
Impact of Impaired Kidney Function on Arrhythmia-Promoting Cardiac Ion Channel Regulation

1
Department for Internal Medicine II, University Medical Center Regensburg, 93053 Regensburg, Germany
2
Institute of Physiology, University of Regensburg, 93053 Regensburg, Germany
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2023, 24(18), 14198; https://doi.org/10.3390/ijms241814198
Submission received: 31 August 2023
/
Revised: 14 September 2023
/
Accepted: 15 September 2023
/
Published: 17 September 2023
(This article belongs to the Special Issue New Insights into Cardiac Ion Channel Regulation 3.0)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Chronic kidney disease (CKD) is associated with a significantly increased risk of cardiovascular events and sudden cardiac death. Although arrhythmias are one of the most common causes of sudden cardiac death in CKD patients, the molecular mechanisms involved in the development of arrhythmias are still poorly understood. In this narrative review, therefore, we summarize the current knowledge on the regulation of cardiac ion channels that contribute to arrhythmia in CKD. We do this by first explaining the excitation–contraction coupling, outlining current translational research approaches, then explaining the main characteristics in CKD patients, such as abnormalities in electrolytes and pH, activation of the autonomic nervous system, and the renin–angiotensin–aldosterone system, as well as current evidence for proarrhythmic properties of uremic toxins. Finally, we discuss the substance class of sodium–glucose co-transporter 2 inhibitors (SGLT2i) on their potential to modify cardiac channel regulation in CKD and, therefore, as a treatment option for arrhythmias.
1. Introduction
Chronic kidney disease (CKD) is defined as a glomerular filtration rate (GFR) < 60 mL/min/1.73 m2 or kidney damage, which can be represented by an albumin-to-creatnine ratio >30 mg/g for more than 3 months [1]. Impaired kidney function increases the prevalence of cardiovascular complications and mortality, especially for arrhythmias. The prevalence and severity of arrhythmias increase with declining kidney function. In patients with end-stage renal disease (ESRD), sudden cardiac death (SCD) accounts for between 40 and 60% of all deaths [2,3,4]. However, the cellular mechanisms of arrhythmia in chronic kidney disease are not fully understood. In this review, we briefly summarize the current knowledge of cardiac ion channel regulation contributing to arrhythmia in chronic kidney disease. First, we briefly describe the most important channels in cardiomyocytes that contribute to excitation-contraction coupling. Then, as summarized in Figure 1, we discuss the impact of changes in electrolytes, pH homeostasis, autonomous nerve system, and renin–angiotensin–aldosterone regulation on ion channel function in CKD patients. Finally, we discuss SGLT2 inhibitors as an evolving strategy for the treatment of arrhythmias in CKD patients.
Excitation-Contraction Coupling in Healthy Cardiomyocytes
Cardiac excitation–contraction coupling (ECC) is based on the activation and inactivation of different ion channels, which causes a calcium-dependent contraction. The excitation is initiated by Na+ influx via voltage-gated fast Na+ channels (NaV1.5), which depolarize the membrane up to +20 mV (phase 0). The positive membrane potential induces the fast inactivation of the NaV1.5 channels and the activation of transient outward K+ channels (KV1.4, KV4.2, KV4.3). This transient outward K current (Ito) results in an early repolarization (phase 1), which is followed by a plateau phase (phase 2). The plateau phase describes a balance between the small K+ current flux and the Ca2+ inward current (ICa) by slow-activated, voltage-gated L-type Ca2+ channels (CaV1.2) working in tandem with the late sodium current (Late INa). This balance can be easily disturbed even by small currents that destabilize the plateau membrane potential, causing arrhythmias. In phase 3, Ca2+ channels progressively reduce their conductance, and slow K+ channels (KV7.1 and Kir2.x channels) activate, which results in the repolarization of the membrane potential [5].
During the cardiac action potential, Ca2+ entry triggers ryanodine receptor (RyR2) opening and, therefore, Ca2+ release from the sarcoplasmic reticulum (SR). Increasing the cytoplasmic Ca2+ level allows Ca2+ to bind to the myofilament protein troponin C, which then results in contraction during systole. During diastole, SR Ca2+-ATPase (SERCA2a) and Na+/Ca2+ exchange (NCX) remove the vast majority of the cytosolic Ca2+, and a tiny fraction of Ca2+ can be removed from the cytosol by the sarcolemmal Ca2+-ATPase and mitochondrial Ca2+-uniporter [5].
Ventricular arrhythmias are the result of electric instability in cardiomyocytes caused by cell membrane hyperexcitability, altered repolarization, or disturbed conduction of the electrical wavefront across the myocardium [6]. There are two basic mechanisms by which arrhythmias occur in the heart: re-entry arrhythmia and triggered activity. Re-entry occurs when an excitation spreads around an obstacle and hits tissue that has been previously excited. The obstacle can be a structural change, such as an infarct scar or a functionally altered tissue. Functional limitations of the conduction velocity or conduction block can result, for example, from a decrease in the conductivity of the cell–cell contact (e.g., Connexin 43) at the intercalated discs or prolonged recovery time, facilitating life-threatening re-entry arrhythmias [7,8].
Triggered activity can be caused by an imbalance of ion currents in a single cardiomyocyte. Depending on the mechanism of occurrence, these imbalances can be classified as early after-depolarizations (EADs) or delayed after-depolarizations (DADs).
EADs occur during the action potential and are often the result of disproportional fast depolarization or attenuated repolarization. In particular, the plateau phase is vulnerable to EADs, as even small imbalances between Ca2+ influx and K+ efflux can cause a new depolarization with SR calcium release. This can lead to life-threatening tachyarrhythmia if it occurs in several cardiomyocytes simultaneously.
DADs are a product of cytosolic or SR calcium overload. SR calcium overload leads to the spontaneous release of calcium sparks in the cytosol, which then activate the sodium–calcium exchanger (NCX) to remove Ca2+ from the cytosol. The NCX imports three Na+ for each Ca2+ excreted, causing a net import of positive charge, and it can thus depolarize the membrane potential. This can again trigger an AP, which leads to a renewed activation of the calcium influx into the cardiomyocytes, which under pathological conditions can trigger a ventricular arrhythmia [6].
2. Translational Approaches to Arrhythmia in CKD
As kidney function declines, the risk of atrial and ventricular arrhythmias increases. A cross-sectional analysis from the Atherosclerosis Risk in Community study (ARIC) showed in >2000 patients with GFR < 60 mL/min/m2 within 2 weeks of cardiac monitoring a high prevalence of ectopic beats in >90% of the patients, ventricular tachycardia (30.2%), and atrial fibrillation (7.4%) [9]. In patients with end-stage renal disease (ESRD) on dialysis, the prevalence of ventricular arrhythmia increased, as 25.4% of patients experienced asystole or bradyarrhythmia, and 23% of the patients had tachyarrhythmia [10]. The bulk of clinical data does not allow differentiation of the specific signaling pathways of cardiac ion channel regulation.
Therefore, in vivo models in rodents and in vitro models with isolated cardiomyocytes have been established. The most common in vivo model for CKD being used to investigate arrhythmia is subtotal nephrectomy (SNx) [11]. The abrupt reduction in the number of glomeruli is accompanied by an accumulation of urinary substances, electrolyte and pH imbalances, and compensatory mechanisms, such as the activation of the renin–angiotensin–aldosterone system (RAAS) and autonomic nervous system (ANS). Further models include adenine feeding, resulting in tubulointerstitial fibrosis, and genetic modification, such as Col4a3 deficiency, leading to progressive glomerulonephritis with microhematuria and proteinuria [12]. Another approach is the incubation of isolated cardiomyocytes with uremic plasma [13,14,15]. Although these approaches showed seminal changes in action potential and dysbalanced calcium handling, explaining the arrhythmogenicity in chronic kidney failure, more sophisticated approaches are needed to distinguish which mechanism or metabolite is involved in ion channel regulation [16] (Figure 2). Therefore, in the following sections, we present the regulation of cardiac ion channels based on four specific changes in patients and models of CKD.
3. Characteristics of Chronic Kidney Disease
3.1. Electrolyte and pH Abnormalities in CKD
The kidneys play an essential role in the regulation of serum potassium levels and pH. In CKD, the capacity of potassium and H+ secretion declines with reduced glomerular filtration rate, resulting in electrolyte and pH imbalance [17]. The most common abnormalities in CKD are hyperkalemia and metabolic acidosis [17].
During hyperkalemia, the physiological gradient of high intracellular and low extracellular potassium is attenuated. According to the Nernst equation, the resting membrane potential depolarizes due to a reduction in the K+ gradient. Depolarization of the membrane potential initially leads to increased excitability due to the membrane potential being closer to the activation potential of cardiac voltage-gated NaV1.5 channels. However, with further depolarization of the membrane potential, the steady-state inactivation of NaV1.5 channels also increases, leading to fewer channels contributing to AP upstroke during every depolarization [18]. This loss of peak Na+ channel function reduces the amplitude of the Na+ peak and conduction velocity in the myocardium, which is electrocardiographically represented by a broad QRS complex in hyperkalemia [19]. Reduced conduction velocity increases the risk that the action potential wave front will excite already recovered tissue, again leading to re-entry arrhythmias. Additionally, the action potential duration of the myocardium decreases in hyperkalemia. This effect can be explained by an increased repolarization reserve, as hyperkalemia facilitates the inward K+ current (IK1), one of the main repolarization currents in phase 3 of the AP. This can be clinically observed as alternans in T-waves on ECG in hyperkalemia [20]. Enhanced IK1 can also induce post-repolarization refractoriness, which prolongs the period of inexcitability of cardiomyocytes [21]. Spatially discordant excitability of cardiomyocytes and APD alternans in amplitude and duration result in conduction velocity alternans that predispose to re-entry arrhythmias [18,22].
The effect on cardiac ion channels in metabolic acidosis is complex. Protons directly inhibit the sodium conductance of the NaV1.5 in cardiomyocytes by changing the electrostatic potential of the channel with the protonation of carboxyl residues. This results in a reduced peak sodium current with reduced conduction velocity [23]. In CKD patients with acidosis, this effect was clinically manifested as the prolongation of the QT interval on ECG, which was corrected by the treatment of the acidosis with bicarbonate administration. Additionally, acidosis was found to destabilize the fast-inactivated state of NaV1.5 and increase persistent INa in isolated canine cardiomyocytes [24,25]. The decreased peak and persistent INa lead to prolonged repolarization, which can trigger calcium release as early after-depolarizations (EADs) [6].
