
Diastereoselective Synthesis of Novel Spiro-Phosphacoumarins and Evaluation of Their Anti-Cancer Activity
 , , , , , ,
, , , , , ,  ,
,  ,
,
Abstract
:
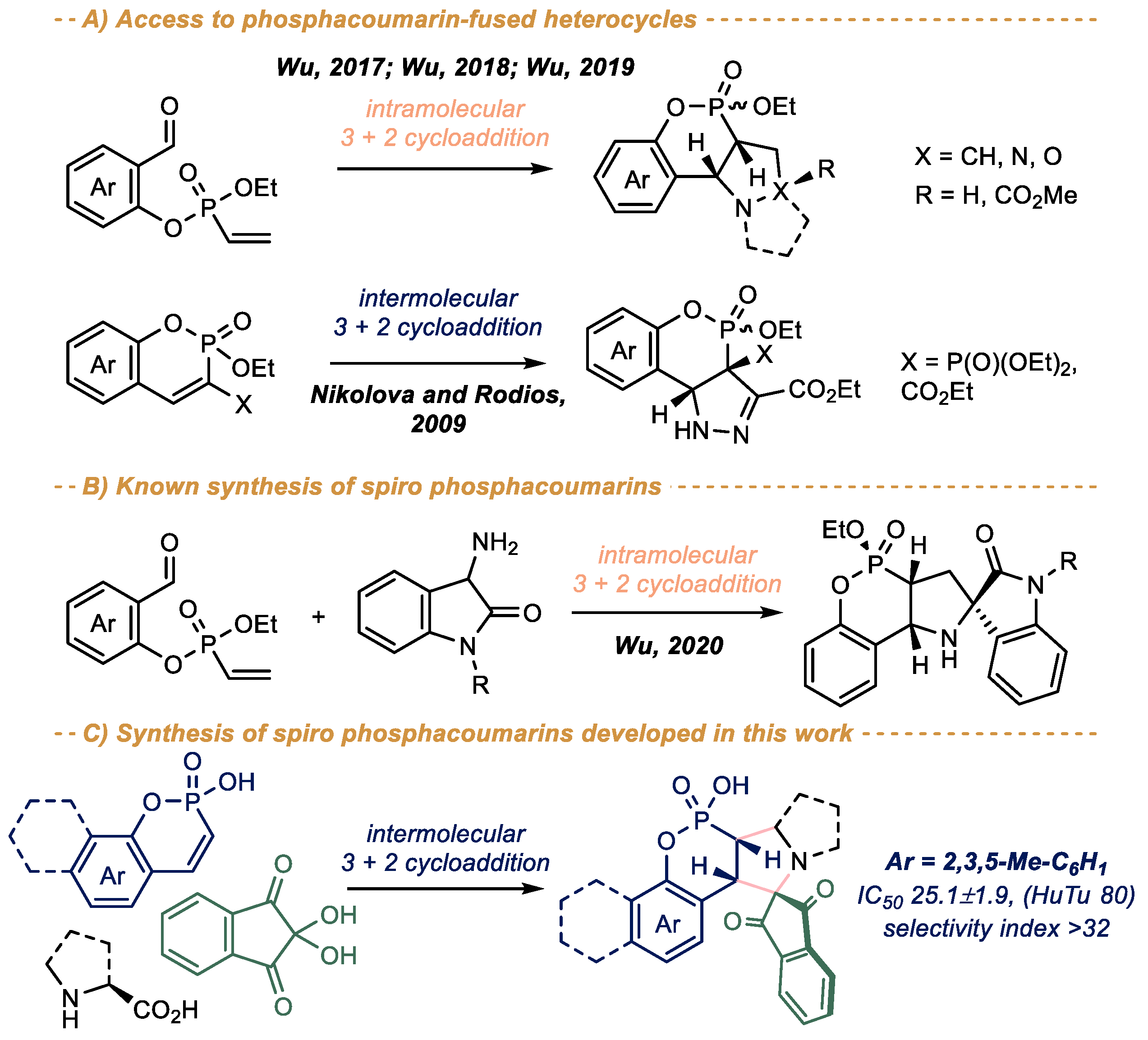
1. Introduction

2. Results and Discussion
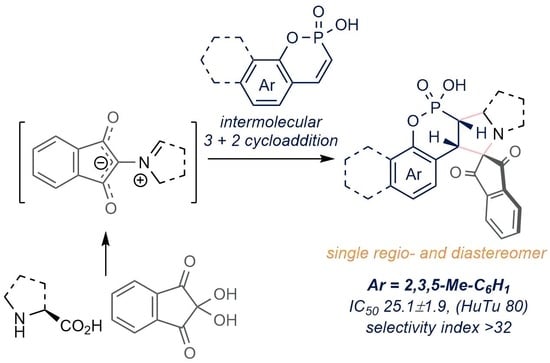
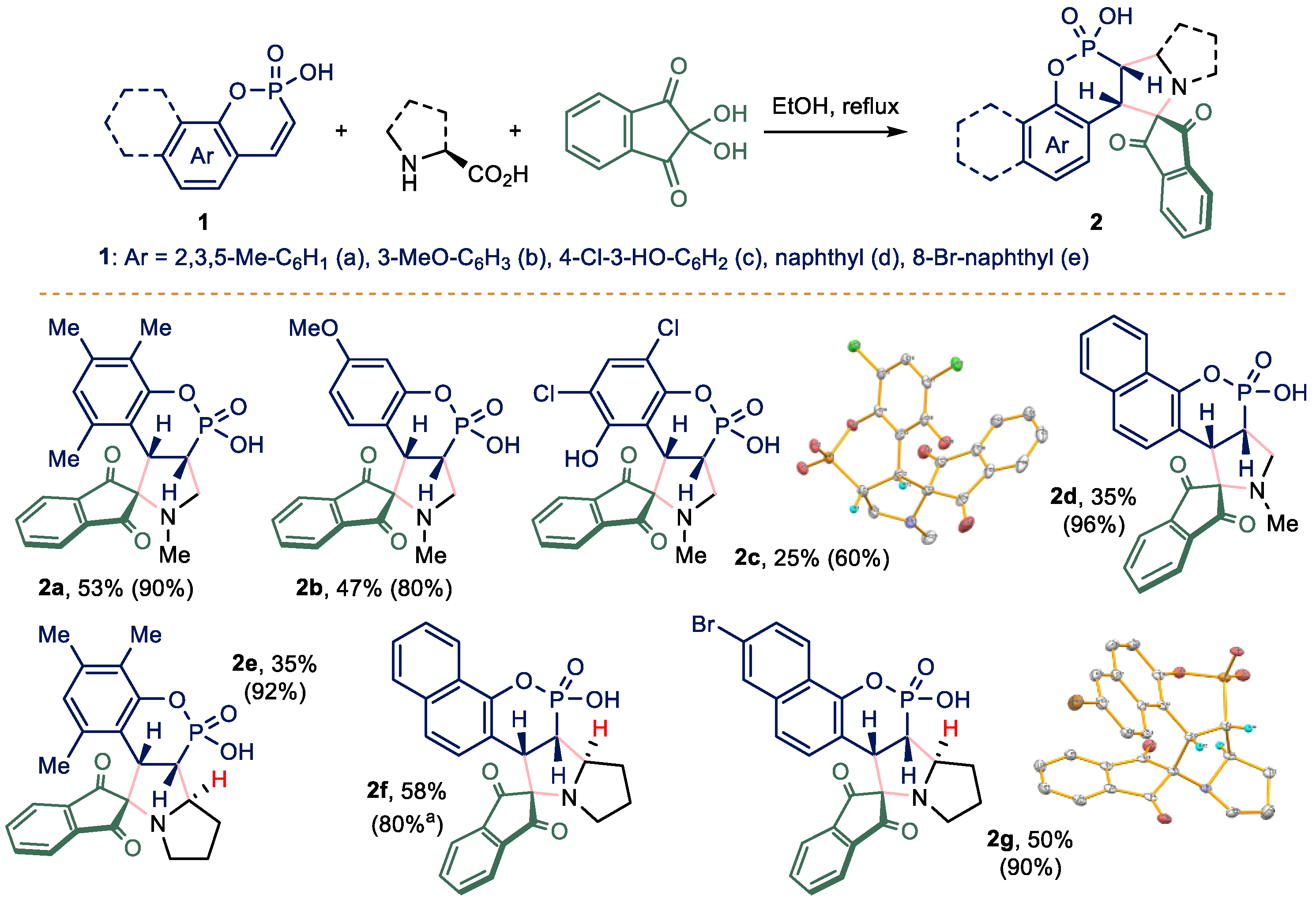
2.1. Chemistry
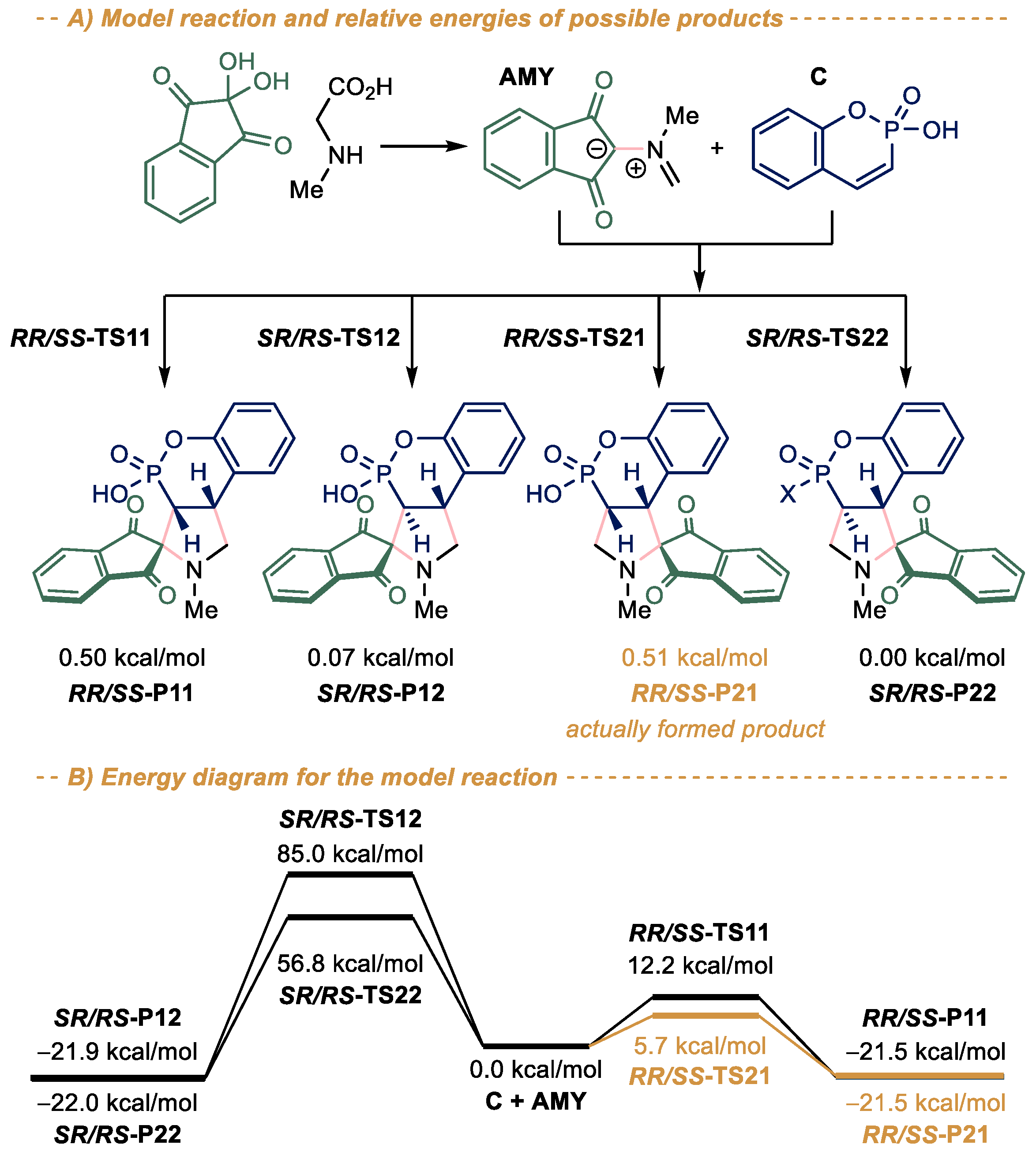
2.2. Quantum Chemistry Studies
2.3. Biological Studies
2.3.1. In Vitro Cytotoxicity
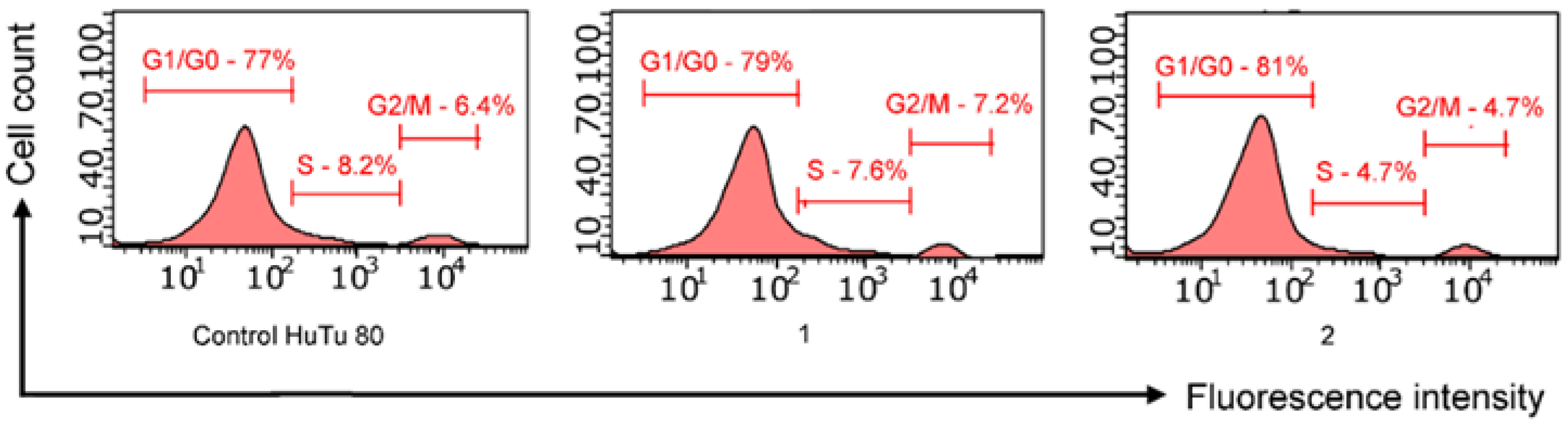
2.3.2. Cell Cycle Analysis
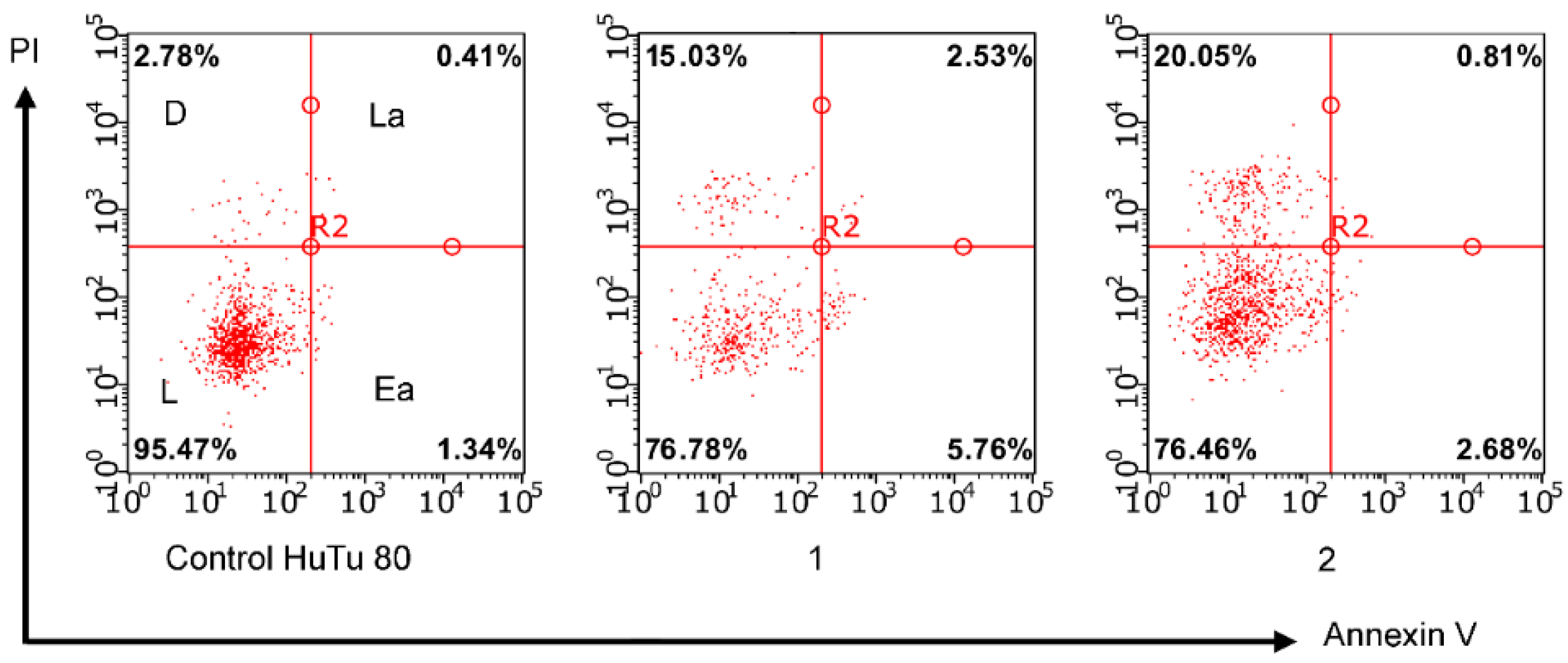
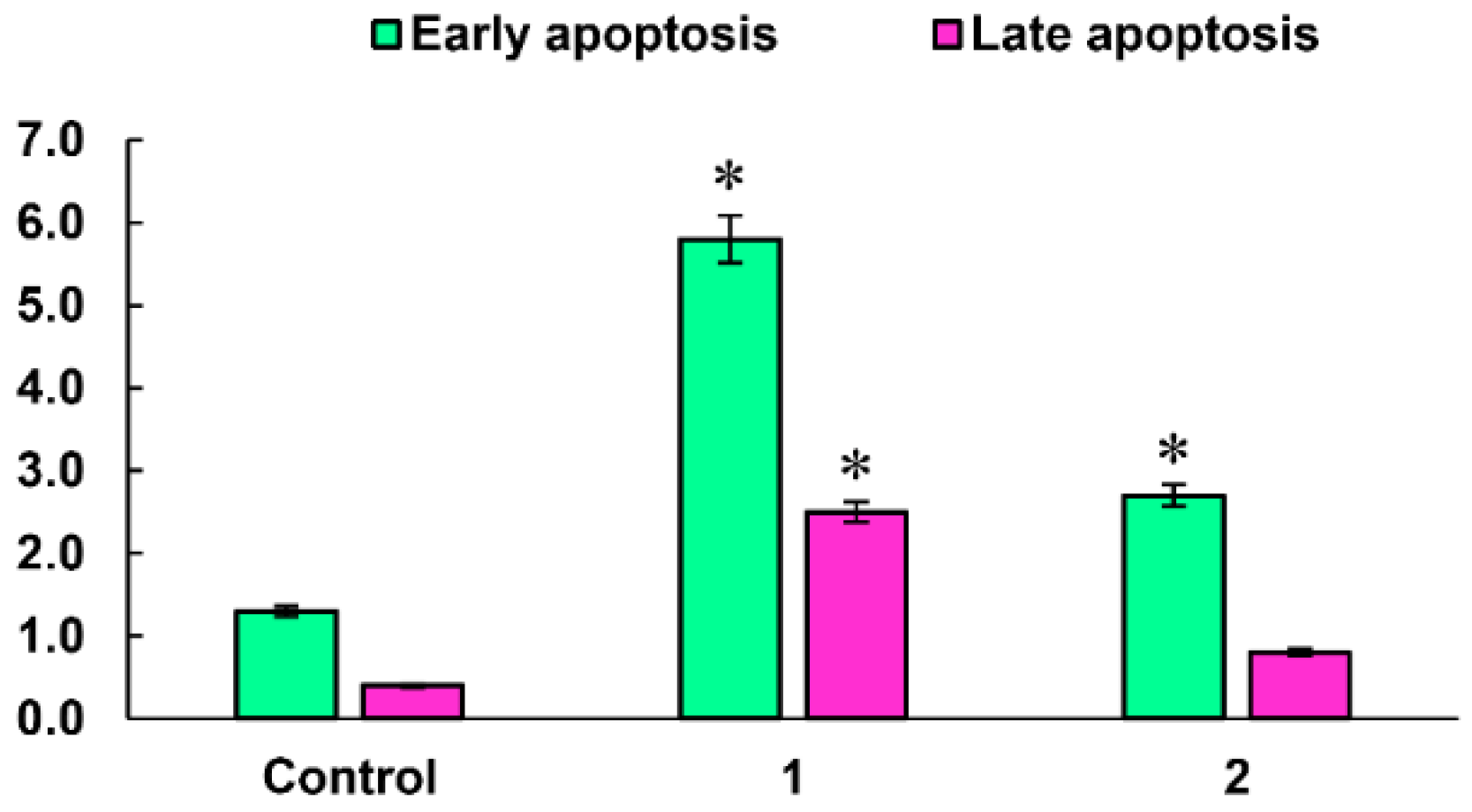
2.3.3. Induction of Apoptotic Effects
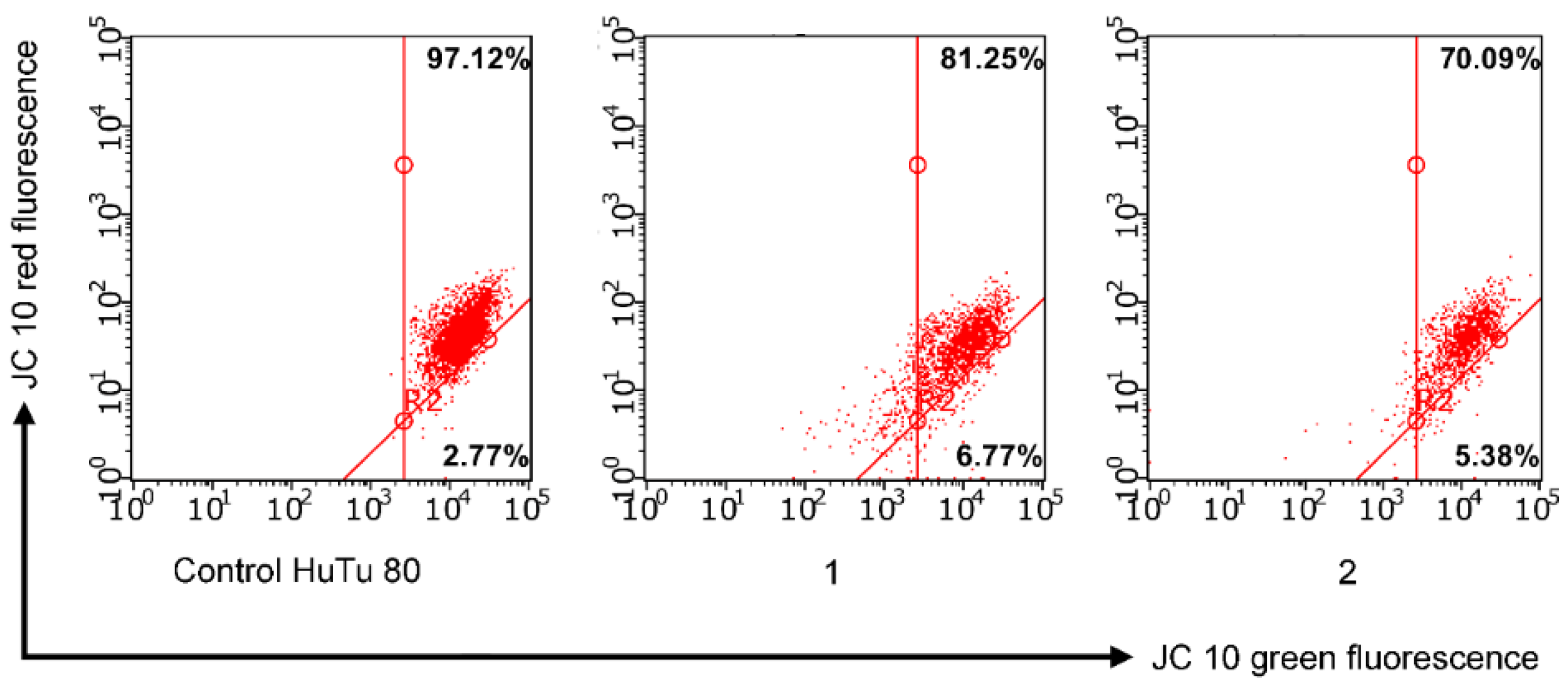
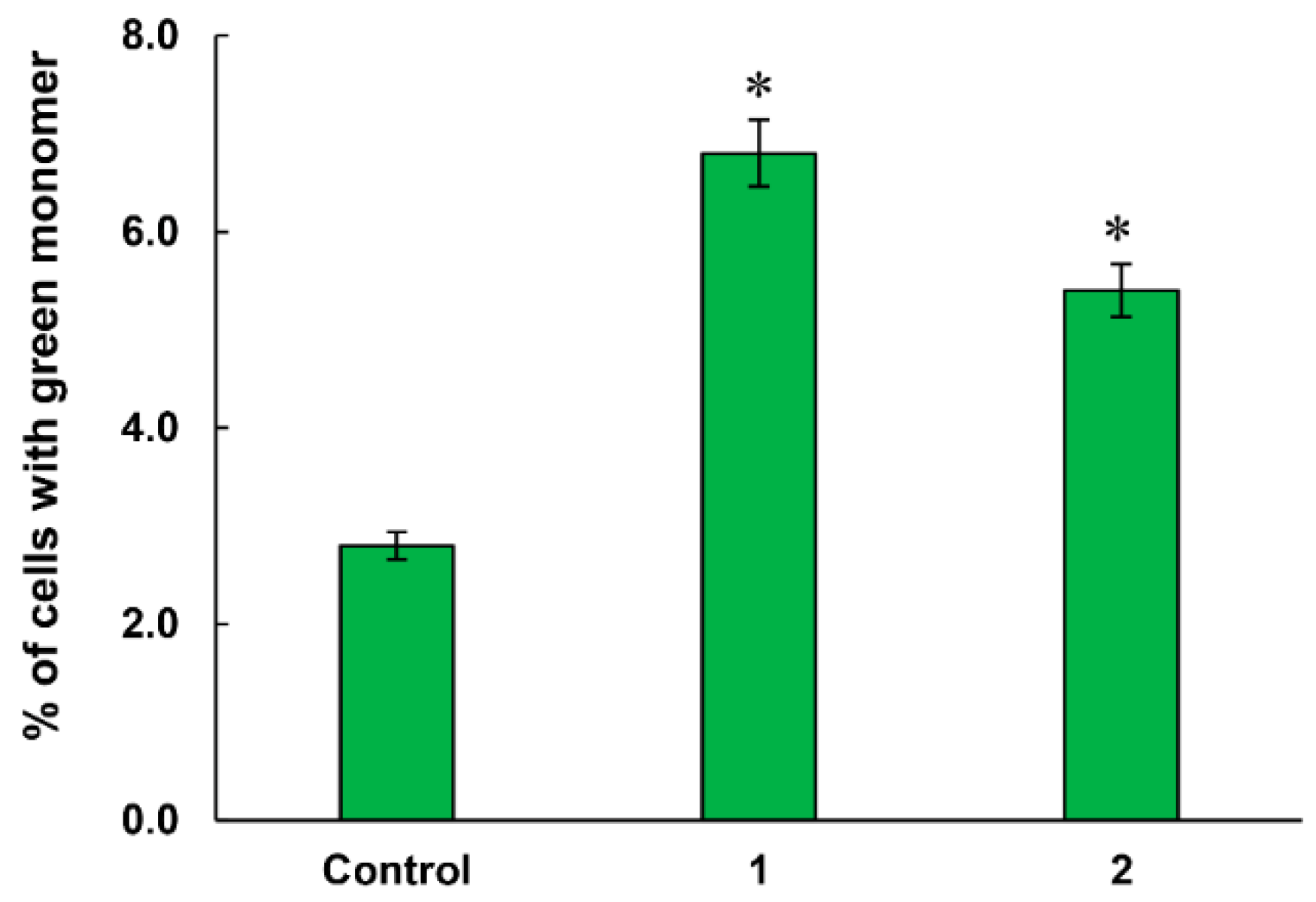
2.3.4. Mitochondrial Membrane Potential
3. Materials and Methods
3.1. Quantum Chemistry Calculations
3.2. Chemistry
3.2.1. General Methods
3.2.2. General Procedure for the Synthesis of Compounds 2
3.3. Biological Studies
3.3.1. Cell Toxicity Assay (MTT-Test)
3.3.2. Induction of Apoptotic Effects by Test Compounds (Flow Cytometry Assay)
3.3.3. Mitochondrial Membrane Potential
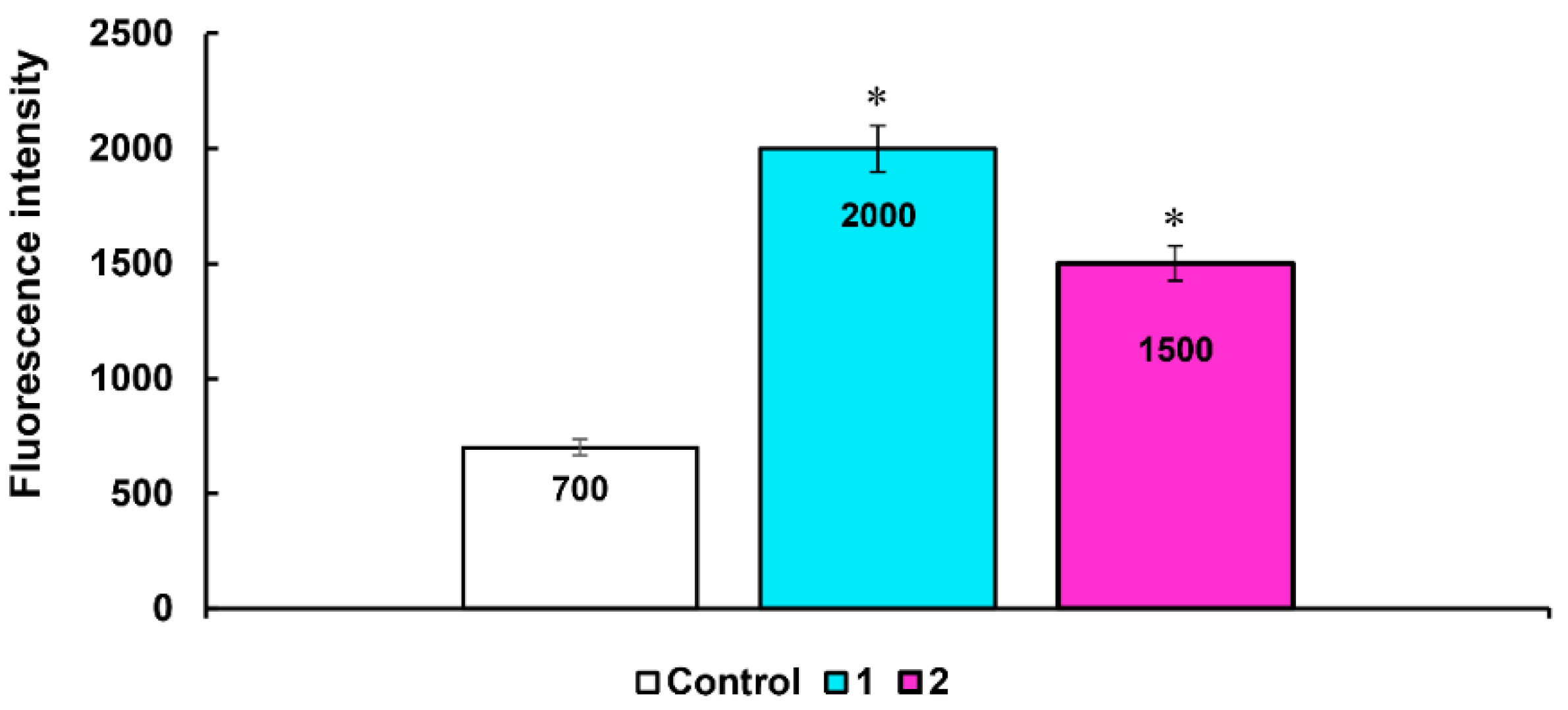
3.3.4. Detection of Intracellular ROS
3.3.5. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Balewski, Ł.; Szulta, S.; Jalińska, A.; Kornicka, A. A Mini-Review: Recent Advances in Coumarin-Metal Complexes with Biological Properties. Front. Chem. 2021, 9, 781779. [Google Scholar] [CrossRef] [PubMed]
