
Anti-Tuberculosis Potential of OJT008 against Active and Multi-Drug-Resistant Mycobacterium Tuberculosis: In Silico and In Vitro Inhibition of Methionine Aminopeptidase
Abstract
:1. Introduction
2. Results and Discussion
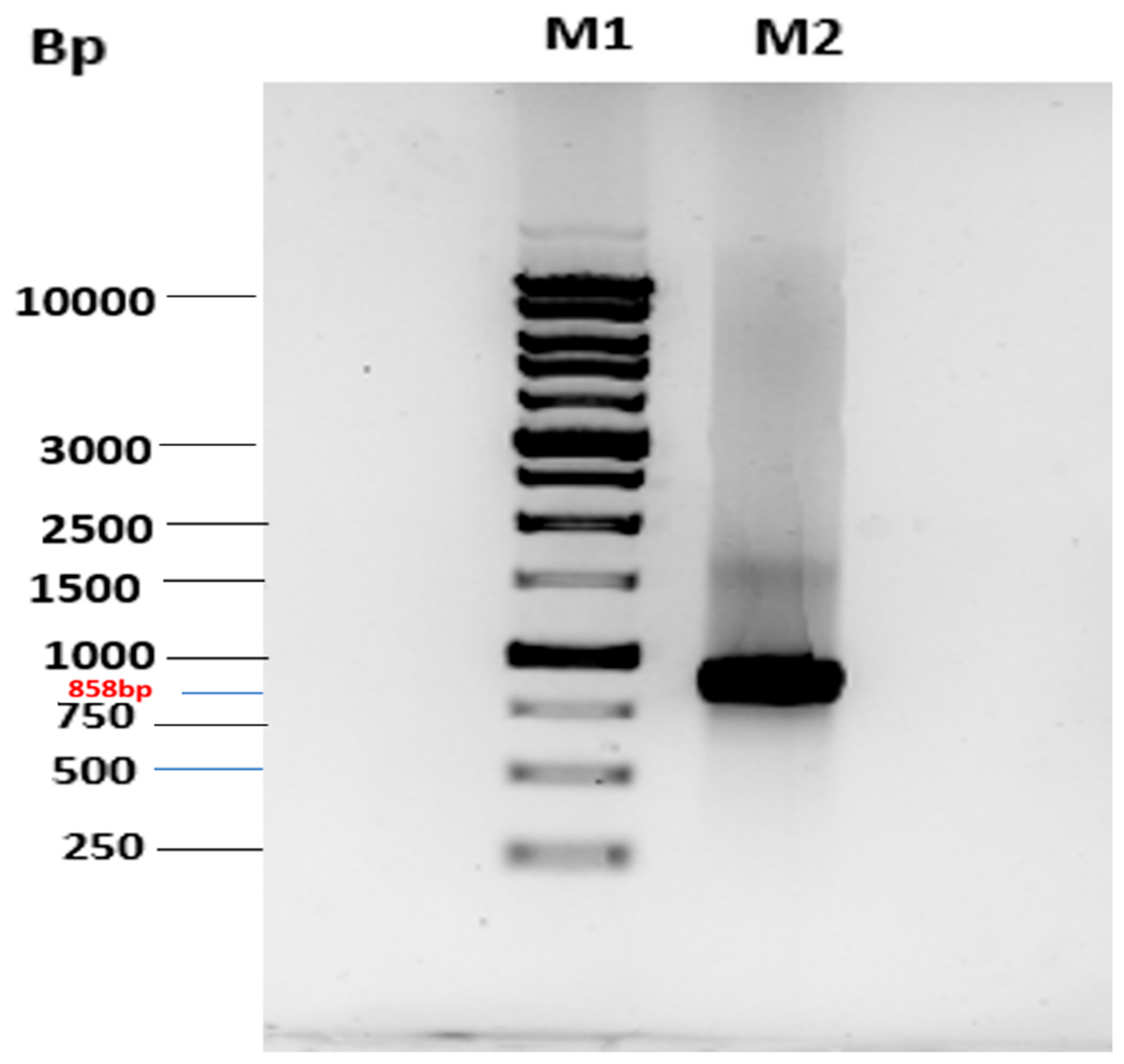
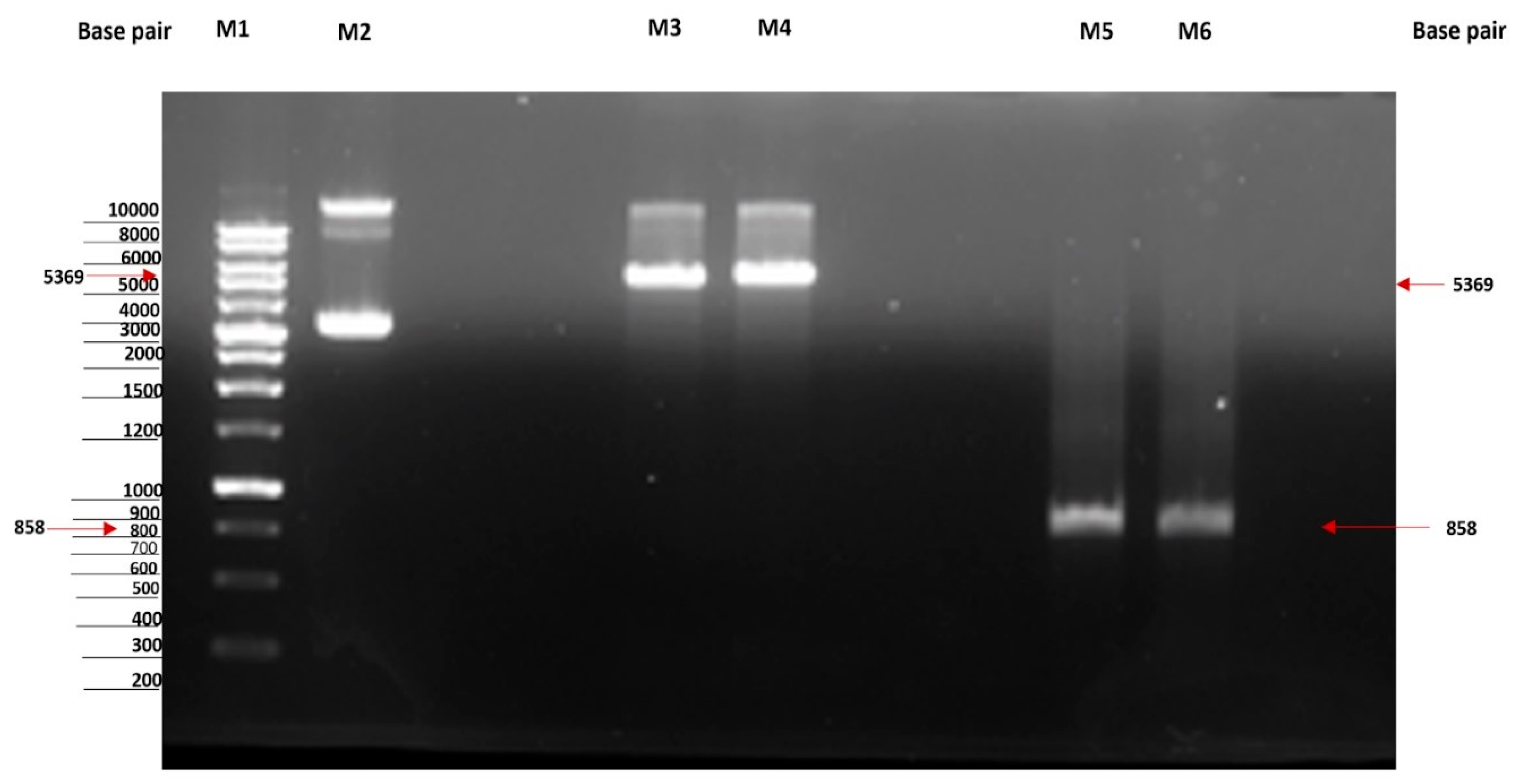
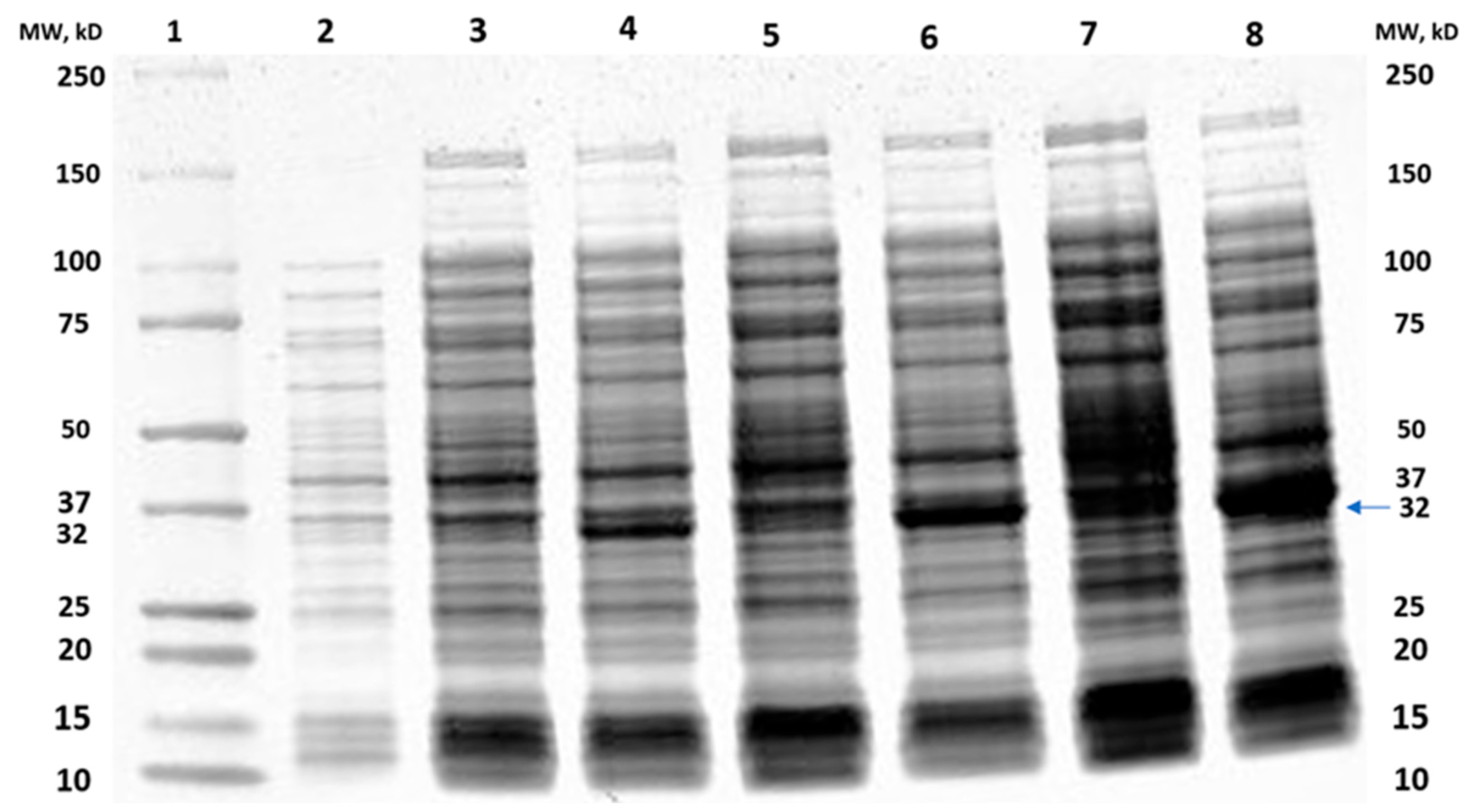
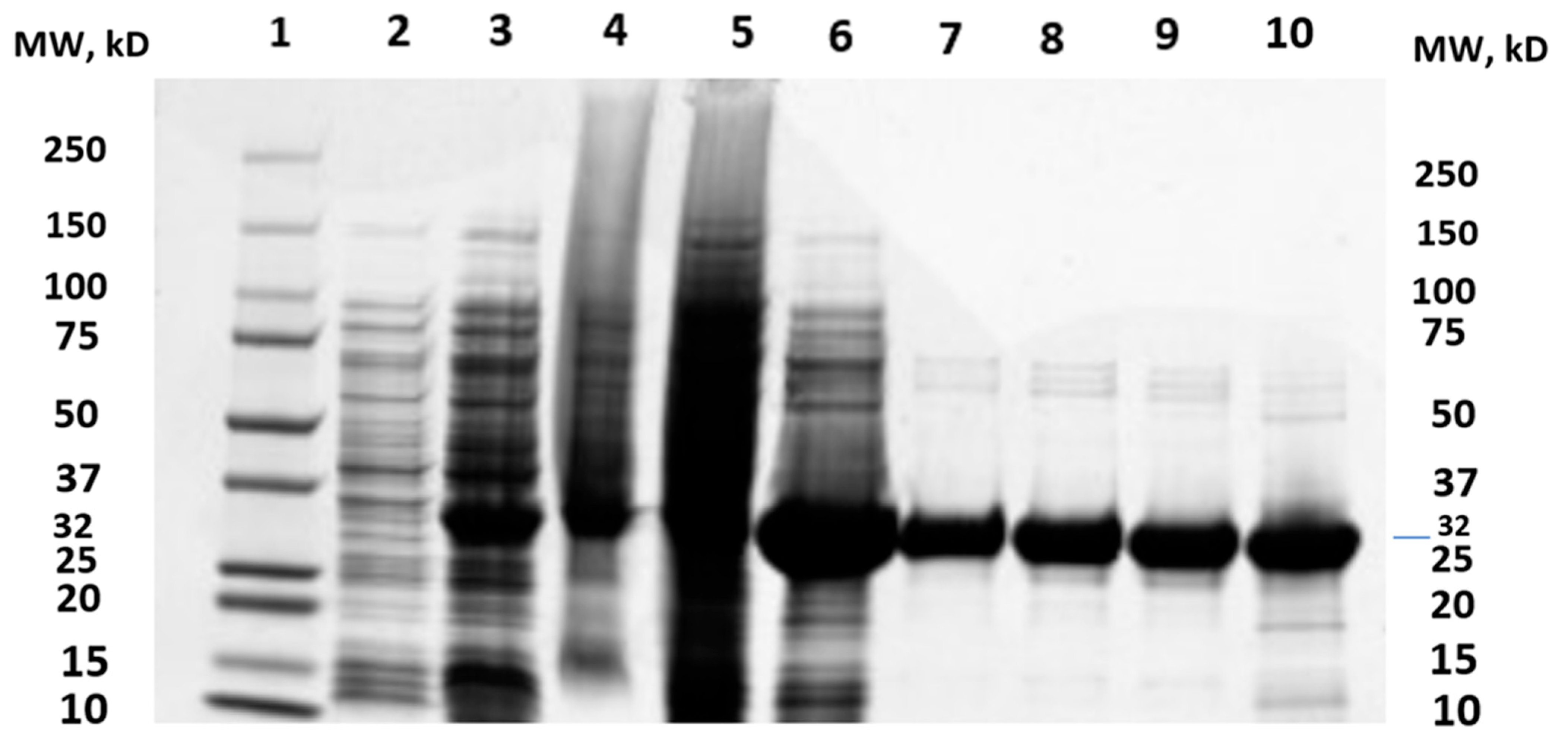