Acidosis is known for reducing inotropy of cardiomyocytes [26,27]. This effect can be explained by molecular experiments in isolated rat ventricular cardiomyocytes that showed desensitized RyRs, resulting in decreased SR Ca2+ release combined with desensitized myofilaments. Additionally, experiments on isolated rat ventricular cardiomyocytes showed that systemic acidosis also reduced cytosolic pH. In these cells, acidosis stimulated acid extrusion via Na+/H+ exchange (NHE), which subsequently increased intracellular Na+ and thereby lowered Ca2+ efflux via NCX [28]. The combination of decreased SR Ca2+ release and low Ca2+ efflux results in SR Ca2+ accumulation, which favors spontaneous Ca2+ release (Ca2+ sparks) during diastole. Although the conductance of NCX is generally reduced by acidosis [29], due to the spatial proximity of NCX and RyRs, the RyRs Ca2+ spark temporarily activates the NCX. Since the NCX exchanges Ca2+ for Na+ in a 1:3 ratio, the activation results in an import of a positive charge with an increase in cytosolic Na+ [30], which can trigger a DAD and thereby initiate an arrhythmic AP [31]. In addition, acidosis can worsen myocardial conductance by inhibiting the conductance of gap junctions, such as connexin 43 [32], which promotes the development of malignant re-entry arrhythmias [33,34].
3.2. Sympathetic Nerve System Activation in CKD
The sympathetic nervous system is an important regulator of blood pressure and heart rhythm, and its force is crucial for appropriate kidney function [35]. Sympathetic innervation of the heart is mainly mediated by norepinephrine and circulating adrenaline targeting β-adrenergic receptors (β-AR). On a molecular level, the stimulation of adrenergic receptors enhances cyclic adenosine monophosphate (cAMP), which enhances the conductance of cardiac ion channels either directly or by phosphorylation by protein kinase A (PKA) [36]. The sympathetic nervous activity is counteracted by the parasympathetic nervous system, which is mainly mediated through acetylcholine binding to muscarinic receptors. While β-AR receptors are present throughout the entire myocardium, muscarinic receptors in the heart are essentially present in the atrium, sinoatrial, and AV node. The activation of muscarinic receptors results in nitric oxide (NO), which stimulates cGMP levels. Increased cGMP levels finally enhance phosphodiesterases that break down cAMP and thereby oppose the sympathetic stimulation of the atrium and conduction system of the heart [37].
In CKD, clinical data and rodent experiments have shown a sympathetic overdrive assessed by muscle sympathetic nerve activity and serum levels of norepinephrine [35,38,39]. Although the mechanisms are not fully elucidated yet, patients with CKD showed a significant increase in inflammation, with oxidative stress leading to a reduced bioavailability of NO [40]. Preclinical and clinical investigations identified a reduced level of endothelial NO as a potent central stimulator of the sympathetic nervous system [35,41]. Additionally, Converse et al. investigated the effect of bilateral nephrectomy in patients and found reduced sympathetic tone compared with patients with ESRD and healthy patients [39]. These findings suggest the direct stimulation of the sympathetic nervous system by the kidneys, which is supported by experiments with renal denervation, which decreased circulating epinephrine levels in a hypertensive pig model [42]. In CKD animal models of subtotal nephrectomy and adenine-induced CKD, renal denervation reduced atrial and ventricular arrhythmogenicity [43,44,45]. Zoccali et al. found that in a cohort of ESRD patients, the plasma level of epinephrine predicted the incidence of cardiovascular events, with arrhythmia being the second-most frequent event recorded [46]. These experiments highlight a direct link between CKD-induced sympathetic overdrive increasing the risk of arrhythmias, which raises the question of the influence of the sympathetic nervous system on the regulation of cardiac ion channels.
The activation of β-AR in cardiomyocytes increases via cAMP-dependent protein kinase A (PKA) phosphorylation of the L-type Ca channel CaV1.2 (LTCC), sarcoplasmic/endoplasmic reticulum Ca2+ ATPase 2 (SERCA2), and phospholamban [47]. This phosphorylation results in increased sarcoplasmic Ca2+ levels, which enhances the systolic Ca2+ transient and positive inotropy. Enhanced ICa would lead to a prolongation of the APD since the plateau phase of the AP is determined by the balance between Ca2+ influx and K+ efflux. First, the PKA-dependent phosphorylation of SERCA2 and PLN counterbalances the increased influx of calcium by increasing the SR Ca2+ reuptake [48]. Furthermore, Bennett et al. found that in guinea pig ventricular myocytes, β-adrenergic stimulation increased the delayed rectifying potassium current (IK), which counteracted the AP prolongation by ICa [49]. This resulted in an accelerated repolarization and thereby shortened the AP [50].
β-AR also triggers spontaneous diastolic Ca2+ release and thus increases cytoplasmic Ca2+ levels, which activates the Na+-Ca2+ exchanger (NCX) [51]. Increased NCX activity leads to Na+ accumulation in cardiomyocytes, which then depolarizes the membrane to the action potential threshold. The more positive the diastolic membrane potential, the more delayed after-depolarizations (DADs) occur, which makes the cardiomyocytes susceptible to atrial and ventricular arrhythmia [52,53,54]. Besides triggered activity, beta-stimulation in rats showed that connexin 43 was redistributed [55], which is known to promote electrical instability and life-threatening arrhythmias [56].
Impaired kidney function was found to reduce parasympathetic stimulation by impaired cardiac vagal stimulation via acetylcholine (ACh) on muscarinic receptors in hemodialysis patients [57]. Muscarinic stimulation leads to hyperpolarization and shortens the APD of the atrium. This happens mainly by reducing cAMP [37] in the atrium and sinoatrial node and increasing the K+ conductance of the G-protein-gated inwardly rectifying potassium (GIRK1 and GIRK4) channels, as measured in isolated guinea pig atrium myocytes with the patch clamp technique [58]. Impaired parasympathetic stimulation could thus explain the tendency toward atrial rhythm disturbances, such as atrial fibrillation, in CKD patients, but ventricular arrhythmias are also present in CKD [9]. Interestingly, Liang et al. localized GIRK1 and GIRK4 in mice and human ventricles with immunohistochemistry and demonstrated that the stimulation of ventricular myocytes with ACh significantly increased the action potential duration in wild-type mice, whereas these results were not observed in GIRK4 knockout mice [59]. Furthermore, the same authors showed that in a family with LongQT syndrome, which causes life-threatening ventricular arrhythmias, the gene coding for GIRK, KCNJ5, was mutated [60]. The authors concluded that GIRKs play an important role in the repolarization of ventricular cells, which may contribute, at least in part, to the development of arrhythmias in CKD patients [59].
3.3. The Renin–Angiotensin–Aldosterone System in CKD
Due to the reduced renal blood flow in CKD and sympathetic nerve stimulation, renin synthesis and secretion are stimulated and thereby activate the renin–angiotensin–aldosterone cascade [61,62]. Since the activation of the RAAS plays a central role in the progression of CKD and congestive heart failure, RAAS blockade is an essential drug target in their treatment.
Angiotensin II is known to regulate several ion channels in cardiomyocytes resulting in changes in the cardiac action potential. In isolated mice ventricular cardiomyocytes, angiotensin II amplified reactive oxygen species accumulation via NADPH oxidase 2 (NOX2), which activates Ca2+/Calmodulin-dependent protein kinase II (CaMKII). CaMKII is known as a central regulator in failing hearts that increases the risk of arrhythmia [63,64]. CaMKII activation resulted in increased peak INa and late INa current, which can cause APD prolongation and EADs [65]. Experimental inhibition of CaMKII in isolated rabbit cardiomyocytes incubated with ROS-generated H2O2 showed a reduction in EADs [66].
Also, in rat ventricular myocytes, angiotensin II and aldosterone both inhibited the Ca2+-independent transient outward K+ current (Ito), which resulted in action potential prolongation, which is known to be proarrhythmogenic [67,68].
While aldosterone increases L-type Ca2+ current in rat ventricular cells by upregulating transcription, the activity of CaV1.2 did not change with acute and chronic incubation of cardiomyocytes with aldosterone [69]. However, angiotensin II additionally seems to induce an increased ICa by increasing the activity of CaV1.2 [70,71,72]. One possible mechanism of the increased activity of the ICa current is the stimulation of the CaMKII. CaMKII facilitates the phosphorylation of LTCC, phospholamban (PLN) at Thr17, and ryanodine (RYR) at site S2814 [73,74]. These hyperphosphorylations increase spontaneous diastolic Ca2+ release, which again can trigger atrial and ventricular arrhythmias [65]. Therefore, the inhibition of CaMKII is a possible target to reduce arrhythmias in heart disease [75].
3.4. Uremic Toxins
With the progression of CKD and GFR decline, compounds accumulate in patients’ blood; under healthy conditions, they are filtered and excreted by the kidneys, the so-called uremic toxins. The large number of uremic toxins can be classified into several chemical groups by their molecular weight, origin, and molecular targets, and some of them are well-established as biomarkers in early-stage chronic kidney disease [76].
In vivo and in vitro experiments have aimed to characterize the impact of uremic conditions on cardiac electrophysiology and identify uremic toxins that have direct alterations of cardiac ion channels.
Indoxylsulfate (IS) is a metabolite of L-tryptophan that accumulates with a declining filtration rate [77]. In patients with CKD, IS has been independently associated with a prolonged QT interval, which increases the risk of arrhythmias [78,79]. On a molecular level, van Ham et al. conducted in vitro experiments with the incubation (48 h) of human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) and immortalized HEK293 cells with IS. They found decreased repolarizing potassium currents (IKr) and lowered protein levels of KV11.1, which subsequently resulted in prolonged action potential duration [80,81]. Furthermore, indoxyl sulfate decreased gap junction intercellular communication by downregulating connexin 43 in cultured neonatal rat cardiomyocytes, leading to increased spontaneous contractions [82]. Another uremic toxin, p-cresyl sulfate, also decreased the potassium channel KV1.2, which, in computational modeling, prolonged the action potential [81]. Chen et al. showed that in pulmonary veins isolated from rabbits, IS induced increased calcium leakage, resulting in an increased occurrence of delayed after-depolarizations [83].
