications of mesenchymal stromal cell−derived small extracellular vesicles. Cytotherapy 2021, 23, 373–380. [Google Scholar] [CrossRef]
- Doeppner, T.R.; Herz, J.; Gorgens, A.; Schlechter, J.; Ludwig, A.K.; Radtke, S.; de Miroschedji, K.; Horn, P.A.; Giebel, B.; Hermann, D.M. Extracellular Vesicles Improve Post−Stroke Neuroregeneration and Prevent Postischemic Immunosuppression. Stem Cells Transl. Med. 2015, 4, 1131–1143. [Google Scholar] [CrossRef]
- Nawaz, M.; Fatima, F.; Vallabhaneni, K.C.; Penfornis, P.; Valadi, H.; Ekstrom, K.; Kholia, S.; Whitt, J.D.; Fernandes, J.D.; Pochampally, R.; et al. Extracellular Vesicles: Evolving Factors in Stem Cell Biology. Stem Cells Int. 2016, 2016, 1073140. [Google Scholar] [CrossRef]
- Baek, G.; Choi, H.; Kim, Y.; Lee, H.C.; Choi, C. Mesenchymal Stem Cell−Derived Extracellular Vesicles as Therapeutics and as a Drug Delivery Platform. Stem Cells Transl. Med. 2019, 8, 880–886. [Google Scholar] [CrossRef]
- Van Balkom, B.W.M.; Gremmels, H.; Giebel, B.; Lim, S.K. Proteomic Signature of Mesenchymal Stromal Cell−Derived Small Extracellular Vesicles. Proteomics 2019, 19, 1800163. [Google Scholar] [CrossRef] [PubMed]
- Witwer, K.W.; Van Balkom, B.W.M.; Bruno, S.; Choo, A.; Dominici, M.; Gimona, M.; Hill, A.F.; De Kleijn, D.; Koh, M.; Lai, R.C.; et al. Defining mesenchymal stromal cell (MSC)−derived small extracellular vesicles for therapeutic applications. J. Extracell. Vesicles 2019, 8, 1609206. [Google Scholar] [CrossRef]
- Toh, W.S.; Zhang, B.I.N.; Lai, R.C.; Lim, S.K. Immune regulatory targets of mesenchymal stromal cell exosomes/small extracellular vesicles in tissue regeneration. Cytotherapy 2018, 20, 1419–1426. [Google Scholar] [CrossRef]
- Lai, R.C.; Tan, S.S.; Teh, B.J.; Sze, S.K.; Arslan, F.; de Kleijn, D.P.; Choo, A.; Lim, S.K. Proteolytic Potential of the MSC Exosome Proteome: Implications for an Exosome−Mediated Delivery of Therapeutic Proteasome. Int. J. Proteom. 2012, 2012, 971907. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Yeo, R.W.Y.; Lai, R.C.; Sim, E.W.K.; Chin, K.C.; Lim, S.K. Mesenchymal stromal cell exosome–enhanced regulatory T−cell production through an antigen−presenting cell–mediated pathway. Cytotherapy 2018, 20, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Yin, Y.; Lai, R.C.; Tan, S.S.; Choo, A.B.H.; Lim, S.K. Mesenchymal Stem Cells Secrete Immunologically Active Exosomes. Stem Cells Dev. 2014, 23, 1233–1244. [Google Scholar] [CrossRef]
- Lai, R.; Yeo, R.; Tan, S.; Zhang, B.; Yin, Y.; Sze, S.; Choo, A.; Lim, S. Mesenchymal Stem Cell Exosomes: The Future MSC−Based Therapy? In Mesenchymal Stem Cell Therapy; Chase, L.G.V., Mohan, C., Eds.; Humana Press: Totowa, NJ, USA, 2013; pp. 39–62. [Google Scholar]
- Arslan, F.; Lai, R.C.; Smeets, M.B.; Akeroyd, L.; Choo, A.; Aguor, E.N.; Timmers, L.; van Rijen, H.V.; Doevendans, P.A.; Pasterkamp, G.; et al. Mesenchymal stem cell−derived exosomes increase ATP levels, decrease oxidative stress and activate PI3K/Akt pathway to enhance myocardial viability and prevent adverse remodeling after myocardial ischemia/reperfusion injury. Stem Cell Res. 2013, 10, 301–312. [Google Scholar] [CrossRef]