- Srikrishna, D.; Godugu, C.; Dubey, P.K. A Review on Pharmacological Properties of Coumarins. Mini-Rev. Med. Chem. 2018, 18, 113–141. [Google Scholar] [CrossRef]
- Annunziata, F.; Pinna, C.; Dallavalle, S.; Tamborini, L.; Pinto, A. An Overview of Coumarin as a Versatile and Readily Accessible Scaffold with Broad-Ranging Biological Activities. Int. J. Mol. Sci. 2020, 21, 4618. [Google Scholar] [CrossRef] [PubMed]
- Kaur, M.; Kohli, S.; Sandhu, S.; Bansal, Y.; Bansal, G. Coumarin: A Promising Scaffold for Anticancer Agents. Anticancer. Agents Med. Chem. 2015, 15, 1032–1048. [Google Scholar] [CrossRef]
- Thakur, A.; Singla, R.; Jaitak, V. Coumarins as anticancer agents: A review on synthetic strategies, mechanism of action and SAR studies. Eur. J. Med. Chem. 2015, 101, 476–495. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Xu, J.; Liu, Y.; Zeng, Y.; Wu, G. A Review on Anti-Tumor Mechanisms of Coumarins. Front. Oncol. 2020, 10, 592853. [Google Scholar] [CrossRef] [PubMed]
- Rawat, A.; Vijaya Bhaskar Reddy, A. Recent advances on anticancer activity of coumarin derivatives. Eur. J. Med. Chem. Reports 2022, 5, 100038. [Google Scholar] [CrossRef]
- Li, X.; Zhang, D.; Pang, H.; Shen, F.; Fu, H.; Jiang, Y.; Zhao, Y. Synthesis of a Diverse Series of Phosphacoumarins with Biological Activity. Org. Lett. 2005, 7, 4919–4922. [Google Scholar] [CrossRef]
- Alexieva, V.; Karanov, E.; Nikolova, R.; Bojilova, A. Plant growth regulating activity of some phosphorus derivatives of coumarin. Bulg. J. Plant Physiol. 1995, 21, 45. [Google Scholar]