Fibroblast growth factor 23 (FGF23) is a hormone for the homeostasis of phosphate and calcitriol synthesized in osteoblasts and osteocytes. In the early stages of CKD, FGF23 increases, with independent associations with atrial arrhythmias and ventricular hypertrophy both increasing mortality in clinical studies [84,85]. FGF-23 alters Ca2+ handling in cardiomyocytes via increased RyR2 and PLB phosphorylation at the PKA and CaMKII sites, which increases SR Ca2+ spark frequency. According to Navarro-Garcia et al., this Ca2+ mishandling induces arrhythmic events, as assessed in mice with electrocardiographic monitoring, with an increased rate of premature ventricular contractions (PVC) after intraperitoneal FGF-23 injection. Additionally, ex vivo FGF-23 perfusion of mice hearts showed a prolonged QT interval [86], and in vitro incubation of ventricular myocytes from mice with FGF-23 increased the number of EADs and DADs [12,87]. The signaling pathway of FGF-23 is mediated by the reduction of AMP-specific 3′,5′-cyclic phosphodiesterase 4B (PDE4B), which leads to cAMP-dependent phosphorylation of calcium RyR2 and PLB. Klotho, a proteohormone counteracting FGF-23, diminished calcium imbalance by reducing diastolic SR Ca2+ release and thereby proarrhythmic events [88].
3.5. SGLT2- Inhibitors: Novel Treatment Strategies for Arrhythmia in CKD?
SGLT2 inhibitors have fundamentally expanded heart failure and CKD therapy in recent years. Due to the previously known mechanism of action, the blockade of SGLT2 in the proximal tubule of the kidney with glycosuria, SGLT2 inhibitors were initially used for diabetes therapy. However, large studies in patients with heart failure independent of ejection fraction and in patients with both diabetic and non-diabetic chronic kidney disease showed a reduction in mortality and cardiovascular complications [89,90,91,92,93,94].
A meta-analysis of 34 clinical trials with more than 100,000 patients included showed a decrease in atrial and ventricular tachyarrhythmias by SGLT2 inhibitors [95,96,97,98]. However, because of the heterogenicity of the experimental groups, no statistical significance was found for a reduced risk of arrhythmias in CKD without HF [95]. Nonetheless, SGLT2 inhibitors were shown in clinical studies and experimental work to act both via the improvement of the characteristics of CKD listed above and directly on the myocardium.
In a meta-analysis of a total of 49,000 patients with type II diabetes, the SGLT2 inhibitor Empagliflozin significantly reduced the risk of severe hyperkalemia (≥6.0 mmol/L, hazard ratio (HR) 0.84, p < 0.001) in patients with an eGFR < 60 mL/min/m2 without increasing the risk of hypokalemia [99]. This mechanism can be explained by the increased natriuretic and diuretic effects of SGLT2 inhibitors [99,100]. Evidence from neurogenic hypertensive mice fed a high-fat diet showed that treatment with the SGLT2 inhibitor Dapagliflozin decreased norepinephrine levels in the same manner as pharmacological sympathetic nerve inhibition did [101]. Although there is no evidence in CKD that Empagliflozin directly inhibits RAAS [102,103], there are clinical data underlining the beneficial effect of the combination of both ACEi and SGLT2i, suggesting a synergistic effect [104].
Besides the indirect effects, there is evidence that SGLT2 inhibitors directly regulate cardiac ion channels (Figure 3). SGLT2 inhibitors are potent correctors of pathological action potential changes. In mouse models, in which the QT interval was prolonged by either myocardial infarction or amitriptyline administration, Empagliflozin reduced the QT interval and significantly reduced EADs [105]. In another mouse model with a prolonged QT time by the induction of metabolic syndrome, Jhuo et al. found that connexin 43 expression was reduced in the ventricle. SGLT2i treatment increased the expression of connexin 43, which was accompanied by a significant shortening of the QT interval, highlighting SGLT2i as a treatment option for malignant re-entry arrhythmias in CKD [7,106]. These findings are in line with data from Dos Santos et al., who demonstrated APD duration normalization by SGLT2i after hypoxia-induced APD prolongation in iPSC [107]. Increased late sodium current (INa) is a well-known mechanism of prolonged APD, with an increased risk of EADs [108] leading to ventricular arrhythmia. Indeed, Mustroph et al. showed that in mice and patients with reduced ejection fraction causing an increased late Na+ current, Empagliflozin inhibited the late Na+ current by inhibition of the CaMKII [109,110]. Additionally, Empagliflozin reduced spontaneous diastolic SR Ca2+ release by the inhibition of RyR2, which was accompanied by a reduction in CaMKII activity [111,112]. All in all, SGLT2i demonstrated antiarrhythmic properties by improving the above-mentioned four mechanisms, which occur in CKD. Additionally, SGLT2i inhibitors seem to inhibit phosphorylation by CaMKII, although the direct mechanisms have not been elucidated yet.
4. Conclusions
Chronic kidney disease is a common and complex disease in which the most frequent causes of death are cardiovascular events such as arrhythmias. In particular, changes in electrolytes, pH, the activation of the autonomic nervous system (ANS), the renin–angiotensin–aldosterone system (RAAS), and proarrhythmogenicity of uremic toxins contribute to arrhythmogenesis in chronic kidney disease. These adaptations to CKD, in addition to predisposing to re-entry tachycardia by conduction velocity alterations, for instance, through altered connexin 43 expression, lead to EADs via action potential prolongation and to DADs via spontaneous calcium release via the activation of the NCX. EADs and DADs increase the risk of arrhythmias. A central role in the regulation of the action potential forming and calcium handling is the activation of Ca2+/Calmodulin-dependent protein kinase II (CaMKII) by reactive oxygen species. This kinase is also the approach for SGLT2 inhibitors, which have already shown potential for reducing atrial and ventricular arrhythmias in experimental work. Further studies are necessary to clearly confirm SGLT2 inhibitors as antiarrhythmic agents.
Author Contributions
Conceptualization, F.S. (Frederick Sinha) and S.W.; writing—original draft preparation, F.S. (Frederick Sinha); writing—review and editing, F.S. (Frederick Sinha), F.S. (Frank Schweda), L.S.M. and S.W.; visualization, F.S. (Frederick Sinha) and S.W. All authors have read and agreed to the published version of the manuscript.
Funding
The authors’ work is supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation), project number 509149993 (TRR374).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Levey, A.S.; Eckardt, K.-U.; Tsukamoto, Y.; Levin, A.; Coresh, J.; Rossert, J.; Zeeuw, D.; de Hostetter, T.H.; Lameire, N.; Eknoyan, G. Definition and classification of chronic kidney disease: A position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int. 2005, 67, 2089–2100. [Google Scholar] [CrossRef] [PubMed]
- Franczyk-Skóra, B.; Gluba-Brzózka, A.; Wranicz, J.K.; Banach, M.; Olszewski, R.; Rysz, J. Sudden cardiac death in CKD patients. Int. Urol. Nephrol. 2015, 47, 971–982. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.J.; Foley, R.N.; Chavers, B.; Gilbertson, D.; Herzog, C.; Johansen, K.; Kasiske, B.; Kutner, N.; Liu, J.; St Peter, W.; et al. United States Renal Data System 2011 Annual Data Report: Atlas of chronic kidney disease & end-stage renal disease in the United States. Am. J. Kidney Dis. 2012, 59, e1–e420. [Google Scholar]
