
Immune System and Brain/Intestinal Barrier Functions in Psychiatric Diseases: Is Sphingosine-1-Phosphate at the Helm?

 ,
,  , ,
, ,
Abstract
:
1. Immune Dysregulation and Barrier Function in Psychiatric Diseases
1.1. Immune Dysregulation in Psychiatric Diseases
1.2. The Brain Barriers in Psychiatric Diseases
1.2.1. Structure and Function of the Brain Barrier
- BBB: constituted by microvascular endothelial cells with apical tight junction complexes lining the cerebral capillaries that traverse the brain and spinal cord. Heterogenic endothelial cells shape the neurovascular complex interacting with neurons, astrocytic endfeet, myocytes, perivascular macrophages, pericytes, and extracellular matrix components [43].
- Arachnoid (or meningeal) barrier: formed by the avascular arachnoid epithelium, featuring tight junctions as a barrier between the cerebrospinal fluid (CSF)-filled subarachnoid space and other meningeal structures.
- Blood-CSF barrier: this barrier, also constituted by epithelial cells with apical tight junctions, separate the blood vessels of the choroid plexus (fenestrated and leaky) from the CSF.
- The fetal CSF-brain barrier: during early development, this barrier exists between the CSF and brain parenchyma during early development. It acts as a functional barrier when junctions interconnect ependymal cells.
- The adult ventricular ependyma: as development progresses, the ependymal cells lose their ability to restrict the passage of larger molecules, such as proteins, between the CSF and the brain.
1.2.2. Brain Barrier Dysfunction in Psychiatric Diseases
- Increased paracellular traffic of macromolecules (albumin, IgG) [56]
- Changes in the expression and function of P-glycoprotein [67] and other transcellular transporters (transferrin receptor (TfR), glucose transporter-1 (GLUT-1), insulin receptor (IR), and low-density lipoprotein receptor (LDLr)), which may impact the access of neuropsychiatric drugs and other promising pharmacological tools (e.g., nanoparticles, viral vectors) to the brain parenchyma through the BBB, as proposed for the treatment of neurological/neurodegenerative diseases [68,69].
1.3. The Intestinal Barrier in Psychiatric Diseases
1.3.1. The Intestinal Barrier in Health and Disease
1.3.2. Intestinal Barrier Dysfunction in Psychiatric Diseases
2. The Sphingosine-1-Phosphate (S1P) Metabolism and Signaling
2.1. S1P Metabolism
2.1.1. S1P Synthesis Enzymes
2.1.2. S1P Degradation Enzymes
2.2. S1P Signaling
3. S1P and the Immune System
4. S1P Signaling in Blood–Brain and Intestinal Barrier Functions
4.1. S1P Signaling in BBB Function
4.2. S1P Signaling in Intestinal Barrier Function
5. Evidence of S1P Role in Psychiatric Diseases
5.1. S1P in MDD
5.2. S1P in Neurodevelopmental Disorders
5.2.1. S1P in SZ
5.2.2. S1P in Autism Spectrum Disorders
5.2.3. S1P in Attention Deficit Hyperactivity Disorder
6. Current and Potential Drugs Targeting S1P in Psychiatric Diseases and IBD
6.1. S1P-Related Drugs for the Treatment of Psychiatric Diseases
6.1.1. S1P-Related Drugs for MDD
6.1.2. S1P-Related Drugs for Neurodevelopmental Disorders
6.2. S1P-Related Drugs for IBD
7. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Howes, O.D.; Thase, M.E.; Pillinger, T. Treatment resistance in psychiatry: State of the art and new directions. Mol. Psychiatry 2022, 27, 58–72. [Google Scholar] [CrossRef]
- Naggan, L.; Robinson, E.; Dinur, E.; Goldenberg, H.; Kozela, E.; Yirmiya, R. Suicide in bipolar disorder patients is associated with hippocampal microglia activation and reduction of lymphocytes-activation gene 3 (LAG3) microglial checkpoint expression. Brain Behav. Immun. 2023, 110, 185–194. [Google Scholar] [CrossRef]
- Runge, K.; Fiebich, B.L.; Kuzior, H.; Rausch, J.; Maier, S.J.; Dersch, R.; Nickel, K.; Domschke, K.; Tebartz van Elst, L.; Endres, D. Altered cytokine levels in the cerebrospinal fluid of adult patients with autism spectrum disorder. J. Psychiatr. Res. 2023, 158, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.J.; Li, S.C.; Li, S.W.; Kuo, H.C.; Lee, S.Y.; Huang, L.H.; Chin, C.Y.; Yang, C.Y. Gut microbiota and plasma cytokine levels in patients with attention-deficit/hyperactivity disorder. Transl. Psychiatry 2022, 12, 76. [Google Scholar] [CrossRef]
- Caso, J.R.; Graell, M.; Navalón, A.; MacDowell, K.S.; Gutiérrez, S.; Soto, M.; Leza, J.C.; Carrasco, J.L.; Marsá, M.D. Dysfunction of inflammatory pathways in adolescent female patients with anorexia nervosa. Prog. Neuropsychopharmacol. Biol. Psychiatry 2020, 96, 109727. [Google Scholar] [CrossRef] [PubMed]
- Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014, 511, 421–427. [Google Scholar] [CrossRef] [Green Version]
- Howard, D.M.; Adams, M.J.; Clarke, T.K.; Hafferty, J.D.; Gibson, J.; Shirali, M.; Coleman, J.R.I.; Hagenaars, S.P.; Ward, J.; Wigmore, E.M.; et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat. Neurosci. 2019, 22, 343–352. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Wang, W.; Zhang, R.; Duan, H.; Tian, X.; Xu, C.; Li, X.; Zhang, D. Multivariate genome-wide association study of depression, cognition, and memory phenotypes and validation analysis identify 12 cross-ethnic variants. Transl. Psychiatry 2022, 12, 304. [Google Scholar] [CrossRef]
- Bioque, M.; Catarina Matias-Martins, A.; Llorca-Bofí, V.; Mezquida, G.; Cuesta, M.J.; Vieta, E.; Amoretti, S.; Lobo, A.; González-Pinto, A.; Moreno, C.; et al. Neutrophil to Lymphocyte Ratio in Patients with a First Episode of Psychosis: A Two-Year Longitudinal Follow-up Study. Schizophr. Bull. 2022, 48, 1327–1335. [Google Scholar] [CrossRef]
- Zulfic, Z.; Weickert, C.S.; Weickert, T.W.; Liu, D.; Myles, N.; Galletly, C. Neutrophil-lymphocyte ratio—A simple, accessible measure of inflammation, morbidity and prognosis in psychiatric disorders? Australas Psychiatry 2020, 28, 454–458. [Google Scholar] [CrossRef]
- Kappelmann, N.; Khandaker, G.M.; Dal, H.; Stochl, J.; Kosidou, K.; Jones, P.B.; Dalman, C.; Karlsson, H. Systemic inflammation and intelligence in early adulthood and subsequent risk of schizophrenia and other non-affective psychoses: A longitudinal cohort and co-relative study. Psychol. Med. 2019, 49, 295–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foley, É.M.; Parkinson, J.T.; Mitchell, R.E.; Turner, L.; Khandaker, G.M. Peripheral blood cellular immunophenotype in depression: A systematic review and meta-analysis. Mol. Psychiatry 2023, 28, 1004–1019. [Google Scholar] [CrossRef] [PubMed]
- Wittenberg, G.M.; Greene, J.; Vértes, P.E.; Drevets, W.C.; Bullmore, E.T. Major Depressive Disorder Is Associated with Differential Expression of Innate Immune and Neutrophil-Related Gene Networks in Peripheral Blood: A Quantitative Review of Whole-Genome Transcriptional Data From Case-Control Studies. Biol. Psychiatry 2020, 88, 625–637. [Google Scholar] [CrossRef] [PubMed]
- Çakici, N.; Sutterland, A.L.; Penninx, B.; Dalm, V.A.; de Haan, L.; van Beveren, N.J.M. Altered peripheral blood compounds in drug-naïve first-episode patients with either schizophrenia or major depressive disorder: A meta-analysis. Brain Behav. Immun. 2020, 88, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Campeau, A.; Mills, R.H.; Stevens, T.; Rossitto, L.A.; Meehan, M.; Dorrestein, P.; Daly, R.; Nguyen, T.T.; Gonzalez, D.J.; Jeste, D.V.; et al. Multi-omics of human plasma reveals molecular features of dysregulated inflammation and accelerated aging in schizophrenia. Mol. Psychiatry 2022, 27, 1217–1225. [Google Scholar] [CrossRef]
- Dudzińska, E.; Szymona, K.; Bogucki, J.; Koch, W.; Cholewińska, E.; Sitarz, R.; Ognik, K. Increased Markers of Oxidative Stress and Positive Correlation Low-Grade Inflammation with Positive Symptoms in the First Episode of Schizophrenia in Drug-Naïve Patients. J. Clin. Med. 2022, 11, 2551. [Google Scholar] [CrossRef]
- Chaudhry, I.B.; Husain, M.O.; Khoso, A.B.; Husain, M.I.; Buch, M.H.; Kiran, T.; Fu, B.; Bassett, P.; Qurashi, I.; Ur Rahman, R.; et al. A randomised clinical trial of methotrexate points to possible efficacy and adaptive immune dysfunction in psychosis. Transl. Psychiatry 2020, 10, 415. [Google Scholar] [CrossRef]
- Nudel, R.; Allesøe, R.L.; Werge, T.; Thompson, W.K.; Rasmussen, S.; Benros, M.E. An immunogenetic investigation of 30 autoimmune and autoinflammatory diseases and their links to psychiatric disorders in a nationwide sample. Immunology 2023, 168, 622–639. [Google Scholar] [CrossRef]
- Enache, D.; Pariante, C.M.; Mondelli, V. Markers of central inflammation in major depressive disorder: A systematic review and meta-analysis of studies examining cerebrospinal fluid, positron emission tomography and post-mortem brain tissue. Brain Behav. Immun. 2019, 81, 24–40. [Google Scholar] [CrossRef]
- Li, H.; Sagar, A.P.; Kéri, S. Microglial markers in the frontal cortex are related to cognitive dysfunctions in major depressive disorder. J. Affect. Disord. 2018, 241, 305–310. [Google Scholar] [CrossRef]
- Hafizi, S.; Tseng, H.H.; Rao, N.; Selvanathan, T.; Kenk, M.; Bazinet, R.P.; Suridjan, I.; Wilson, A.A.; Meyer, J.H.; Remington, G.; et al. Imaging Microglial Activation in Untreated First-Episode Psychosis: A PET Study with [(18)F]FEPPA. Am. J. Psychiatry 2017, 174, 118–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, J.H.; Cervenka, S.; Kim, M.J.; Kreisl, W.C.; Henter, I.D.; Innis, R.B. Neuroinflammation in psychiatric disorders: PET imaging and promising new targets. Lancet Psychiatry 2020, 7, 1064–1074. [Google Scholar] [CrossRef] [PubMed]
- Felger, J.C.; Li, Z.; Haroon, E.; Woolwine, B.J.; Jung, M.Y.; Hu, X.; Miller, A.H. Inflammation is associated with decreased functional connectivity within corticostriatal reward circuitry in depression. Mol. Psychiatry 2016, 21, 1358–1365. [Google Scholar] [CrossRef] [Green Version]
- Su, K.P.; Lai, H.C.; Peng, C.Y.; Su, W.P.; Chang, J.P.; Pariante, C.M. Interferon-alpha-induced depression: Comparisons between early- and late-onset subgroups and with patients with major depressive disorder. Brain Behav. Immun. 2019, 80, 512–518. [Google Scholar] [CrossRef]
- Zhou, C.; Peng, S.; Lin, A.; Jiang, A.; Peng, Y.; Gu, T.; Liu, Z.; Cheng, Q.; Zhang, J.; Luo, P. Psychiatric disorders associated with immune checkpoint inhibitors: A pharmacovigilance analysis of the FDA Adverse Event Reporting System (FAERS) database. EClinicalMedicine 2023, 59, 101967. [Google Scholar] [CrossRef]
- Sandiego, C.M.; Gallezot, J.D.; Pittman, B.; Nabulsi, N.; Lim, K.; Lin, S.F.; Matuskey, D.; Lee, J.Y.; O’Connor, K.C.; Huang, Y.; et al. Imaging robust microglial activation after lipopolysaccharide administration in humans with PET. Proc. Natl. Acad. Sci. USA 2015, 112, 12468–12473. [Google Scholar] [CrossRef] [PubMed]
- Cattane, N.; Vernon, A.C.; Borsini, A.; Scassellati, C.; Endres, D.; Capuron, L.; Tamouza, R.; Benros, M.E.; Leza, J.C.; Pariante, C.M.; et al. Preclinical animal models of mental illnesses to translate findings from the bench to the bedside: Molecular brain mechanisms and peripheral biomarkers associated to early life stress or immune challenges. Eur. Neuropsychopharmacol. 2022, 58, 55–79. [Google Scholar] [CrossRef]
