
Complementary Sets of Autoantibodies Induced by SARS-CoV-2, Adenovirus and Bacterial Antigens Cross-React with Human Blood Protein Antigens in COVID-19 Coagulopathies

Abstract
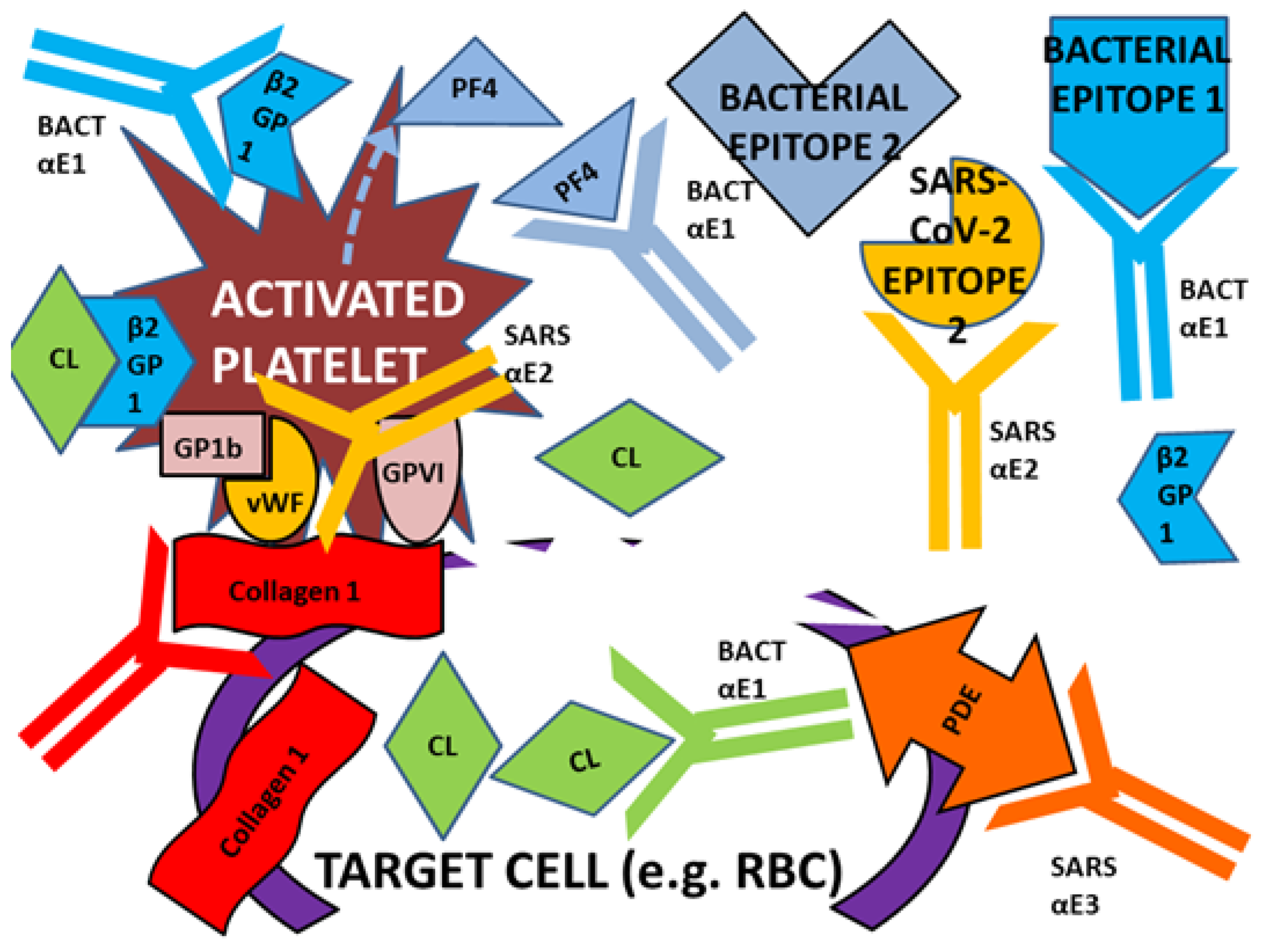
:1. Introduction
2. Results
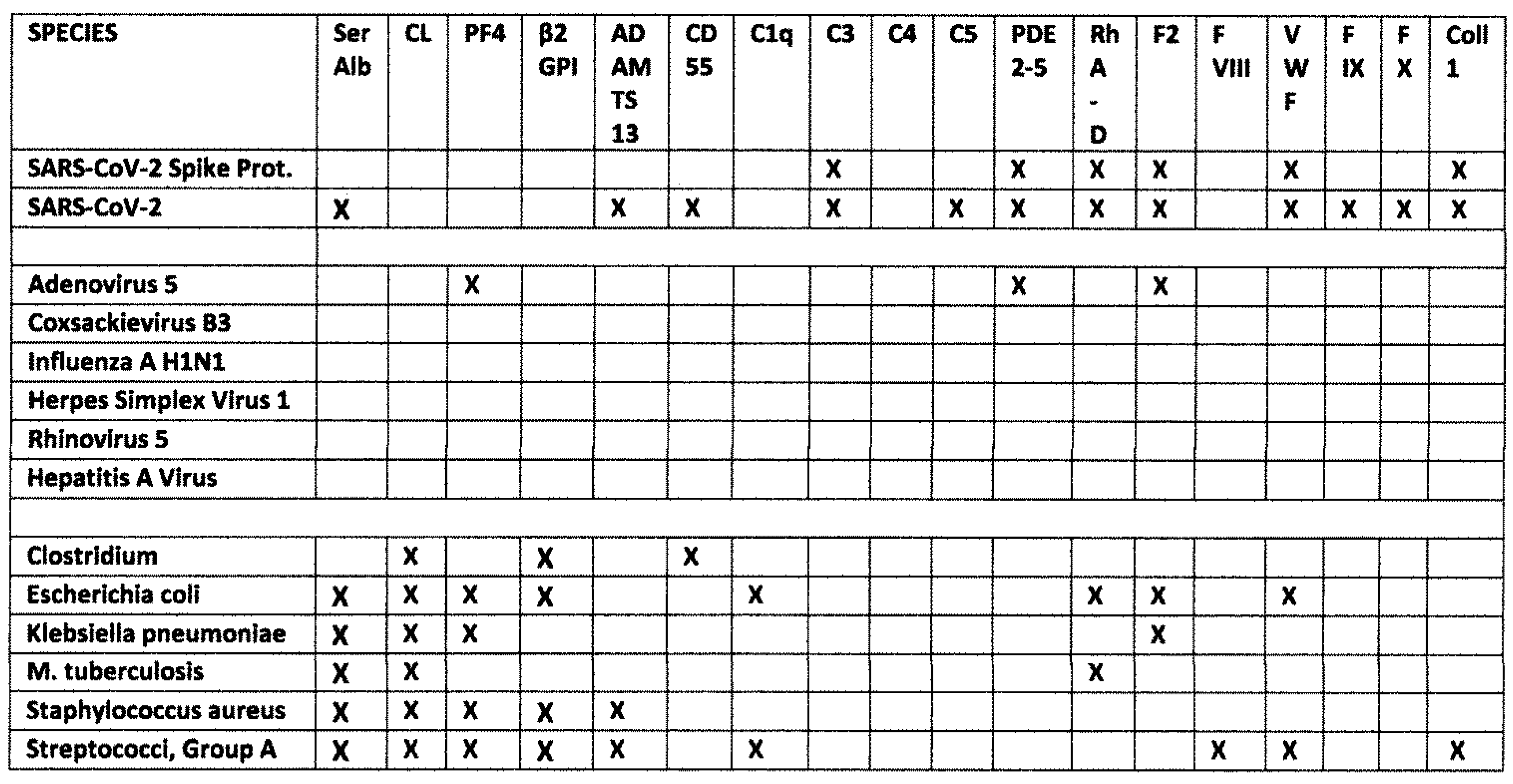
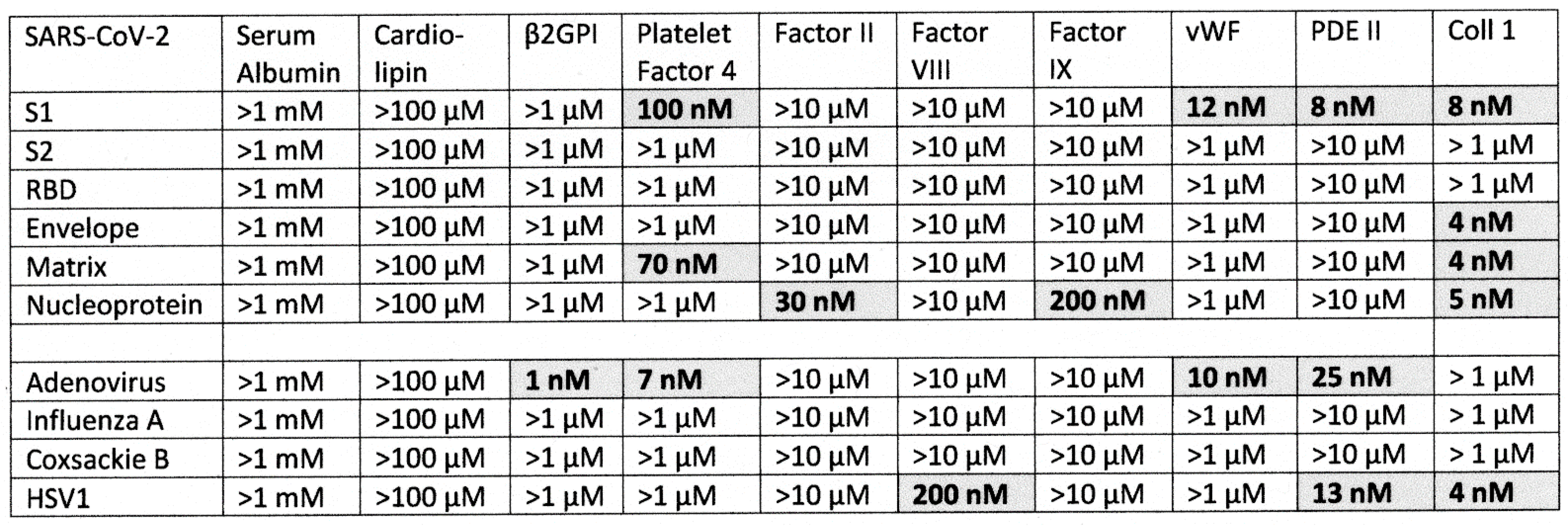
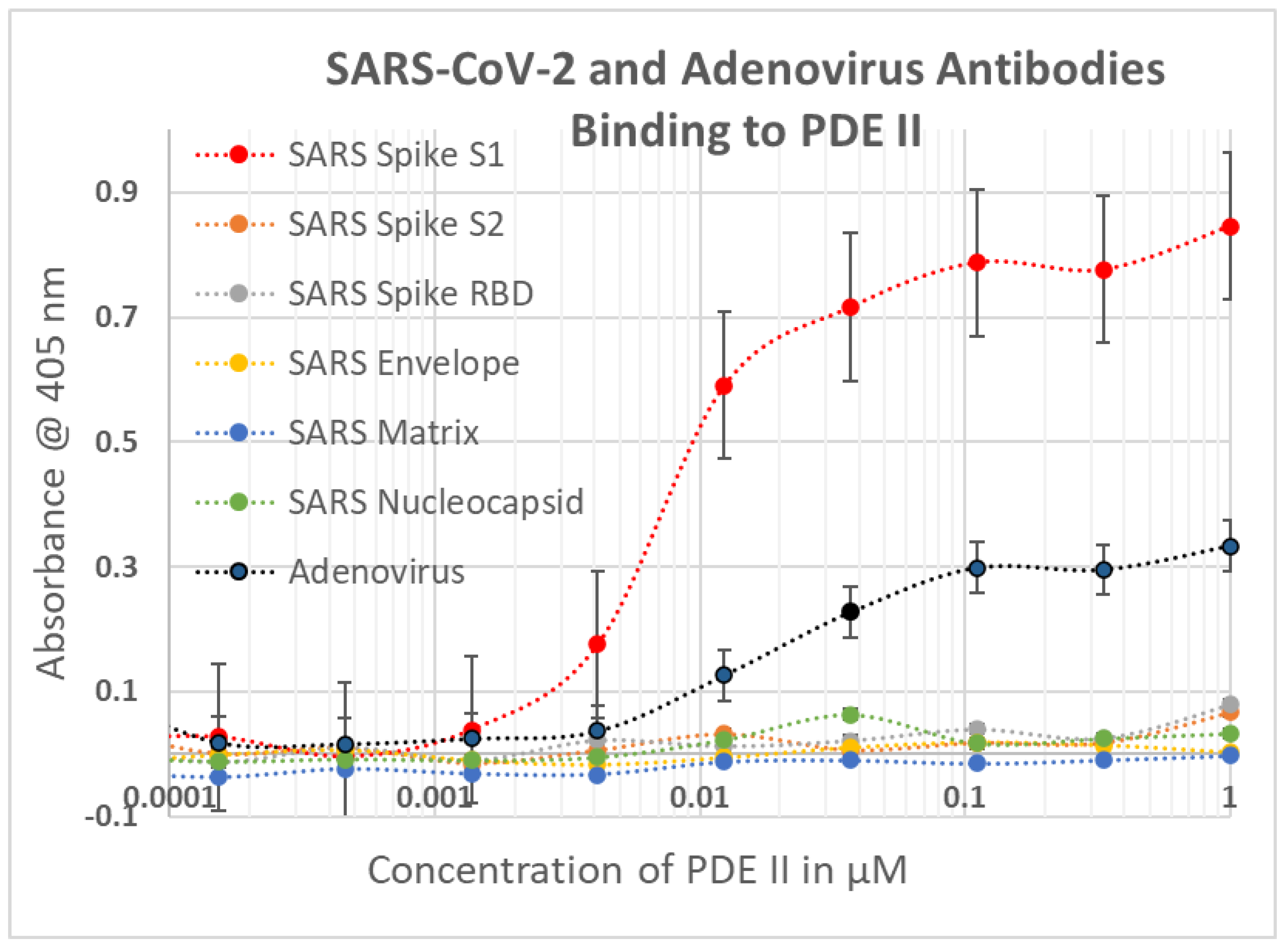
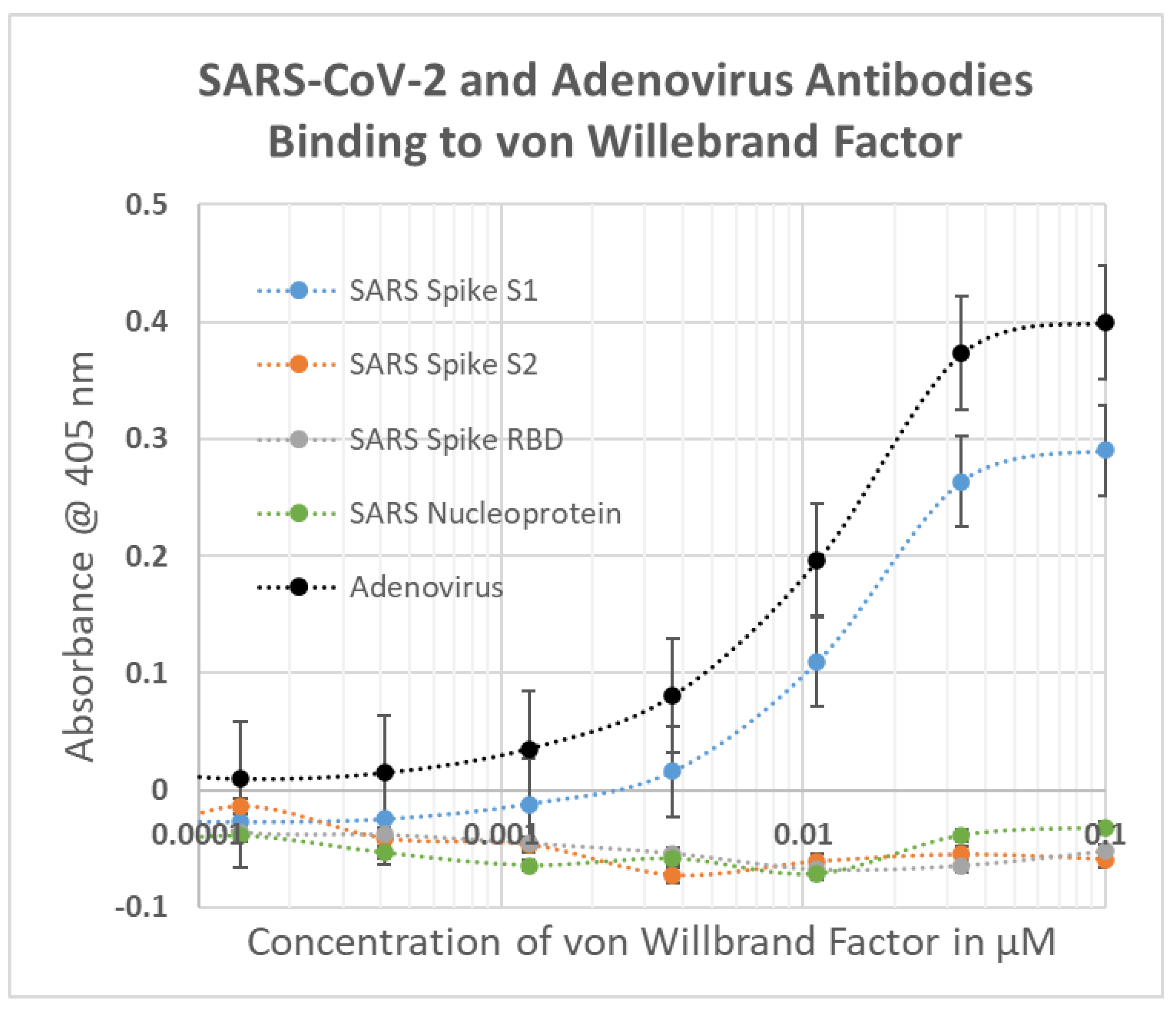
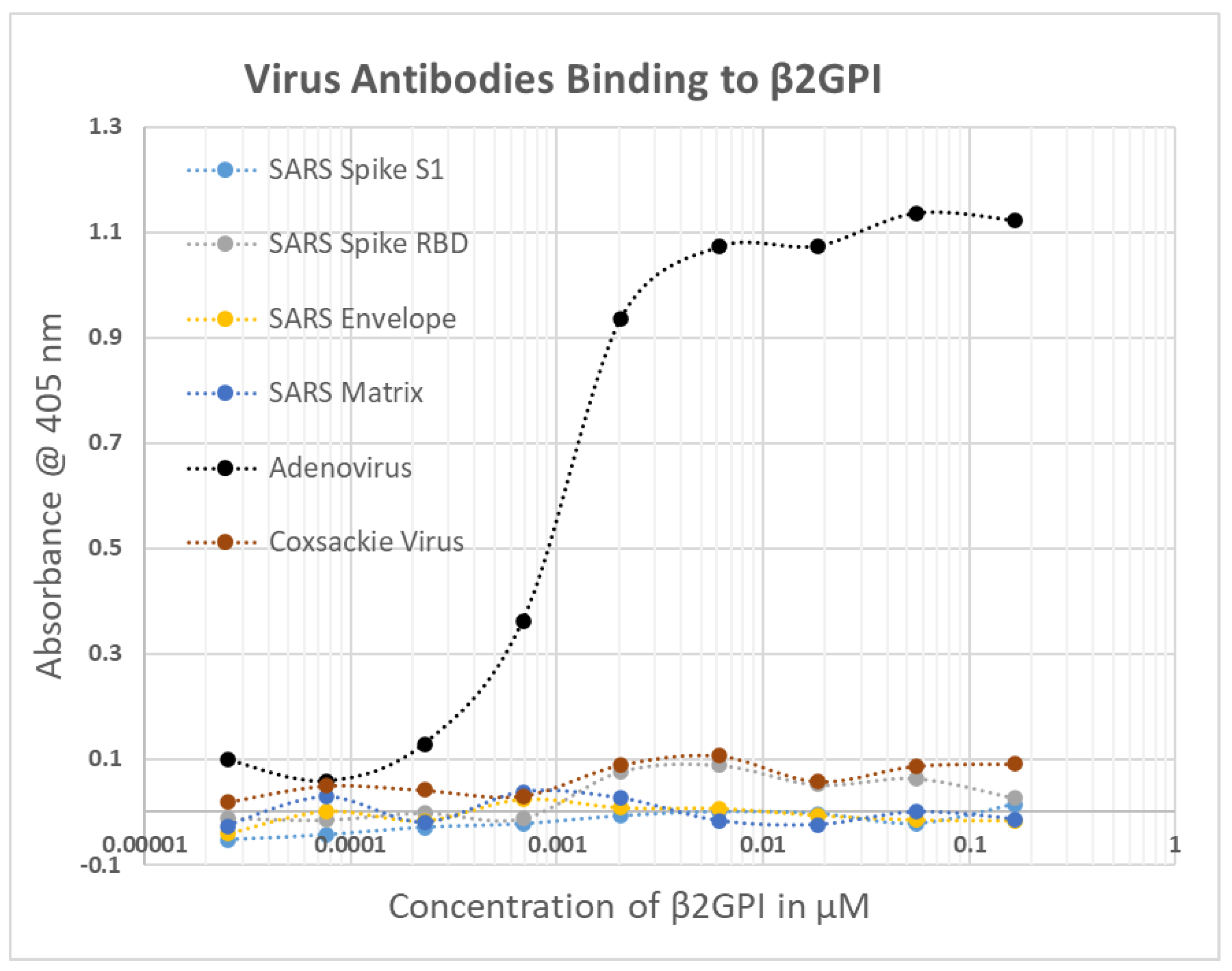
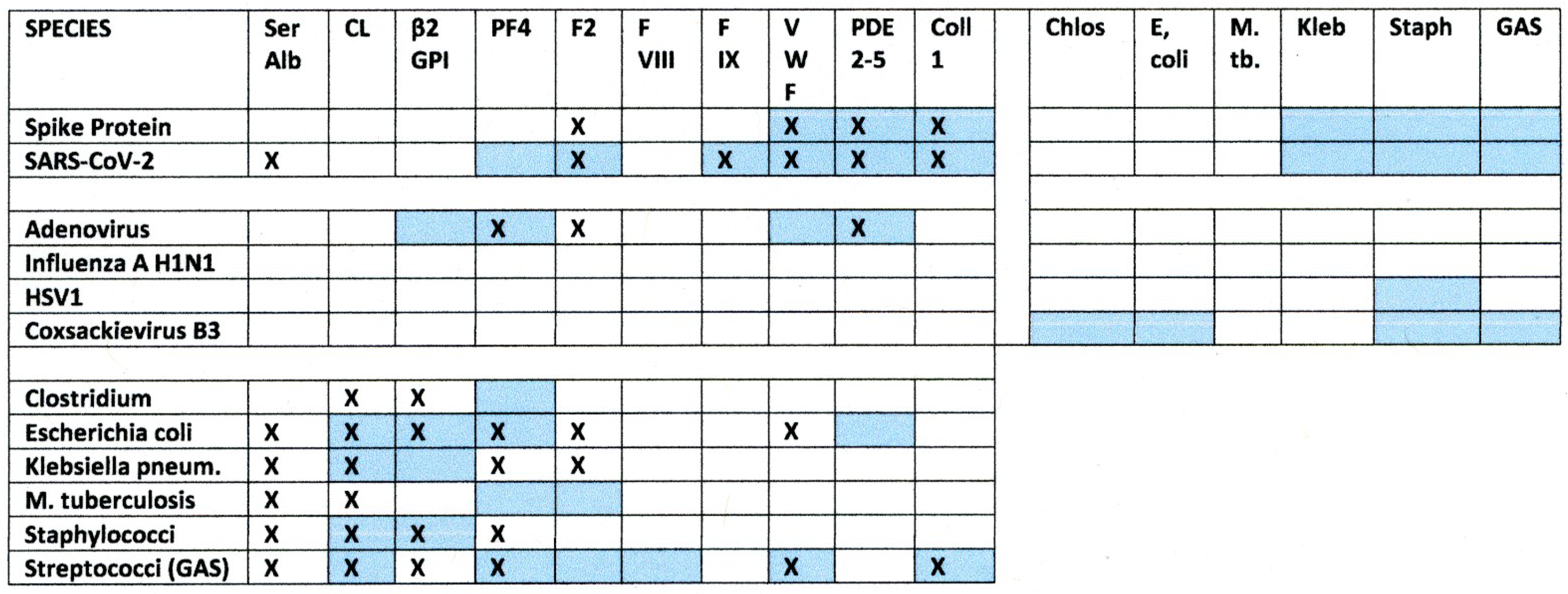
2.1. SARS-CoV-2, Adenovirus and Control Virus Antibody Binding to Blood Proteins
2.2. Determining Significance of Antibody Binding Constants
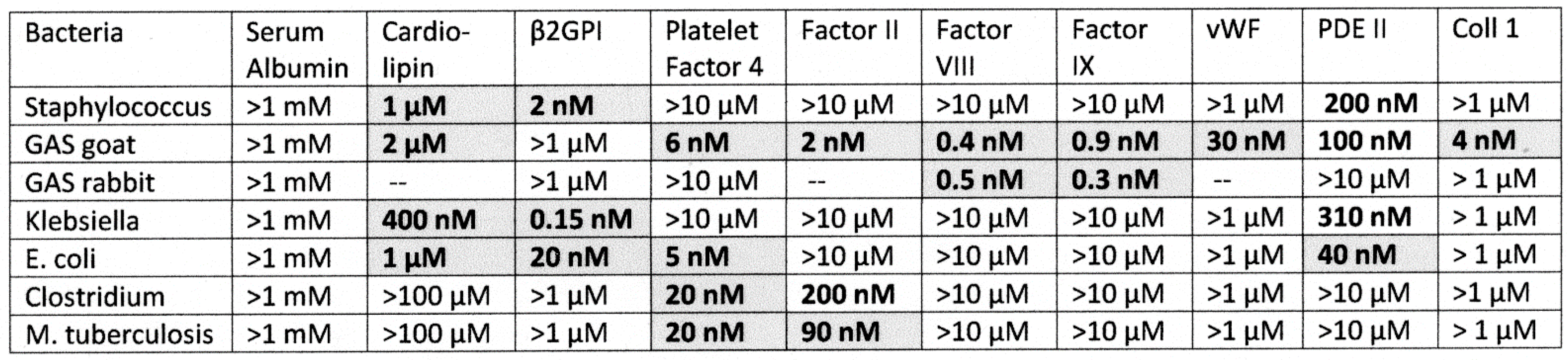
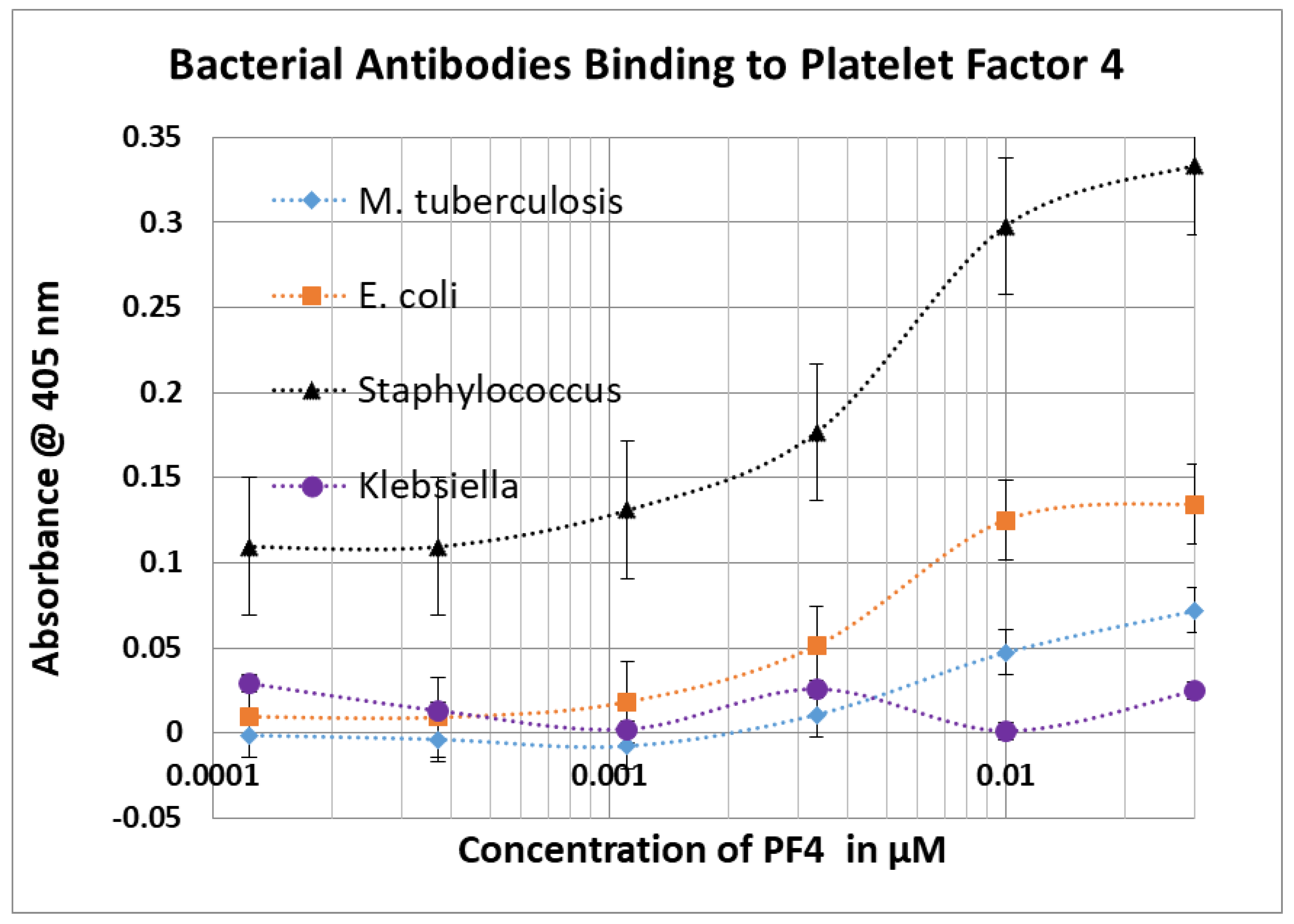
2.3. Bacterial Antibody Binding to Blood Proteins
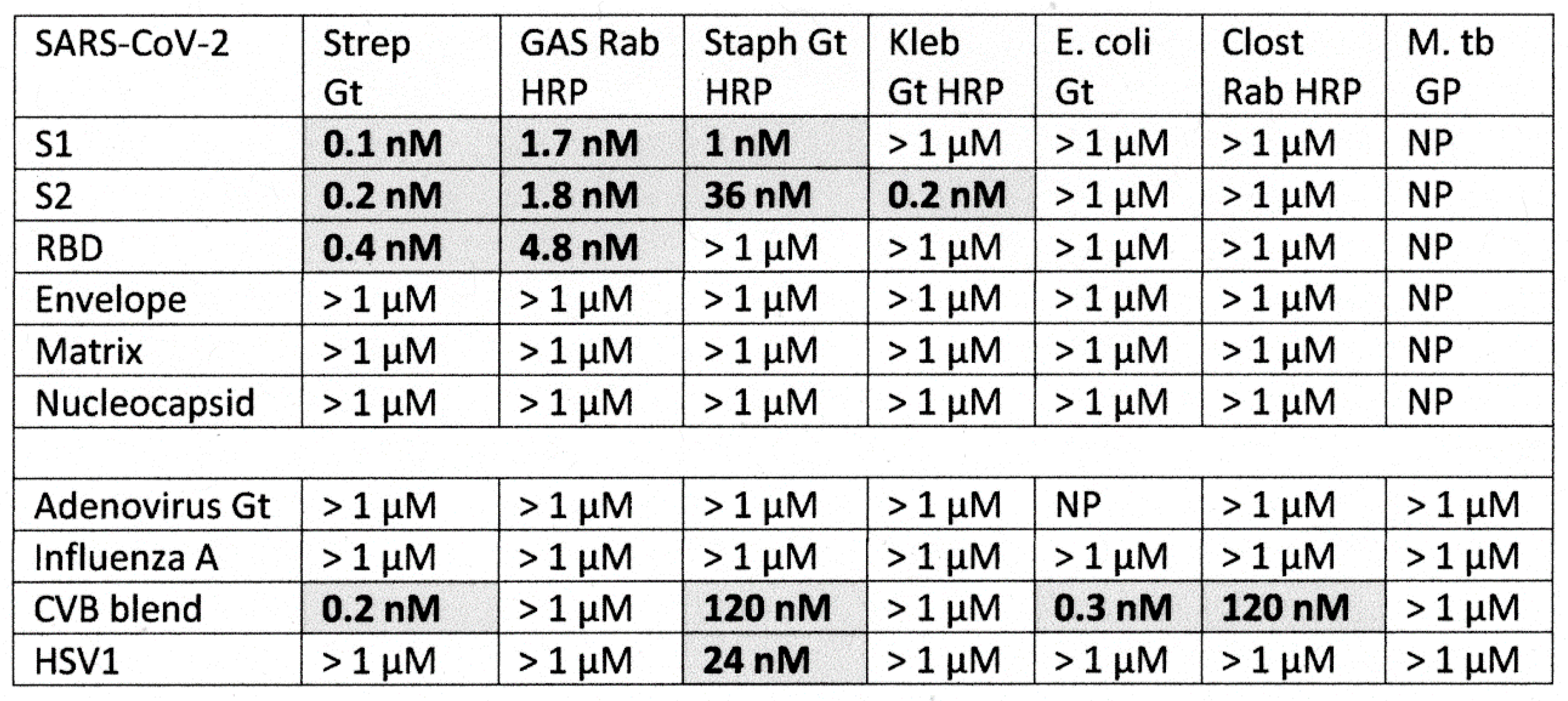
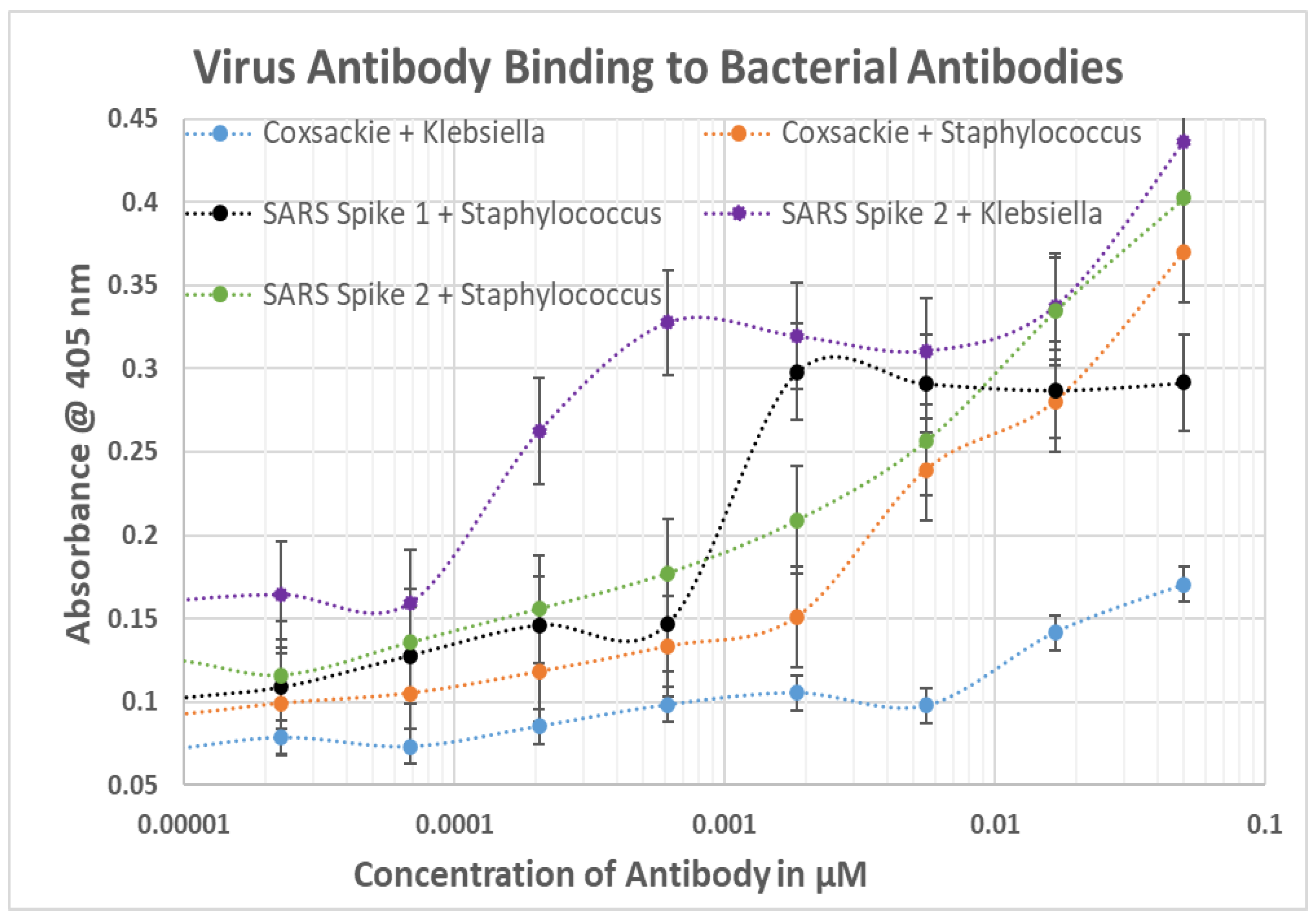
2.4. DA-ELISA Results: Virus Polyclonal Antibody Binding to Bacterial Polyclonal Antibodies
2.5. Results Summary
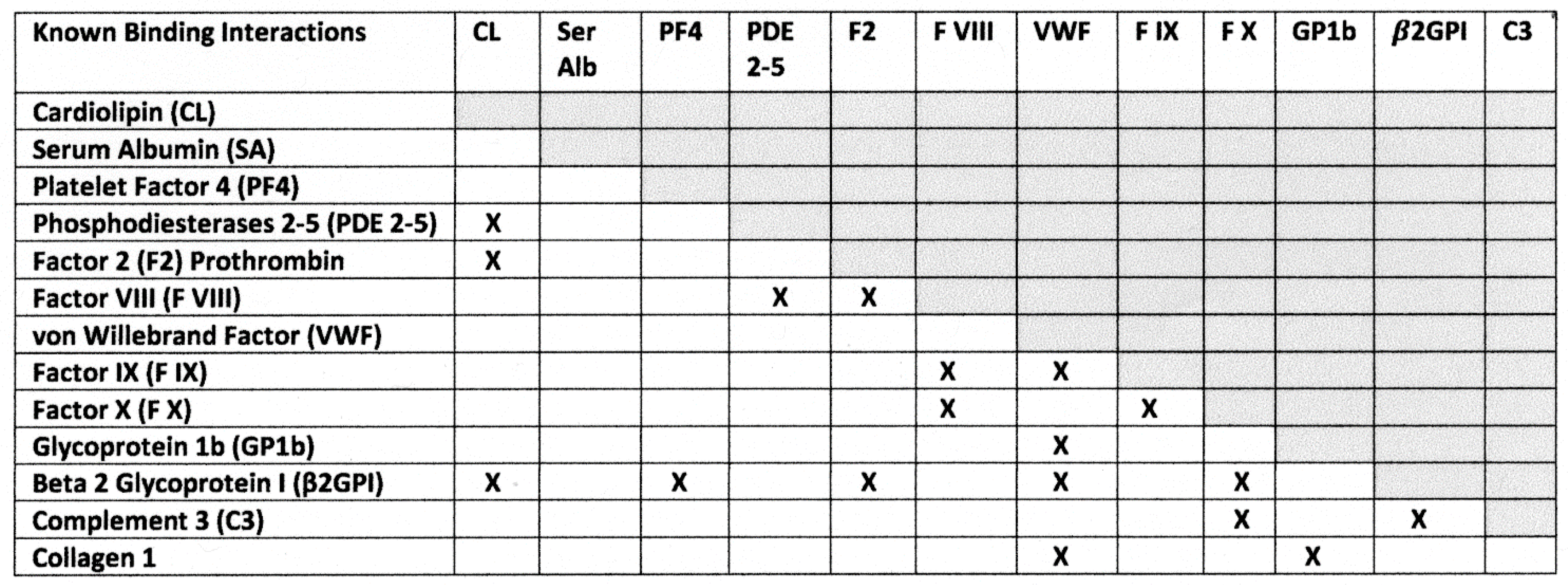
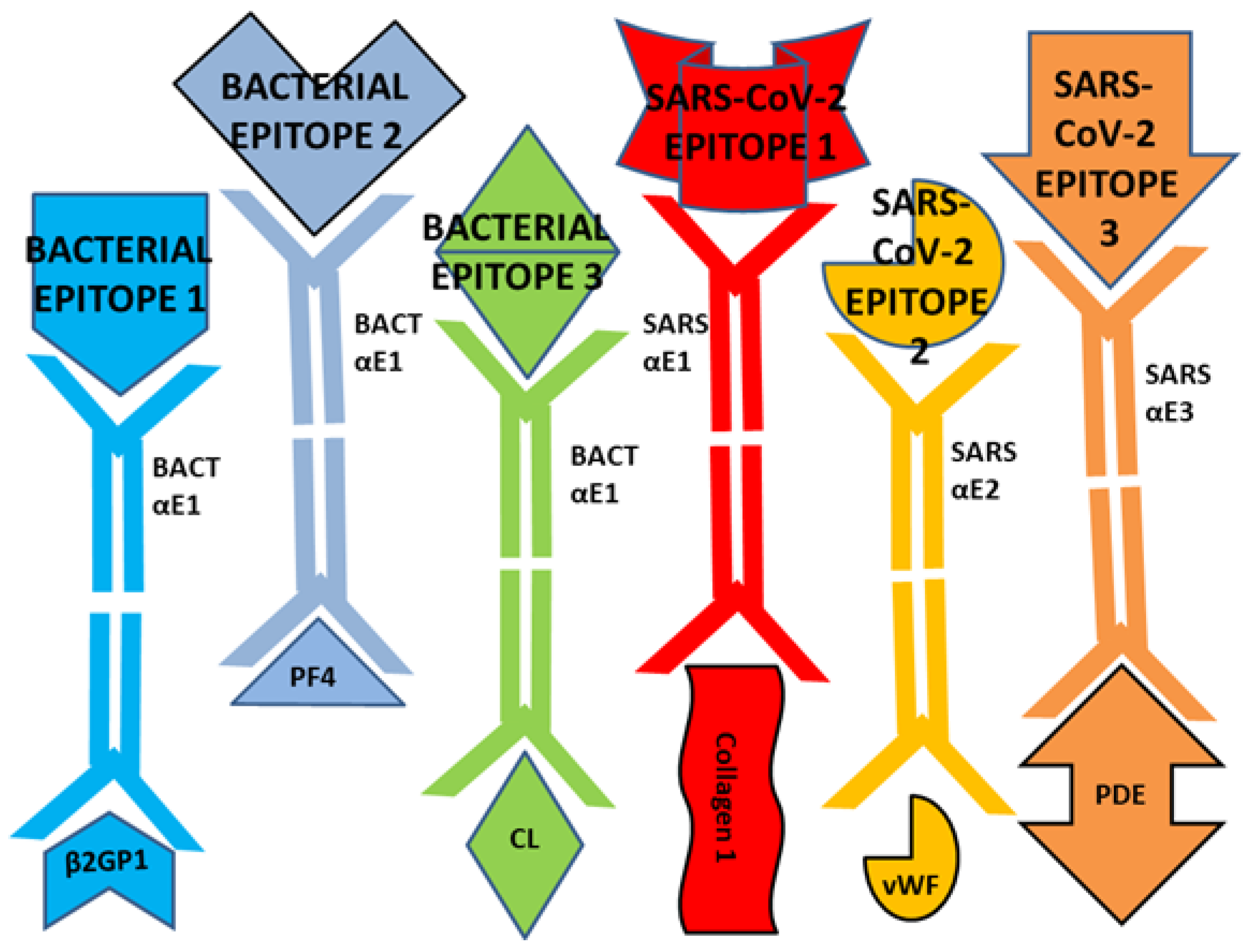
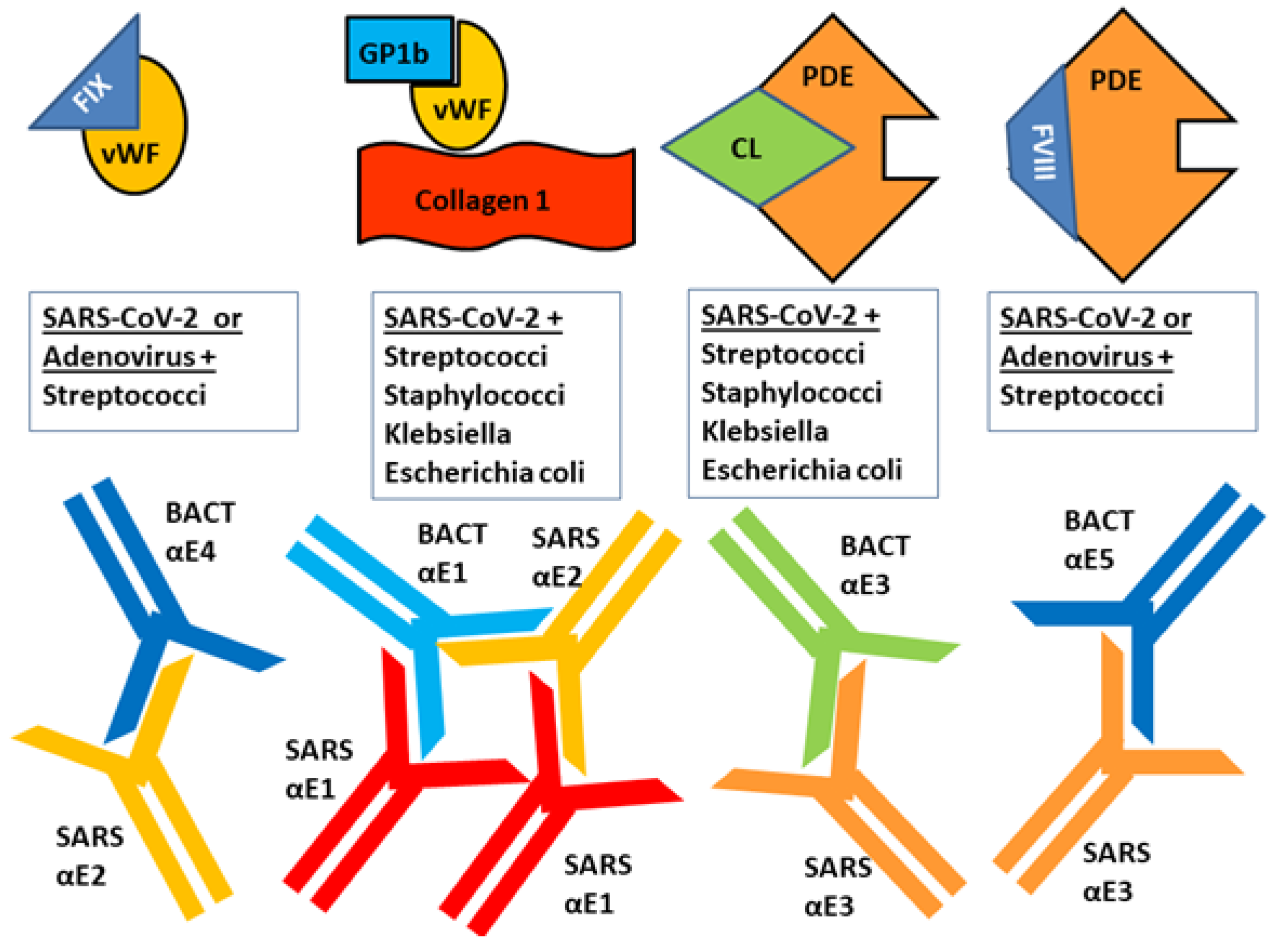
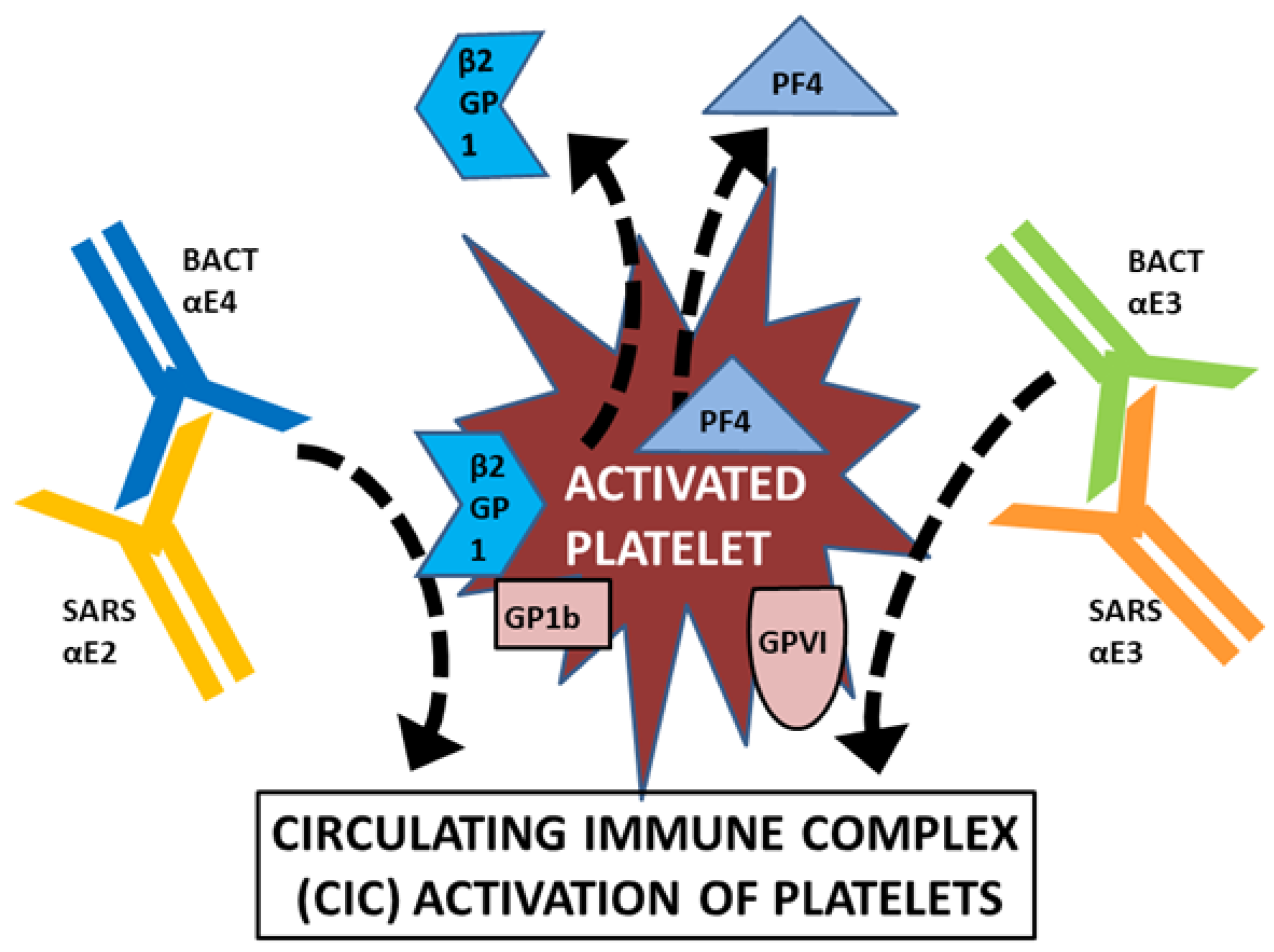
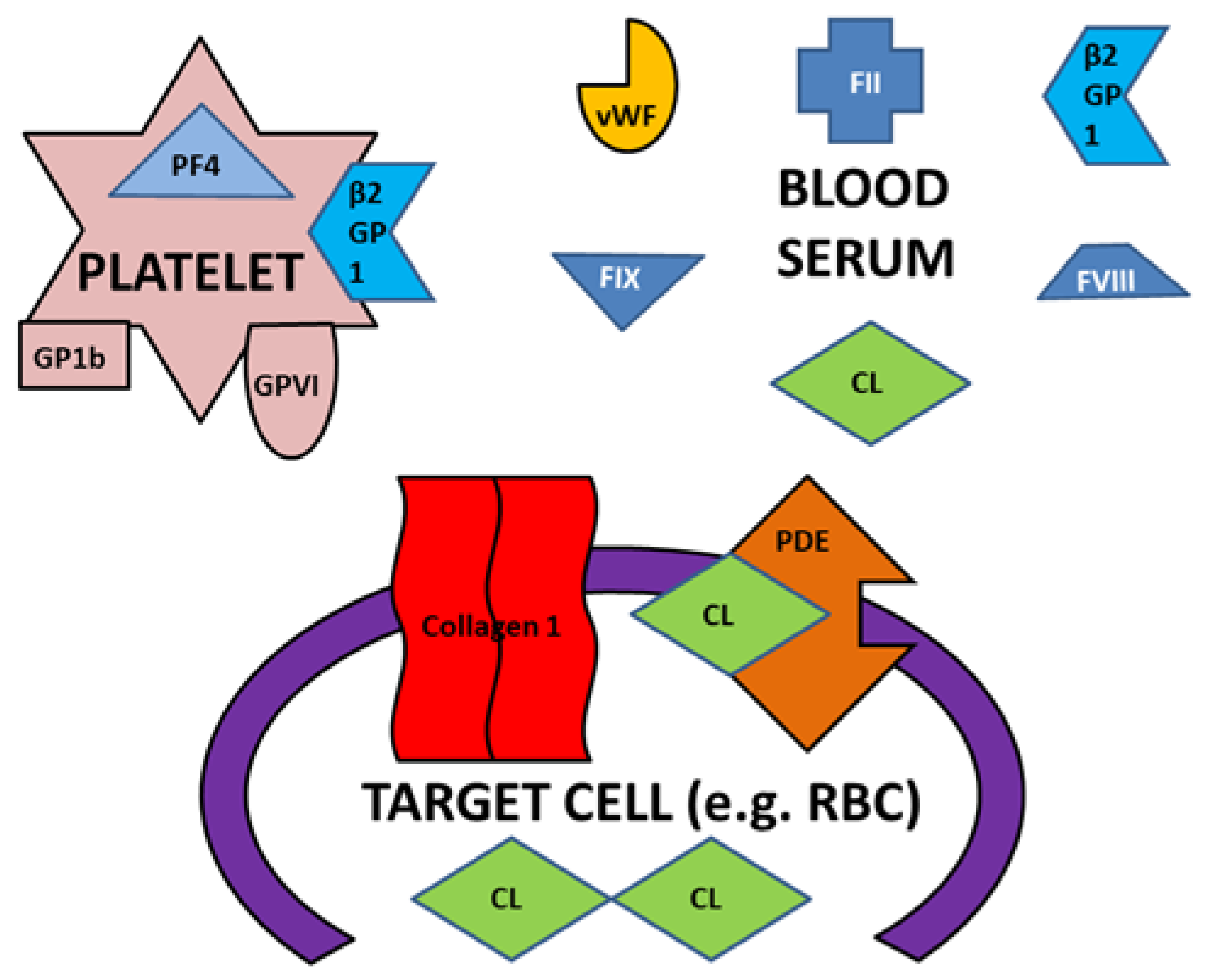
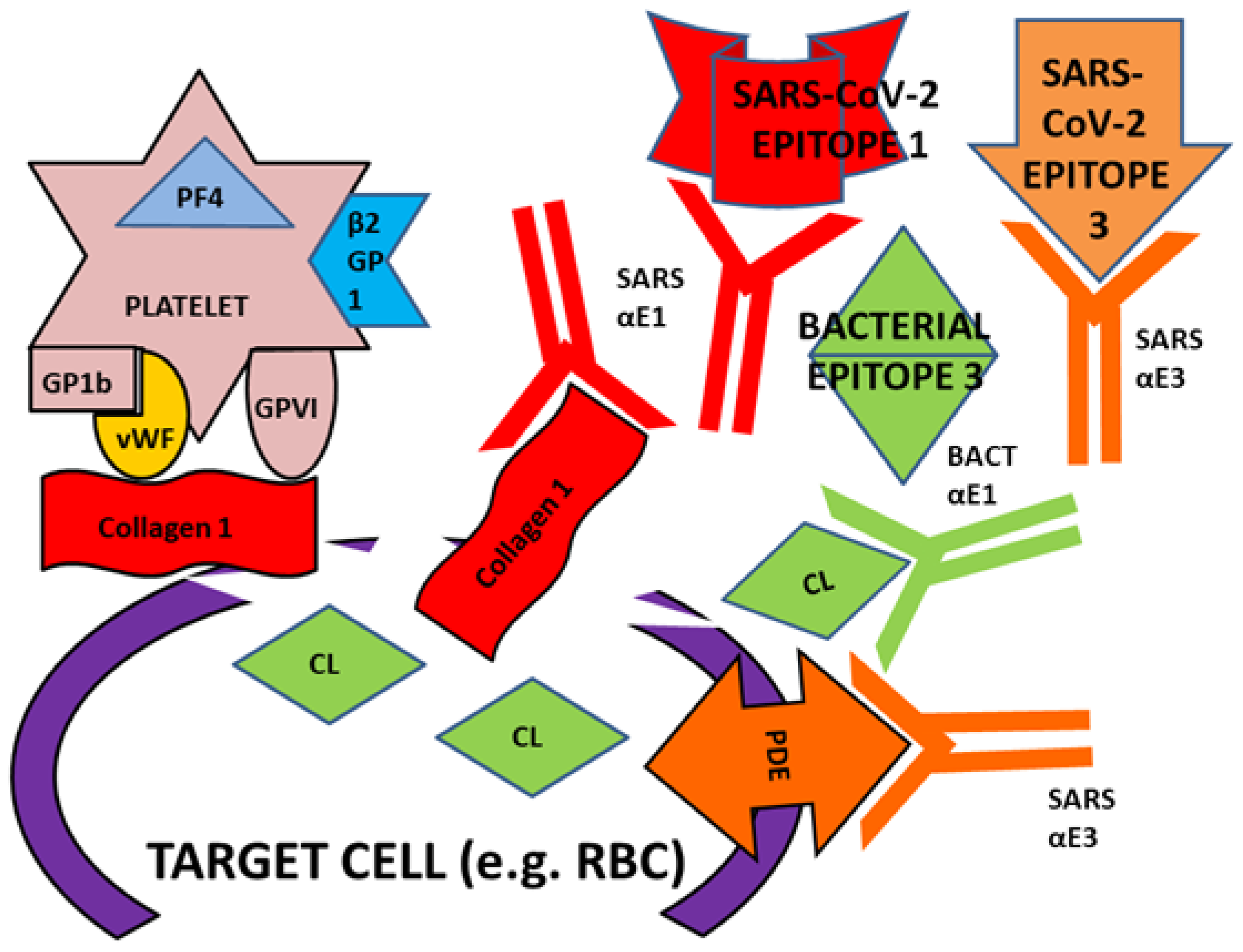
3. Discussion
4. Materials and Methods
4.1. Enzyme-Linked Immunosorbent Assay
4.2. Double Antibody ELISA
4.3. Antibodies
4.4. Proteins
4.5. Statistics
4.6. Methodological Limitations
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan; China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Zhang, J.-J.; Dong, X.; Cao, Y.-Y.; Yuan, Y.-D.; Yang, Y.-B.; Yan, Y.-Q.; Akdis, C.A.; Gao, Y.-D. Clinical characteristics of 140 patients infected with SARS-CoV-2 in Wuhan, China. Allergy 2020, 75, 1730–1741. [Google Scholar] [CrossRef]
- Connors, J.M.; Levy, J.H. COVID-19 and its implications for thrombosis and anticoagulation. Blood 2020, 135, 2033–2040. [Google Scholar] [CrossRef] [PubMed]
- Lund, L.C.; Hallas, J.; Nielsen, H.; Koch, A.; Mogensen, S.H.; Brun, N.C.; Christiansen, C.F.; Thomsen, R.W.; Pottegård, A. Post-acute effects of SARS-CoV-2 infection in individuals not requiring hospital admission: A Danish population-based cohort study. Lancet Infect. Dis. 2021, 21, 1373–1382. [Google Scholar] [CrossRef]