- Genovesi, S.; Boriani, G.; Covic, A.; Vernooij, R.W.M.; Combe, C.; Burlacu, A.; Davenport, A.; Kanbay, M.; Kirmizis, D.; Schneditz, D.; et al. Sudden cardiac death in dialysis patients: Different causes and management strategies. Nephrol. Dial. Transplant. 2021, 36, 396–405. [Google Scholar] [CrossRef]
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef]
- Wagner, S.; Maier, L.S.; Bers, D.M. Role of sodium and calcium dysregulation in tachyarrhythmias in sudden cardiac death. Circ. Res. 2015, 116, 1956–1970. [Google Scholar] [CrossRef]
- Andelova, K.; Egan Benova, T.; Szeiffova Bacova, B.; Sykora, M.; Prado, N.J.; Diez, E.R.; Hlivak, P.; Tribulova, N. Cardiac Connexin-43 Hemichannels and Pannexin1 Channels: Provocative Antiarrhythmic Targets. Int. J. Mol. Sci. 2020, 22, 260. [Google Scholar] [CrossRef]
- Danik, S.B.; Liu, F.; Zhang, J.; Suk, H.J.; Morley, G.E.; Fishman, G.I.; Gutstein, D.E. Modulation of cardiac gap junction expression and arrhythmic susceptibility. Circ. Res. 2004, 95, 1035–1041. [Google Scholar] [CrossRef]
- Kim, E.D.; Soliman, E.Z.; Coresh, J.; Matsushita, K.; Chen, L.Y. Two-Week Burden of Arrhythmias across CKD Severity in a Large Community-Based Cohort: The ARIC Study. J. Am. Soc. Nephrol. 2021, 32, 629–638. [Google Scholar] [CrossRef]
- Rautavaara, J.; Kerola, T.; Kaartinen, K.; Vilpakka, M.; Aitkoski, A.; Anttonen, O.; Ahvonen, J.; Koistinen, J.; Vääräniemi, K.; Miettinen, M.; et al. Asystole episodes and bradycardia in patients with end-stage renal disease. Nephrol. Dial. Transplant. 2022, 37, 575–583. [Google Scholar] [CrossRef]
- van Ham, W.B.; Cornelissen, C.M.; van Veen, T.A.B. Uremic toxins in chronic kidney disease highlight a fundamental gap in understanding their detrimental effects on cardiac electrophysiology and arrhythmogenesis. Acta Physiol. 2022, 236, e13888. [Google Scholar] [CrossRef]
- Touchberry, C.D.; Green, T.M.; Tchikrizov, V.; Mannix, J.E.; Mao, T.F.; Carney, B.W.; Girgis, M.; Vincent, R.J.; Wetmore, L.A.; Dawn, B.; et al. FGF23 is a novel regulator of intracellular calcium and cardiac contractility in addition to cardiac hypertrophy. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E863–E873. [Google Scholar] [CrossRef]
- Donohoe, P.; McMahon, A.C.; Walgama, O.V.; Bertaso, F.; Dockrell, M.E.; Cramp, H.A.; Mullen, A.M.; Shattock, M.J.; Hendry, B.M.; James, A.F. L-type calcium current of isolated rat cardiac myocytes in experimental uraemia. Nephrol. Dial. Transplant. 2000, 15, 791–798. [Google Scholar] [CrossRef]
- McMahon, A.C.; Vescovo, G.; Dalla Libera, L.; Wynne, D.G.; Fluck, R.J.; Harding, S.E.; Raine, A.E. Contractile dysfunction of isolated ventricular myocytes in experimental uraemia. Exp. Nephrol. 1996, 4, 144–150. [Google Scholar]
- McMahon, A.C.; Greenwald, S.E.; Dodd, S.M.; Hurst, M.J.; Raine, A.E.G. Prolonged calcium transients and myocardial remodelling in early experimental uraemia. Nephrol. Dial. Transplant. 2002, 17, 759–764. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.; Omran, E.; Periyasamy, S.M.; Nadoor, J.; Priyadarshi, A.; Willey, J.C.; Malhotra, D.; Xie, Z.; Shapiro, J.I. Effect of chronic renal failure on cardiac contractile function, calcium cycling, and gene expression of proteins important for calcium homeostasis in the rat. J. Am. Soc. Nephrol. 2003, 14, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Einhorn, L.M.; Zhan, M.; van Hsu, D.; Walker, L.D.; Moen, M.F.; Seliger, S.L.; Weir, M.R.; Fink, J.C. The frequency of hyperkalemia and its significance in chronic kidney disease. Arch. Intern. Med. 2009, 169, 1156–1162. [Google Scholar] [CrossRef]
- Weiss, J.N.; Qu, Z.; Shivkumar, K. Electrophysiology of Hypokalemia and Hyperkalemia. Circ. Arrhythm. Electrophysiol. 2017, 10, e004667. [Google Scholar] [CrossRef]
- Parham, W.A.; Mehdirad, A.A.; Biermann, K.M.; Fredman, C.S. Hyperkalemia Revisited. Tex. Heart Inst. J. 2006, 33, 40–47. [Google Scholar] [PubMed]
- Pastore, J.M.; Girouard, S.D.; Laurita, K.R.; Akar, F.G.; Rosenbaum, D.S. Mechanism linking T-wave alternans to the genesis of cardiac fibrillation. Circulation 1999, 99, 1385–1394. [Google Scholar] [CrossRef]
- Hegyi, B.; Chen-Izu, Y.; Izu, L.T.; Bányász, T. Altered K+ current profiles underlie cardiac action potential shortening in hyperkalemia and β-adrenergic stimulation. Can. J. Physiol. Pharmacol. 2019, 97, 773–780. [Google Scholar] [CrossRef]
- Han, B.; Trew, M.L.; Zgierski-Johnston, C.M. Cardiac Conduction Velocity, Remodeling and Arrhythmogenesis. Cells 2021, 10, 2923. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.K.; Peters, C.H.; Allard, C.R.; Claydon, T.W.; Ruben, P.C. Proton sensors in the pore domain of the cardiac voltage-gated sodium channel. J. Biol. Chem. 2013, 288, 4782–4791. [Google Scholar] [CrossRef] [PubMed]
- Murphy, L.; Renodin, D.; Antzelevitch, C.; Di Diego, J.M.; Cordeiro, J.M. Extracellular proton depression of peak and late Na+ current in the canine left ventricle. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H936–H944. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.K.; Peters, C.H.; Tolhurst, S.A.; Claydon, T.W.; Ruben, P.C. Extracellular proton modulation of the cardiac voltage-gated sodium channel, Nav1.5. Biophys. J. 2011, 101, 2147–2156. [Google Scholar] [CrossRef] [PubMed]
- Fabiato, A.; Fabiato, F. Effects of pH on the myofilaments and the sarcoplasmic reticulum of skinned cells from cardiace and skeletal muscles. J. Physiol. 1978, 276, 233–255. [Google Scholar] [CrossRef]
- Orchard, C.H.; Kentish, J.C. Effects of changes of pH on the contractile function of cardiac muscle. Am. J. Physiol. 1990, 258, C967–C981. [Google Scholar] [CrossRef]
- Choi, H.S.; Trafford, A.W.; Orchard, C.H.; Eisner, D.A. The effect of acidosis on systolic Ca2+ and sarcoplasmic reticulum calcium content in isolated rat ventricular myocytes. J. Physiol. 2000, 529 Pt 3, 661–668. [Google Scholar] [CrossRef]
- Li, L.; Watanabe, Y.; Matsuoka, I.; Kimura, J. Acidic preconditioning inhibits Na+/H+ and Na+/Ca2+ exchanger interaction via PKCepsilon in guinea-pig ventricular myocytes. J. Pharmacol. Sci. 2008, 107, 309–316. [Google Scholar] [CrossRef]
- Kreitmeier, K.G.; Tarnowski, D.; Nanadikar, M.S.; Baier, M.J.; Wagner, S.; Katschinski, D.M.; Maier, L.S.; Sag, C.M. CaMKIIδ Met281/282 oxidation is not required for recovery of calcium transients during acidosis. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H1199–H1212. [Google Scholar] [CrossRef]
- Bögeholz, N.; Pauls, P.; Bauer, B.K.; Schulte, J.S.; Dechering, D.G.; Frommeyer, G.; Kirchhefer, U.; Goldhaber, J.I.; Müller, F.U.; Eckardt, L.; et al. Suppression of Early and Late Afterdepolarizations by Heterozygous Knockout of the Na+/Ca2+ Exchanger in a Murine Model. Circ. Arrhythm. Electrophysiol. 2015, 8, 1210–1218. [Google Scholar] [CrossRef] [PubMed]
- Ek-Vitorín, J.F.; Calero, G.; Morley, G.E.; Coombs, W.; Taffet, S.M.; Delmar, M. PH regulation of connexin43: Molecular analysis of the gating particle. Biophys. J. 1996, 71, 1273–1284. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Sinovas, A.; Sánchez, J.A.; Valls-Lacalle, L.; Consegal, M.; Ferreira-González, I. Connexins in the Heart: Regulation, Function and Involvement in Cardiac Disease. Int. J. Mol. Sci. 2021, 22, 4413. [Google Scholar] [CrossRef]