- Kim, C.-E.; Ryu, T.; Kim, S.; Lee, K.; Lee, C.-H.; Lee, P.H. Gold-Catalyzed Hydroarylation of Aryl Alkynylphosphonates for the Synthesis of Phosphacoumarins. Adv. Synth. Catal. 2013, 355, 2873–2883. [Google Scholar] [CrossRef]
- Hariri, M.; Darvish, F.; Mengue Me Ndong, K.-P.; Sechet, N.; Chacktas, G.; Boosaliki, H.; Tran Do, M.L.; Mwande-Maguene, G.; Lebibi, J.; Burilov, A.R.; et al. Gold-Catalyzed Access to Isophosphinoline 2-Oxides. J. Org. Chem. 2021, 86, 7813–7824. [Google Scholar] [CrossRef] [PubMed]
- Hariri, M.; Darvish, F.; Ndong, K.-P.M.M.; Babouri, R.; Babouri, R.; Mwande-Maguene, G.; Burilov, A.R.; Licznar-Fajardo, P.; Pirat, J.-L.; Ayad, T.; et al. Biologically relevant surrogates of coumarins: 2-phenyl H-isophosphinoline 2-oxides with antibacterial activity. GSC Biol. Pharm. Sci. 2021, 16, 283–296. [Google Scholar] [CrossRef]
- Balašova, A.; Žalubovskis, R. Synthetic Methods toward Phosphacoumarins (microreview). Chem. Heterocycl. Compd. 2022, 58, 310–312. [Google Scholar] [CrossRef]
- Koleva, A.I.; Petkova-Yankova, N.I.; Nikolova, R.D. Synthesis and Chemical Properties of 3-Phosphono-coumarins and 1,2-Benzoxaphosphorins as Precursors for Bioactive Compounds. Molecules 2019, 24, 2030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, T.; Wang, Q.; Kong, D.; Wu, M. Diastereoselective catalyst-free construction of isoxazolidine-cis-fused phospha dihydrocoumarins via an intramolecular Nitrone–Vinylphosphonate dipolar cycloaddition. Tetrahedron Lett. 2019, 60, 150913. [Google Scholar] [CrossRef]