- Taquet, M.; Husain, M.; Geddes, J.R.; Luciano, S.; Harrison, P.J. Cerebral venous thrombosis and portal vein thrombosis: A retrospective cohort study of 537,913 COVID-19 cases. EClinicalMedicine 2021, 39, 101061. [Google Scholar] [CrossRef]
- Abou-Ismail, M.Y.; Diamond, A.; Kapoor, S.; Arafah, Y.; Nayak, L. The hypercoagulable state in COVID-19: Incidence, pathophysiology, and management. Thromb. Res. 2020, 194, 101–115. [Google Scholar] [CrossRef]
- Taha, M.; Samavati, L. Antiphospholipid antibodies in COVID-19: A meta-analysis and systematic review. RMD Open 2021, 7, e001580. [Google Scholar] [CrossRef]
- Najim, M.; Rahhal, A.; Khir, F.; Aljundi, A.H.; Abu Yousef, S.; Ibrahim, F.; Amer, A.; Mohamed, A.S.; Saleh, S.; Alfaridi, D.; et al. Prevalence and clinical significance of antiphospholipid antibodies in patients with coronavirus disease 2019 admitted to intensive care units: A prospective observational study. Rheumatol. Int. 2021, 41, 1243–1252. [Google Scholar] [CrossRef]
- Hendrickson, K.W.; Knox, D.B.; Bledsoe, J.R.; Peltan, I.D.; Jacobs, J.R.; Lloyd, J.F.; Dean, N.C.; Woller, S.C.; Brown, S.M. Comparative frequency of venous thromboembolism in patients admitted to the hospital with SARS-CoV-2 infection vs. community-acquired pneumonia. Ann. Am. Thorac. Soc. 2022, 19, 1233–1235. [Google Scholar] [CrossRef]
- Llitjos, J.F.; Leclerc, M.; Chochois, C.; Monsallier, J.M.; Ramakers, M.; Auvray, M.; Merouani, K. High incidence of venous thromboembolic events in anticoagulated severe COVID-19 patients. J. Thromb. Haemost. 2020, 18, 1743–1746. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; Banerjee, M. Immune thrombocytopenia secondary to COVID-19: A systematic review. SN Compr. Clin. Med. 2020, 2, 2048–2058. [Google Scholar] [CrossRef]
- Burkhard-Koren, N.M.; Haberecker, M.; Maccio, U.; Ruschitzka, F.; Schuepbach, R.A.; Zinkernagel, A.S.; Hardmeier, T.; Varga, Z.; Moch, H. Higher prevalence of pulmonary macrothrombi in SARS-CoV-2 than in influenza A: Autopsy results from ‘Spanish flu’ 1918/1919 in Switzerland to Coronavirus disease 2019. J. Pathol. Clin. Res. 2021, 7, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Wygrecka, M.; Birnhuber, A.; Seeliger, B.; Michalick, L.; Pak, O.; Schultz, A.S.; Schramm, F.; Zacharias, M.; Gorkiewicz, G.; David, S.; et al. Altered fibrin clot structure and dysregulated fibrinolysis contribute to thrombosis risk in severe COVID-19. Blood Adv. 2022, 6, 1074–1087. [Google Scholar] [CrossRef]