- Salameh, A.; Dhein, S. Pharmacology of gap junctions. New pharmacological targets for treatment of arrhythmia, seizure and cancer? Biochim. Biophys. Acta 2005, 1719, 36–58. [Google Scholar] [CrossRef] [PubMed]
- Ewen, S.; Ukena, C.; Linz, D.; Schmieder, R.E.; Böhm, M.; Mahfoud, F. The sympathetic nervous system in chronic kidney disease. Curr. Hypertens. Rep. 2013, 15, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Cohn, J.N. Sympathetic nervous system activity and the heart. Am. J. Hypertens. 1989, 2, 353S–356S. [Google Scholar]
- Myers, R.W.; Pearlman, A.S.; Hyman, R.M.; Goldstein, R.A.; Kent, K.M.; Goldstein, R.E.; Epstein, S.E. Beneficial effects of vagal stimulation and bradycardia during experimental acute myocardial ischemia. Circulation 1974, 49, 943–947. [Google Scholar] [CrossRef]
- Koomans, H.A.; Blankestijn, P.J.; Joles, J.A. Sympathetic hyperactivity in chronic renal failure: A wake-up call. J. Am. Soc. Nephrol. 2004, 15, 524–537. [Google Scholar] [CrossRef]
- Converse, R.L.; Jacobsen, T.N.; Toto, R.D.; Jost, C.M.; Cosentino, F.; Fouad-Tarazi, F.; Victor, R.G. Sympathetic overactivity in patients with chronic renal failure. N. Engl. J. Med. 1992, 327, 1912–1918. [Google Scholar] [CrossRef]
- Baylis, C. Nitric oxide deficiency in chronic kidney disease. Am. J. Physiol. Renal Physiol. 2008, 294, F1–F9. [Google Scholar] [CrossRef]
- Grassi, G.; Seravalle, G.; Ghiadoni, L.; Tripepi, G.; Bruno, R.M.; Mancia, G.; Zoccali, C. Sympathetic nerve traffic and asymmetric dimethylarginine in chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2011, 6, 2620–2627. [Google Scholar] [CrossRef] [PubMed]
- Mahfoud, F.; Moon, L.B.; Pipenhagen, C.A.; Jensen, J.A.; Pathak, A.; Papademetriou, V.; Ewen, S.; Linz, D.; Böhm, M. Catheter-based radio-frequency renal nerve denervation lowers blood pressure in obese hypertensive swine model. J. Hypertens. 2016, 34, 1854–1862. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.-H.; Lo, L.-W.; Chou, Y.-H.; Lin, W.-L.; Tsai, T.-Y.; Cheng, W.-H.; Yamada, S.; Chen, S.-A. Renal denervation prevents myocardial structural remodeling and arrhythmogenicity in a chronic kidney disease rabbit model. Heart Rhythm. 2021, 18, 1596–1604. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Shi, L.; Cui, X.; Yu, Y.; Qi, T.; Chen, C.; Tang, X. Renal denervation decreases susceptibility of the heart to ventricular fibrillation in a canine model of chronic kidney disease. Exp. Physiol. 2017, 102, 1414–1423. [Google Scholar] [CrossRef] [PubMed]
- Hohl, M.; Selejan, S.-R.; Wintrich, J.; Lehnert, U.; Speer, T.; Schneider, C.; Mauz, M.; Markwirth, P.; Wong, D.W.L.; Boor, P.; et al. Renal Denervation Prevents Atrial Arrhythmogenic Substrate Development in CKD. Circ. Res. 2022, 130, 814–828. [Google Scholar] [CrossRef] [PubMed]
- Zoccali, C.; Mallamaci, F.; Parlongo, S.; Cutrupi, S.; Benedetto, F.A.; Tripepi, G.; Bonanno, G.; Rapisarda, F.; Fatuzzo, P.; Seminara, G.; et al. Plasma norepinephrine predicts survival and incident cardiovascular events in patients with end-stage renal disease. Circulation 2002, 105, 1354–1359. [Google Scholar] [CrossRef]
- Sperelakis, N. Regulation of calcium slow channels of heart by cyclic nucleotides and effects of ischemia. Adv. Pharmacol. 1994, 31, 1–24. [Google Scholar]
- Chu, G.; Lester, J.W.; Young, K.B.; Luo, W.; Zhai, J.; Kranias, E.G. A single site (Ser16) phosphorylation in phospholamban is sufficient in mediating its maximal cardiac responses to beta -agonists. J. Biol. Chem. 2000, 275, 38938–38943. [Google Scholar] [CrossRef]
- Bennett, P.B.; Begenisich, T.B. Catecholamines modulate the delayed rectifying potassium current (IK) in guinea pig ventricular myocytes. Pflugers Arch. 1987, 410, 217–219. [Google Scholar] [CrossRef]
- Taggart, P.; Sutton, P.; Chalabi, Z.; Boyett, M.R.; Simon, R.; Elliott, D.; Gill, J.S. Effect of adrenergic stimulation on action potential duration restitution in humans. Circulation 2003, 107, 285–289. [Google Scholar] [CrossRef]
- Gutierrez, D.A.; Fernandez-Tenorio, M.; Ogrodnik, J.; Niggli, E. NO-dependent CaMKII activation during β-adrenergic stimulation of cardiac muscle. Cardiovasc. Res. 2013, 100, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Myles, R.C.; Wang, L.; Kang, C.; Bers, D.M.; Ripplinger, C.M. Local β-adrenergic stimulation overcomes source-sink mismatch to generate focal arrhythmia. Circ. Res. 2012, 110, 1454–1464. [Google Scholar] [CrossRef] [PubMed]
- Herring, N.; Kalla, M.; Paterson, D.J. The autonomic nervous system and cardiac arrhythmias: Current concepts and emerging therapies. Nat. Rev. Cardiol. 2019, 16, 707–726. [Google Scholar] [CrossRef] [PubMed]
- Shiferaw, Y.; Aistrup, G.L.; Wasserstrom, J.A. Intracellular Ca2+ waves, afterdepolarizations, and triggered arrhythmias. Cardiovasc. Res. 2012, 95, 265–268. [Google Scholar] [CrossRef]
- Szeiffova Bacova, B.; Viczenczova, C.; Andelova, K.; Sykora, M.; Chaudagar, K.; Barancik, M.; Adamcova, M.; Knezl, V.; Egan Benova, T.; Weismann, P.; et al. Antiarrhythmic Effects of Melatonin and Omega-3 Are Linked with Protection of Myocardial Cx43 Topology and Suppression of Fibrosis in Catecholamine Stressed Normotensive and Hypertensive Rats. Antioxidants 2020, 9, 546. [Google Scholar] [CrossRef]
- Peters, N.S. New insights into myocardial arrhythmogenesis: Distribution of gap-junctional coupling in normal, ischaemic and hypertrophied human hearts. Clin. Sci. 1996, 90, 447–452. [Google Scholar] [CrossRef]
- Mircoli, L.; Rivera, R.; Bonforte, G.; Fedele, L.; Genovesi, S.; Surian, M.; Ferrari, A.U. Influence of left ventricular mass, uremia and hypertension on vagal tachycardic reserve. J. Hypertens. 2003, 21, 1547–1553. [Google Scholar] [CrossRef]
- Belardinelli, L.; Isenberg, G. Isolated atrial myocytes: Adenosine and acetylcholine increase potassium conductance. Am. J. Physiol. 1983, 244, H734–H737. [Google Scholar] [CrossRef]
- Liang, B.; Nissen, J.D.; Laursen, M.; Wang, X.; Skibsbye, L.; Hearing, M.C.; Andersen, M.N.; Rasmussen, H.B.; Wickman, K.; Grunnet, M.; et al. G-protein-coupled inward rectifier potassium current contributes to ventricular repolarization. Cardiovasc. Res. 2014, 101, 175–184. [Google Scholar] [CrossRef]
- Yang, Y.; Yang, Y.; Liang, B.; Liu, J.; Li, J.; Grunnet, M.; Olesen, S.-P.; Rasmussen, H.B.; Ellinor, P.T.; Gao, L.; et al. Identification of a Kir3.4 mutation in congenital long QT syndrome. Am. J. Hum. Genet. 2010, 86, 872–880. [Google Scholar] [CrossRef]
- Schweda, F.; Kurtz, A. Regulation of renin release by local and systemic factors. Rev. Physiol. Biochem. Pharmacol. 2011, 161, 1–44. [Google Scholar] [PubMed]
- Schweda, F.; Friis, U.; Wagner, C.; Skott, O.; Kurtz, A. Renin release. Physiology 2007, 22, 310–319. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, P.D.; Purohit, A.; Hund, T.J.; Anderson, M.E. Calmodulin-dependent protein kinase II: Linking heart failure and arrhythmias. Circ. Res. 2012, 110, 1661–1677. [Google Scholar] [CrossRef] [PubMed]
- Palomeque, J.; Rueda, O.V.; Sapia, L.; Valverde, C.A.; Salas, M.; Petroff, M.V.; Mattiazzi, A. Angiotensin II-induced oxidative stress resets the Ca2+ dependence of Ca2+-calmodulin protein kinase II and promotes a death pathway conserved across different species. Circ. Res. 2009, 105, 1204–1212. [Google Scholar] [CrossRef]