- Zhang, B.; Lai, R.C.; Sim, W.K.; Choo, A.B.H.; Lane, E.B.; Lim, S.K. Topical Application of Mesenchymal Stem Cell Exosomes Alleviates the Imiquimod Induced Psoriasis−Like Inflammation. Int. J. Mol. Sci. 2021, 22, 720. [Google Scholar] [CrossRef]
- Loh, J.T.; Zhang, B.; Teo, J.K.H.; Lai, R.C.; Choo, A.B.H.; Lam, K.-P.; Lim, S.K. Mechanism for the attenuation of neutrophil and complement hyperactivity by MSC exosomes. Cytotherapy 2022, 24, 711–719. [Google Scholar] [CrossRef]
- Loinard, C.; Ribault, A.; Lhomme, B.; Benderitter, M.; Flamant, S.; Paul, S.; Dubois, V.; Lai, R.C.; Lim, S.K.; Tamarat, R. HuMSC−EV induce monocyte/macrophage mobilization to orchestrate neovascularization in wound healing process following radiation injury. Cell Death Discov. 2023, 9, 38. [Google Scholar] [CrossRef]
- Zhang, S.; Chuah, S.J.; Lai, R.C.; Hui, J.H.P.; Lim, S.K.; Toh, W.S. MSC exosomes mediate cartilage repair by enhancing proliferation, attenuating apoptosis and modulating immune reactivity. Biomaterials 2018, 156, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Chuah, S.J.; Yong, C.W.; Teo, K.Y.W.; Chew, J.R.J.; Cheow, Y.A.; Zhang, S.; Wong, R.C.W.; Lim, A.A.T.; Lim, S.K.; Toh, W.S. Mesenchymal stromal cell−derived small extracellular vesicles modulate macrophage polarization and enhance angio−osteogenesis to promote bone healing. Genes Dis. 2022, 9, 841–844. [Google Scholar] [CrossRef] [PubMed]
- Boutilier, A.J.; Elsawa, S.F. Macrophage Polarization States in the Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 6995. [Google Scholar] [CrossRef] [PubMed]
- Grunhut, J.; Wang, W.; Aykut, B.; Gakhal, I.; Torres−Hernandez, A.; Miller, G. Macrophages in Nonalcoholic Steatohepatitis: Friend or Foe? Eur. Med. J. Hepatol. 2018, 6, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Heymann, F.; Trautwein, C.; Tacke, F. Monocytes and macrophages as cellular targets in liver fibrosis. Inflamm. Allergy Drug Targets 2009, 8, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Chen, L.; Lv, G. Interleukin−6 Receptor Blockade can Increase the Risk of Nonalcoholic Fatty Liver Disease: Indications From Mendelian Randomization. Front. Pharmacol. 2022, 13, 905936. [Google Scholar] [CrossRef]
- Nasir, G.A.; Mohsin, S.; Khan, M.; Shams, S.; Ali, G.; Khan, S.N.; Riazuddin, S. Mesenchymal stem cells and Interleukin−6 attenuate liver fibrosis in mice. J. Transl. Med. 2013, 11, 78. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Itoh, Y.; Yokomizo, C.; Nishimura, T.; Niimi, T.; Fujii, H.; Okanoue, T.; Yoshikawa, T. Blockade of interleukin−6 signaling enhances hepatic steatosis but improves liver injury in methionine choline−deficient diet−fed mice. Lab. Investig. 2010, 90, 1169–1178. [Google Scholar] [CrossRef]
- Saito, K.; Uebanso, T.; Maekawa, K.; Ishikawa, M.; Taguchi, R.; Nammo, T.; Nishimaki−Mogami, T.; Udagawa, H.; Fujii, M.; Shibazaki, Y.; et al. Characterization of hepatic lipid profiles in a mouse model with nonalcoholic steatohepatitis and subsequent fibrosis. Sci. Rep. 2015, 5, 12466. [Google Scholar] [CrossRef]