- Park, Y.S. Thrombosis and severe acute respiratory syndrome coronavirus 2 vaccines: Vaccine-induced immune thrombotic thrombocytopenia. Clin. Exp. Pediatr. 2021, 64, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Cines, D.B.; Bussel, J.B. SARS-CoV-2 vaccine-induced immune thrombotic thrombocytopenia. N. Engl. J. Med. 2021, 384, 2254–2256. [Google Scholar] [CrossRef]
- Gemmati, D.; Bramanti, B.; Serino, M.L.; Secchiero, P.; Zauli, G.; Tisato, V. COVID-19 and individual genetic susceptibility/receptivity: Role of ACE1/ACE2 genes.; immunity.; inflammation and coagulation. might the double x-chromosome in females be protective against SARS-CoV-2 compared to the single X-chromosome in males? Int. J. Mol. Sci. 2020, 21, 3474. [Google Scholar] [CrossRef]
- Henry, B.M.; Vikse, J.; Benoit, S.; Favaloro, E.J.; Lippi, G. Hyperinflammation and derangement of renin-angiotensin-aldosterone system in COVID-19: A novel hypothesis for clinically suspected hypercoagulopathy and microvascular immunothrombosis. Clin. Chim. Acta 2020, 507, 167–173. [Google Scholar] [CrossRef]
- Manne, B.K.; Denorme, F.; Middleton, E.A.; Portier, I.; Rowley, J.W.; Stubben, C.; Petrey, A.C.; Tolley, N.D.; Guo, L.; Cody, M.; et al. Platelet gene expression and function in patients with COVID-19. Blood 2020, 136, 1317–1329. [Google Scholar] [CrossRef] [PubMed]
- Kamel, M.H.; Yin, W.; Zavaro, C.; Francis, J.M.; Chitalia, V.C. Hyperthrombotic milieu in COVID-19 patients. Cells 2020, 9, 2392. [Google Scholar] [CrossRef]
- Greinacher, A.; Selleng, K.; Mayerle, J.; Palankar, R.; Wesche, J.; Reiche, S.; Aebischer, A.; Warkentin, T.E.; Muenchhoff, M.; Hellmuth, J.C.; et al. Anti-Platelet Factor 4 antibodies causing VITT do not cross-react with SARS-CoV-2 spike protein. Blood 2021, 138, 1269–1277. [Google Scholar] [CrossRef]
- Greinacher, A.; Selleng, K.; Palankar, R.; Wesche, J.; Handtke, S.; Wolff, M.; Aurich, K.; Lalk, M.; Methling, K.; Völker, U.; et al. Insights in ChAdOx1 nCov-19 vaccine-induced immune thrombotic thrombocytopenia (VITT). Blood 2021, 138, 2256–2268. [Google Scholar] [CrossRef] [PubMed]