- Wagner, S.; Dantz, C.; Flebbe, H.; Azizian, A.; Sag, C.M.; Engels, S.; Möllencamp, J.; Dybkova, N.; Islam, T.; Shah, A.M.; et al. NADPH oxidase 2 mediates angiotensin II-dependent cellular arrhythmias via PKA and CaMKII. J. Mol. Cell Cardiol. 2014, 75, 206–215. [Google Scholar] [CrossRef]
- Zhao, Z.; Fefelova, N.; Shanmugam, M.; Bishara, P.; Babu, G.J.; Xie, L.-H. Angiotensin II induces afterdepolarizations via reactive oxygen species and calmodulin kinase II signaling. J. Mol. Cell Cardiol. 2011, 50, 128–136. [Google Scholar] [CrossRef]
- Caballero, R.; Gómez, R.; Moreno, I.; Nuñez, L.; González, T.; Arias, C.; Guizy, M.; Valenzuela, C.; Tamargo, J.; Delpón, E. Interaction of angiotensin II with the angiotensin type 2 receptor inhibits the cardiac transient outward potassium current. Cardiovasc. Res. 2004, 62, 86–95. [Google Scholar] [CrossRef]
- Matsuda, H.; Kurata, Y.; Imanishi, S.; Sato, R.; Shibamoto, T. Effects of angiotensin II on sustained outward currents in rat ventricular myocytes. Pflugers Arch. 2004, 448, 54–62. [Google Scholar] [CrossRef]
- Bénitah, J.P.; Vassort, G. Aldosterone upregulates Ca(2+) current in adult rat cardiomyocytes. Circ. Res. 1999, 85, 1139–1145. [Google Scholar] [CrossRef]
- Kashihara, T.; Nakada, T.; Kojima, K.; Takeshita, T.; Yamada, M. Angiotensin II activates CaV 1.2 Ca2+ channels through β-arrestin2 and casein kinase 2 in mouse immature cardiomyocytes. J. Physiol. 2017, 595, 4207–4225. [Google Scholar] [CrossRef]
- Mello WC, de. Intracellular angiotensin II regulates the inward calcium current in cardiac myocytes. Hypertension 1998, 32, 976–982. [Google Scholar] [CrossRef] [PubMed]
- Kaibara, M.; Mitarai, S.; Yano, K.; Kameyama, M. Involvement of Na(+)-H+ antiporter in regulation of L-type Ca2+ channel current by angiotensin II in rabbit ventricular myocytes. Circ. Res. 1994, 75, 1121–1125. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M.; Morotti, S. Ca(2+) current facilitation is CaMKII-dependent and has arrhythmogenic consequences. Front. Pharmacol. 2014, 5, 144. [Google Scholar] [CrossRef] [PubMed]
- Mattiazzi, A.; Kranias, E.G. The role of CaMKII regulation of phospholamban activity in heart disease. Front. Pharmacol. 2014, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Mustroph, J.; Neef, S.; Maier, L.S. CaMKII as a target for arrhythmia suppression. Pharmacol. Ther. 2017, 176, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; Smet, R.; de Glorieux, G.; Argilés, A.; Baurmeister, U.; Brunet, P.; Clark, W.; Cohen, G.; Deyn PP de Deppisch, R.; Descamps-Latscha, B.; et al. Review on uremic toxins: Classification, concentration, and interindividual variability. Kidney Int. 2003, 63, 1934–1943. [Google Scholar] [CrossRef]
- Berg, A.H.; Kumar, S.; Karumanchi, S.A. Indoxyl sulfate in uremia: An old idea with updated concepts. J. Clin. Invest. 2022, 132, e155860. [Google Scholar] [CrossRef]
- Aoki, K.; Teshima, Y.; Kondo, H.; Saito, S.; Fukui, A.; Fukunaga, N.; Nawata, T.; Shimada, T.; Takahashi, N.; Shibata, H. Role of Indoxyl Sulfate as a Predisposing Factor for Atrial Fibrillation in Renal Dysfunction. J. Am. Heart Assoc. 2015, 4, e002023. [Google Scholar] [CrossRef]
- Tang, W.-H.; Wang, C.-P.; Chung, F.-M.; Huang, L.L.H.; Yu, T.-H.; Hung, W.-C.; Lu, L.-F.; Chen, P.-Y.; Luo, C.-H.; Lee, K.-T.; et al. Uremic retention solute indoxyl sulfate level is associated with prolonged QTc interval in early CKD patients. PLoS ONE 2015, 10, e0119545. [Google Scholar] [CrossRef]
- van Ham, W.B.; Cornelissen, C.M.; Polyakova, E.; van der Voorn, S.M.; Ligtermoet, M.L.; Monshouwer-Kloots, J.; Vos, M.A.; Bossu, A.; van Rooij, E.; van der Heyden, M.A.G.; et al. Pro-Arrhythmic Potential of Accumulated Uremic Toxins Is Mediated via Vulnerability of Action Potential Repolarization. Int. J. Mol. Sci. 2023, 24, 5373. [Google Scholar] [CrossRef]
- Tsai, I.-T.; Hsu, C.-C.; Hung, W.-C.; Wang, C.-P.; Yu, T.-H.; Houng, J.-Y.; Lee, K.-T.; Tang, W.-H. The Arrhythmogenic Effect of Protein-Bound Uremic Toxin p-Cresylsulfate: An In Vitro Study. Acta Cardiol. Sin. 2019, 35, 641–648. [Google Scholar] [PubMed]
- Changchien, C.-Y.; Sung, M.-H.; Chang, H.-H.; Tsai, W.-C.; Peng, Y.-S.; Chen, Y. Uremic toxin indoxyl sulfate suppresses myocardial Cx43 assembly and expression via JNK activation. Chem. Biol. Interact. 2020, 319, 108979. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-T.; Chen, Y.-C.; Hsieh, M.-H.; Huang, S.-Y.; Kao, Y.-H.; Chen, Y.-A.; Lin, Y.-K.; Chen, S.-A.; Chen, Y.-J. The uremic toxin indoxyl sulfate increases pulmonary vein and atrial arrhythmogenesis. J. Cardiovasc. Electrophysiol. 2015, 26, 203–210. [Google Scholar] [CrossRef]
- Mehta, R.; Cai, X.; Lee, J.; Scialla, J.J.; Bansal, N.; Sondheimer, J.H.; Chen, J.; Hamm, L.L.; Ricardo, A.C.; Navaneethan, S.D.; et al. Association of Fibroblast Growth Factor 23 with Atrial Fibrillation in Chronic Kidney Disease, From the Chronic Renal Insufficiency Cohort Study. JAMA Cardiol. 2016, 1, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.B.; Ning, H.; Boer IH de Kestenbaum, B.; Lima, J.A.C.; Mehta, R.; Allen, N.B.; Shah, S.J.; Lloyd-Jones, D.M. Fibroblast Growth Factor 23 and Long-Term Cardiac Function: The Multi-Ethnic Study of Atherosclerosis. Circ. Cardiovasc. Imaging 2020, 13, e011925. [Google Scholar] [CrossRef] [PubMed]
- Graves, J.M.; Vallejo, J.A.; Hamill, C.S.; Wang, D.; Ahuja, R.; Patel, S.; Faul, C.; Wacker, M.J. Fibroblast growth factor 23 (FGF23) induces ventricular arrhythmias and prolongs QTc interval in mice in an FGF receptor 4-dependent manner. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H2283–H2294. [Google Scholar] [CrossRef]
- Navarro-García, J.A.; Rueda, A.; Romero-García, T.; Aceves-Ripoll, J.; Rodríguez-Sánchez, E.; González-Lafuente, L.; Zaragoza, C.; Fernández-Velasco, M.; Kuro, O.M.; Ruilope, L.M.; et al. Enhanced Klotho availability protects against cardiac dysfunction induced by uraemic cardiomyopathy by regulating Ca2+ handling. Br. J. Pharmacol. 2020, 177, 4701–4719. [Google Scholar] [CrossRef]
- Lindner, M.; Mehel, H.; David, A.; Leroy, C.; Burtin, M.; Friedlander, G.; Terzi, F.; Mika, D.; Fischmeister, R.; Prié, D. Fibroblast growth factor 23 decreases PDE4 expression in heart increasing the risk of cardiac arrhythmia; Klotho opposes these effects. Basic. Res. Cardiol. 2020, 115, 51. [Google Scholar] [CrossRef]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Solomon, S.D.; McMurray, J.J.V.; Claggett, B.; Boer RA de DeMets, D.; Hernandez, A.F.; Inzucchi, S.E.; Kosiborod, M.N.; Lam, C.S.P.; Martinez, F.; Shah, S.J.; et al. Dapagliflozin in Heart Failure with Mildly Reduced or Preserved Ejection Fraction. N. Engl. J. Med. 2022, 387, 1089–1098. [Google Scholar] [CrossRef]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and Renal Outcomes with Empagliflozin in Heart Failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef] [PubMed]
- Heerspink, H.J.L.; Stefánsson, B.V.; Correa-Rotter, R.; Chertow, G.M.; Greene, T.; Hou, F.-F.; Mann, J.F.E.; McMurray, J.J.V.; Lindberg, M.; Rossing, P.; et al. Dapagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2020, 383, 1436–1446. [Google Scholar] [CrossRef] [PubMed]