- Martin-Hernandez, D.; Caso, J.R.; Javier Meana, J.; Callado, L.F.; Madrigal, J.L.M.; Garcia-Bueno, B.; Leza, J.C. Intracellular inflammatory and antioxidant pathways in postmortem frontal cortex of subjects with major depression: Effect of antidepressants. J. Neuroinflamm. 2018, 15, 251. [Google Scholar] [CrossRef]
- Tendilla-Beltrán, H.; Meneses-Prado, S.; Vázquez-Roque, R.A.; Tapia-Rodríguez, M.; Vázquez-Hernández, A.J.; Coatl-Cuaya, H.; Martín-Hernández, D.; MacDowell, K.S.; Garcés-Ramírez, L.; Leza, J.C.; et al. Risperidone Ameliorates Prefrontal Cortex Neural Atrophy and Oxidative/Nitrosative Stress in Brain and Peripheral Blood of Rats with Neonatal Ventral Hippocampus Lesion. J. Neurosci. 2019, 39, 8584–8599. [Google Scholar] [CrossRef]
- Köhler-Forsberg, O.; Lydholm, C.N.; Hjorthøj, C.; Nordentoft, M.; Mors, O.; Benros, M.E. Efficacy of anti-inflammatory treatment on major depressive disorder or depressive symptoms: Meta-analysis of clinical trials. Acta Psychiatr. Scand. 2019, 139, 404–419. [Google Scholar] [CrossRef]
- Jeppesen, R.; Christensen, R.H.B.; Pedersen, E.M.J.; Nordentoft, M.; Hjorthøj, C.; Köhler-Forsberg, O.; Benros, M.E. Efficacy and safety of anti-inflammatory agents in treatment of psychotic disorders—A comprehensive systematic review and meta-analysis. Brain Behav. Immun. 2020, 90, 364–380. [Google Scholar] [CrossRef] [PubMed]
- Bai, S.; Guo, W.; Feng, Y.; Deng, H.; Li, G.; Nie, H.; Guo, G.; Yu, H.; Ma, Y.; Wang, J.; et al. Efficacy and safety of anti-inflammatory agents for the treatment of major depressive disorder: A systematic review and meta-analysis of randomised controlled trials. J. Neurol. Neurosurg. Psychiatry 2020, 91, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Khandaker, G.M.; Pearson, R.M.; Zammit, S.; Lewis, G.; Jones, P.B. Association of serum interleukin 6 and C-reactive protein in childhood with depression and psychosis in young adult life: A population-based longitudinal study. JAMA Psychiatry 2014, 71, 1121–1128. [Google Scholar] [CrossRef] [Green Version]
- Wium-Andersen, M.K.; Ørsted, D.D.; Nordestgaard, B.G. Elevated C-reactive protein associated with late- and very-late-onset schizophrenia in the general population: A prospective study. Schizophr. Bull. 2014, 40, 1117–1127. [Google Scholar] [CrossRef] [Green Version]
- Ishida, I.; Ogura, J.; Aizawa, E.; Ota, M.; Hidese, S.; Yomogida, Y.; Matsuo, J.; Yoshida, S.; Kunugi, H. Gut permeability and its clinical relevance in schizophrenia. Neuropsychopharmacol. Rep. 2022, 42, 70–76. [Google Scholar] [CrossRef]
- Ohlsson, L.; Gustafsson, A.; Lavant, E.; Suneson, K.; Brundin, L.; Westrin, Å.; Ljunggren, L.; Lindqvist, D. Leaky gut biomarkers in depression and suicidal behavior. Acta Psychiatr. Scand. 2019, 139, 185–193. [Google Scholar] [CrossRef] [Green Version]
- Iordache, M.M.; Tocia, C.; Aschie, M.; Dumitru, A.; Manea, M.; Cozaru, G.C.; Petcu, L.; Vlad, S.E.; Dumitru, E.; Chisoi, A. Intestinal Permeability and Depression in Patients with Inflammatory Bowel Disease. J. Clin. Med. 2022, 11, 5121. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; Guo, P.; Li, Y.; Liu, L.; Yan, R.; Liu, S.; Wang, S.; Xue, F.; Zhou, X.; Yuan, Z. Role of the Gut-Brain Axis in the Shared Genetic Etiology Between Gastrointestinal Tract Diseases and Psychiatric Disorders: A Genome-Wide Pleiotropic Analysis. JAMA Psychiatry 2023, 80, 360–370. [Google Scholar] [CrossRef]
- Ponferrada, A.; Caso, J.R.; Alou, L.; Colón, A.; Sevillano, D.; Moro, M.A.; Lizasoain, I.; Menchén, P.; Gómez-Lus, M.L.; Lorenzo, P.; et al. The role of PPARgamma on restoration of colonic homeostasis after experimental stress-induced inflammation and dysfunction. Gastroenterology 2007, 132, 1791–1803. [Google Scholar] [CrossRef]
- Cai, H.Q.; Catts, V.S.; Webster, M.J.; Galletly, C.; Liu, D.; O’Donnell, M.; Weickert, T.W.; Weickert, C.S. Increased macrophages and changed brain endothelial cell gene expression in the frontal cortex of people with schizophrenia displaying inflammation. Mol. Psychiatry 2020, 25, 761–775. [Google Scholar] [CrossRef] [Green Version]
- Kadry, H.; Noorani, B.; Cucullo, L. A blood-brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS 2020, 17, 69. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Kaur, C.; Ling, E.A. The circumventricular organs. Histol. Histopathol. 2017, 32, 879–892. [Google Scholar] [CrossRef] [PubMed]
- Luissint, A.C.; Artus, C.; Glacial, F.; Ganeshamoorthy, K.; Couraud, P.O. Tight junctions at the blood brain barrier: Physiological architecture and disease-associated dysregulation. Fluids Barriers CNS 2012, 9, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatovic, S.M.; Johnson, A.M.; Keep, R.F.; Andjelkovic, A.V. Junctional proteins of the blood-brain barrier: New insights into function and dysfunction. Tissue Barriers 2016, 4, e1154641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barar, J.; Rafi, M.A.; Pourseif, M.M.; Omidi, Y. Blood-brain barrier transport machineries and targeted therapy of brain diseases. Bioimpacts 2016, 6, 225–248. [Google Scholar] [CrossRef] [Green Version]
- Ayloo, S.; Gu, C. Transcytosis at the blood-brain barrier. Curr. Opin. Neurobiol. 2019, 57, 32–38. [Google Scholar] [CrossRef]
- Jessen, N.A.; Munk, A.S.; Lundgaard, I.; Nedergaard, M. The Glymphatic System: A Beginner’s Guide. Neurochem. Res. 2015, 40, 2583–2599. [Google Scholar] [CrossRef] [Green Version]
- Bernardo-Castro, S.; Sousa, J.A.; Brás, A.; Cecília, C.; Rodrigues, B.; Almendra, L.; Machado, C.; Santo, G.; Silva, F.; Ferreira, L.; et al. Pathophysiology of Blood-Brain Barrier Permeability Throughout the Different Stages of Ischemic Stroke and Its Implication on Hemorrhagic Transformation and Recovery. Front. Neurol. 2020, 11, 594672. [Google Scholar] [CrossRef]
- Cash, A.; Theus, M.H. Mechanisms of Blood-Brain Barrier Dysfunction in Traumatic Brain Injury. Int. J. Mol. Sci. 2020, 21, 3344. [Google Scholar] [CrossRef]
- Byrne, J.F.; Healy, C.; Mongan, D.; Susai, S.R.; Zammit, S.; Föcking, M.; Cannon, M.; Cotter, D.R. Transdiagnostic inflammatory subgroups among psychiatric disorders and their relevance to role functioning: A nested case-control study of the ALSPAC cohort. Transl. Psychiatry 2022, 12, 377. [Google Scholar] [CrossRef] [PubMed]
- Turkheimer, F.E.; Veronese, M.; Mondelli, V.; Cash, D.; Pariante, C.M. Sickness behaviour and depression: An updated model of peripheral-central immunity interactions. Brain Behav. Immun. 2023, 111, 202–210. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Greene, C.; Munnich, A.; Campbell, M. The CLDN5 gene at the blood-brain barrier in health and disease. Fluids Barriers CNS 2023, 20, 22. [Google Scholar] [CrossRef]
- Greene, C.; Hanley, N.; Campbell, M. Blood-brain barrier associated tight junction disruption is a hallmark feature of major psychiatric disorders. Transl. Psychiatry 2020, 10, 373. [Google Scholar] [CrossRef] [PubMed]
- Kealy, J.; Greene, C.; Campbell, M. Blood-brain barrier regulation in psychiatric disorders. Neurosci. Lett. 2020, 726, 133664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeppesen, R.; Orlovska-Waast, S.; Sørensen, N.V.; Christensen, R.H.B.; Benros, M.E. Cerebrospinal Fluid and Blood Biomarkers of Neuroinflammation and Blood-Brain Barrier in Psychotic Disorders and Individually Matched Healthy Controls. Schizophr. Bull. 2022, 48, 1206–1216. [Google Scholar] [CrossRef]
- Jaudon, F.; Thalhammer, A.; Cingolani, L.A. Integrin adhesion in brain assembly: From molecular structure to neuropsychiatric disorders. Eur. J. Neurosci. 2021, 53, 3831–3850. [Google Scholar] [CrossRef]
- Redies, C.; Hertel, N.; Hübner, C.A. Cadherins and neuropsychiatric disorders. Brain Res. 2012, 1470, 130–144. [Google Scholar] [CrossRef]
- Schlaaff, K.; Dobrowolny, H.; Frodl, T.; Mawrin, C.; Gos, T.; Steiner, J.; Bogerts, B. Increased densities of T and B lymphocytes indicate neuroinflammation in subgroups of schizophrenia and mood disorder patients. Brain Behav. Immun. 2020, 88, 497–506. [Google Scholar] [CrossRef]
- Michetti, F.; D’Ambrosi, N.; Toesca, A.; Puglisi, M.A.; Serrano, A.; Marchese, E.; Corvino, V.; Geloso, M.C. The S100B story: From biomarker to active factor in neural injury. J. Neurochem. 2019, 148, 168–187. [Google Scholar] [CrossRef] [Green Version]
- Chong, Z.Z.; Changyaleket, B.; Xu, H.; Dull, R.O.; Schwartz, D.E. Identifying S100B as a Biomarker and a Therapeutic Target For Brain Injury and Multiple Diseases. Curr. Med. Chem. 2016, 23, 1571–1596. [Google Scholar] [CrossRef] [PubMed]
- Torres-Platas, S.G.; Cruceanu, C.; Chen, G.G.; Turecki, G.; Mechawar, N. Evidence for increased microglial priming and macrophage recruitment in the dorsal anterior cingulate white matter of depressed suicides. Brain Behav. Immun. 2014, 42, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Puvogel, S.; Alsema, A.; Kracht, L.; Webster, M.J.; Weickert, C.S.; Sommer, I.E.C.; Eggen, B.J.L. Single-nucleus RNA sequencing of midbrain blood-brain barrier cells in schizophrenia reveals subtle transcriptional changes with overall preservation of cellular proportions and phenotypes. Mol. Psychiatry 2022, 27, 4731–4740. [Google Scholar] [CrossRef] [PubMed]
- Treccani, G.; Schlegelmilch, A.L.; Schultz, N.; Herzog, D.P.; Bessa, J.M.; Sotiropoulos, I.; Müller, M.B.; Wennström, M. Hippocampal NG2+ pericytes in chronically stressed rats and depressed patients: A quantitative study. Stress 2021, 24, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues-Neves, A.C.; Ambrósio, A.F.; Gomes, C.A. Microglia sequelae: Brain signature of innate immunity in schizophrenia. Transl. Psychiatry 2022, 12, 493. [Google Scholar] [CrossRef]
- Rahimian, R.; Wakid, M.; O’Leary, L.A.; Mechawar, N. The emerging tale of microglia in psychiatric disorders. Neurosci. Biobehav. Rev. 2021, 131, 1–29. [Google Scholar] [CrossRef]
- De Klerk, O.L.; Bosker, F.J.; Luurtsema, G.; Nolte, I.M.; Dierckx, R.; Den Boer, J.A.; Potschka, H. The role of p-glycoprotein in psychiatric disorders: A reliable guard of the brain? Cent. Nerv. Syst. Agents Med. Chem. 2011, 11, 197–209. [Google Scholar] [CrossRef]
- Han, L.; Jiang, C. Evolution of blood-brain barrier in brain diseases and related systemic nanoscale brain-targeting drug delivery strategies. Acta Pharm. Sin. B 2021, 11, 2306–2325. [Google Scholar] [CrossRef]
- Gonçalves, R.A.; De Felice, F.G. The crosstalk between brain and periphery: Implications for brain health and disease. Neuropharmacology 2021, 197, 108728. [Google Scholar] [CrossRef]
- Segawa, K.; Blumenthal, Y.; Yamawaki, Y.; Ohtsuki, G. A Destruction Model of the Vascular and Lymphatic Systems in the Emergence of Psychiatric Symptoms. Biology 2021, 10, 34. [Google Scholar] [CrossRef]
- Turner, J.R. Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 2009, 9, 799–809. [Google Scholar] [CrossRef]
- Bhat, A.A.; Uppada, S.; Achkar, I.W.; Hashem, S.; Yadav, S.K.; Shanmugakonar, M.; Al-Naemi, H.A.; Haris, M.; Uddin, S. Tight Junction Proteins and Signaling Pathways in Cancer and Inflammation: A Functional Crosstalk. Front. Physiol. 2018, 9, 1942. [Google Scholar] [CrossRef] [Green Version]
- Madara, J.L. Loosening tight junctions. Lessons from the intestine. J. Clin. Investig. 1989, 83, 1089–1094. [Google Scholar] [CrossRef] [PubMed]