- Fraser, D.D.; Patterson, E.K.; Slessarev, M.; Gill, S.E.; Martin, C.; Daley, M.; Miller, M.R.; Patel, M.A.; Dos Santos, C.C.; Bosma, K.J.; et al. Endothelial injury and glycocalyx degradation in critically Ill coronavirus disease 2019 patients: Implications for microvascular platelet aggregation. Crit. Care Explor. 2020, 2, e0194. [Google Scholar] [CrossRef] [PubMed]
- Dotan, A.; Muller, S.; Kanduc, D.; David, P.; Halpert, G.; Shoenfeld, Y. The SARS-CoV-2 as an instrumental trigger of autoimmunity. Autoimmun. Rev. 2021, 20, 102792. [Google Scholar] [CrossRef]
- Dotan, A.; Shoenfeld, Y. Perspectives on vaccine induced thrombotic thrombocytopenia. J. Autoimmun. 2021, 121, 102663. [Google Scholar] [CrossRef]
- Xiao, M.; Zhang, Y.; Zhang, S.; Qin, X.; Xia, P.; Cao, W.; Jiang, W.; Chen, H.; Ding, X.; Zhao, H.; et al. Antiphospholipid antibodies in critically ill patients with COVID-19. Arthritis Rheumatol. 2020, 72, 1998–2004. [Google Scholar] [CrossRef] [PubMed]
- Borghi, M.O.; Beltagy, A.; Garrafa, E.; Curreli, D.; Cecchini, G.; Bodio, C.; Grossi, C.; Blengino, S.; Tincani, A.; Franceschini, F.; et al. Anti-phospholipid antibodies in COVID-19 are different from those detectable in the anti-phospholipid syndrome. Front. Immunol. 2020, 11, 584241. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Estes, S.K.; Ali, R.A.; Gandhi, A.A.; Yalavarthi, S.; Shi, H.; Sule, G.; Gockman, K.; Madison, J.A.; Zuo, M.; et al. Prothrombotic autoantibodies in serum from patients hospitalized with COVID-19. Sci. Transl Med. 2020, 12, eabd3876. [Google Scholar] [CrossRef]
- Grobler, C.; Maphumulo, S.C.; Grobbelaar, L.M.; Grobler, C.; Maphumulo, S.C.; Grobbelaar, L.M.; Bredenkamp, J.C.; Laubscher, G.J.; Lourens, P.J.; Steenkamp, J.; et al. COVID-19: The rollercoaster of fibrin(ogen), D-dimer, von Willebrand Factor, P-selectin and their interactions with endothelial cells, platelets and erythrocytes. Int. J. Mol. Sci. 2020, 21, 5168. [Google Scholar] [CrossRef]
- Favaloro, E.J.; Henry, B.M.; Lippi, G. Is lupus anticoagulant a significant feature of COVID-19? A critical appraisal of the literature. Semin. Thromb. Hemost. 2021, 48, 055–071. [Google Scholar] [CrossRef]
- Dragonetti, D.; Guarini, G.; Pizzuti, M. Detection of anti-heparin-PF4 complex antibodies in COVID-19 patients on heparin therapy. Blood Transfus. 2020, 18, 328. [Google Scholar] [CrossRef]