- Bruno, S.; Pasquino, C.; Herrera Sanchez, M.B.; Tapparo, M.; Figliolini, F.; Grange, C.; Chiabotto, G.; Cedrino, M.; Deregibus, M.C.; Tetta, C.; et al. HLSC−Derived Extracellular Vesicles Attenuate Liver Fibrosis and Inflammation in a Murine Model of Non−alcoholic Steatohepatitis. Mol. Ther. 2020, 28, 479–489. [Google Scholar] [CrossRef]
- Ohara, M.; Ohnishi, S.; Hosono, H.; Yamamoto, K.; Yuyama, K.; Nakamura, H.; Fu, Q.; Maehara, O.; Suda, G.; Sakamoto, N. Extracellular Vesicles from Amnion−Derived Mesenchymal Stem Cells Ameliorate Hepatic Inflammation and Fibrosis in Rats. Stem Cells Int. 2018, 2018, 3212643. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Tsuchiya, A.; Takeuchi, S.; Nojiri, S.; Yoshida, T.; Ogawa, M.; Itoh, M.; Takamura, M.; Suganami, T.; Ogawa, Y.; et al. Development of a non−alcoholic steatohepatitis model with rapid accumulation of fibrosis, and its treatment using mesenchymal stem cells and their small extracellular vesicles. Regen. Ther. 2020, 14, 252–261. [Google Scholar] [CrossRef]
- Nakagami, H.; Kiomy Osako, M.; Nakagami, F.; Shimosato, T.; Minobe, N.; Moritani, T.; Shimamura, M.; Miyake, T.; Shimizu, H.; Takeya, Y.; et al. Prevention and regression of non−alcoholic steatohepatitis (NASH) in a rat model by metabosartan, telmisartan. Int. J. Mol. Med. 2010, 26, 477–481. [Google Scholar] [CrossRef]
- Ikewaki, N.; Levy, G.A.; Kurosawa, G.; Iwasaki, M.; Dedeepiya, V.D.; Vaddi, S.; Senthilkumar, R.; Preethy, S.; Abraham, S.J.K. Hepatoprotective Effects of Aureobasidium pullulans Derived β 1,3–1,6 Glucans in a Murine Model of Non−alcoholic Steatohepatitis. J. Clin. Exp. Hepatol. 2022, 12, 1428–1437. [Google Scholar] [CrossRef] [PubMed]
- Tomizawa, M.; Kawanabe, Y.; Shinozaki, F.; Sato, S.; Motoyoshi, Y.; Sugiyama, T.; Yamamoto, S.; Sueishi, M. Triglyceride is strongly associated with nonalcoholic fatty liver disease among markers of hyperlipidemia and diabetes. Biomed. Rep. 2014, 2, 633–636. [Google Scholar] [CrossRef]
- Alisi, A.; Carpino, G.; Oliveira, F.L.; Panera, N.; Nobili, V.; Gaudio, E. The Role of Tissue Macrophage−Mediated Inflammation on NAFLD Pathogenesis and Its Clinical Implications. Mediat. Inflamm. 2017, 2017, 8162421. [Google Scholar] [CrossRef] [PubMed]
- Hirsova, P.; Bamidele, A.O.; Wang, H.; Povero, D.; Revelo, X.S. Emerging Roles of T Cells in the Pathogenesis of Nonalcoholic Steatohepatitis and Hepatocellular Carcinoma. Front. Endocrinol. 2021, 12, 760860. [Google Scholar] [CrossRef]
- Barrow, F.; Khan, S.; Wang, H.; Revelo, X.S. The Emerging Role of B Cells in the Pathogenesis of NAFLD. Hepatology 2021, 74, 2277–2286. [Google Scholar] [CrossRef]
- Arelaki, S.; Koletsa, T.; Sinakos, E.; Papadopoulos, V.; Arvanitakis, K.; Skendros, P.; Akriviadis, E.; Ritis, K.; Germanidis, G.; Hytiroglou, P. Neutrophil extracellular traps enriched with IL−1β and IL−17A participate in the hepatic inflammatory process of patients with non−alcoholic steatohepatitis. Virchows Arch. 2022, 481, 455–465. [Google Scholar] [CrossRef]
- Guo, Z.; Fan, X.; Yao, J.; Tomlinson, S.; Yuan, G.; He, S. The role of complement in nonalcoholic fatty liver disease. Front. Immunol. 2022, 13. [Google Scholar] [CrossRef]