- Turpin, W.; Lee, S.H.; Raygoza Garay, J.A.; Madsen, K.L.; Meddings, J.B.; Bedrani, L.; Power, N.; Espin-Garcia, O.; Xu, W.; Smith, M.I.; et al. Increased Intestinal Permeability Is Associated with Later Development of Crohn’s Disease. Gastroenterology 2020, 159, 2092–2100.e5. [Google Scholar] [CrossRef] [PubMed]
- López-Cauce, B.; Puerto, M.; García, J.J.; Ponce-Alonso, M.; Becerra-Aparicio, F.; Del Campo, R.; Peligros, I.; Fernández-Aceñero, M.J.; Gómez-Navarro, Y.; Lara, J.M.; et al. Akkermansia deficiency and mucin depletion are implicated in intestinal barrier dysfunction as earlier event in the development of inflammation in interleukin-10-deficient mice. Front. Microbiol. 2022, 13, 1083884. [Google Scholar] [CrossRef] [PubMed]
- Caso, J.R.; Leza, J.C.; Menchen, L. The effects of physical and psychological stress on the gastro-intestinal tract: Lessons from animal models. Curr. Mol. Med. 2008, 8, 299–312. [Google Scholar] [PubMed]
- Bernstein, C.N.; Singh, S.; Graff, L.A.; Walker, J.R.; Miller, N.; Cheang, M. A prospective population-based study of triggers of symptomatic flares in IBD. Am. J. Gastroenterol. 2010, 105, 1994–2002. [Google Scholar] [CrossRef]
- Barberio, B.; Zamani, M.; Black, C.J.; Savarino, E.V.; Ford, A.C. Prevalence of symptoms of anxiety and depression in patients with inflammatory bowel disease: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2021, 6, 359–370. [Google Scholar] [CrossRef]
- García-Álvarez, L.; García-Portilla, M.P.; Caso, J.R.; de la Fuente-Tomás, L.; González-Blanco, L.; Sáiz Martínez, P.; Leza, J.C.; Bobes, J. Early versus late stage schizophrenia. What markers make the difference? World J. Biol. Psychiatry 2019, 20, 159–165. [Google Scholar] [CrossRef]
- Leza, J.C.; García-Bueno, B.; Bioque, M.; Arango, C.; Parellada, M.; Do, K.; O’Donnell, P.; Bernardo, M. Inflammation in schizophrenia: A question of balance. Neurosci. Biobehav. Rev. 2015, 55, 612–626. [Google Scholar] [CrossRef]
- Morrens, M.; Overloop, C.; Coppens, V.; Loots, E.; Van Den Noortgate, M.; Vandenameele, S.; Leboyer, M.; De Picker, L. The relationship between immune and cognitive dysfunction in mood and psychotic disorder: A systematic review and a meta-analysis. Mol. Psychiatry 2022, 27, 3237–3246. [Google Scholar] [CrossRef] [PubMed]
- Mongan, D.; Raj Susai, S.; Föcking, M.; Byrne, J.F.; Zammit, S.; Cannon, M.; Cotter, D.R. Associations between plasma inflammatory markers and psychotic disorder, depressive disorder and generalised anxiety disorder in early adulthood: A nested case-control study. Brain Behav. Immun. 2023, 111, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Severance, E.G.; Gressitt, K.L.; Stallings, C.R.; Origoni, A.E.; Khushalani, S.; Leweke, F.M.; Dickerson, F.B.; Yolken, R.H. Discordant patterns of bacterial translocation markers and implications for innate immune imbalances in schizophrenia. Schizophr. Res. 2013, 148, 130–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maes, M.; Kubera, M.; Leunis, J.C.; Berk, M. Increased IgA and IgM responses against gut commensals in chronic depression: Further evidence for increased bacterial translocation or leaky gut. J. Affect. Disord. 2012, 141, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Caso, J.R.; MacDowell, K.S.; González-Pinto, A.; García, S.; de Diego-Adeliño, J.; Carceller-Sindreu, M.; Sarramea, F.; Caballero-Villarraso, J.; Gracia-García, P.; De la Cámara, C.; et al. Gut microbiota, innate immune pathways, and inflammatory control mechanisms in patients with major depressive disorder. Transl. Psychiatry 2021, 11, 645. [Google Scholar] [CrossRef]
- Maes, M.; Kanchanatawan, B.; Sirivichayakul, S.; Carvalho, A.F. In Schizophrenia, Increased Plasma IgM/IgA Responses to Gut Commensal Bacteria Are Associated with Negative Symptoms, Neurocognitive Impairments, and the Deficit Phenotype. Neurotox. Res. 2019, 35, 684–698. [Google Scholar] [CrossRef]
- Martín-Hernández, D.; Caso, J.; Bris, Á.; Maus, S.; Madrigal, J.; García-Bueno, B.; MacDowell, K.; Alou, L.; Gómez-Lus, M.; Leza, J. Bacterial translocation affects intracellular neuroinflammatory pathways in a depression-like model in rats. Neuropharmacology 2016, 103, 122–133. [Google Scholar] [CrossRef]
- Martín-Hernández, D.; Gutiérrez, I.L.; González-Prieto, M.; MacDowell, K.S.; Robledo-Montaña, J.; Tendilla-Beltrán, H.; Calleja-Rodríguez, N.; Bris, G.; Ulecia-Morón, C.; Moreno, B.; et al. Sphk2 deletion is involved in structural abnormalities and Th17 response but does not aggravate colon inflammation induced by sub-chronic stress. Sci. Rep. 2022, 12, 4073. [Google Scholar] [CrossRef]
- Gárate, I.; García-Bueno, B.; Madrigal, J.L.M.; Bravo, L.; Berrocoso, E.; Caso, J.R.; Mico, J.A.; Leza, J.C. Origin and consequences of brain Toll-like receptor 4 pathway stimulation in an experimental model of depression. J. Neuroinflamm. 2011, 8, 151. [Google Scholar] [CrossRef] [Green Version]
- Hashioka, S.; Inoue, K.; Miyaoka, T.; Hayashida, M.; Wake, R.; Oh-Nishi, A.; Inagaki, M. The Possible Causal Link of Periodontitis to Neuropsychiatric Disorders: More Than Psychosocial Mechanisms. Int. J. Mol. Sci. 2019, 20, 3723. [Google Scholar] [CrossRef]
- Serrats, J.; Schiltz, J.C.; García-Bueno, B.; van Rooijen, N.; Reyes, T.M.; Sawchenko, P.E. Dual roles for perivascular macrophages in immune-to-brain signaling. Neuron 2010, 65, 94–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bravo, G.; Cedeño, R.R.; Casadevall, M.P.; Ramió-Torrentà, L. Sphingosine-1-Phosphate (S1P) and S1P Signaling Pathway Modulators, from Current Insights to Future Perspectives. Cells 2022, 11, 2058. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, S.; Milstien, S. The outs and the ins of sphingosine-1-phosphate in immunity. Nat. Rev. Immunol. 2011, 11, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, S.; Maczis, M.A.; Maceyka, M.; Milstien, S. New insights into functions of the sphingosine-1-phosphate transporter SPNS2. J. Lipid Res. 2019, 60, 484–489. [Google Scholar] [CrossRef] [Green Version]
- Nagahashi, M.; Takabe, K.; Terracina, K.P.; Soma, D.; Hirose, Y.; Kobayashi, T.; Matsuda, Y.; Wakai, T. Sphingosine-1-phosphate transporters as targets for cancer therapy. BioMed Res. Int. 2014, 2014, 651727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ihlefeld, K.; Vienken, H.; Claas, R.F.; Blankenbach, K.; Rudowski, A.; ter Braak, M.; Koch, A.; Van Veldhoven, P.P.; Pfeilschifter, J.; Meyer zu Heringdorf, D. Upregulation of ABC transporters contributes to chemoresistance of sphingosine 1-phosphate lyase-deficient fibroblasts. J. Lipid Res. 2015, 56, 60–69. [Google Scholar] [CrossRef] [Green Version]
- Vu, T.M.; Ishizu, A.N.; Foo, J.C.; Toh, X.R.; Zhang, F.; Whee, D.M.; Torta, F.; Cazenave-Gassiot, A.; Matsumura, T.; Kim, S.; et al. Mfsd2b is essential for the sphingosine-1-phosphate export in erythrocytes and platelets. Nature 2017, 550, 524–528. [Google Scholar] [CrossRef]
- Ouyang, J.; Shu, Z.; Chen, S.; Xiang, H.; Lu, H. The role of sphingosine 1-phosphate and its receptors in cardiovascular diseases. J. Cell. Mol. Med. 2020, 24, 10290–10301. [Google Scholar] [CrossRef]
- Wollny, T.; Wątek, M.; Durnaś, B.; Niemirowicz, K.; Piktel, E.; Żendzian-Piotrowska, M.; Góźdź, S.; Bucki, R. Sphingosine-1-Phosphate Metabolism and Its Role in the Development of Inflammatory Bowel Disease. Int. J. Mol. Sci. 2017, 18, 741. [Google Scholar] [CrossRef] [Green Version]
- Brinkmann, V.; Billich, A.; Baumruker, T.; Heining, P.; Schmouder, R.; Francis, G.; Aradhye, S.; Burtin, P. Fingolimod (FTY720): Discovery and development of an oral drug to treat multiple sclerosis. Nat. Rev. Drug Discov. 2010, 9, 883–897. [Google Scholar] [CrossRef]
- Bartke, N.; Hannun, Y.A. Bioactive sphingolipids: Metabolism and function. J. Lipid Res. 2009, 50, S91–S96. [Google Scholar] [CrossRef] [Green Version]
- Ueda, N. A Rheostat of Ceramide and Sphingosine-1-Phosphate as a Determinant of Oxidative Stress-Mediated Kidney Injury. Int. J. Mol. Sci. 2022, 23, 4010. [Google Scholar] [CrossRef]
- Parveen, F.; Bender, D.; Law, S.H.; Mishra, V.K.; Chen, C.C.; Ke, L.Y. Role of Ceramidases in Sphingolipid Metabolism and Human Diseases. Cells 2019, 8, 1573. [Google Scholar] [CrossRef] [Green Version]
- Hatoum, D.; Haddadi, N.; Lin, Y.; Nassif, N.T.; McGowan, E.M. Mammalian sphingosine kinase (SphK) isoenzymes and isoform expression: Challenges for SphK as an oncotarget. Oncotarget 2017, 8, 36898–36929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canals, D.; Clarke, C.J. Compartmentalization of Sphingolipid metabolism: Implications for signaling and therapy. Pharmacol. Ther. 2022, 232, 108005. [Google Scholar] [CrossRef] [PubMed]
- Cartier, A.; Hla, T. Sphingosine 1-phosphate: Lipid signaling in pathology and therapy. Science 2019, 366, eaar5551. [Google Scholar] [CrossRef] [PubMed]
- Kohama, T.; Olivera, A.; Edsall, L.; Nagiec, M.M.; Dickson, R.; Spiegel, S. Molecular cloning and functional characterization of murine sphingosine kinase. J. Biol. Chem. 1998, 273, 23722–23728. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Sugiura, M.; Nava, V.E.; Edsall, L.C.; Kono, K.; Poulton, S.; Milstien, S.; Kohama, T.; Spiegel, S. Molecular cloning and functional characterization of a novel mammalian sphingosine kinase type 2 isoform. J. Biol. Chem. 2000, 275, 19513–19520. [Google Scholar] [CrossRef] [Green Version]
- Maceyka, M.; Sankala, H.; Hait, N.C.; Le Stunff, H.; Liu, H.; Toman, R.; Collier, C.; Zhang, M.; Satin, L.S.; Merrill, A.H., Jr.; et al. SphK1 and SphK2, sphingosine kinase isoenzymes with opposing functions in sphingolipid metabolism. J. Biol. Chem. 2005, 280, 37118–37129. [Google Scholar] [CrossRef] [Green Version]
- Olivera, A.; Kohama, T.; Edsall, L.; Nava, V.; Cuvillier, O.; Poulton, S.; Spiegel, S. Sphingosine kinase expression increases intracellular sphingosine-1-phosphate and promotes cell growth and survival. J. Cell Biol. 1999, 147, 545–558. [Google Scholar] [CrossRef]
- Qiao, H.; Jiang, T.; Mu, P.; Chen, X.; Wen, X.; Hu, Z.; Tang, S.; Wen, J.; Deng, Y. Cell fate determined by the activation balance between PKR and SPHK1. Cell Death Differ. 2021, 28, 401–418. [Google Scholar] [CrossRef] [PubMed]
- Shida, D.; Takabe, K.; Kapitonov, D.; Milstien, S.; Spiegel, S. Targeting SphK1 as a new strategy against cancer. Curr. Drug Targets 2008, 9, 662–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, R.D.R.; Veerman, B.E.P.; Oh, J.; Tate, R.J.; Torta, F.; Cunningham, M.R.; Adams, D.R.; Pyne, S.; Pyne, N.J. A new model for regulation of sphingosine kinase 1 translocation to the plasma membrane in breast cancer cells. J. Biol. Chem. 2021, 296, 100674. [Google Scholar] [CrossRef]
- Dominguez, G.; Maddelein, M.L.; Pucelle, M.; Nicaise, Y.; Maurage, C.A.; Duyckaerts, C.; Cuvillier, O.; Delisle, M.B. Neuronal sphingosine kinase 2 subcellular localization is altered in Alzheimer’s disease brain. Acta Neuropathol. Commun. 2018, 6, 25. [Google Scholar] [CrossRef] [PubMed]
- Fyrst, H.; Saba, J.D. An update on sphingosine-1-phosphate and other sphingolipid mediators. Nat. Chem. Biol. 2010, 6, 489–497. [Google Scholar] [CrossRef]