2.1. Cloning of MtMetAP1c and Gene Purification and Characterization of MtMetAP1c
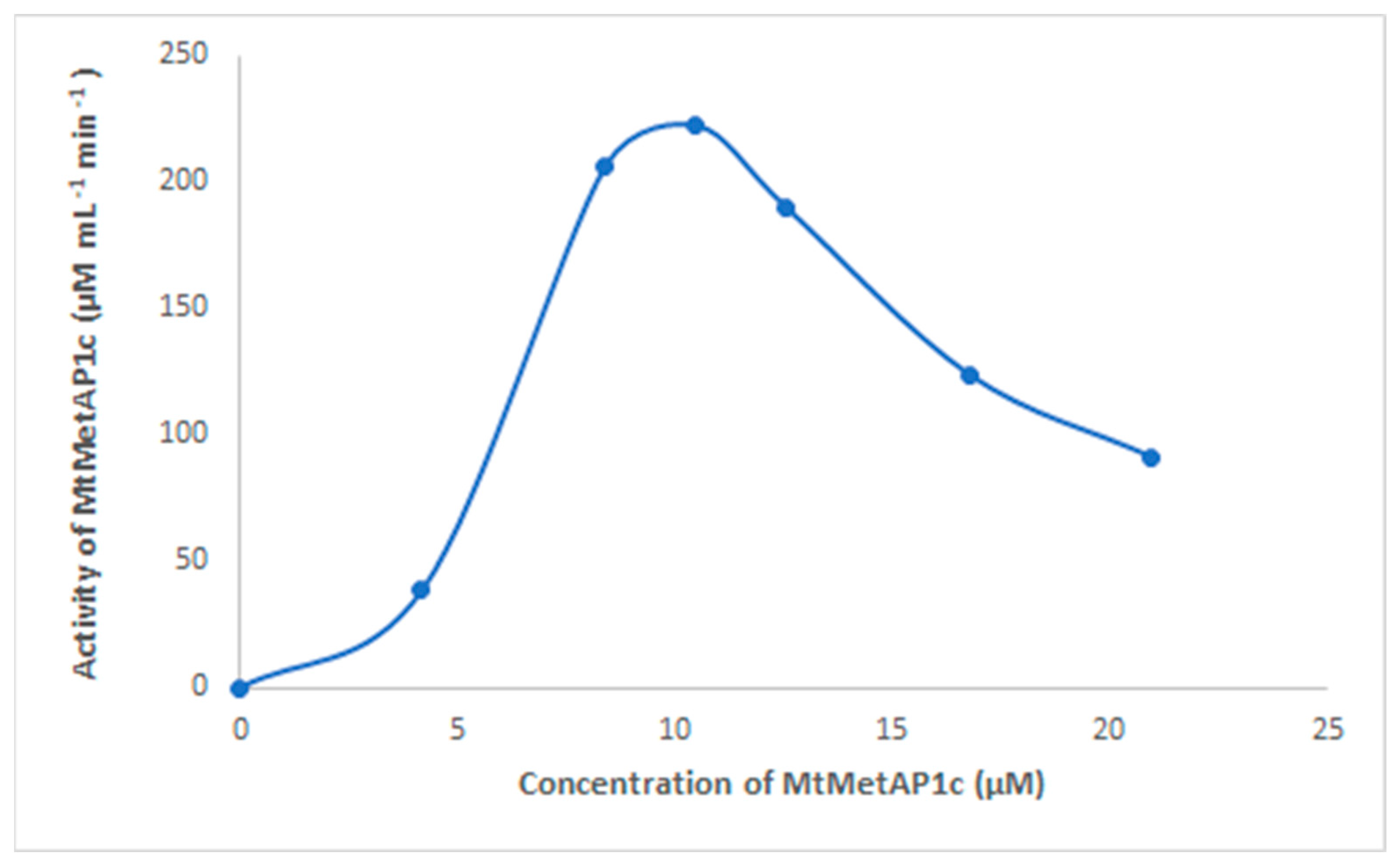
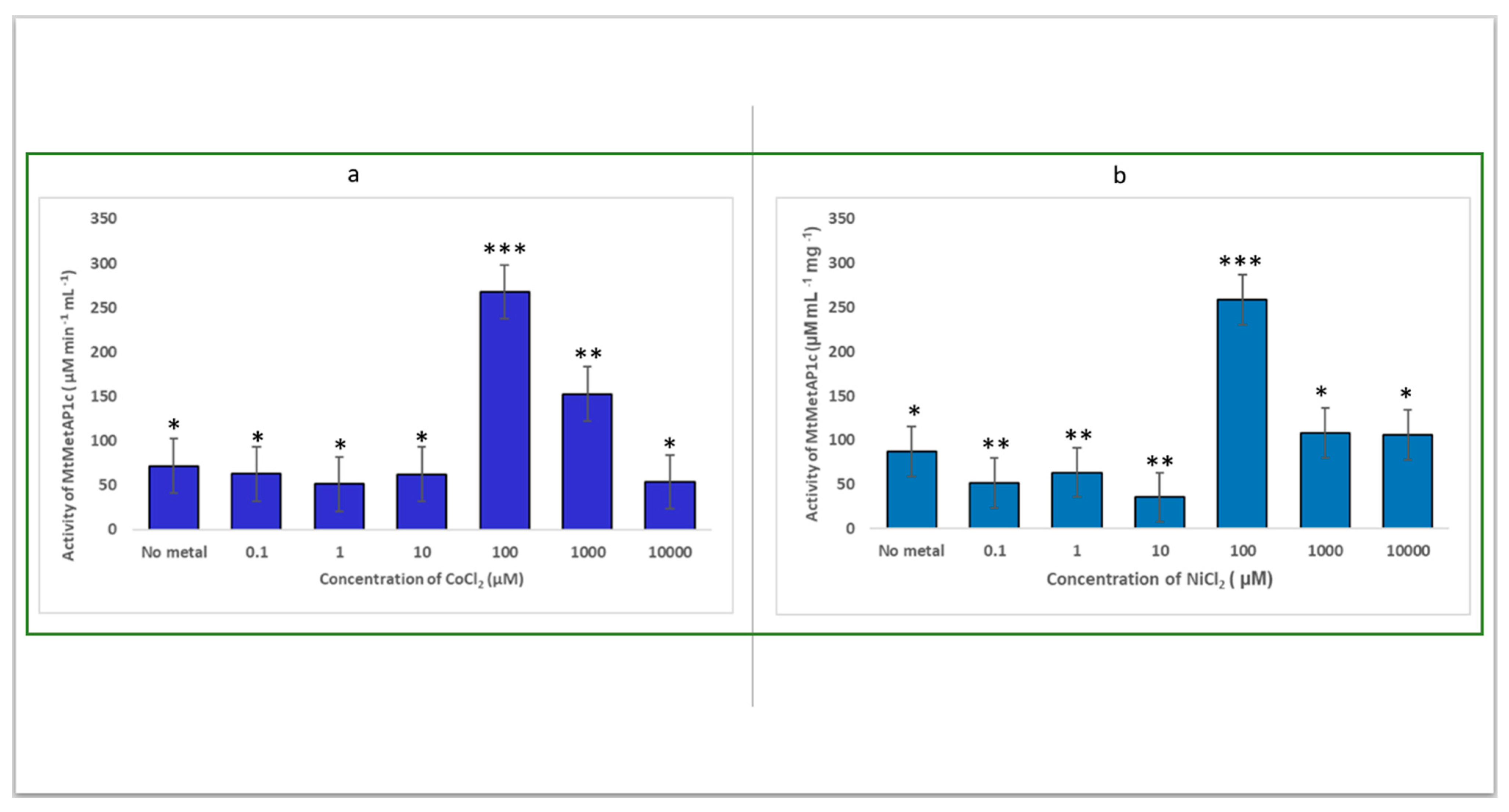
2.2. Determination of the Catalytic Effect of the Divalent Metal of Recombinant MtMetAP1c Activity
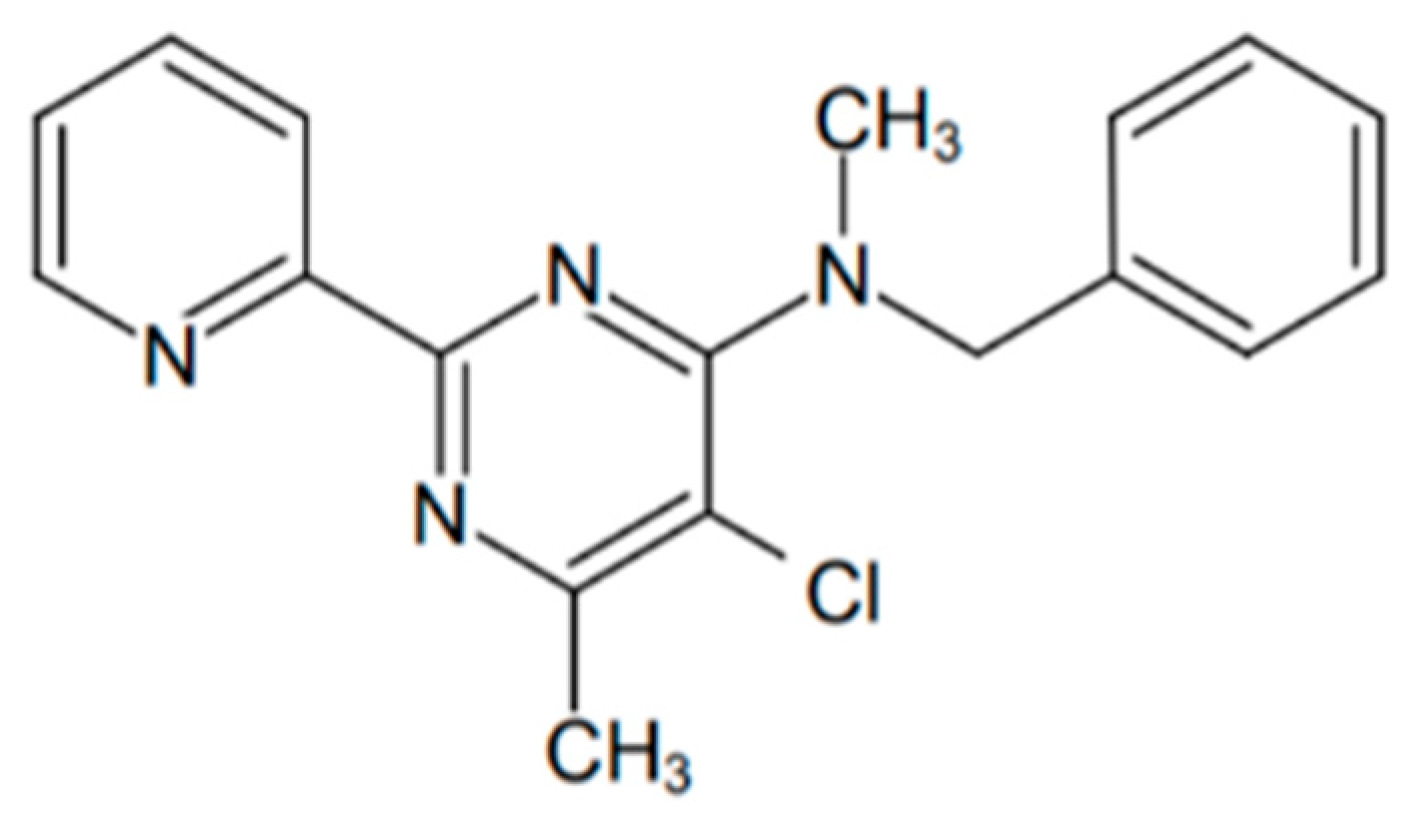
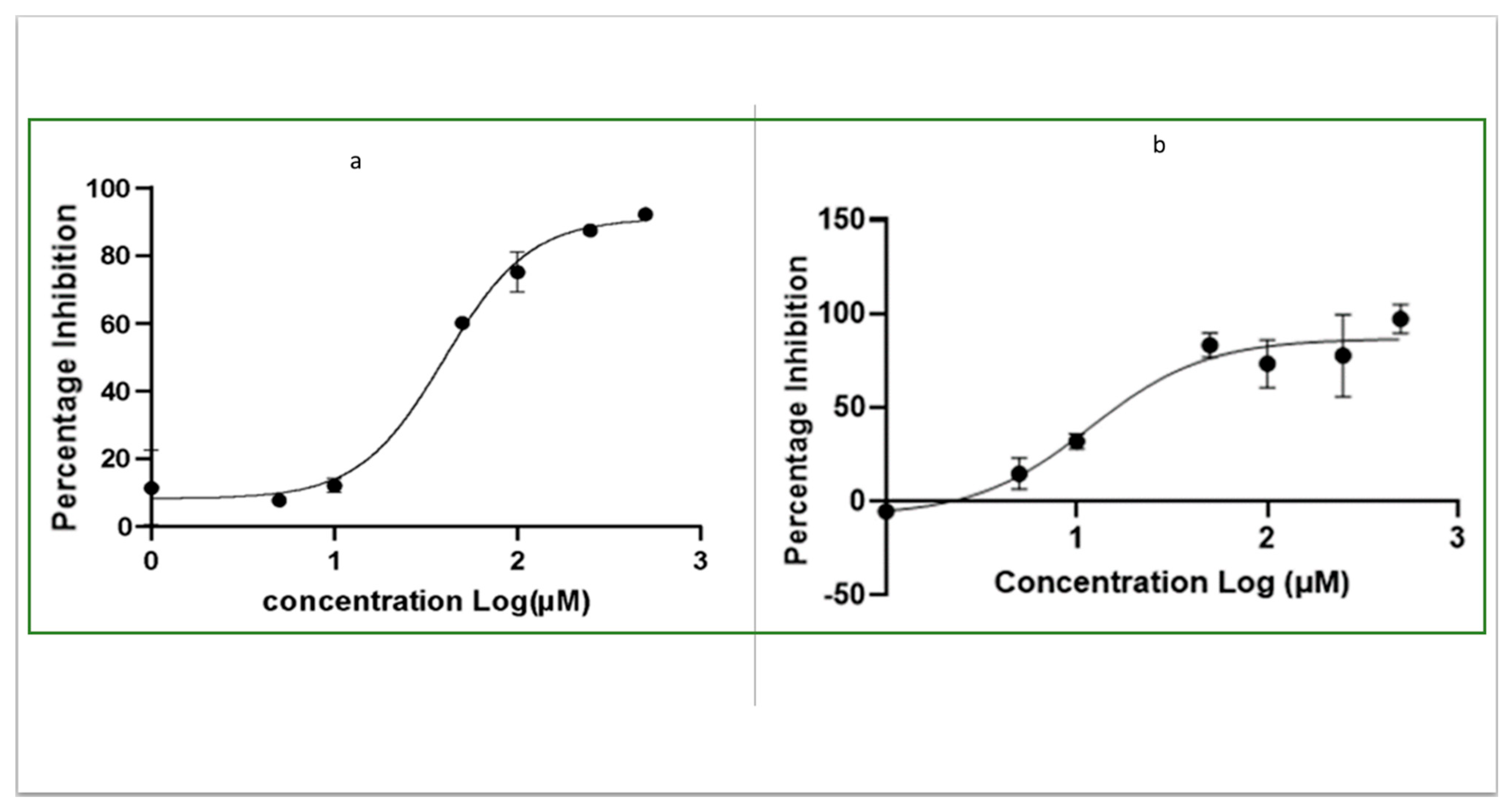