- Chen, T.S.; Arslan, F.; Yin, Y.; Tan, S.S.; Lai, R.C.; Choo, A.B.; Padmanabhan, J.; Lee, C.N.; de Kleijn, D.P.; Lim, S.K. Enabling a robust scalable manufacturing process for therapeutic exosomes through oncogenic immortalization of human ESC−derived MSCs. J. Transl. Med. 2011, 9, 47. [Google Scholar] [CrossRef] [PubMed]
- Lai, R.C.; Arslan, F.; Tan, S.S.; Tan, B.; Choo, A.; Lee, M.M.; Chen, T.S.; Teh, B.J.; Eng, J.K.; Sidik, H.; et al. Derivation and characterization of human fetal MSCs: An alternative cell source for large−scale production of cardioprotective microparticles. J. Mol. Cell Cardiol. 2010, 48, 1215–1224. [Google Scholar] [CrossRef] [PubMed]
- Sze, S.K.; de Kleijn, D.P.; Lai, R.C.; Tan, E.K.W.; Zhao, H.; Yeo, K.S.; Low, T.Y.; Lian, Q.; Lee, C.N.; Mitchell, W. Elucidating the secretion proteome of human embryonic stem cell−derived mesenchymal stem cells. Mol. Cell Proteom. 2007, 6, 1680–1689. [Google Scholar] [CrossRef] [PubMed]
- Hansen, H.H.; Feigh, M.; Veidal, S.S.; Rigbolt, K.T.; Vrang, N.; Fosgerau, K. Mouse models of nonalcoholic steatohepatitis in preclinical drug development. Drug Discov. Today 2017, 22, 1707–1718. [Google Scholar] [CrossRef]
- Savari, F.; Mard, S.A.; Badavi, M.; Rezaie, A.; Gharib−Naseri, M.K. A new method to induce nonalcoholic steatohepatitis (NASH) in mice. BMC Gastroenterol. 2019, 19, 125. [Google Scholar] [CrossRef] [PubMed]
- Fujii, M.; Shibazaki, Y.; Wakamatsu, K.; Honda, Y.; Kawauchi, Y.; Suzuki, K.; Arumugam, S.; Watanabe, K.; Ichida, T.; Asakura, H.; et al. A murine model for non−alcoholic steatohepatitis showing evidence of association between diabetes and hepatocellular carcinoma. Med. Mol. Morphol. 2013, 46, 141–152. [Google Scholar] [CrossRef]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp−Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- Roszer, T. Understanding the Mysterious M2 Macrophage through Activation Markers and Effector Mechanisms. Mediat. Inflamm. 2015, 2015, 816460. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Item | Extent | Score |
|---|---|---|
| Steatosis | Steatosis at 50−fold magnification | |
| <5% | 0 | |
| 5–33% | 1 | |
| >33–66% | 2 | |
| >66% | 3 | |
| Lobular inflammation | Estimation of inflammatory foci | |
| No foci | 0 | |
| <2 foci/200× | 1 | |
| 2–4 foci/200× | 2 | |
| >4 foci/200× | 3 | |
| Ballooning | Estimation of number of ballooning cells | |
| None | 0 | |
| Few balloon cells | 1 | |
| Many cells/prominent ballooning | 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, B.; Zhang, B.; Lai, R.C.; Sim, W.K.; Lam, K.P.; Lim, S.K. MSC−sEV Treatment Polarizes Pro−Fibrotic M2 Macrophages without Exacerbating Liver Fibrosis in NASH. Int. J. Mol. Sci. 2023, 24, 8092. https://doi.org/10.3390/ijms24098092
Zhang B, Zhang B, Lai RC, Sim WK, Lam KP, Lim SK. MSC−sEV Treatment Polarizes Pro−Fibrotic M2 Macrophages without Exacerbating Liver Fibrosis in NASH. International Journal of Molecular Sciences. 2023; 24(9):8092. https://doi.org/10.3390/ijms24098092
Chicago/Turabian StyleZhang, Bin, Biyan Zhang, Ruenn Chai Lai, Wei Kian Sim, Kong Peng Lam, and Sai Kiang Lim. 2023. "MSC−sEV Treatment Polarizes Pro−Fibrotic M2 Macrophages without Exacerbating Liver Fibrosis in NASH" International Journal of Molecular Sciences 24, no. 9: 8092. https://doi.org/10.3390/ijms24098092