- Nishino, S.; Yamashita, H.; Tamori, M.; Mashimo, M.; Yamagata, K.; Nakamura, H.; Murayama, T. Translocation and activation of sphingosine kinase 1 by ceramide-1-phosphate. J. Cell. Biochem. 2019, 120, 5396–5408. [Google Scholar] [CrossRef]
- Mohammed, S.; Vineetha, N.S.; James, S.; Aparna, J.S.; Babu Lankadasari, M.; Maeda, T.; Ghosh, A.; Saha, S.; Li, Q.Z.; Spiegel, S.; et al. Regulatory role of SphK1 in TLR7/9-dependent type I interferon response and autoimmunity. FASEB J. 2020, 34, 4329–4347. [Google Scholar] [CrossRef]
- Fakhr, Y.; Koshti, S.; Habibyan, Y.B.; Webster, K.; Hemmings, D.G. Tumor Necrosis Factor-α Induces a Preeclamptic-like Phenotype in Placental Villi via Sphingosine Kinase 1 Activation. Int. J. Mol. Sci. 2022, 23, 3750. [Google Scholar] [CrossRef]
- Diaz Escarcega, R.; McCullough, L.D.; Tsvetkov, A.S. The Functional Role of Sphingosine Kinase 2. Front. Mol. Biosci. 2021, 8, 683767. [Google Scholar] [CrossRef]
- Ikeda, M.; Kihara, A.; Igarashi, Y. Sphingosine-1-phosphate lyase SPL is an endoplasmic reticulum-resident, integral membrane protein with the pyridoxal 5’-phosphate binding domain exposed to the cytosol. Biochem. Biophys. Res. Commun. 2004, 325, 338–343. [Google Scholar] [CrossRef]
- Engel, N.; Adamus, A.; Frank, M.; Kraft, K.; Kühn, J.; Müller, P.; Nebe, B.; Kasten, A.; Seitz, G. First evidence of SGPL1 expression in the cell membrane silencing the extracellular S1P siren in mammary epithelial cells. PLoS ONE 2018, 13, e0196854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maharaj, A.; Williams, J.; Bradshaw, T.; Güran, T.; Braslavsky, D.; Casas, J.; Chan, L.F.; Metherell, L.A.; Prasad, R. Sphingosine-1-phosphate lyase (SGPL1) deficiency is associated with mitochondrial dysfunction. J. Steroid Biochem. Mol. Biol. 2020, 202, 105730. [Google Scholar] [CrossRef] [PubMed]
- Kihara, A.; Sano, T.; Iwaki, S.; Igarashi, Y. Transmembrane topology of sphingoid long-chain base-1-phosphate phosphatase, Lcb3p. Genes Cells 2003, 8, 525–535. [Google Scholar] [CrossRef]
- Mechtcheriakova, D.; Wlachos, A.; Sobanov, J.; Kopp, T.; Reuschel, R.; Bornancin, F.; Cai, R.; Zemann, B.; Urtz, N.; Stingl, G.; et al. Sphingosine 1-phosphate phosphatase 2 is induced during inflammatory responses. Cell. Signal. 2007, 19, 748–760. [Google Scholar] [CrossRef] [PubMed]
- Mandala, S.M. Sphingosine-1-phosphate phosphatases. Prostaglandins Other Lipid Mediat. 2001, 64, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Le Stunff, H.; Giussani, P.; Maceyka, M.; Lépine, S.; Milstien, S.; Spiegel, S. Recycling of sphingosine is regulated by the concerted actions of sphingosine-1-phosphate phosphohydrolase 1 and sphingosine kinase 2. J. Biol. Chem. 2007, 282, 34372–34380. [Google Scholar] [CrossRef] [Green Version]
- Quinville, B.M.; Deschenes, N.M.; Ryckman, A.E.; Walia, J.S. A Comprehensive Review: Sphingolipid Metabolism and Implications of Disruption in Sphingolipid Homeostasis. Int. J. Mol. Sci. 2021, 22, 5793. [Google Scholar] [CrossRef]
- Mendelson, K.; Evans, T.; Hla, T. Sphingosine 1-phosphate signalling. Development 2014, 141, 5–9. [Google Scholar] [CrossRef] [Green Version]
- Rivera, J.; Proia, R.L.; Olivera, A. The alliance of sphingosine-1-phosphate and its receptors in immunity. Nat. Rev. Immunol. 2008, 8, 753–763. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, T.; Hla, T. Structural and functional characteristics of S1P receptors. J. Cell. Biochem. 2004, 92, 913–922. [Google Scholar] [CrossRef]
- Sugimoto, N.; Takuwa, N.; Okamoto, H.; Sakurada, S.; Takuwa, Y. Inhibitory and stimulatory regulation of Rac and cell motility by the G12/13-Rho and Gi pathways integrated downstream of a single G protein-coupled sphingosine-1-phosphate receptor isoform. Mol. Cell. Biol. 2003, 23, 1534–1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pappu, R.; Schwab, S.R.; Cornelissen, I.; Pereira, J.P.; Regard, J.B.; Xu, Y.; Camerer, E.; Zheng, Y.W.; Huang, Y.; Cyster, J.G.; et al. Promotion of lymphocyte egress into blood and lymph by distinct sources of sphingosine-1-phosphate. Science 2007, 316, 295–298. [Google Scholar] [CrossRef]
- Kumar, A.; Saba, J.D. Regulation of Immune Cell Migration by Sphingosine-1-Phosphate. Cell. Mol. Biol. (OMICS) 2015, 61, 121. [Google Scholar] [PubMed]
- Jozefczuk, E.; Guzik, T.J.; Siedlinski, M. Significance of sphingosine-1-phosphate in cardiovascular physiology and pathology. Pharmacol. Res. 2020, 156, 104793. [Google Scholar] [CrossRef] [PubMed]
- Zhiyi, Z.; Chenqi, Z. Sphingosine-1-Phosphate and Rheumatoid Arthritis: Pathological Implications and Potential Therapeutic Targets. In Innovative Rheumatology; Hiroaki, M., Ed.; IntechOpen: Rijeka, Croatia, 2013. [Google Scholar]
- Techarang, T.; Jariyapong, P.; Punsawad, C. Role of sphingosine kinase and sphingosine-1-phosphate receptor in the liver pathology of mice infected with Plasmodium berghei ANKA. PLoS ONE 2022, 17, e0266055. [Google Scholar] [CrossRef]
- McGinley, M.P.; Cohen, J.A. Sphingosine 1-phosphate receptor modulators in multiple sclerosis and other conditions. Lancet 2021, 398, 1184–1194. [Google Scholar] [CrossRef]
- Chua, X.Y.; Ho, L.T.Y.; Xiang, P.; Chew, W.S.; Lam, B.W.S.; Chen, C.P.; Ong, W.Y.; Lai, M.K.P.; Herr, D.R. Preclinical and Clinical Evidence for the Involvement of Sphingosine 1-Phosphate Signaling in the Pathophysiology of Vascular Cognitive Impairment. Neuromol. Med. 2021, 23, 47–67. [Google Scholar] [CrossRef] [PubMed]
- Blaho, V.A.; Galvani, S.; Engelbrecht, E.; Liu, C.; Swendeman, S.L.; Kono, M.; Proia, R.L.; Steinman, L.; Han, M.H.; Hla, T. HDL-bound sphingosine-1-phosphate restrains lymphopoiesis and neuroinflammation. Nature 2015, 523, 342–346. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Kawabori, M.; Houkin, K. FTY720 (Fingolimod) Ameliorates Brain Injury through Multiple Mechanisms and is a Strong Candidate for Stroke Treatment. Curr. Med. Chem. 2020, 27, 2979–2993. [Google Scholar] [CrossRef]
- Blaho, V.A.; Hla, T. An update on the biology of sphingosine 1-phosphate receptors. J. Lipid Res. 2014, 55, 1596–1608. [Google Scholar] [CrossRef] [Green Version]
- Gorlino, C.V.; Ranocchia, R.P.; Harman, M.F.; García, I.A.; Crespo, M.I.; Morón, G.; Maletto, B.A.; Pistoresi-Palencia, M.C. Neutrophils exhibit differential requirements for homing molecules in their lymphatic and blood trafficking into draining lymph nodes. J. Immunol. 2014, 193, 1966–1974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walzer, T.; Chiossone, L.; Chaix, J.; Calver, A.; Carozzo, C.; Garrigue-Antar, L.; Jacques, Y.; Baratin, M.; Tomasello, E.; Vivier, E. Natural killer cell trafficking in vivo requires a dedicated sphingosine 1-phosphate receptor. Nat. Immunol. 2007, 8, 1337–1344. [Google Scholar] [CrossRef] [PubMed]
- Chiba, K.; Yanagawa, Y.; Masubuchi, Y.; Kataoka, H.; Kawaguchi, T.; Ohtsuki, M.; Hoshino, Y. FTY720, a novel immunosuppressant, induces sequestration of circulating mature lymphocytes by acceleration of lymphocyte homing in rats. I. FTY720 selectively decreases the number of circulating mature lymphocytes by acceleration of lymphocyte homing. J. Immunol. 1998, 160, 5037–5044. [Google Scholar] [CrossRef]
- Mandala, S.; Hajdu, R.; Bergstrom, J.; Quackenbush, E.; Xie, J.; Milligan, J.; Thornton, R.; Shei, G.J.; Card, D.; Keohane, C.; et al. Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science 2002, 296, 346–349. [Google Scholar] [CrossRef] [PubMed]
- Matloubian, M.; Lo, C.G.; Cinamon, G.; Lesneski, M.J.; Xu, Y.; Brinkmann, V.; Allende, M.L.; Proia, R.L.; Cyster, J.G. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 2004, 427, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Baeyens, A.A.L.; Schwab, S.R. Finding a Way Out: S1P Signaling and Immune Cell Migration. Annu. Rev. Immunol. 2020, 38, 759–784. [Google Scholar] [CrossRef]
- Hallisey, V.M.; Schwab, S.R. Get me out of here: Sphingosine 1-phosphate signaling and T cell exit from tissues during an immune response. Immunol. Rev. 2023. [Google Scholar] [CrossRef]
- Healy, L.M.; Antel, J.P. Sphingosine-1-Phosphate Receptors in the Central Nervous and Immune Systems. Curr. Drug Targets 2016, 17, 1841–1850. [Google Scholar] [CrossRef]
- Tsai, H.C.; Nguyen, K.; Hashemi, E.; Engleman, E.; Hla, T.; Han, M.H. Myeloid sphingosine-1-phosphate receptor 1 is important for CNS autoimmunity and neuroinflammation. J. Autoimmun. 2019, 105, 102290. [Google Scholar] [CrossRef]
- Weichand, B.; Weis, N.; Weigert, A.; Grossmann, N.; Levkau, B.; Brüne, B. Apoptotic cells enhance sphingosine-1-phosphate receptor 1 dependent macrophage migration. Eur. J. Immunol. 2013, 43, 3306–3313. [Google Scholar] [CrossRef]
- Oo, M.L.; Chang, S.H.; Thangada, S.; Wu, M.T.; Rezaul, K.; Blaho, V.; Hwang, S.I.; Han, D.K.; Hla, T. Engagement of S1P₁-degradative mechanisms leads to vascular leak in mice. J. Clin. Investig. 2011, 121, 2290–2300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Igawa, S.; Ohzono, A.; Pham, P.; Wang, Z.; Nakatsuji, T.; Dokoshi, T.; Di Nardo, A. Sphingosine 1-Phosphate Receptor 2 Is Central to Maintaining Epidermal Barrier Homeostasis. J. Investig. Dermatol. 2020, 141, 1188–1197. [Google Scholar] [CrossRef] [PubMed]
- Nussbaum, C.; Bannenberg, S.; Keul, P.; Gräler, M.H.; Gonçalves-de-Albuquerque, C.F.; Korhonen, H.; von Wnuck Lipinski, K.; Heusch, G.; de Castro Faria Neto, H.C.; Rohwedder, I.; et al. Sphingosine-1-phosphate receptor 3 promotes leukocyte rolling by mobilizing endothelial P-selectin. Nat. Commun. 2015, 6, 6416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryan, A.M.; Del Poeta, M. Sphingosine-1-phosphate receptors and innate immunity. Cell. Microbiol. 2018, 20, e12836. [Google Scholar] [CrossRef] [Green Version]
- Olesch, C.; Ringel, C.; Brüne, B.; Weigert, A. Beyond Immune Cell Migration: The Emerging Role of the Sphingosine-1-phosphate Receptor S1PR4 as a Modulator of Innate Immune Cell Activation. Mediat. Inflamm. 2017, 2017, 6059203. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Tian, Z.; Lu, Y.; Huang, Z.; Fan, Y.; Li, B.; Zheng, H.; Wu, X.; Wang, M.; Zhao, J.; et al. Sphingosine-1-Phosphate Receptor 4 Attenuates Neutrophilic Airway Inflammation in Experimental Asthma via Repressing Proinflammatory Macrophage Activation. Int. J. Biol. Sci. 2023, 19, 1597–1615. [Google Scholar] [CrossRef]
- Im, D.S.; Clemens, J.; Macdonald, T.L.; Lynch, K.R. Characterization of the human and mouse sphingosine 1-phosphate receptor, S1P5 (Edg-8): Structure-activity relationship of sphingosine1-phosphate receptors. Biochemistry 2001, 40, 14053–14060. [Google Scholar] [CrossRef]
- Debien, E.; Mayol, K.; Biajoux, V.; Daussy, C.; De Aguero, M.G.; Taillardet, M.; Dagany, N.; Brinza, L.; Henry, T.; Dubois, B.; et al. S1PR5 is pivotal for the homeostasis of patrolling monocytes. Eur. J. Immunol. 2013, 43, 1667–1675. [Google Scholar] [CrossRef] [Green Version]
- Chun, J.; Kihara, Y.; Jonnalagadda, D.; Blaho, V.A. Fingolimod: Lessons Learned and New Opportunities for Treating Multiple Sclerosis and Other Disorders. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 149–170. [Google Scholar] [CrossRef]
- Cohan, S.L.; Benedict, R.H.B.; Cree, B.A.C.; DeLuca, J.; Hua, L.H.; Chun, J. The Two Sides of Siponimod: Evidence for Brain and Immune Mechanisms in Multiple Sclerosis. CNS Drugs 2022, 36, 703–719. [Google Scholar] [CrossRef]