- Anker, S.D.; Butler, J.; Filippatos, G.; Ferreira, J.P.; Bocchi, E.; Böhm, M.; Brunner-La Rocca, H.-P.; Choi, D.-J.; Chopra, V.; Chuquiure-Valenzuela, E.; et al. Empagliflozin in Heart Failure with a Preserved Ejection Fraction. N. Engl. J. Med. 2021, 385, 1451–1461. [Google Scholar] [CrossRef]
- Aklilu, A.M.; Kumar, S.; Yamamoto, Y.; Moledina, D.G.; Sinha, F.; Testani, J.M.; Wilson, F.P. Outcomes Associated with Sodium-Glucose Cotransporter-2 Inhibitor Use in Acute Heart Failure Hospitalizations Complicated by Acute Kidney Injury. Kidney360 2023. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-L.; Lip, G.Y.H.; Feng, Q.; Fei, Y.; Tse, Y.-K.; Wu, M.-Z.; Ren, Q.-W.; Tse, H.-F.; Cheung, B.-M.Y.; Yiu, K.-H. Sodium-glucose cotransporter 2 inhibitors (SGLT2i) and cardiac arrhythmias: A systematic review and meta-analysis. Cardiovasc. Diabetol. 2021, 20, 100. [Google Scholar] [CrossRef]
- Fernandes, G.C.; Fernandes, A.; Cardoso, R.; Penalver, J.; Knijnik, L.; Mitrani, R.D.; Myerburg, R.J.; Goldberger, J.J. Association of SGLT2 inhibitors with arrhythmias and sudden cardiac death in patients with type 2 diabetes or heart failure: A meta-analysis of 34 randomized controlled trials. Heart Rhythm. 2021, 18, 1098–1105. [Google Scholar] [CrossRef]
- Sfairopoulos, D.; Zhang, N.; Wang, Y.; Chen, Z.; Letsas, K.P.; Tse, G.; Li, G.; Lip, G.Y.H.; Liu, T.; Korantzopoulos, P. Association between sodium-glucose cotransporter-2 inhibitors and risk of sudden cardiac death or ventricular arrhythmias: A meta-analysis of randomized controlled trials. Europace 2022, 24, 20–30. [Google Scholar] [CrossRef]
- Oates, C.P.; Santos-Gallego, C.G.; Smith, A.; Basyal, B.; Moss, N.; Kawamura, I.; Musikantow, D.R.; Turagam, M.K.; Miller, M.A.; Whang, W.; et al. SGLT2 inhibitors reduce sudden cardiac death risk in heart failure: Meta-analysis of randomized clinical trials. J. Cardiovasc. Electrophysiol. 2023, 34, 1277–1285. [Google Scholar] [CrossRef]
- Neuen, B.L.; Oshima, M.; Agarwal, R.; Arnott, C.; Cherney, D.Z.; Edwards, R.; Langkilde, A.M.; Mahaffey, K.W.; McGuire, D.K.; Neal, B.; et al. Sodium-Glucose Cotransporter 2 Inhibitors and Risk of Hyperkalemia in People with Type 2 Diabetes: A Meta-Analysis of Individual Participant Data From Randomized, Controlled Trials. Circulation 2022, 145, 1460–1470. [Google Scholar] [CrossRef]
- Sinha, F.; Federlein, A.; Biesold, A.; Schwarzfischer, M.; Krieger, K.; Schweda, F.; Tauber, P. Empagliflozin increases kidney weight due to increased cell size in the proximal tubule S3 segment and the collecting duct. Front. Pharmacol. 2023, 14, 1118358. [Google Scholar] [CrossRef]
- Herat, L.Y.; Magno, A.L.; Rudnicka, C.; Hricova, J.; Carnagarin, R.; Ward, N.C.; Arcambal, A.; Kiuchi, M.G.; Head, G.A.; Schlaich, M.P.; et al. SGLT2 Inhibitor-Induced Sympathoinhibition: A Novel Mechanism for Cardiorenal Protection. JACC Basic. Transl. Sci. 2020, 5, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Kravtsova, O.; Bohovyk, R.; Levchenko, V.; Palygin, O.; Klemens, C.A.; Rieg, T.; Staruschenko, A. SGLT2 inhibition effect on salt-induced hypertension, RAAS, and Na+ transport in Dahl SS rats. Am. J. Physiol. Renal Physiol. 2022, 322, F692–F707. [Google Scholar] [CrossRef] [PubMed]
- Tauber, P.; Sinha, F.; Berger, R.S.; Gronwald, W.; Dettmer, K.; Kuhn, M.; Trum, M.; Maier, L.S.; Wagner, S.; Schweda, F. Empagliflozin Reduces Renal Hyperfiltration in Response to Uninephrectomy, but Is Not Nephroprotective in UNx/DOCA/Salt Mouse Models. Front. Pharmacol. 2021, 12, 761855. [Google Scholar] [CrossRef] [PubMed]
- Lytvyn, Y.; Kimura, K.; Peter, N.; Lai, V.; Tse, J.; Cham, L.; Perkins, B.A.; Soleymanlou, N.; Cherney, D.Z.I. Renal and Vascular Effects of Combined SGLT2 and Angiotensin-Converting Enzyme Inhibition. Circulation 2022, 146, 450–462. [Google Scholar] [CrossRef]
- Xue, G.; Yang, X.; Zhan, G.; Wang, X.; Gao, J.; Zhao, Y.; Wang, X.; Li, J.; Pan, Z.; Xia, Y. Sodium-Glucose cotransporter 2 inhibitor empagliflozin decreases ventricular arrhythmia susceptibility by alleviating electrophysiological remodeling post-myocardial-infarction in mice. Front. Pharmacol. 2022, 13, 988408. [Google Scholar] [CrossRef]
- Jhuo, S.-J.; Liu, I.-H.; Tasi, W.-C.; Chou, T.-W.; Lin, Y.-H.; Wu, B.-N.; Lee, K.-T.; Lai, W.-T. Characteristics of Ventricular Electrophysiological Substrates in Metabolic Mice Treated with Empagliflozin. Int. J. Mol. Sci. 2021, 22, 6105. [Google Scholar] [CrossRef]
- Silva Dos Santos, D.; Turaça, L.T.; Da Coutinho, K.C.S.; Barbosa, R.A.Q.; Polidoro, J.Z.; Kasai-Brunswick, T.H.; Campos de Carvalho, A.C.; Girardi, A.C.C. Empagliflozin reduces arrhythmogenic effects in rat neonatal and human iPSC-derived cardiomyocytes and improves cytosolic calcium handling at least partially independent of NHE1. Sci. Rep. 2023, 13, 8689. [Google Scholar] [CrossRef]
- Maier, L.S.; Sossalla, S. The late Na current as a therapeutic target: Where are we? J. Mol. Cell Cardiol. 2013, 61, 44–50. [Google Scholar] [CrossRef]
- Mustroph, J.; Wagemann, O.; Lücht, C.M.; Trum, M.; Hammer, K.P.; Sag, C.M.; Lebek, S.; Tarnowski, D.; Reinders, J.; Perbellini, F.; et al. Empagliflozin reduces Ca/calmodulin-dependent kinase II activity in isolated ventricular cardiomyocytes. ESC Heart Fail. 2018, 5, 642–648. [Google Scholar] [CrossRef]
- Mustroph, J.; Baier, M.J.; Pabel, S.; Stehle, T.; Trum, M.; Provaznik, Z.; Mohler, P.J.; Musa, H.; Hund, T.J.; Sossalla, S.; et al. Empagliflozin Inhibits Cardiac Late Sodium Current by Ca/Calmodulin-Dependent Kinase II. Circulation 2022, 146, 1259–1261. [Google Scholar] [CrossRef]
- Mustroph, J.; Wagemann, O.; Trum, M.; Lebek, S.; Tarnowski, D.; Reinders, J.; Schmid, C.; Schopka, S.; Hilker, M.; Graf, B.; et al. 3145Empagliflozin potently reduces sarcoplasmic Ca leak and increases Ca transient amplitude of human failing ventricular cardiomyocytes. Eur. Heart J. 2018, 39, ehy563.3145. [Google Scholar] [CrossRef]
- Baris, V.; Gedikli, E.; Dincsoy, A.B.; Erdem, A. Empagliflozin significantly prevents QTc prolongation due to amitriptyline intoxication via intracellular calcium regulation. Eur. Heart J. 2021, 42, 1–5. [Google Scholar] [CrossRef]
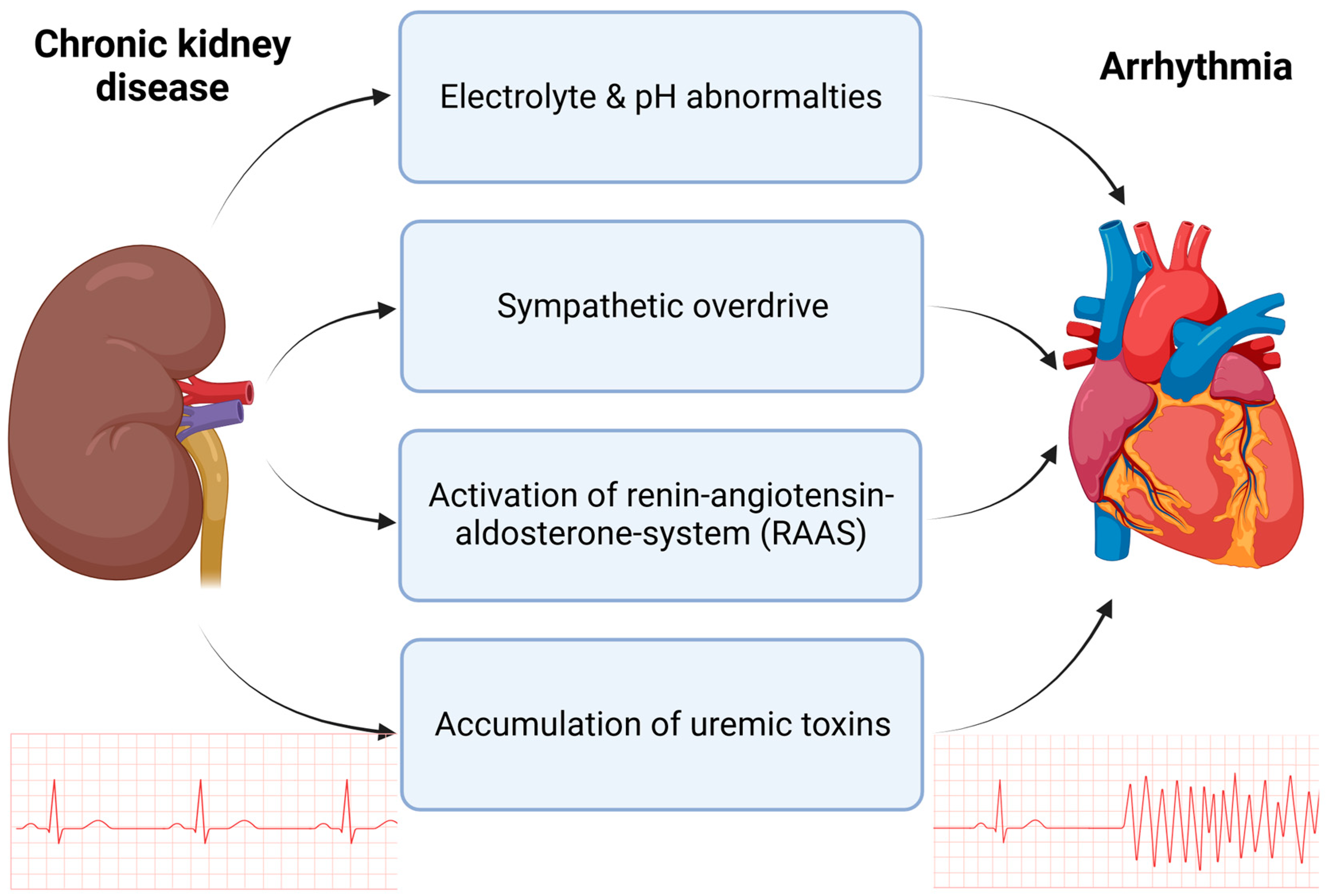
Figure 1.
Mechanisms in CKD contributing to increased arrhythmogenesis. Chronic kidney disease is accompanied by electrolyte and pH abnormalities, such as hyperkalemia and metabolic acidosis. Additionally, in patients with CKD, an overdrive of the sympathetic nervous system is overserved as well as chronic upregulation of the renin–angiotensin–aldosterone system (RAAS). The accumulation of uremic toxins, such as indoxylsulfate (IS) and fibroblast growth factor (FGF-23), contribute to arrhythmia in CKD. Created with BioRender.com, accessed on 31 August 2023.
Figure 1.
Mechanisms in CKD contributing to increased arrhythmogenesis. Chronic kidney disease is accompanied by electrolyte and pH abnormalities, such as hyperkalemia and metabolic acidosis. Additionally, in patients with CKD, an overdrive of the sympathetic nervous system is overserved as well as chronic upregulation of the renin–angiotensin–aldosterone system (RAAS). The accumulation of uremic toxins, such as indoxylsulfate (IS) and fibroblast growth factor (FGF-23), contribute to arrhythmia in CKD. Created with BioRender.com, accessed on 31 August 2023.

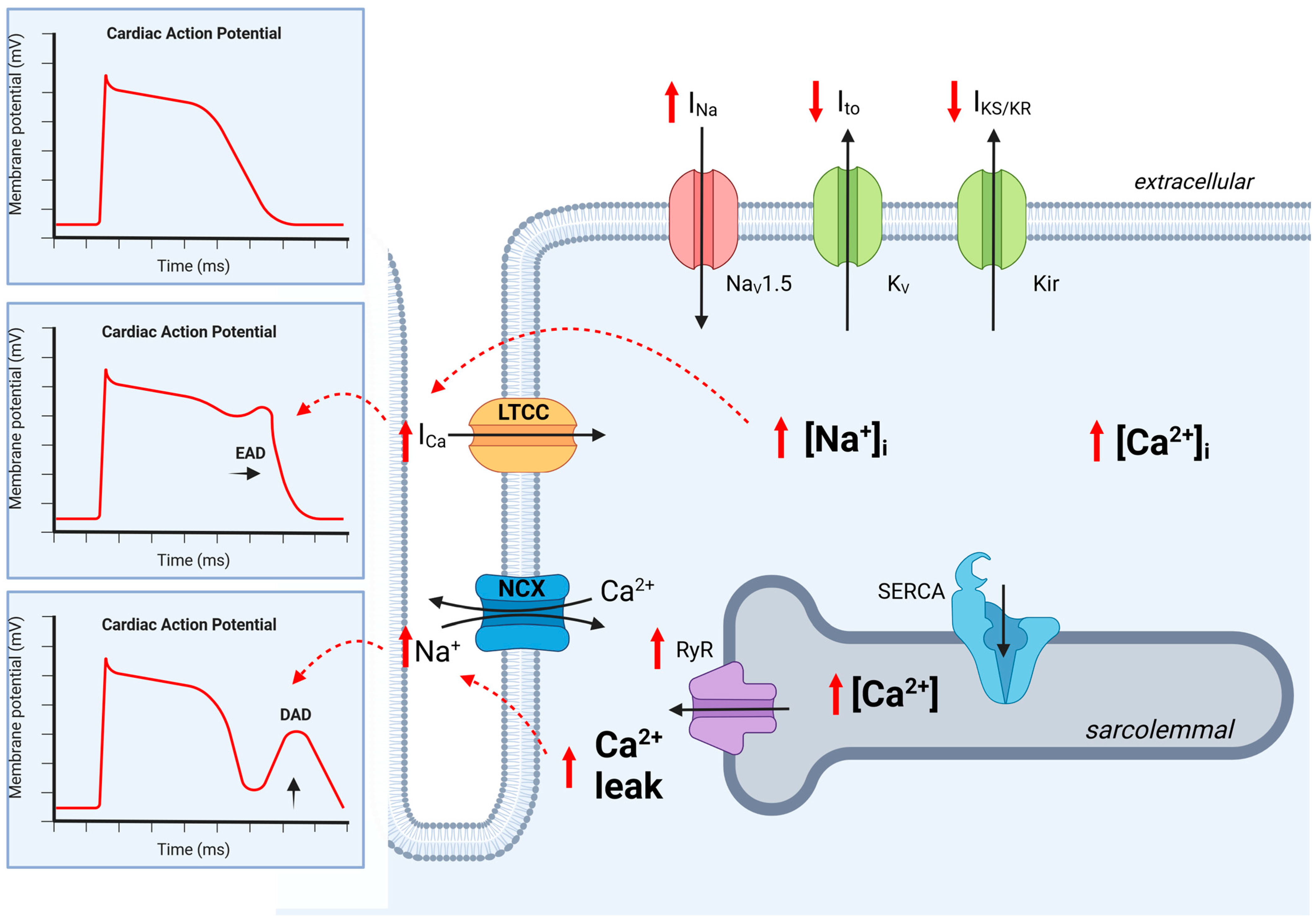
Figure 2.