- Jiang, J.; Wu, M.; Zhu, Z.; Kong, D. Catalyst-Free Intramolecular 1,3-Dipolar Cycloaddition of Ethyl (2-Formylphenyl) Vinylphosphonates: A Highly Stereoselective Access to Phosphadihydrocoumarin-Fused Pyrrolizidines/Pyrrolidines. Synthesis 2017, 49, 3731–3739. [Google Scholar] [CrossRef]
- Wu, M.; Jiang, J.; Zhu, Z.; Wang, Q.; Kong, D. One-Pot Catalyst-Free Domino Condensation/Intramolecular 1,3-Dipolar Cycloaddition: Highly Stereoselective Access to Phosphadihydrocoumarin-Fused N,N-Bicyclic Pyrazolidin-3-ones. Synthesis 2018, 50, 139–145. [Google Scholar] [CrossRef]
- Petkova, N.I.; Nikolova, R.D.; Bojilova, A.G.; Rodios, N.A.; Kopf, J. Synthesis of heterocyclic methylenebisphosphonates by 1,3-dipolar cycloaddition of ethyl diazoacetate to 1,2-benzoxaphosphorin-3-phosphonates. Tetrahedron 2009, 65, 1639–1647. [Google Scholar] [CrossRef]
- Huang, T.; Liu, L.; Wang, Q.; Wu, M.; Kong, D. 1,3-Dipolar Cycloaddition of 3-Amino Oxindole-Based Azomethine Ylides and O-Vinylphosphonylated Salicylaldehydes for Diastereoselective Synthesis of Oxindole Spiro-P,N-polycyclic Heterocycles. Synthesis 2020, 52, 1387–1397. [Google Scholar] [CrossRef]
- Li, D.-F.; Gu, Y.; Zhang, J.-R.; Liu, K.; Zhao, L.-M. Diastereoselective Construction of Spiro-furo[3,2-c]benzopyranoxindoles through a Cu(OTf) 2/AcOH Cooperative Promoted Bicyclization Reaction. J. Org. Chem. 2019, 84, 879–887. [Google Scholar] [CrossRef]