- van Echten-Deckert, G. The role of sphingosine 1-phosphate metabolism in brain health and disease. Pharmacol. Ther. 2023, 244, 108381. [Google Scholar] [CrossRef]
- Karunakaran, I.; Alam, S.; Jayagopi, S.; Frohberger, S.J.; Hansen, J.N.; Kuehlwein, J.; Hölbling, B.V.; Schumak, B.; Hübner, M.P.; Gräler, M.H.; et al. Neural sphingosine 1-phosphate accumulation activates microglia and links impaired autophagy and inflammation. Glia 2019, 67, 1859–1872. [Google Scholar] [CrossRef] [PubMed]
- Navone, S.E.; Campanella, R.; Guarnaccia, L.; Ouellet, J.A.; Locatelli, M.; Cordiglieri, C.; Gualtierotti, R.; Gaudino, C.; Ciniglio Appiani, G.; Luzzi, S.; et al. Inflammatory interactions between degenerated intervertebral discs and microglia: Implication of sphingosine-1-phosphate signaling. J. Orthop. Res. 2021, 39, 1479–1495. [Google Scholar] [CrossRef] [PubMed]
- Arseni, L.; Sharma, R.; Mack, N.; Nagalla, D.; Ohl, S.; Hielscher, T.; Singhal, M.; Pilz, R.; Augustin, H.; Sandhoff, R.; et al. Sphingosine-1-Phosphate Recruits Macrophages and Microglia and Induces a Pro-Tumorigenic Phenotype That Favors Glioma Progression. Cancers 2023, 15, 479. [Google Scholar] [CrossRef]
- Montarolo, F.; Martire, S.; Marnetto, F.; Valentino, P.; Valverde, S.; Capobianco, M.A.; Bertolotto, A. The Selective Agonist for Sphingosine-1-Phosphate Receptors Siponimod Increases the Expression Level of NR4A Genes in Microglia Cell Line. Curr. Issues Mol. Biol. 2022, 44, 1247–1256. [Google Scholar] [CrossRef]
- Sood, A.; Fernandes, V.; Preeti, K.; Khatri, D.K.; Singh, S.B. Sphingosine 1 phosphate lyase inhibition rescues cognition in diabetic mice by promoting anti-inflammatory microglia. Behav. Brain Res. 2023, 446, 114415. [Google Scholar] [CrossRef]
- Chen, Z.; Doyle, T.M.; Luongo, L.; Largent-Milnes, T.M.; Giancotti, L.A.; Kolar, G.; Squillace, S.; Boccella, S.; Walker, J.K.; Pendleton, A.; et al. Sphingosine-1-phosphate receptor 1 activation in astrocytes contributes to neuropathic pain. Proc. Natl. Acad. Sci. USA 2019, 116, 10557–10562. [Google Scholar] [CrossRef] [Green Version]
- Giordana, M.T.; Cavalla, P.; Uccelli, A.; Laroni, A.; Bandini, F.; Vercellino, M.; Mancardi, G. Overexpression of sphingosine-1-phosphate receptors on reactive astrocytes drives neuropathology of multiple sclerosis rebound after fingolimod discontinuation. Mult. Scler. J. 2018, 24, 1133–1137. [Google Scholar] [CrossRef]
- Rothhammer, V.; Kenison, J.E.; Tjon, E.; Takenaka, M.C.; de Lima, K.A.; Borucki, D.M.; Chao, C.C.; Wilz, A.; Blain, M.; Healy, L.; et al. Sphingosine 1-phosphate receptor modulation suppresses pathogenic astrocyte activation and chronic progressive CNS inflammation. Proc. Natl. Acad. Sci. USA 2017, 114, 2012–2017. [Google Scholar] [CrossRef]
- Shirakawa, H.; Katsumoto, R.; Iida, S.; Miyake, T.; Higuchi, T.; Nagashima, T.; Nagayasu, K.; Nakagawa, T.; Kaneko, S. Sphingosine-1-phosphate induces Ca(2+) signaling and CXCL1 release via TRPC6 channel in astrocytes. Glia 2017, 65, 1005–1016. [Google Scholar] [CrossRef] [Green Version]
- O’Sullivan, S.A.; O’Sullivan, C.; Healy, L.M.; Dev, K.K.; Sheridan, G.K. Sphingosine 1-phosphate receptors regulate TLR4-induced CXCL5 release from astrocytes and microglia. J. Neurochem. 2018, 144, 736–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaire, B.P.; Choi, J.W. Critical Roles of Lysophospholipid Receptors in Activation of Neuroglia and Their Neuroinflammatory Responses. Int. J. Mol. Sci. 2021, 22, 7864. [Google Scholar] [CrossRef] [PubMed]
- Bu, Y.; Wu, H.; Deng, R.; Wang, Y. Therapeutic Potential of SphK1 Inhibitors Based on Abnormal Expression of SphK1 in Inflammatory Immune Related-Diseases. Front. Pharmacol. 2021, 12, 733387. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Yang, Y.; Zhan, X.; Huang, Y.; Fu, Y.; Zhang, Z.; Liu, H.; Zhang, L.; Li, Y.; Wen, Q.; et al. Vitamin B6 prevents excessive inflammation by reducing accumulation of sphingosine-1-phosphate in a sphingosine-1-phosphate lyase-dependent manner. J. Cell. Mol. Med. 2020, 24, 13129–13138. [Google Scholar] [CrossRef]
- Hahn, C.; Tyka, K.; Saba, J.D.; Lenzen, S.; Gurgul-Convey, E. Overexpression of sphingosine-1-phosphate lyase protects insulin-secreting cells against cytokine toxicity. J. Biol. Chem. 2017, 292, 20292–20304. [Google Scholar] [CrossRef] [Green Version]
- Degagné, E.; Pandurangan, A.; Bandhuvula, P.; Kumar, A.; Eltanawy, A.; Zhang, M.; Yoshinaga, Y.; Nefedov, M.; de Jong, P.J.; Fong, L.G.; et al. Sphingosine-1-phosphate lyase downregulation promotes colon carcinogenesis through STAT3-activated microRNAs. J. Clin. Investig. 2014, 124, 5368–5384. [Google Scholar] [CrossRef] [Green Version]
- Stepanovska, B.; Lange, A.I.; Schwalm, S.; Pfeilschifter, J.; Coldewey, S.M.; Huwiler, A. Downregulation of S1P Lyase Improves Barrier Function in Human Cerebral Microvascular Endothelial Cells Following an Inflammatory Challenge. Int. J. Mol. Sci. 2020, 21, 1240. [Google Scholar] [CrossRef] [Green Version]
- Schwiebs, A.; Herrero San Juan, M.; Schmidt, K.G.; Wiercinska, E.; Anlauf, M.; Ottenlinger, F.; Thomas, D.; Elwakeel, E.; Weigert, A.; Farin, H.F.; et al. Cancer-induced inflammation and inflammation-induced cancer in colon: A role for S1P lyase. Oncogene 2019, 38, 4788–4803. [Google Scholar] [CrossRef]
- Huang, W.C.; Liang, J.; Nagahashi, M.; Avni, D.; Yamada, A.; Maceyka, M.; Wolen, A.R.; Kordula, T.; Milstien, S.; Takabe, K.; et al. Sphingosine-1-phosphate phosphatase 2 promotes disruption of mucosal integrity, and contributes to ulcerative colitis in mice and humans. FASEB J. 2016, 30, 2945–2958. [Google Scholar] [CrossRef] [Green Version]
- Prager, B.; Spampinato, S.F.; Ransohoff, R.M. Sphingosine 1-phosphate signaling at the blood-brain barrier. Trends Mol. Med. 2015, 21, 354–363. [Google Scholar] [CrossRef]
- Kiyozuka, K.; Zhao, X.; Konishi, A.; Minamishima, Y.A.; Obinata, H. Apolipoprotein M supports S1P production and conservation and mediates prolonged Akt activation via S1PR1 and S1PR3. J. Biochem. 2023, mvad037. [Google Scholar] [CrossRef] [PubMed]
- Mathiesen Janiurek, M.; Soylu-Kucharz, R.; Christoffersen, C.; Kucharz, K.; Lauritzen, M. Apolipoprotein M-bound sphingosine-1-phosphate regulates blood-brain barrier paracellular permeability and transcytosis. eLife 2019, 8, e49405. [Google Scholar] [CrossRef] [PubMed]
- Wilkerson, B.A.; Grass, G.D.; Wing, S.B.; Argraves, W.S.; Argraves, K.M. Sphingosine 1-phosphate (S1P) carrier-dependent regulation of endothelial barrier: High density lipoprotein (HDL)-S1P prolongs endothelial barrier enhancement as compared with albumin-S1P via effects on levels, trafficking, and signaling of S1P1. J. Biol. Chem. 2012, 287, 44645–44653. [Google Scholar] [CrossRef] [Green Version]
- Spampinato, S.F.; Merlo, S.; Sano, Y.; Kanda, T.; Sortino, M.A. Protective effect of the sphingosine-1 phosphate receptor agonist siponimod on disrupted blood brain barrier function. Biochem. Pharmacol. 2021, 186, 114465. [Google Scholar] [CrossRef] [PubMed]
- Yanagida, K.; Liu, C.H.; Faraco, G.; Galvani, S.; Smith, H.K.; Burg, N.; Anrather, J.; Sanchez, T.; Iadecola, C.; Hla, T. Size-selective opening of the blood-brain barrier by targeting endothelial sphingosine 1-phosphate receptor 1. Proc. Natl. Acad. Sci. USA 2017, 114, 4531–4536. [Google Scholar] [CrossRef]
- Cannon, R.E.; Peart, J.C.; Hawkins, B.T.; Campos, C.R.; Miller, D.S. Targeting blood-brain barrier sphingolipid signaling reduces basal P-glycoprotein activity and improves drug delivery to the brain. Proc. Natl. Acad. Sci. USA 2012, 109, 15930–15935. [Google Scholar] [CrossRef]
- Cartwright, T.A.; Campos, C.R.; Cannon, R.E.; Miller, D.S. Mrp1 is essential for sphingolipid signaling to p-glycoprotein in mouse blood-brain and blood-spinal cord barriers. J. Cereb. Blood Flow Metab. 2013, 33, 381–388. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zheng, Y.; Wang, F.; Zhong, J.; Zhao, T.; Xie, Q.; Zhu, T.; Ma, F.; Tang, Q.; Zhou, B.; et al. Mfsd2a and Spns2 are essential for sphingosine-1-phosphate transport in the formation and maintenance of the blood-brain barrier. Sci. Adv. 2020, 6, eaay8627. [Google Scholar] [CrossRef]
- Zhu, C.; Lee, J.Y.; Woo, J.Z.; Xu, L.; Nguyenla, X.; Yamashiro, L.H.; Ji, F.; Biering, S.B.; Van Dis, E.; Gonzalez, F.; et al. An intranasal ASO therapeutic targeting SARS-CoV-2. Nat. Commun. 2022, 13, 4503. [Google Scholar] [CrossRef]
- Nitzsche, A.; Poittevin, M.; Benarab, A.; Bonnin, P.; Faraco, G.; Uchida, H.; Favre, J.; Garcia-Bonilla, L.; Garcia, M.C.L.; Léger, P.L.; et al. Endothelial S1P. Circ. Res. 2021, 128, 363–382. [Google Scholar] [CrossRef]
- Pitteri, M.; Magliozzi, R.; Bajrami, A.; Camera, V.; Calabrese, M. Potential neuroprotective effect of Fingolimod in multiple sclerosis and its association with clinical variables. Expert Opin. Pharmacother. 2018, 19, 387–395. [Google Scholar] [CrossRef]
- Leo, A.; Citraro, R.; Marra, R.; Palma, E.; Paola, E.D.D.; Constanti, A.; De Sarro, G.; Russo, E. The Sphingosine 1-Phosphate Signaling Pathway in Epilepsy: A Possible Role for the Immunomodulator Drug Fingolimod in Epilepsy Treatment. CNS Neurol. Disord. Drug Targets 2017, 16, 311–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, S.F.; Bowen, J.D.; Reder, A.T. The Direct Effects of Fingolimod in the Central Nervous System: Implications for Relapsing Multiple Sclerosis. CNS Drugs 2016, 30, 135–147. [Google Scholar] [CrossRef] [Green Version]
- Gaire, B.P.; Lee, C.H.; Sapkota, A.; Lee, S.Y.; Chun, J.; Cho, H.J.; Nam, T.G.; Choi, J.W. Identification of Sphingosine 1-Phosphate Receptor Subtype 1 (S1P. Mol. Neurobiol. 2018, 55, 2320–2332. [Google Scholar] [CrossRef]
- Mandeville, E.T.; Li, W.; Quinto-Alemany, D.; Zhang, F.; Esposito, E.; Nakano, T.; Mandeville, J.B.; Lee, J.; Park, J.H.; Arai, K.; et al. Fingolimod Does Not Reduce Infarction After Focal Cerebral Ischemia in Mice During Active or Inactive Circadian Phases. Stroke 2022, 53, 3741–3750. [Google Scholar] [CrossRef] [PubMed]
- Xiang, P.; Chew, W.S.; Seow, W.L.; Lam, B.W.S.; Ong, W.Y.; Herr, D.R. The S1P. Neurochem. Int. 2021, 146, 105018. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Dai, L.; Mu, J.; Wang, X.; Hong, Y.; Zhu, C.; Jin, L.; Li, S. S1PR2 antagonist alleviates oxidative stress-enhanced brain endothelial permeability by attenuating p38 and Erk1/2-dependent cPLA. Cell. Signal. 2019, 53, 151–161. [Google Scholar] [CrossRef]
- Seyedsadr, M.S.; Weinmann, O.; Amorim, A.; Ineichen, B.V.; Egger, M.; Mirnajafi-Zadeh, J.; Becher, B.; Javan, M.; Schwab, M.E. Inactivation of sphingosine-1-phosphate receptor 2 (S1PR2) decreases demyelination and enhances remyelination in animal models of multiple sclerosis. Neurobiol. Dis. 2019, 124, 189–201. [Google Scholar] [CrossRef]
- Sapkota, A.; Gaire, B.P.; Kang, M.G.; Choi, J.W. S1P. Sci. Rep. 2019, 9, 12106. [Google Scholar] [CrossRef] [Green Version]
- Wan, Y.; Jin, H.J.; Zhu, Y.Y.; Fang, Z.; Mao, L.; He, Q.; Xia, Y.P.; Li, M.; Li, Y.; Chen, X.; et al. MicroRNA-149-5p regulates blood-brain barrier permeability after transient middle cerebral artery occlusion in rats by targeting S1PR2 of pericytes. FASEB J. 2018, 32, 3133–3148. [Google Scholar] [CrossRef] [Green Version]