Ion channel regulation in cardiomyocytes in chronic kidney disease. The regulation of ion channels leads to abnormalities in cardiac action potential. In CKD, Na+ influx is disturbed and prolonged as late INa, resulting in increased cytoplasmic Na+ levels [Na+]I and increased Ca2+ influx (ICa) via the L-type calcium channel (LTCC). This results in a prolonged action potential with the reactivation of the LTCC, causing an early after-depolarization (EAD). Additionally, an increased sarcolemmal Ca2+ concentration increases Ca2+ leak, which then activates NCX, resulting in delayed after-depolarization (DAD). CKD leads to the downregulation of K+ efflux, which prolongs the action potential duration. Dashed red lines indicate activation. Kir, K+ inward rectifier; KV, K+ efflux; NaV1.5, Na+ influx; LTCC, L-type calcium channel; NCX, Na+-Ca2+ exchanger; RyR, ryanodine receptor; SERCA, sarcoplasmic reticulum, EAD, early after-depolarization; DAD, delayed after-depolarization, black arrow, Ion current; red arrow, regulation by CKD. Created with BioRender.com, accessed on 31 August 2023.
Figure 2.
Ion channel regulation in cardiomyocytes in chronic kidney disease. The regulation of ion channels leads to abnormalities in cardiac action potential. In CKD, Na+ influx is disturbed and prolonged as late INa, resulting in increased cytoplasmic Na+ levels [Na+]I and increased Ca2+ influx (ICa) via the L-type calcium channel (LTCC). This results in a prolonged action potential with the reactivation of the LTCC, causing an early after-depolarization (EAD). Additionally, an increased sarcolemmal Ca2+ concentration increases Ca2+ leak, which then activates NCX, resulting in delayed after-depolarization (DAD). CKD leads to the downregulation of K+ efflux, which prolongs the action potential duration. Dashed red lines indicate activation. Kir, K+ inward rectifier; KV, K+ efflux; NaV1.5, Na+ influx; LTCC, L-type calcium channel; NCX, Na+-Ca2+ exchanger; RyR, ryanodine receptor; SERCA, sarcoplasmic reticulum, EAD, early after-depolarization; DAD, delayed after-depolarization, black arrow, Ion current; red arrow, regulation by CKD. Created with BioRender.com, accessed on 31 August 2023.

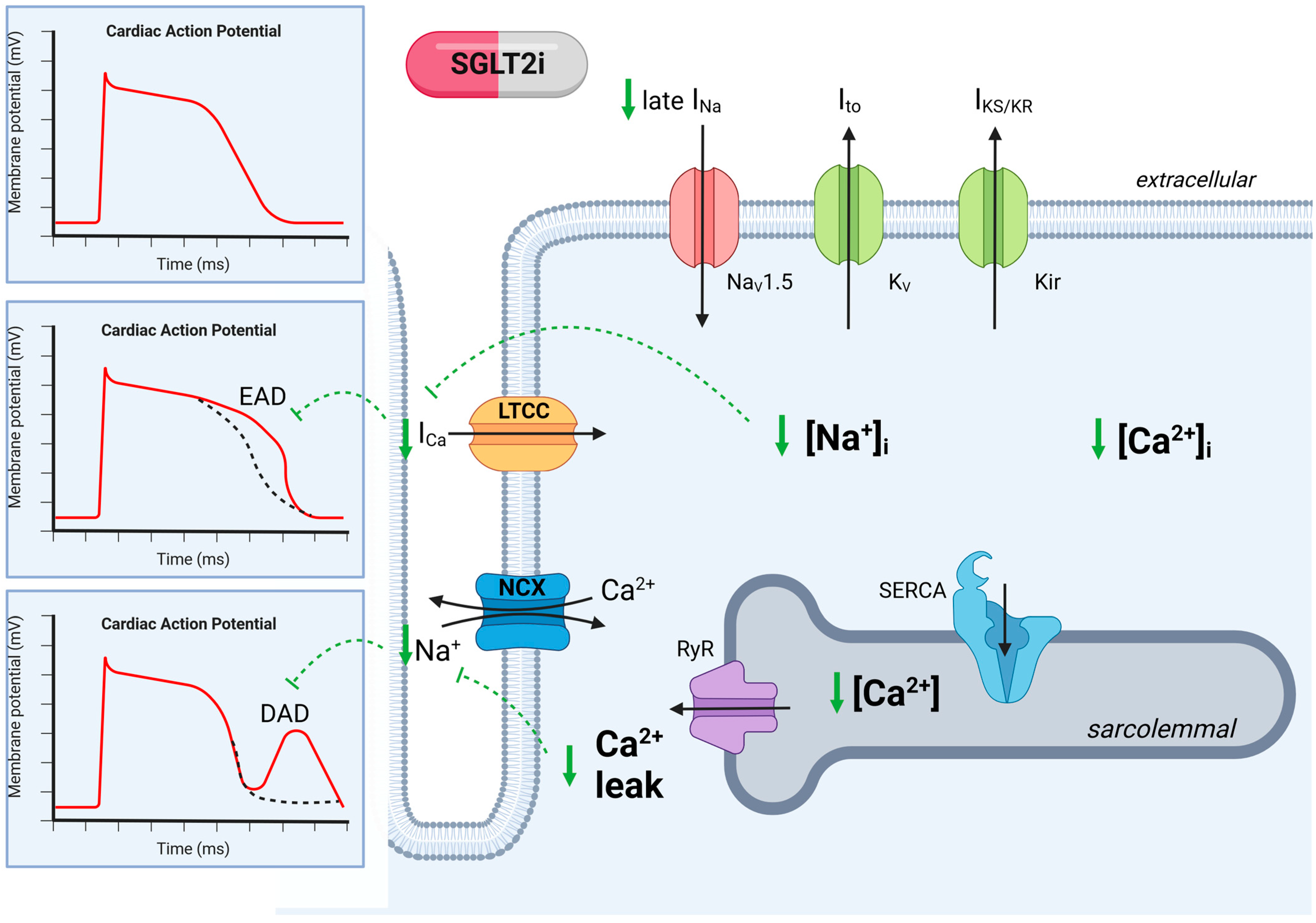
Figure 3.
Ion channel regulation in cardiomyocytes in chronic kidney disease with SGLT2 inhibition. Regulation of ion channels leads to abnormalities in cardiac action potential, which is reversed by SGLT2i. SGLT2i reduces Na+ influx and late Na+ (INa), which decreases Na+ overload and thereby reduces Ca2+ influx, which decreases the risk of action potential prolongation. SGLT2i decreases the sarcolemmal Ca2+ concentration, which reduces spontaneous Ca2+ leakage. The reduction in Ca2+ leak decreases the risk of delayed after-polarization (DAD). Dashed green lines indicate SGLT2i inhibition. Kir, K+ inward rectifier; KV, K+ efflux; NaV1.5, Na+ influx; LTCC, L-type calcium channel; NCX, Na+-Ca2+ exchanger; RyR, Ryanodin receptor; SERCA, sarcoplasmic reticulum; EAD, early after-depolarization; DAD, delayed after-depolarization. Created with BioRender.com, accessed on 31 August 2023.
Figure 3.
Ion channel regulation in cardiomyocytes in chronic kidney disease with SGLT2 inhibition. Regulation of ion channels leads to abnormalities in cardiac action potential, which is reversed by SGLT2i. SGLT2i reduces Na+ influx and late Na+ (INa), which decreases Na+ overload and thereby reduces Ca2+ influx, which decreases the risk of action potential prolongation. SGLT2i decreases the sarcolemmal Ca2+ concentration, which reduces spontaneous Ca2+ leakage. The reduction in Ca2+ leak decreases the risk of delayed after-polarization (DAD). Dashed green lines indicate SGLT2i inhibition. Kir, K+ inward rectifier; KV, K+ efflux; NaV1.5, Na+ influx; LTCC, L-type calcium channel; NCX, Na+-Ca2+ exchanger; RyR, Ryanodin receptor; SERCA, sarcoplasmic reticulum; EAD, early after-depolarization; DAD, delayed after-depolarization. Created with BioRender.com, accessed on 31 August 2023.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sinha, F.; Schweda, F.; Maier, L.S.; Wagner, S. Impact of Impaired Kidney Function on Arrhythmia-Promoting Cardiac Ion Channel Regulation. Int. J. Mol. Sci. 2023, 24, 14198. https://doi.org/10.3390/ijms241814198
AMA Style
Sinha F, Schweda F, Maier LS, Wagner S. Impact of Impaired Kidney Function on Arrhythmia-Promoting Cardiac Ion Channel Regulation. International Journal of Molecular Sciences. 2023; 24(18):14198. https://doi.org/10.3390/ijms241814198
Chicago/Turabian StyleSinha, Frederick, Frank Schweda, Lars S. Maier, and Stefan Wagner. 2023. "Impact of Impaired Kidney Function on Arrhythmia-Promoting Cardiac Ion Channel Regulation" International Journal of Molecular Sciences 24, no. 18: 14198. https://doi.org/10.3390/ijms241814198
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.