- Kowalczyk-Dworak, D.; Albrecht, Ł. α,β-Unsaturated butenolides in an organocatalytic doubly annulative cascade for the preparation of 3,4-dihydrocoumarins. Org. Biomol. Chem. 2019, 17, 2624–2628. [Google Scholar] [CrossRef]
- Ming, Y.-C.; Lv, X.-J.; Liu, M.; Liu, Y.-K. Synthesis of Chiral Polycyclic Tetrahydrocarbazoles by Enantioselective Aminocatalytic Double Activation of 2-Hydroxycinnamaldehydes with Dienals. Org. Lett. 2021, 23, 6515–6519. [Google Scholar] [CrossRef]
- Hussain, M.; Niu, C.; Wang, G.-W. Palladium-catalyzed synthesis of [60]fullerene-fused furochromenones and further electrochemical functionalization. Org. Chem. Front. 2020, 7, 1249–1254. [Google Scholar] [CrossRef]
- Sadykova, Y.M.; Sadikova, L.M.; Zalaltdinova, A.V.; Sultanova, Z.N.; Burilov, A.R.; Pudovik, M.A. 2H-Benzo[e]-1,2-oxaphosphorine Related Heterocycles as Precursors for the Synthesis of Unsymmetrical Bicyclic Phosphonates. Russ. J. Gen. Chem. 2018, 88, 1941–1943. [Google Scholar] [CrossRef]
- Sadykova, Y.M.; Zalaltdinova, A.V.; Smailov, A.K.; Trofimova, L.M.; Voronina, J.K.; Burilov, A.R.; Pudovik, M.A. Synthesis of unsymmetrical cage phosphonates from heterocyclic systems based on 2H-1,2-benzoxaphosphinine. Chem. Heterocycl. Compd. 2020, 56, 1605–1610. [Google Scholar] [CrossRef]
- Zalaltdinova, A.V.; Sadykova, Y.M.; Smailov, A.K.; Trofimova, L.M.; Burilov, A.R.; Pudovik, M.A. New intramolecular cyclization of 2 H-benzo[e]-1,2-oxaphosphorinine derivatives—A way to the synthesis of unsymmetrical cage phosphonates. Phosphorus. Sulfur. Silicon Relat. Elem. 2022, 197, 549–550. [Google Scholar] [CrossRef]