- Brodard, J.; Kremer Hovinga, J.A.; Fontana, P.; Studt, J.D.; Gruel, Y.; Greinacher, A. COVID-19 patients often show high-titer non-platelet-activating anti-PF4/heparin IgG antibodies. J. Thromb. Haemost. 2021, 19, 1294–1298. [Google Scholar] [CrossRef] [PubMed]
- Greinacher, A.; Thiele, T.; Warkentin, T.E.; Weisser, K.; Kyrle, P.A.; Eichinger, S. Thrombotic thrombocytopenia after ChAdOx1 nCov-19 vaccination. N. Engl. J. Med. 2021, 384, 2092–2101. [Google Scholar] [CrossRef] [PubMed]
- Scully, M.; Singh, D.; Lown, R.; Poles, A.; Solomon, T.; Levi, M.; Goldblatt, D.; Kotoucek, P.; Thomas, W.; Lester, W. Pathologic antibodies to platelet factor 4 after ChAdOx1 nCoV-19 vaccination. N. Engl. J. Med. 2021, 384, 2202–2211. [Google Scholar] [CrossRef] [PubMed]
- Cristiano, A.; Fortunati, V.; Cherubini, F.; Bernardini, S.; Nuccetelli, M. Anti-phospholipids antibodies and immune complexes in COVID-19 patients: A putative role in disease course for anti-annexin-V antibodies. Clin. Rheumatol. 2021, 40, 2939–2945. [Google Scholar] [CrossRef]
- Emmenegger, M.; Kumar, S.S.; Emmenegger, V.; Malinauskas, T.; Buettner, T.; Rose, L.; Schierack, P.; Sprinzl, M.F.; Sommer, C.J.; Lackner, K.J.; et al. Anti-prothrombin autoantibodies enriched after infection with SARS-CoV-2 and influenced by strength of antibody response against SARS-CoV-2 proteins. PLoS Pathog. 2021, 17, e1010118. [Google Scholar] [CrossRef]
- Favaloro, E.J.; Henry, B.M.; Lippi, G. The complicated relationships of heparin-induced thrombocytopenia and platelet factor 4 antibodies with COVID-19. Int. J. Lab. Hematol. 2021, 43, 547–558. [Google Scholar] [CrossRef]
- Frydman, G.H.; Streiff, M.B.; Connors, J.M.; Piazza, G. The potential role of coagulation Factor Xa in the pathophysiology of COVID-19: A role for anticoagulants as multimodal therapeutic agents. TH Open 2020, 4, e288–e299. [Google Scholar] [CrossRef] [PubMed]
- Mancini, I.; Baronciani, L.; Artoni, A.; Colpani, P.; Biganzoli, M.; Cozzi, G.; Novembrino, C.; Boscolo Anzoletti, M.; De Zan, V.; Pagliari, M.T.; et al. The ADAMTS13-von Willebrand factor axis in COVID-19 patients. J. Thromb. Haemost. 2021, 19, 513–521. [Google Scholar] [CrossRef]
- Kanduc, D. Thromboses and hemostasis disorders associated with COVID-19: The possible causal role of cross-reactivity and immunological imprinting. Glob. Med. Genet. 2021, 8, 162–170. [Google Scholar] [CrossRef]