2.3. Determination of IC50 of OJT008 for MtMetAP1c Inhibition and Minimum Inhibitory Concentrations (MICs) of OJT008 in Replicating and Multi-Drug-Resistant Mtb
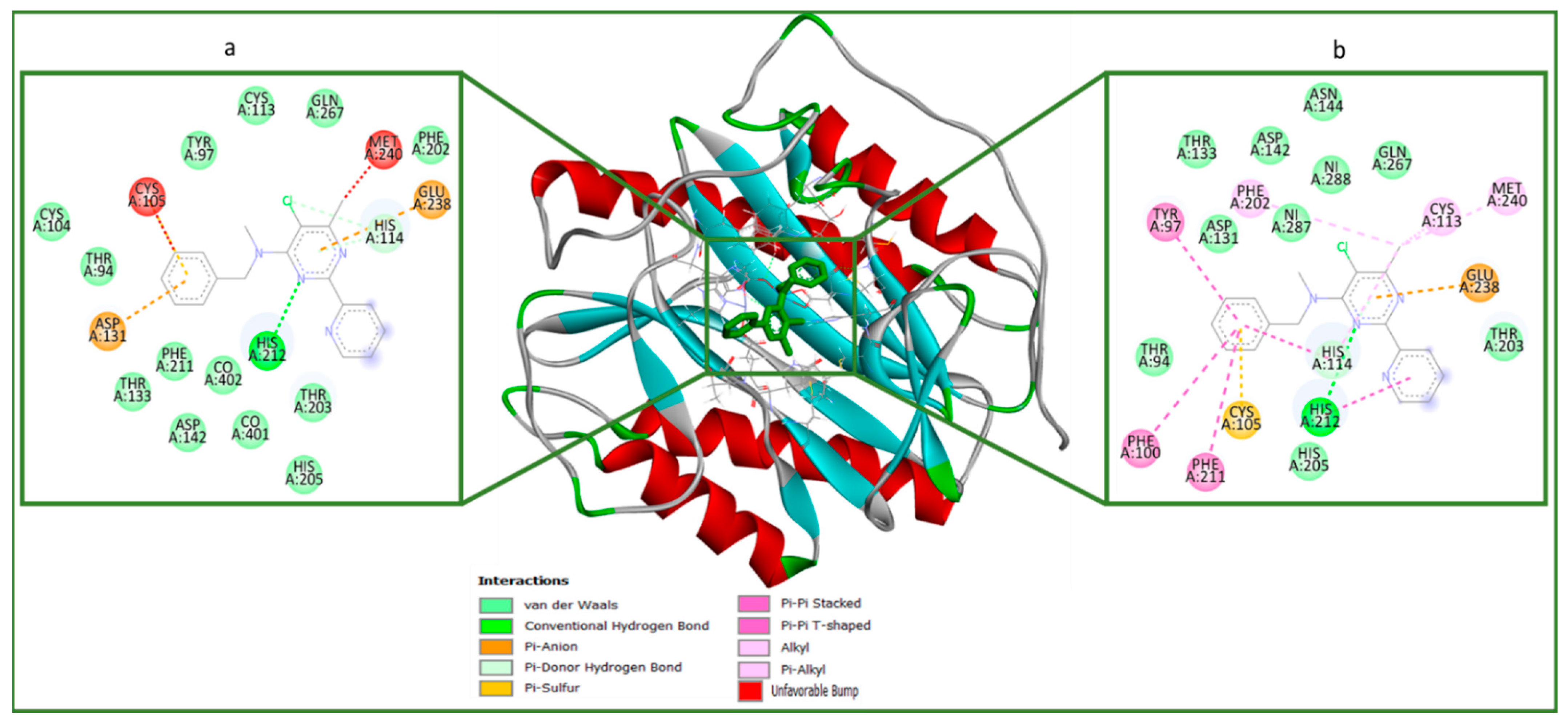
2.4. Molecular Docking (In Silico) Analysis
3. Materials and Method
3.1. Materials
3.2. Method
Cloning of MtMetAP1c Gene and Expression of MtMetAP1c Protein
3.3. Determination of the Catalytic Effect of the Divalent Metal of Recombinant MtMetAP1c Activity
3.4. Determination of IC50 of OJT008 for MtMetAP1c Inhibition
3.5. Determination of Minimum Inhibitory Concentration of OJT008 in Replicating Mtb
3.6. Determination of Minimum Inhibitory Concentration of OJT008 in Multi-Drug-Resistant Mtb
3.7. Molecular Docking
MtMetAP1c Acquisition and Preparation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sakula, A.; Robert, K. Centenary of the discovery of the tubercle bacillus. Thorax 1882, 4, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.; Neumann, C. Streptococcus agalactiae. Int. J. Syst. Bacteriol. 1896, 30, 367. [Google Scholar]
- World Health Organization. Global Tuberculosis Report 2022; World Health Organization: Geneva, Switzerland, 2022; Licence: CC BY-NC-SA 3.0 IGO. [Google Scholar]
- World Health Organization. World Health Organization Global Tuberculosis Report 2020; World Health Organization: Geneva, Switzerland, 2020; Available online: https://www.who.int/teams/global-tuberculosis-programme/tb-reports (accessed on 21 April 2021)ISBN 978-92-4-001313-1.