- Das, S. Recent applications of ninhydrin in multicomponent reactions. RSC Adv. 2020, 10, 18875–18906. [Google Scholar] [CrossRef] [PubMed]
- Filatov, A.S.; Wang, S.; Khoroshilova, O.V.; Lozovskiy, S.V.; Larina, A.G.; Boitsov, V.M.; Stepakov, A.V. Stereo- and Regioselective 1,3-Dipolar Cycloaddition of the Stable Ninhydrin-Derived Azomethine Ylide to Cyclopropenes: Trapping of Unstable Cyclopropene Dipolarophiles. J. Org. Chem. 2019, 84, 7017–7036. [Google Scholar] [CrossRef]
- Peña-Morán, O.; Villarreal, M.; Álvarez-Berber, L.; Meneses-Acosta, A.; Rodríguez-López, V. Cytotoxicity, Post-Treatment Recovery, and Selectivity Analysis of Naturally Occurring Podophyllotoxins from Bursera fagaroides var. fagaroides on Breast Cancer Cell Lines. Molecules 2016, 21, 1013. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self—Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian—Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Petersson, G.A.; Bennett, A.; Tensfeldt, T.G.; Al-Laham, M.A.; Shirley, W.A.; Mantzaris, J. A complete basis set model chemistry. I. The total energies of closed-shell atoms and hydrides of the first-row elements. J. Chem. Phys. 1988, 89, 2193–2218. [Google Scholar] [CrossRef]
- 35. Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Revision B.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Bursch, M.; Mewes, J.-M.; Hansen, A.; Grimme, S. Best-Practice DFT Protocols for Basic Molecular Computational Chemistry. Angew. Chem. Int. Ed. 2022, 61, e202205735. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Design of Density Functionals That Are Broadly Accurate for Thermochemistry, Thermochemical Kinetics, and Nonbonded Interactions. J. Phys. Chem. A 2005, 109, 5656. [Google Scholar] [CrossRef]
- Rappoport, D.; Furche, F. Property-optimized Gaussian basis sets for molecular response calculations. J. Chem. Phys. 2010, 133, 134105. [Google Scholar] [CrossRef] [Green Version]
- Li, Y. Energy Diagram Plotter (CDXML). Available online: https://github.com/liyuanhe211/Energy_Diagram_Plotter_CDXML (accessed on 10 September 2022).
- Sheldrick, G.M. SADABS, Program for Empirical Absorption Correction of Area Detector Data; University of Gottingen: Gottingen, Germany, 1997. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3. [Google Scholar] [CrossRef] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sadykova, Y.M.; Sadikova, L.M.; Badrtdinova, A.R.; Dobrynin, A.B.; Burilov, A.R.; Pudovik, M.A. Condensation of 2-Ethoxyvinylphosphonic Acid Dichloroanhydride with 2,3,5-Trimethylphenol. Novel Method for Preparation of Phosphacoumarins. Phosphorus. Sulfur. Silicon Relat. Elem. 2015, 190, 2267–2272. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cmpd | Cancer Cell Line | Normal Cell Line | |||||
|---|---|---|---|---|---|---|---|
| M-HeLa | MCF-7 | HuTu 80 | Chang Liver | ||||
| IC50, µM | SI | IC50, µM | SI | IC50, µM | SI | IC50, µM | |
| 2a | 52.6 ± 4.1 | >15 | 82.3 ± 7.5 | >10 | 25.1 ± 1.9 | >32 | >800 |
| 2b | 59.7 ± 4.6 | 1 | >100 | ns | 53.4 ± 4.2 | 1.2 | 62.0 ± 5.5 |