- Root-Bernstein, R. COVID-19 coagulopathies: Human blood proteins mimic SARS-CoV-2 virus.; vaccine proteins and bacterial co-infections inducing autoimmunity: Combinations of bacteria and SARS-CoV-2 synergize to induce autoantibodies targeting cardiolipin, cardiolipin-binding proteins, platelet factor 4, prothrombin, and coagulation factors. Bioessays 2021, 43, e2100158. [Google Scholar] [CrossRef]
- Mir, T.H. Thrombotic microangiopathy (aHUS/iTTP) reported so far in Covid-19 patients: The virus alone or an omnium gatherum of mechanisms and etiologies? Crit. Rev. Oncol. Hematol. 2021, 162, 103347. [Google Scholar] [CrossRef] [PubMed]
- Root-Bernstein, R. Innate receptor activation patterns involving TLR and NLR synergisms in COVID-19; ALI/ARDS and sepsis cytokine storms: A review and model making novel predictions and therapeutic suggestions. Int. J. Mol. Sci. 2021, 22, 2108. [Google Scholar] [CrossRef]
- Cambier, S.; Metzemaekers, M.; de Carvalho, A.C.; Nooyens, A.; Jacobs, C.; Vanderbeke, L.; Malengier-Devlies, B.; Gouwy, M.; Heylen, E.; Meersseman, P.; et al. Atypical response to bacterial coinfection and persistent neutrophilic bronchoalveolar inflammation distinguish critical COVID-19 from influenza. JCI Insight 2022, 7, e155055. [Google Scholar] [CrossRef] [PubMed]
- Keikha, M.; Karbalaei, M. Potential association between bacterial infections and ischemic stroke based on fifty case-control studies: A systematic review and meta-analysis. New Microbes New Infect. 2022, 47, 100980. [Google Scholar] [CrossRef] [PubMed]
- Budzyński, J.; Wiśniewska, J.; Ciecierski, M.; Kędzia, A. Association between bacterial infection and peripheral vascular disease: A review. Int. J. Angiol. 2016, 25, 3–13. [Google Scholar]
- Lucchese, G.; Flöel, A.; Stahl, B. Cross-reactivity as a mechanism linking infections to stroke. Front. Neurol. 2019, 10, 469. [Google Scholar] [CrossRef]
- Blank, M.; Krause, I.; Fridkin, M.; Keller, N.; Kopolovic, J.; Goldberg, I.; Tobar, A.; Shoenfeld, Y. Bacterial induction of autoantibodies to β2-glycoprotein-I accounts for the infectious etiology of antiphospholipid syndrome. J. Clin. Investig. 2002, 109, 797–804. [Google Scholar] [CrossRef]