- Yue, X.; Jin, H.; Liu, H.; Rosenberg, A.J.; Klein, R.S.; Tu, Z. A potent and selective C-11 labeled PET tracer for imaging sphingosine-1-phosphate receptor 2 in the CNS demonstrates sexually dimorphic expression. Org. Biomol. Chem. 2015, 13, 7928–7939. [Google Scholar] [CrossRef] [Green Version]
- Cruz-Orengo, L.; Daniels, B.P.; Dorsey, D.; Basak, S.A.; Grajales-Reyes, J.G.; McCandless, E.E.; Piccio, L.; Schmidt, R.E.; Cross, A.H.; Crosby, S.D.; et al. Enhanced sphingosine-1-phosphate receptor 2 expression underlies female CNS autoimmunity susceptibility. J. Clin. Investig. 2014, 124, 2571–2584. [Google Scholar] [CrossRef] [Green Version]
- Dusaban, S.S.; Chun, J.; Rosen, H.; Purcell, N.H.; Brown, J.H. Sphingosine 1-phosphate receptor 3 and RhoA signaling mediate inflammatory gene expression in astrocytes. J. Neuroinflamm. 2017, 14, 111. [Google Scholar] [CrossRef] [Green Version]
- Murakami, A.; Takasugi, H.; Ohnuma, S.; Koide, Y.; Sakurai, A.; Takeda, S.; Hasegawa, T.; Sasamori, J.; Konno, T.; Hayashi, K.; et al. Sphingosine 1-phosphate (S1P) regulates vascular contraction via S1P3 receptor: Investigation based on a new S1P3 receptor antagonist. Mol. Pharmacol. 2010, 77, 704–713. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Gao, Q.; Wang, F.; Peng, Q.; Wang, G.; Wei, Q.; Lei, S.; Zhao, S.; Zhang, L.; Guo, F. Sphingosine-1-phosphate receptor 3 is implicated in BBB injury via the CCL2-CCR2 axis following acute intracerebral hemorrhage. CNS Neurosci. Ther. 2021, 27, 674–686. [Google Scholar] [CrossRef] [PubMed]
- Gaire, B.P.; Song, M.R.; Choi, J.W. Sphingosine 1-phosphate receptor subtype 3 (S1P. J. Neuroinflamm. 2018, 15, 284. [Google Scholar] [CrossRef] [Green Version]
- Hansen, L.; Lohfink, N.; Vutukuri, R.; Kestner, R.I.; Trautmann, S.; Hecht, M.; Wagner, P.V.; Spitzer, D.; Khel, M.I.; Macas, J.; et al. Endothelial Sphingosine-1-Phosphate Receptor 4 Regulates Blood-Brain Barrier Permeability and Promotes a Homeostatic Endothelial Phenotype. J. Neurosci. 2022, 42, 1908–1929. [Google Scholar] [CrossRef]
- van Doorn, R.; Lopes Pinheiro, M.A.; Kooij, G.; Lakeman, K.; van het Hof, B.; van der Pol, S.M.; Geerts, D.; van Horssen, J.; van der Valk, P.; van der Kam, E.; et al. Sphingosine 1-phosphate receptor 5 mediates the immune quiescence of the human brain endothelial barrier. J. Neuroinflamm. 2012, 9, 133. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Zhang, X.; Liu, Y.; Li, Z.; Wei, R.; Zhang, Y.; Zhang, R.; Khan, S.; Yong, V.W.; Xue, M. Neuroprotection by Ozanimod Following Intracerebral Hemorrhage in Mice. Front. Mol. Neurosci. 2022, 15, 927150. [Google Scholar] [CrossRef]
- Di Pardo, A.; Castaldo, S.; Amico, E.; Pepe, G.; Marracino, F.; Capocci, L.; Giovannelli, A.; Madonna, M.; van Bergeijk, J.; Buttari, F.; et al. Stimulation of S1PR5 with A-971432, a selective agonist, preserves blood-brain barrier integrity and exerts therapeutic effect in an animal model of Huntington’s disease. Hum. Mol. Genet. 2018, 27, 2490–2501. [Google Scholar] [CrossRef] [Green Version]
- Lima, S.; Milstien, S.; Spiegel, S. Sphingosine and Sphingosine Kinase 1 Involvement in Endocytic Membrane Trafficking. J. Biol. Chem. 2017, 292, 3074–3088. [Google Scholar] [CrossRef] [Green Version]
- Wacker, B.K.; Freie, A.B.; Perfater, J.L.; Gidday, J.M. Junctional protein regulation by sphingosine kinase 2 contributes to blood-brain barrier protection in hypoxic preconditioning-induced cerebral ischemic tolerance. J. Cereb. Blood Flow Metab. 2012, 32, 1014–1023. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhong, W.; Cheng, Q.; Xiao, C.; Xu, J.; Su, Z.; Su, H.; Liu, X. Histone methyltransferase Smyd2 contributes to blood-brain barrier breakdown in stroke. Clin. Transl. Med. 2022, 12, e761. [Google Scholar] [CrossRef]
- Foster, C.A.; Mechtcheriakova, D.; Storch, M.K.; Balatoni, B.; Howard, L.M.; Bornancin, F.; Wlachos, A.; Sobanov, J.; Kinnunen, A.; Baumruker, T. FTY720 rescue therapy in the dark agouti rat model of experimental autoimmune encephalomyelitis: Expression of central nervous system genes and reversal of blood-brain-barrier damage. Brain Pathol. 2009, 19, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Greenspon, J.; Li, R.; Xiao, L.; Rao, J.N.; Sun, R.; Strauch, E.D.; Shea-Donohue, T.; Wang, J.Y.; Turner, D.J. Sphingosine-1-phosphate regulates the expression of adherens junction protein E-cadherin and enhances intestinal epithelial cell barrier function. Dig. Dis. Sci. 2011, 56, 1342–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenhoffer, G.T.; Loftus, P.D.; Yoshigi, M.; Otsuna, H.; Chien, C.B.; Morcos, P.A.; Rosenblatt, J. Crowding induces live cell extrusion to maintain homeostatic cell numbers in epithelia. Nature 2012, 484, 546–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, T.; Lin, R.; Jin, S.; Chen, R.; Xue, H.; Ye, H.; Huang, Z. The Sphingosine-1-Phosphate/Sphingosine-1-Phosphate Receptor 2 Axis in Intestinal Epithelial Cells Regulates Intestinal Barrier Function During Intestinal Epithelial Cells-CD4+T-Cell Interactions. Cell. Physiol. Biochem. 2018, 48, 1188–1200. [Google Scholar] [CrossRef]
- Dong, J.; Wang, H.; Zhao, J.; Sun, J.; Zhang, T.; Zuo, L.; Zhu, W.; Gong, J.; Li, Y.; Gu, L.; et al. SEW2871 protects from experimental colitis through reduced epithelial cell apoptosis and improved barrier function in interleukin-10 gene-deficient mice. Immunol. Res. 2015, 61, 303–311. [Google Scholar] [CrossRef]
- Montrose, D.C.; Scherl, E.J.; Bosworth, B.P.; Zhou, X.K.; Jung, B.; Dannenberg, A.J.; Hla, T. S1P₁ localizes to the colonic vasculature in ulcerative colitis and maintains blood vessel integrity. J. Lipid Res. 2013, 54, 843–851. [Google Scholar] [CrossRef] [Green Version]
- Karuppuchamy, T.; Behrens, E.H.; González-Cabrera, P.; Sarkisyan, G.; Gima, L.; Boyer, J.D.; Bamias, G.; Jedlicka, P.; Veny, M.; Clark, D.; et al. Sphingosine-1-phosphate receptor-1 (S1P(1)) is expressed by lymphocytes, dendritic cells, and endothelium and modulated during inflammatory bowel disease. Mucosal Immunol. 2017, 10, 162–171. [Google Scholar] [CrossRef] [Green Version]
- Lépine, J.P.; Briley, M. The increasing burden of depression. Neuropsychiatr. Dis. Treat. 2011, 7, 3–7. [Google Scholar] [CrossRef] [Green Version]
- Bauer, M.E.; Teixeira, A.L. Inflammation in psychiatric disorders: What comes first? Ann. N. Y. Acad. Sci. 2019, 1437, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Valkanova, V.; Ebmeier, K.P.; Allan, C.L. CRP, IL-6 and depression: A systematic review and meta-analysis of longitudinal studies. J. Affect. Disord. 2013, 150, 736–744. [Google Scholar] [CrossRef] [PubMed]
- Chatzikonstantinou, S.; Poulidou, V.; Arnaoutoglou, M.; Kazis, D.; Heliopoulos, I.; Grigoriadis, N.; Boziki, M. Signaling through the S1P-S1PR Axis in the Gut, the Immune and the Central Nervous System in Multiple Sclerosis: Implication for Pathogenesis and Treatment. Cells 2021, 10, 3217. [Google Scholar] [CrossRef] [PubMed]
- Willner, P. The chronic mild stress (CMS) model of depression: History, evaluation and usage. Neurobiol. Stress 2016, 6, 78–93. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Gan, X.; Zhou, H.; Zhou, H.; Pu, S.; Long, X.; Ren, C.; Feng, T.; Tang, H. Fingolimod suppressed the chronic unpredictable mild stress-induced depressive-like behaviors via affecting microglial and NLRP3 inflammasome activation. Life Sci. 2020, 263, 118582. [Google Scholar] [CrossRef]
- Shan, B.; Ai, Z.; Zeng, S.; Song, Y.; Song, J.; Zeng, Q.; Liao, Z.; Wang, T.; Huang, C.; Su, D. Gut microbiome-derived lactate promotes to anxiety-like behaviors through GPR81 receptor-mediated lipid metabolism pathway. Psychoneuroendocrinology 2020, 117, 104699. [Google Scholar] [CrossRef]
- Martín-Hernández, D.; Martínez, M.; Robledo-Montaña, J.; Muñoz-López, M.; Virto, L.; Ambrosio, N.; Marín, M.J.; Montero, E.; Herrera, D.; Sanz, M.; et al. Neuroinflammation related to the blood-brain barrier and sphingosine-1-phosphate in a pre-clinical model of periodontal diseases and depression in rats. J. Clin. Periodontol. 2023, 50, 642–656. [Google Scholar] [CrossRef]
- Corbett, B.F.; Luz, S.; Arner, J.; Pearson-Leary, J.; Sengupta, A.; Taylor, D.; Gehrman, P.; Ross, R.; Bhatnagar, S. Sphingosine-1-phosphate receptor 3 in the medial prefrontal cortex promotes stress resilience by reducing inflammatory processes. Nat. Commun. 2019, 10, 3146. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Desse, S.; Martinez, A.; Worthen, R.J.; Jope, R.S.; Beurel, E. TNFα disrupts blood brain barrier integrity to maintain prolonged depressive-like behavior in mice. Brain Behav. Immun. 2018, 69, 556–567. [Google Scholar] [CrossRef]
- Miller, A.H.; Raison, C.L. The role of inflammation in depression: From evolutionary imperative to modern treatment target. Nat. Rev. Immunol. 2016, 16, 22–34. [Google Scholar] [CrossRef] [Green Version]
- Parenti, I.; Rabaneda, L.G.; Schoen, H.; Novarino, G. Neurodevelopmental Disorders: From Genetics to Functional Pathways. Trends Neurosci. 2020, 43, 608–621. [Google Scholar] [CrossRef]
- Reemst, K.; Noctor, S.C.; Lucassen, P.J.; Hol, E.M. The Indispensable Roles of Microglia and Astrocytes during Brain Development. Front. Hum. Neurosci. 2016, 10, 566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Tilborg, E.; de Theije, C.G.M.; van Hal, M.; Wagenaar, N.; de Vries, L.S.; Benders, M.J.; Rowitch, D.H.; Nijboer, C.H. Origin and dynamics of oligodendrocytes in the developing brain: Implications for perinatal white matter injury. Glia 2018, 66, 221–238. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.J.; O’Donovan, M.C. Schizophrenia and the neurodevelopmental continuum:evidence from genomics. World Psychiatry 2017, 16, 227–235. [Google Scholar] [CrossRef] [Green Version]
- Barendse, M.E.A.; Lara, G.A.; Guyer, A.E.; Swartz, J.R.; Taylor, S.L.; Shirtcliff, E.A.; Lamb, S.T.; Miller, C.; Ng, J.; Yu, G.; et al. Sex and pubertal influences on the neurodevelopmental underpinnings of schizophrenia: A case for longitudinal research on adolescents. Schizophr. Res. 2023, 252, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Cetin-Karayumak, S.; Di Biase, M.A.; Chunga, N.; Reid, B.; Somes, N.; Lyall, A.E.; Kelly, S.; Solgun, B.; Pasternak, O.; Vangel, M.; et al. White matter abnormalities across the lifespan of schizophrenia: A harmonized multi-site diffusion MRI study. Mol. Psychiatry 2020, 25, 3208–3219. [Google Scholar] [CrossRef]
- Fessel, J. Abnormal oligodendrocyte function in schizophrenia explains the long latent interval in some patients. Transl. Psychiatry 2022, 12, 120. [Google Scholar] [CrossRef]
- Roggeri, A.; Schepers, M.; Tiane, A.; Rombaut, B.; van Veggel, L.; Hellings, N.; Prickaerts, J.; Pittaluga, A.; Vanmierlo, T. Sphingosine-1-Phosphate Receptor Modulators and Oligodendroglial Cells: Beyond Immunomodulation. Int. J. Mol. Sci. 2020, 21, 7537. [Google Scholar] [CrossRef]
- Esaki, K.; Balan, S.; Iwayama, Y.; Shimamoto-Mitsuyama, C.; Hirabayashi, Y.; Dean, B.; Yoshikawa, T. Evidence for Altered Metabolism of Sphingosine-1-Phosphate in the Corpus Callosum of Patients with Schizophrenia. Schizophr. Bull. 2020, 46, 1172–1181. [Google Scholar] [CrossRef]