- Fauci, A.S. Multidrug-Resistant and Extensively Drug-Resistant Tuberculosis: The National Institute of Allergy and Infectious Diseases Research Agenda and Recommendations for Priority Research. J. Infect. Dis. 2008, 197, 1493–1498. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.T.; Brosch, R.; Parkhill, J.; Garnier, T.; Churcher, C.; Harris, D.; Gordon, S.V.; Eiglmeier, K.; Gas, S.; Barry, C.E. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998, 393, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Vernon, A.; Fielding, K.; Savic, R.; Dodd, L.; Nahid, P. The importance of adherence in tuberculosis treatment clinical trials and its relevance in explanatory and pragmatic trials. PLoS Med. 2019, 16, e1002884. [Google Scholar] [CrossRef]
- Olaleye, O.; Raghunand, T.R.; Bhat, S.; He, J.; Tyagi, S.; Lamichhane, G.; Gu, P.; Zhou, J.; Zhang, Y.; Grosset, J.; et al. Methionine aminopeptidases from Mycobacterium tuberculosis as novel antimycobacterial targets. Chem. Biol. 2010, 17, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.Y.; McGary, E.C.; Chang, S. Methionine aminopeptidase gene of Escherichia coli is essential for cell growth. J. Bacteriol. 1989, 171, 4071–4072. [Google Scholar] [CrossRef] [PubMed]
- Lowther, W.T.; Matthews, B.W. Structure and function of the methionine aminopeptidases. Biochim. Biophys. Acta 2000, 1477, 157–167. [Google Scholar] [CrossRef]
- Giglione, C.; Boularot, A.; Meinnel, T. Protein N-terminal methionine excision. Cell Mol. Life Sci. 2004, 61, 1455–1474. [Google Scholar] [CrossRef]
- Mark, D.V.; Peter, B.S.; John, F.H. Methionine in and out of proteins: Targets for drug design. Curr. Med. Chem. 2002, 9, 385–409. [Google Scholar]
- Sherman, F.J.W.; Stewart, S.T. Methionine or not methionine at the beginning of a protein. Bioessays 1985, 3, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Solbiati, J.A.; Chapman-Smith, A.; Miller, J.L.; Miller, C.G.; Cronan, J.E., Jr. Processing of the N termini of nascent polypeptide chains requires deformylation prior to methionine removal. J. Mol. Biol. 1999, 290, 607–614. [Google Scholar] [CrossRef]
- Travis, R.H.; Phumvadee, W.; Bart, L.S.; Timothy, J.H. Advances in bacterial methionine aminopeptidase inhibition. Curr. Top. Med. Chem. 2016, 16, 397–414. [Google Scholar]
- Carmela, G.; Olivier, V.; Thierry, M. Control of protein life-span by N-terminal methionine excision. EMBO J. 2003, 22, 13–23. [Google Scholar]
- Jieyi, W.; George, S.S.; Pingping, L.; Megumi, K.; Chang, P.; David, A.E.; Andrew, S.; Jennifer, B.; Rick, L.; Jack, H. Physiologically relevant metal cofactor for methionine aminopeptidase-2 is manganese. Biochemistry 2003, 42, 5035–5042. [Google Scholar]
- Sergio, C.C.; Wen-Long, W.; Qi-Zhuang, Y. FE(II) is the native cofactor for Escherichia coli methionine aminopeptidase. J. Biol. Chem. 2008, 283, 26879–26885. [Google Scholar]
- Arfin, S.M.; Kendall, R.L.; Hall, L.; Weaver, L.H.; Stewart, A.E.; Matthews, B.W.; Bradshaw, R.A. Eukaryotic methionyl aminopeptidases: Two classes of cobalt-dependent enzymes. Proc. Natl. Acad. Sci. USA 1995, 92, 7714–7718. [Google Scholar] [CrossRef]
- Addlagatta, A.; Quillin, M.L.; Omotoso, O.; Liu, J.O.; Matthews, B.W. Identification of an SH3-binding motif in a new class of methionine aminopeptidases from Mycobacterium tuberculosis suggests a mode of interaction with the ribosome. Biochemistry 2005, 44, 7166–7174. [Google Scholar] [CrossRef]
- Bala, S.C.; Haque, N.; Pillalamarri, V.; Reddi, R.; Kashyap, R.; Marapaka, A.K.; Addlagatta, A. Discovery of a new class of type 1 methionine aminopeptidases that have relaxed substrate specificity. Int. J. Biol. Macromol. 2019, 129, 523–529. [Google Scholar] [CrossRef]
- Pavitra, K.; Monica, M.; Sangaralingam, K.; Pradip, K.C. Amino-terminal extension present in the methionine aminopeptidase type 1c of Mycobacterium tuberculosis is indispensable for its activity. BMC Biochem. 2011, 12, 35. [Google Scholar]
- Miller, C.G.; Kukral, A.M.; Miller, J.L.; Movva, N.R. pepM is an essential gene in Salmonella typhimurium. J. Bacteriol. 1989, 171, 5215–5217. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, S.; Hu, Z.; Zhang, L.; Wang, H. Expression and characterization of two functional methionine aminopeptidases from Mycobacterium tuberculosis H37Rv. Curr. Microbiol. 2009, 59, 520–525. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro-Guimarães, M.L.; Pessolani, M.C. Comparative genomics of mycobacterial proteases. Microb Pathog. 2007, 43, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Griffin, J.E.; Gawronski, J.D.; DeJesus, M.A.; Ioerger, T.R.; Akerley, B.J.; Sassetti, C.M. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 2011, 7, e1002251. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.P.; Chai, S.C.; Ye, Q.Z. Catalysis and inhibition of Mycobacterium tuberculosis methionine aminopeptidase. J. Med. Chem. 2010, 53, 1329. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.P.; Ye, Q.Z. Expression and characterization of Mycobacterium tuberculosis methionine aminopeptidase type 1a. Bioorg. Med. Chem. Lett. 2010, 20, 2776–2779. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.P.; Yuan, X.H.; Yuan, H.; Wang, W.L.; Wan, B.; Franzblau, S.G.; Ye, Q.Z. Inhibition of Mycobacterium tuberculosis methionine aminopeptidases by bengamide derivatives. Chem. Med. Chem. 2011, 6, 1041–1048. [Google Scholar] [CrossRef]