- Loof, T.G.; Deicke, C.; Medina, E. The role of coagulation/fibrinolysis during Streptococcus pyogenes infection. Front. Cell. Infect. Microbiol. 2014, 4, 128. [Google Scholar] [CrossRef]
- Rokkam, V.R.P.; Kutti Sridharan, G.; Vegunta, R.; Vegunta, R.; Boregowda, U.; Mohan, B.P. Clostridium difficile and COVID-19: Novel risk factors for acute portal vein thrombosis. Case Rep. Vasc. Med. 2021, 2021, 8832638. [Google Scholar] [CrossRef]
- Khanna, S.; Kraft, C.S. The interplay of SARS-CoV-2 and Clostridioides difficile infection. Future Microbiol. 2021, 16, 439–443. [Google Scholar] [CrossRef]
- Di Micco, P.; Imparato, M.; Lubrano, G.; Iannuzzo, D.; Fontanella, L.; Improta, L.; Poggiano, M.R.; Salzano, C.; Rodolico, A.; Fontanella, A. Resolution of disseminated intravascular coagulation in a patient with COVID-19 and associated sepsis-induced neutropenia. Medicina 2021, 57, 106. [Google Scholar] [CrossRef] [PubMed]
- Harmon, M.B.A.; Heijnen, N.F.L.; de Bruin, S.; Sperna Weiland, N.H.; Meijers, J.C.M.; de Boer, A.M.; Schultz, M.J.; Horn, J.; Juffermans, N.P. Induced normothermia ameliorates the procoagulant host response in human endotoxaemia. Br. J. Anaesth. 2021, 126, 1111–1118. [Google Scholar] [CrossRef] [PubMed]
- Tome, J.; Maselli, D.B.; Im, R.; Amdahl, M.B.; Pfeifle, D.; Hagen, C.; Halland, M. A case of hemolytic uremic syndrome caused by Shiga toxin-producing Escherichia coli after pericardiectomy. Clin. J. Gastroenterol. 2022, 15, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Perinkulam Sathyanarayanan, S.; Ahmed, M.; Hericks, A. Purpura fulminans due to Streptococcus pneumoniae bacteraemia in an unsplectomised immunocompetent adult without primary hypocomplementaemia. BMJ Case Rep. 2022, 15, e249514. [Google Scholar] [CrossRef] [PubMed]
- Kopenhagen, A.; Ramming, I.; Camp, B.; Hammerschmidt, S.; Fulde, M.; Müsken, M.; Steinert, M.; Bergmann, S. Streptococcus pneumoniae affects endothelial cell migration in microfluidic circulation. Front. Microbiol. 2022, 13, 852036. [Google Scholar] [CrossRef] [PubMed]
- Meini, S.; Sozio, E.; Bertolino, G.; Sbrana, F.; Ripoli, A.; Pallotto, C.; Viaggi, B.; Andreini, R.; Attanasio, V.; Rescigno, C.; et al. D-dimer as biomarker for early prediction of clinical outcomes in patients with severe invasive infections due to Streptococcus pneumoniae and Neisseria meningitidis. Front. Med. 2021, 8, 627830. [Google Scholar] [CrossRef]