- Chand, G.B.; Jiang, H.; Miller, J.P.; Rhodes, C.H.; Tu, Z.; Wong, D.F. Differential Sphingosine-1-Phosphate Receptor-1 Protein Expression in the Dorsolateral Prefrontal Cortex Between Schizophrenia Type 1 and Type 2. Front. Psychiatry 2022, 13, 827981. [Google Scholar] [CrossRef]
- Mariaselvam, C.M.; Wu, C.-L.; Boukouaci, W.; Richard, J.-R.; Barau, C.; Le Corvoisier, P.; Group, O.S. The Complement C4 Genetic Diversity in First Episode Psychosis of the OPTiMiSE Cohort. Schizophr. Bull. Open 2021, 2, sgab003. [Google Scholar] [CrossRef]
- Rey, R.; Suaud-Chagny, M.F.; Bohec, A.L.; Dorey, J.M.; d’Amato, T.; Tamouza, R.; Leboyer, M. Overexpression of complement component C4 in the dorsolateral prefrontal cortex, parietal cortex, superior temporal gyrus and associative striatum of patients with schizophrenia. Brain Behav. Immun. 2020, 90, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Soteros, B.M.; Sia, G.M. Complement and microglia dependent synapse elimination in brain development. WIREs Mech. Dis. 2022, 14, e1545. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Chung, W.S. Glial Control of Synapse Number in Healthy and Diseased Brain. Front. Cell. Neurosci. 2019, 13, 42. [Google Scholar] [CrossRef] [PubMed]
- Riganti, L.; Antonucci, F.; Gabrielli, M.; Prada, I.; Giussani, P.; Viani, P.; Valtorta, F.; Menna, E.; Matteoli, M.; Verderio, C. Sphingosine-1-Phosphate (S1P) Impacts Presynaptic Functions by Regulating Synapsin I Localization in the Presynaptic Compartment. J. Neurosci. 2016, 36, 4624–4634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osimo, E.F.; Beck, K.; Reis Marques, T.; Howes, O.D. Synaptic loss in schizophrenia: A meta-analysis and systematic review of synaptic protein and mRNA measures. Mol. Psychiatry 2019, 24, 549–561. [Google Scholar] [CrossRef]
- Pépin, É.; Jalinier, T.; Lemieux, G.L.; Massicotte, G.; Cyr, M. Sphingosine-1-Phosphate Receptors Modulators Decrease Signs of Neuroinflammation and Prevent Parkinson’s Disease Symptoms in the 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine Mouse Model. Front. Pharmacol. 2020, 11, 77. [Google Scholar] [CrossRef] [Green Version]
- McCutcheon, R.A.; Abi-Dargham, A.; Howes, O.D. Schizophrenia, Dopamine and the Striatum: From Biology to Symptoms. Trends Neurosci. 2019, 42, 205–220. [Google Scholar] [CrossRef] [Green Version]
- Spampinato, S.F.; Obermeier, B.; Cotleur, A.; Love, A.; Takeshita, Y.; Sano, Y.; Kanda, T.; Ransohoff, R.M. Sphingosine 1 Phosphate at the Blood Brain Barrier: Can the Modulation of S1P Receptor 1 Influence the Response of Endothelial Cells and Astrocytes to Inflammatory Stimuli? PLoS ONE 2015, 10, e0133392. [Google Scholar] [CrossRef] [Green Version]
- Notter, T. Astrocytes in schizophrenia. Brain Neurosci. Adv. 2021, 5, 23982128211009148. [Google Scholar] [CrossRef] [PubMed]
- Batiuk, M.Y.; Martirosyan, A.; Wahis, J.; de Vin, F.; Marneffe, C.; Kusserow, C.; Koeppen, J.; Viana, J.F.; Oliveira, J.F.; Voet, T.; et al. Identification of region-specific astrocyte subtypes at single cell resolution. Nat. Commun. 2020, 11, 1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldwaser, E.L.; Swanson, R.L.; Arroyo, E.J.; Venkataraman, V.; Kosciuk, M.C.; Nagele, R.G.; Hong, L.E.; Acharya, N.K. A Preliminary Report: The Hippocampus and Surrounding Temporal Cortex of Patients with Schizophrenia Have Impaired Blood-Brain Barrier. Front. Hum. Neurosci. 2022, 16, 836980. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, B.; Wu, C.; Wang, J.; Sun, M. Autism Spectrum Disorder: Neurodevelopmental Risk Factors, Biological Mechanism, and Precision Therapy. Int. J. Mol. Sci. 2023, 24, 1819. [Google Scholar] [CrossRef]
- Wang, H.; Liang, S.; Wang, M.; Gao, J.; Sun, C.; Wang, J.; Xia, W.; Wu, S.; Sumner, S.J.; Zhang, F.; et al. Potential serum biomarkers from a metabolomics study of autism. J. Psychiatry Neurosci. 2016, 41, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Kurochkin, I.; Khrameeva, E.; Tkachev, A.; Stepanova, V.; Vanyushkina, A.; Stekolshchikova, E.; Li, Q.; Zubkov, D.; Shichkova, P.; Halene, T.; et al. Metabolome signature of autism in the human prefrontal cortex. Commun. Biol. 2019, 2, 234. [Google Scholar] [CrossRef] [Green Version]
- Almandil, N.B.; Alismail, M.A.; Alsuwat, H.S.; AlSulaiman, A.; AbdulAzeez, S.; Borgio, J.F. Exome-wide analysis identify multiple variations in olfactory receptor genes (OR12D2 and OR5V1) associated with autism spectrum disorder in Saudi females. Front. Med. 2023, 10, 1051039. [Google Scholar] [CrossRef]
- Wu, H.; Zhang, Q.; Gao, J.; Sun, C.; Wang, J.; Xia, W.; Cao, Y.; Hao, Y.; Wu, L. Modulation of sphingosine 1-phosphate (S1P) attenuates spatial learning and memory impairments in the valproic acid rat model of autism. Psychopharmacology 2018, 235, 873–886. [Google Scholar] [CrossRef]
- Endo, N.; Makinodan, M.; Somayama, N.; Komori, T.; Kishimoto, T.; Nishi, M. Characterization of behavioral phenotypes in the BTBR T(+) Itpr3(tf)/J mouse model of autism spectrum disorder under social housing conditions using the multiple animal positioning system. Exp. Anim. 2019, 68, 319–330. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Cerdeño, V. Dendrite and spine modifications in autism and related neurodevelopmental disorders in patients and animal models. Dev. Neurobiol. 2017, 77, 393–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koevoet, D.; Deschamps, P.K.H.; Kenemans, J.L. Catecholaminergic and cholinergic neuromodulation in autism spectrum disorder: A comparison to attention-deficit hyperactivity disorder. Front. Neurosci. 2022, 16, 1078586. [Google Scholar] [CrossRef]
- Henriquez-Henriquez, M.; Acosta, M.T.; Martinez, A.F.; Vélez, J.I.; Lopera, F.; Pineda, D.; Palacio, J.D.; Quiroga, T.; Worgall, T.S.; Deckelbaum, R.J.; et al. Mutations in sphingolipid metabolism genes are associated with ADHD. Transl. Psychiatry 2020, 10, 231. [Google Scholar] [CrossRef] [PubMed]
- Henríquez-Henríquez, M.P.; Solari, S.; Quiroga, T.; Kim, B.I.; Deckelbaum, R.J.; Worgall, T.S. Low serum sphingolipids in children with attention deficit-hyperactivity disorder. Front. Neurosci. 2015, 9, 300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunkhorst-Kanaan, N.; Trautmann, S.; Schreiber, Y.; Thomas, D.; Kittel-Schneider, S.; Gurke, R.; Geisslinger, G.; Reif, A.; Tegeder, I. Sphingolipid and Endocannabinoid Profiles in Adult Attention Deficit Hyperactivity Disorder. Biomedicines 2021, 9, 1173. [Google Scholar] [CrossRef] [PubMed]
- Gergely, P.; Nuesslein-Hildesheim, B.; Guerini, D.; Brinkmann, V.; Traebert, M.; Bruns, C.; Pan, S.; Gray, N.S.; Hinterding, K.; Cooke, N.G.; et al. The selective sphingosine 1-phosphate receptor modulator BAF312 redirects lymphocyte distribution and has species-specific effects on heart rate. Br. J. Pharmacol. 2012, 167, 1035–1047. [Google Scholar] [CrossRef] [Green Version]
- Scott, F.L.; Clemons, B.; Brooks, J.; Brahmachary, E.; Powell, R.; Dedman, H.; Desale, H.G.; Timony, G.A.; Martinborough, E.; Rosen, H.; et al. Ozanimod (RPC1063) is a potent sphingosine-1-phosphate receptor-1 (S1P1) and receptor-5 (S1P5) agonist with autoimmune disease-modifying activity. Br. J. Pharmacol. 2016, 173, 1778–1792. [Google Scholar] [CrossRef] [Green Version]
- Buzard, D.J.; Kim, S.H.; Lopez, L.; Kawasaki, A.; Zhu, X.; Moody, J.; Thoresen, L.; Calderon, I.; Ullman, B.; Han, S.; et al. Discovery of APD334: Design of a Clinical Stage Functional Antagonist of the Sphingosine-1-phosphate-1 Receptor. ACS Med. Chem. Lett. 2014, 5, 1313–1317. [Google Scholar] [CrossRef] [Green Version]
- Mike, E.V.; Makinde, H.M.; Der, E.; Stock, A.; Gulinello, M.; Gadhvi, G.T.; Winter, D.R.; Cuda, C.M.; Putterman, C. Neuropsychiatric Systemic Lupus Erythematosus Is Dependent on Sphingosine-1-Phosphate Signaling. Front. Immunol. 2018, 9, 2189. [Google Scholar] [CrossRef] [Green Version]
- Hunter, S.F.; Agius, M.; Miller, D.M.; Cutter, G.; Barbato, L.; McCague, K.; Meng, X.; Agashivala, N.; Chin, P.; Hollander, E. Impact of a switch to fingolimod on depressive symptoms in patients with relapsing multiple sclerosis: An analysis from the EPOC (Evaluate Patient OutComes) trial. J. Neurol. Sci. 2016, 365, 190–198. [Google Scholar] [CrossRef]
- Kornhuber, J.; Medlin, A.; Bleich, S.; Jendrossek, V.; Henkel, A.W.; Wiltfang, J.; Gulbins, E. High activity of acid sphingomyelinase in major depression. J. Neural Transm. 2005, 112, 1583–1590. [Google Scholar] [CrossRef]
- Kornhuber, J.; Tripal, P.; Reichel, M.; Mühle, C.; Rhein, C.; Muehlbacher, M.; Groemer, T.W.; Gulbins, E. Functional Inhibitors of Acid Sphingomyelinase (FIASMAs): A novel pharmacological group of drugs with broad clinical applications. Cell. Physiol. Biochem. 2010, 26, 9–20. [Google Scholar] [CrossRef]
- Li, S.; Sakurai, K.; Ohgidani, M.; Kato, T.A.; Hikida, T. Ameliorative effects of Fingolimod (FTY720) on microglial activation and psychosis-related behavior in short term cuprizone exposed mice. Mol. Brain 2023, 16, 59. [Google Scholar] [CrossRef]
- Chun, J.; Hartung, H.-P. Mechanism of Action of Oral Fingolimod (FTY720) in Multiple Sclerosis. Clin. Neuropharmacol. 2010, 33, 91–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francis, M.M.; Hummer, T.A.; Liffick, E.; Vohs, J.L.; Mehdiyoun, N.F.; Visco, A.C.; Yang, Z.; Kovacs, R.J.; Zhang, Y.; Breier, A. Effects of fingolimod, a sphingosine-1-phosphate (S1P) receptor agonist, on white matter microstructure, cognition and symptoms in schizophrenia. Brain Imaging Behav. 2021, 15, 1802–1814. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Brieland, J.K.; Kim, J.H.; Chen, Y.J.; O’Neal, J.; O’Neil, S.P.; Tu, T.W.; Trinkaus, K.; Song, S.K. Diffusion tensor imaging detects treatment effects of FTY720 in experimental autoimmune encephalomyelitis mice. NMR Biomed. 2013, 26, 1742–1750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurevich, M.; Waknin, R.; Stone, E.; Achiron, A. Fingolimod-improved axonal and myelin integrity of white matter tracts associated with multiple sclerosis-related functional impairments. CNS Neurosci. Ther. 2018, 24, 412–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coelho, R.P.; Payne, S.G.; Bittman, R.; Spiegel, S.; Sato-Bigbee, C. The immunomodulator FTY720 has a direct cytoprotective effect in oligodendrocyte progenitors. J. Pharmacol. Exp. Ther. 2007, 323, 626–635. [Google Scholar] [CrossRef] [Green Version]
- Jung, C.G.; Kim, H.J.; Miron, V.E.; Cook, S.; Kennedy, T.E.; Foster, C.A.; Antel, J.P.; Soliven, B. Functional consequences of S1P receptor modulation in rat oligodendroglial lineage cells. Glia 2007, 55, 1656–1667. [Google Scholar] [CrossRef]
- Miron, V.E.; Jung, C.G.; Kim, H.J.; Kennedy, T.E.; Soliven, B.; Antel, J.P. FTY720 modulates human oligodendrocyte progenitor process extension and survival. Ann. Neurol. 2008, 63, 61–71. [Google Scholar] [CrossRef]
- Miron, V.E.; Ludwin, S.K.; Darlington, P.J.; Jarjour, A.A.; Soliven, B.; Kennedy, T.E.; Antel, J.P. Fingolimod (FTY720) enhances remyelination following demyelination of organotypic cerebellar slices. Am. J. Pathol. 2010, 176, 2682–2694. [Google Scholar] [CrossRef]