| 2c | >100 | ns | 77.6 ± 6.2 | ns | 100 ± 8.4 | ns | >100 |
| 2d | 60 ± 5.4 | ns | 92.2 ± 8.3 | ns | 82.6 ± 7.6 | ns | 57.0 ± 4.3 |
| 2e | >100 | ns | >100 | ns | >100 | ns | >100 |
| 5-fluorouracil | 62.0 ± 4.7 | 1.4 | 16.7 ± 1.3 | 5 | 65.2 ± 5.6 | 1.3 | 86.3 ± 6.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sennikova, V.V.; Zalaltdinova, A.V.; Sadykova, Y.M.; Khamatgalimov, A.R.; Gazizov, A.S.; Voloshina, A.D.; Lyubina, A.P.; Amerhanova, S.K.; Voronina, J.K.; Chugunova, E.A.; et al. Diastereoselective Synthesis of Novel Spiro-Phosphacoumarins and Evaluation of Their Anti-Cancer Activity. Int. J. Mol. Sci. 2022, 23, 14348. https://doi.org/10.3390/ijms232214348
Sennikova VV, Zalaltdinova AV, Sadykova YM, Khamatgalimov AR, Gazizov AS, Voloshina AD, Lyubina AP, Amerhanova SK, Voronina JK, Chugunova EA, et al. Diastereoselective Synthesis of Novel Spiro-Phosphacoumarins and Evaluation of Their Anti-Cancer Activity. International Journal of Molecular Sciences. 2022; 23(22):14348. https://doi.org/10.3390/ijms232214348
Chicago/Turabian StyleSennikova, Valeriia V., Alena V. Zalaltdinova, Yulia M. Sadykova, Ayrat R. Khamatgalimov, Almir S. Gazizov, Alexandra D. Voloshina, Anna P. Lyubina, Syumbelya K. Amerhanova, Julia K. Voronina, Elena A. Chugunova, and et al. 2022. "Diastereoselective Synthesis of Novel Spiro-Phosphacoumarins and Evaluation of Their Anti-Cancer Activity" International Journal of Molecular Sciences 23, no. 22: 14348. https://doi.org/10.3390/ijms232214348