- Miriam, V.; Patrick, S.; Tali-Haviv, R.; Pradip, K.C.; Bernhard, G.; Daniel, B. MapB Protein is the Essential Methionine Aminopeptidase in Mycobacterium tuberculosis. Cells 2019, 8, 393. [Google Scholar]
- Rodriguez, F.; John, S.F.; Iniguez, E.; Montalvo, S.; Michael, K.; White, L.; Liang, D.; Olaleye, O.A.; Maldonado, R.A. In Vitro and In Vivo Characterization of Potent Antileishmanial Methionine Aminopeptidase 1 Inhibitors. Antimicrob Agents Chemother. 2020, 64, e01422-19. [Google Scholar] [CrossRef]
- D’souza, V.M.; Holz, R.C. The methionyl aminopeptidase from Escherichia coli can function as an iron (II) enzyme. Biochemistry 1999, 38, 11079–11085. [Google Scholar]
- Walker, K.W.; Bradshaw, R.A. Yeast methionine aminopeptidase I can utilize either Zn2+ or Co2+ as a cofactor: A case of mistaken identity? Protein Sci. 1998, 7, 2684–2687. [Google Scholar] [CrossRef] [PubMed]
- Todd, W.L.; Brian, W.M. Metalloaminopeptidases: Common functional themes in disparate structural surroundings. Chem. Rev. 2002, 102, 4581–4608. [Google Scholar]
- Shode, F.O.; Idowu, A.; Uhomoibhi, O.J.; Sabiu, S. Repurposing drugs and identification of inhibitors of integral proteins (spike protein and main protease) of SARS-CoV-2. J. Biomolecul. Struct. Dynam. 2021, 40, 6587–6602. [Google Scholar] [CrossRef]
- Idowu, K.; Ramharack, P.; Nlooto, M.; Gordon, M. Molecular dynamic mechanism(s) of inhibition of bioactive antiviral phytochemical compounds targeting cytochrome P450 3A4 and P-glycoprotein. J. Biomolec. Struct. Dyn. 2020, 40, 1037–1047. [Google Scholar]
- Sinyani, A.; Idowu, K.; Shunmugam, L.; Kumalo, H.M.; Rene, K. A molecular dynamics perspective into estrogen receptor inhibition by selective flavonoids as alternative therapeutic options. J. Biomol. Struct. Dyn. 2022, 41, 4093–4105. [Google Scholar] [CrossRef]
- Sabiu, S.; Idowu, K. An insight on the nature of biochemical interactions between glycyrrhizin, myricetin and CYP3A4 isoform. J. Food Biochem. 2021, 2, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Addlagatta, A.; Hu, X.; Liu, J.O.; Matthews, B.W. Structural Basis for the Functional Differences between Type I and Type II Human Methionine Aminopeptidases. Biochemistry 2005, 44, 14741–14749. [Google Scholar] [CrossRef]
- Zhou, Y.; Guo, X.C.; Yi, T.; Yoshimoto, T.; Pei, D. Two continuous spectrophotometric assays for methionine aminopeptidase. Anal. Biochem. 2000, 280, 159–165. [Google Scholar] [CrossRef]
- Olaleye, O.; Raghunand, T.R.; Bhat, S.; Chong, C.; Gu, P.; Zhou, J.; Zhang, Y.; Bishai, W.R.; Liu, J.O. Characterization of clioquinol and analogues as novel inhibitors of methionine aminopeptidases from Mycobacterium tuberculosis. Tuberculosis 2011, 91, S61–S65. [Google Scholar] [CrossRef]
- Yang, Z.; Lasker, K.; Schneidman-Duhovny, D.; Webb, B.; Huang, C.C.; Pettersen, E.F.; Goddard, T.D.; Meng, E.C.; Sali, A.; Ferrin, T.E. UCSF chimera, MODELLER, and IMP: An integrated modeling system. J. Struct. Biol. 2012, 179, 269–278. [Google Scholar] [CrossRef]
- Uhomoibhi, J.O.; Shode, F.O.; Kehinde, I.A.; Sabiu, S. Molecular modelling identification of phytocompounds from selected African botanicals as promising therapeutics against druggable human host cell targets of SARS-CoV-2. J. Mol. Graph. Model. 2022, 114, 108185. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeerschd, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef]
- Idowu, A.K.; Egbeyemi, A.; Kaur, M.; Onyenaka, C.; Adebusuyi, T.; Olaleye, O.A. Inhibitory mechanism of Clioquinol and Its Derivatives at the Exopeptidase site of Human Angiotensin-Converting Enzyme-2 and Receptor Binding Domain of SARS-CoV-2 Viral Spike protein. J. Biomol. Str. Dyn. 2022, 41, 2992–3001. [Google Scholar]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | IC50 (µM) | |
|---|---|---|
| MtMetAP1c + 100 µM CoCl2 | MtMetAP1c + 100 µM NiCl2 | |
| 0JT008 | 11.81 ± 1.30 | 40.12 ± 1.12 |
| Inhibitor | MIC (µg/mL) | |
|---|---|---|
| Replicating Mtb (CDC 1551) | Multi-Drug-Resistant Mtb (HN 3409) | |
| Isoniazid | 0.25 | ND |
| Kanamycin | ND | 5.00 |
| 0JT008 | <0.063 | <0.063 |
| Co-Enzyme | ||
|---|---|---|
| MtMetAP1c + CoCl2 | MtMetAP1c + Ni Cl2 | |
| Docking Scores (Kcal/mol) | −6.52 | −7.01 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Onyenaka, C.; Idowu, K.A.; Ha, N.P.; Graviss, E.A.; Olaleye, O.A. Anti-Tuberculosis Potential of OJT008 against Active and Multi-Drug-Resistant Mycobacterium Tuberculosis: In Silico and In Vitro Inhibition of Methionine Aminopeptidase. Int. J. Mol. Sci. 2023, 24, 17142. https://doi.org/10.3390/ijms242417142
Onyenaka C, Idowu KA, Ha NP, Graviss EA, Olaleye OA. Anti-Tuberculosis Potential of OJT008 against Active and Multi-Drug-Resistant Mycobacterium Tuberculosis: In Silico and In Vitro Inhibition of Methionine Aminopeptidase. International Journal of Molecular Sciences. 2023; 24(24):17142. https://doi.org/10.3390/ijms242417142
Chicago/Turabian StyleOnyenaka, Collins, Kehinde A. Idowu, Ngan P. Ha, Edward A. Graviss, and Omonike A. Olaleye. 2023. "Anti-Tuberculosis Potential of OJT008 against Active and Multi-Drug-Resistant Mycobacterium Tuberculosis: In Silico and In Vitro Inhibition of Methionine Aminopeptidase" International Journal of Molecular Sciences 24, no. 24: 17142. https://doi.org/10.3390/ijms242417142