- Zhuo, C.; Zhao, F.; Tian, H.; Chen, J.; Li, Q.; Yang, L.; Ping, J.; Li, R.; Wang, L.; Xu, Y.; et al. Acid sphingomyelinase/ceramide system in schizophrenia: Implications for therapeutic intervention as a potential novel target. Transl. Psychiatry 2022, 12, 260. [Google Scholar] [CrossRef] [PubMed]
- Ameis, S.H.; Catani, M. Altered white matter connectivity as a neural substrate for social impairment in Autism Spectrum Disorder. Cortex 2015, 62, 158–181. [Google Scholar] [CrossRef]
- Hong, S.J.; Hyung, B.; Paquola, C.; Bernhardt, B.C. The Superficial White Matter in Autism and Its Role in Connectivity Anomalies and Symptom Severity. Cereb. Cortex 2019, 29, 4415–4425. [Google Scholar] [CrossRef]
- Naegelin, Y.; Kuhle, J.; Schädelin, S.; Datta, A.N.; Magon, S.; Amann, M.; Barro, C.; Ramelli, G.P.; Heesom, K.; Barde, Y.A.; et al. Fingolimod in children with Rett syndrome: The FINGORETT study. Orphanet J. Rare Dis. 2021, 16, 19. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, A.; Spiombi, E.; Frasca, A.; Landsberger, N.; Zagrebelsky, M.; Korte, M. Fingolimod Modulates Dendritic Architecture in a BDNF-Dependent Manner. Int. J. Mol. Sci. 2020, 21, 3079. [Google Scholar] [CrossRef] [PubMed]
- Penzes, P.; Cahill, M.E.; Jones, K.A.; VanLeeuwen, J.E.; Woolfrey, K.M. Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci. 2011, 14, 285–293. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Shi, Y.; Li, X.; Yang, Y.; Zhang, X.; Xu, L.; Ma, Z.; Wang, J.; Fan, L.; Wu, L. Proteomic-Based Approach Reveals the Involvement of Apolipoprotein A-I in Related Phenotypes of Autism Spectrum Disorder in the BTBR Mouse Model. Int. J. Mol. Sci. 2022, 23, 15290. [Google Scholar] [CrossRef]
- Choden, T.; Cohen, N.A.; Rubin, D.T. Sphingosine-1 Phosphate Receptor Modulators: The Next Wave of Oral Therapies in Inflammatory Bowel Disease. Gastroenterol. Hepatol. 2022, 18, 265–271. [Google Scholar]
- Sandborn, W.J.; Feagan, B.G.; Wolf, D.C.; D’Haens, G.; Vermeire, S.; Hanauer, S.B.; Ghosh, S.; Smith, H.; Cravets, M.; Frohna, P.A.; et al. Ozanimod Induction and Maintenance Treatment for Ulcerative Colitis. N. Engl. J. Med. 2016, 374, 1754–1762. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Feagan, B.G.; Hanauer, S.; Vermeire, S.; Ghosh, S.; Liu, W.J.; Petersen, A.; Charles, L.; Huang, V.; Usiskin, K.; et al. Long-Term Efficacy and Safety of Ozanimod in Moderately to Severely Active Ulcerative Colitis: Results From the Open-Label Extension of the Randomized, Phase 2 TOUCHSTONE Study. J. Crohns Colitis 2021, 15, 1120–1129. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Feagan, B.G.; D’Haens, G.; Wolf, D.C.; Jovanovic, I.; Hanauer, S.B.; Ghosh, S.; Petersen, A.; Hua, S.Y.; Lee, J.H.; et al. Ozanimod as Induction and Maintenance Therapy for Ulcerative Colitis. N. Engl. J. Med. 2021, 385, 1280–1291. [Google Scholar] [CrossRef] [PubMed]
- Sandborn, W.J.; Vermeire, S.; Peyrin-Biroulet, L.; Dubinsky, M.C.; Panes, J.; Yarur, A.; Ritter, T.; Baert, F.; Schreiber, S.; Sloan, S.; et al. Etrasimod as induction and maintenance therapy for ulcerative colitis (ELEVATE): Two randomised, double-blind, placebo-controlled, phase 3 studies. Lancet 2023, 401, 1159–1171. [Google Scholar] [CrossRef] [PubMed]
- Kiyomi, K.; Caroline, L.; Lisette, A.; Kye, G.; John, G. P045 Effect of Etrasimod on Circulating Lymphocyte Subsets: Data From a Randomized Phase 1 Study in Healthy Japanese and Caucasian Men. Am. J. Gastroenterol. 2020, 115, S12. [Google Scholar] [CrossRef] [PubMed]
- Peyrin-Biroulet, L.; Christopher, R.; Behan, D.; Lassen, C. Modulation of sphingosine-1-phosphate in inflammatory bowel disease. Autoimmun. Rev. 2017, 16, 495–503. [Google Scholar] [CrossRef] [PubMed]




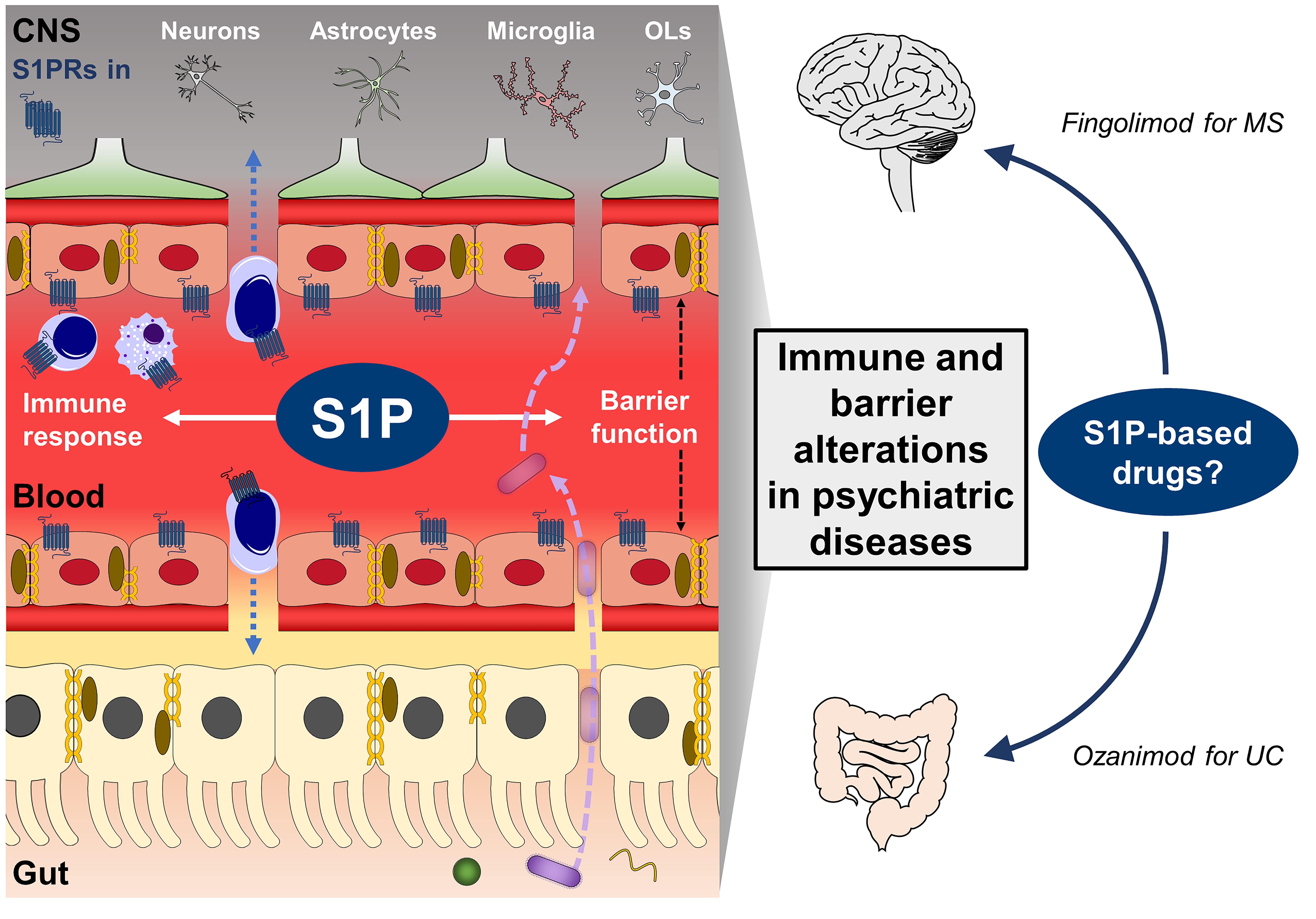{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Authors and Year | Type of Study | Main Results |
|---|---|---|
| Major depressive disorder (MDD) | ||
| Martín-Hernández et al., 2023 [229] | Preclinical (periodontitis + CMS) | S1P signaling modulation, decreased BBB-related proteins, and increased neuroinflammation |
| Shan et al., 2020 [228] | Preclinical (CMS) | S1PR2 was lower in the hippocampus, negatively correlated with symptom severity, and reduced TNF-α levels. |
| Corbett et al., 2019 [230] | Preclinical (chronic social defeat) Clinical (PTSD) | S1PR3 in the medial-PFC was elevated in resilient rats. S1PR3 was reduced in the blood of PTSD patients |
| Cheng et al., 2018 [231] | Preclinical (learned helplessness) | Fingolimod and TNF-α inhibition enhanced BBB integrity and ameliorate depressive behavior |
| Mike et al., 2018 [269] | Preclinical (neuropsychiatric lupus) | Fingolimod attenuated depressive-like behavior, neuroinflammation, and BBB permeability |
| Hunter et al., 2016 [270] | Clinical (MS) | Fingolimod improved depressive symptoms compared to other treatments in relapsing-remitting MS patients |
| Kornhuber et al., 2005 [271] | Clinical | Acid sphingomyelinase (ASM), which produces the S1P precursor ceramide, was increased in MDD patients |
| Neurodevelopmental disorders | ||
| Schizophrenia (SZ) | ||
| Li et al., 2023 [273] | Preclinical (cuprizone) | Fingolimod improved psychotic-behavior and decreased neuroinflammation. |
| Chand et al., 2022 [242] | Clinical | S1PR1 was higher in the dorsolateral-PFC of Type 2 SZ patients |
| Francis et al., 2021 [275] | Clinical | Fingolimod reduced circulating lymphocytes and improved white matter microstructure, but had no effects on symptoms in SZ patients |
| Esaki et al., 2020 [241] | Clinical | S1P was decreased in the corpus callosum of SZ patients |
| Patnaik et al., 2020 [286] | Preclinical (in vitro) | Fingolimod increased BDNF and promoted spinogenesis |
| Pépin et al., 2020 [249] | Preclinical (MPTP—PD model) | S1PR1 agonism ameliorated loss of dopaminergic neurons and motor deficits |
| Riganti et al., 2016 [247] | Preclinical | S1P induced synapsin I mobilization from synapses |
| Spampinato et al., 2015 [251] | Preclinical (in vitro) | Fingolimod reduced the immune-induced BBB damage |
| Miron et al., 2010 [281] | Preclinical (in vitro) | Fingolimod induced remyelination after demyelination |
| Miron et al., 2008 [280] | Preclinical (in vitro) | Fingolimod promoted extension and survival of oligodendrocytes |
| Coelho et al., 2007 [278] | Preclinical (in vitro) | Fingolimod exerted a cytoprotective effect and stimulation of remyelination on oligodendrocytes |
| Jung et al., 2007 [279] | Preclinical (in vitro) | Fingolimod improved the survival of rat oligodendrocytes |
| Autism spectrum disorders (ASD) | ||
| Almandil et al., 2023 [258] | Clinical | Single nucleotide polymorphisms (SNPs) in the Sphk1 gene were associated with ASD in females |
| Li et al., 2022 [288] | Preclinical (BTBR mouse strain) | SphK blocker SKI-II recovered behavioral and molecular impairments |
| Naegelin et al., 2021 [285] | Clinical (Rett syndrome) | Fingolimod did not show behavioral and biochemical effects in children with RS |
| Kurochkin et al., 2019 [257] | Clinical | S1P levels were unaltered in the PFC of ASD patients |
| Gurevich et al., 2018 [277] | Clinical (MS) | Fingolimod improved white matter microstructure in MS patients with impaired pyramidal function |
| Wu et al., 2018 [259] | Preclinical (valproic acid) | S1P was decreased in the hippocampus and serum and there were memory impairments |
| Wang et al., 2016 [256] | Clinical | S1P serum levels were reduced in ASD patients |
| Wang et al., 2013 [276] | Preclinical (EAE—MS model) | Fingolimod ameliorated white matter microstructure |
| Attention deficit hyperactivity disorder (ADHD) | ||
| Brunkhorst-Kanaan et al., 2021 [265] | Clinical | S1P plasma levels were increased in ADHD adults |
| Henriquez-Henriquez et al., 2020 [263] | Clinical | Alterations in ceramide synthesis genes, a precursor of S1P in ADHD patients |
| Henriquez-Henriquez et al., 2015 [264] | Clinical | Lower levels of sphingomyelins in ADHD children |
| Authors and Year | Type of Study | Main Results |
|---|---|---|
| Inflammatory bowel diseases (IBD) | ||
| Sandborn et al., 2023 [293] | Clinical (UC) | Etrasimod was effective and safe as an induction and maintenance therapy in moderate to severe UC patients. |
| Martín-Hernández et al., 2022 [88] | Preclinical (sub-chronic stress) | Stress increased S1P, dysregulated immune responses, and enhanced intestinal permeability. Sphk2−/− mice presented a cytokine-expression profile towards a boosted Th17 response, lower expression of claudins, and structural abnormalities in the colon. |
| Sandborn et al., 2021 [291] | Clinical (UC) | Ozanimod exerted long-term (2 years) benefits and was safe (4 years) in moderate to severe UC patients |
| Sandborn et al., 2021 [292] | Clinical (UC) | Ozanimod was effective and safe as an induction and maintenance therapy in moderate to severe UC patients. |
| Kiyomi et al., 2020 [294] | Clinical | Etrasimod reduced circulating adaptive but not innate immune cells in healthy individuals. |
| Chen et al., 2018 [218] | Preclinical (DSS-induced colitis) | Intestinal barrier damage was higher in S1PR2−/−. S1P/S1PR2 axis mediated CD4+T cell activation via ERK and MHC-II in IECs |
| Karuppuchamy et al. 2017 [221] | Preclinical (DSS-induced colitis) | Chronic inflammation modulated S1PR1 expression and tissue S1P levels. |
| Dong et al., 2015 [219] | Preclinical (IL-10−/− mice) | S1PR1 agonism improved barrier function by restoring TJ proteins and suppressing IEC apoptosis |
| Montrose et al., 2013 [220] | Preclinical (DSS-induced colitis) Clinical (UC) | S1PR1 deletion enhanced vascular permeability and bleeding in mice. Patients with active UC showed overexpression of S1PR1 and increased vascular density in inflamed the colon mucosa. |
| Eisenhoffer et al., 2012 [217] | Preclinical (in vitro) | S1PR2 receptor and ROCK controlled IEC extrusion, critical for a healthy intestinal barrier function |
| Greenspon et al., 2011 [216] | Preclinical (in vitro) | S1P increased E-cadherin levels, enhancing barrier function in cultured IECs |
| Sandbornd et al., 2016 [290] | Clinical (UC) | Ozanimod exerted a slightly higher rate of clinical response and remission (preliminary trial) in moderate to severe UC patients. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martín-Hernández, D.; Muñoz-López, M.; Tendilla-Beltrán, H.; Caso, J.R.; García-Bueno, B.; Menchén, L.; Leza, J.C. Immune System and Brain/Intestinal Barrier Functions in Psychiatric Diseases: Is Sphingosine-1-Phosphate at the Helm? Int. J. Mol. Sci. 2023, 24, 12634. https://doi.org/10.3390/ijms241612634
Martín-Hernández D, Muñoz-López M, Tendilla-Beltrán H, Caso JR, García-Bueno B, Menchén L, Leza JC. Immune System and Brain/Intestinal Barrier Functions in Psychiatric Diseases: Is Sphingosine-1-Phosphate at the Helm? International Journal of Molecular Sciences. 2023; 24(16):12634. https://doi.org/10.3390/ijms241612634
Chicago/Turabian StyleMartín-Hernández, David, Marina Muñoz-López, Hiram Tendilla-Beltrán, Javier R. Caso, Borja García-Bueno, Luis Menchén, and Juan C. Leza. 2023. "Immune System and Brain/Intestinal Barrier Functions in Psychiatric Diseases: Is Sphingosine-1-Phosphate at the Helm?" International Journal of Molecular Sciences 24, no. 16: 12634. https://doi.org/10.3390/ijms241612634