- Walsh, L.F.; Sherbuk, J.E.; Wispelwey, B. Pneumococcal induced thrombotic thrombocytopenic purpura with features of purpura fulminans. BMJ Case Rep. 2021, 14, e235580. [Google Scholar] [CrossRef]
- Djurdjevic, N.; Taweesedt, P.T.; Paulson, M.; LaNou, A.; Radovanovic, M.; Patel, J.N.; Veselinovic, M.; McDermott, W.R.; Dumic, I. Septic shock and purpura fulminans due to Streptococcus pneumoniae bacteremia in an unvaccinated immunocompetent adult: Case report and review. Am. J. Case Rep. 2020, 21, e923266. [Google Scholar] [CrossRef]
- Meyers, S.; Crescente, M.; Verhamme, P.; Martinod, K. Staphylococcus aureus and neutrophil extracellular traps: The master manipulator meets its match in immunothrombosis. Arterioscler. Thromb. Vasc. Biol. 2022, 42, 261–276. [Google Scholar] [CrossRef]
- Grapsa, J.; Blauth, C.; Chandrashekhar, Y.S.; Prendergast, B.; Erb, B., Jr.; Mack, M.; Fuster, V. Staphylococcus aureus infective endocarditis: JACC Patient Pathways. JACC Case Rep. 2021, 4, 1–12. [Google Scholar] [CrossRef]
- Khalaf, S.A.; Mansour, A.; Perveze, I.; Fender, B.; Walker, D.R.; Dandachi, D. Staphylococcus lugdunensis as cause of septic pericarditis. Mo. Med. 2021, 118, 552–555. [Google Scholar] [PubMed]
- Westall, F.C.; Root-Bernstein, R. Cause and prevention of postinfectious and postvaccinal neuropathies in light of a new theory of autoimmunity. Lancet 1986, 2, 251–252. [Google Scholar] [CrossRef]
- Root-Bernstein, R.; Couturier, J. Antigenic complementarity in the origins of autoimmunity: A general theory illustrated with a case study of idiopathic thrombocytopenia purpura. Clin. Dev. Immunol. 2006, 13, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Gaebler, C.; Wang, Z.; Lorenzi, J.; Muecksch, F.; Finkin, S.; Tokuyama, M.; Cho, A.; Jankovic, M.; Schaefer-Babajew, D.; Oliveira, T.Y.; et al. Evolution of antibody immunity to SARS-CoV-2. Nature 2021, 591, 639–644. [Google Scholar] [CrossRef]
- Deguchi, H.; Fernandez, J.A.; Hackeng, T.M.; Banka, C.L.; Griffin, J.H. Cardiolipin is a normal component of human plasma lipoproteins. Proc. Natl. Acad. Sci. USA 2000, 97, 1743–1748. [Google Scholar] [CrossRef]
- McNally, T.; Mackie, I.J.; Machin, S.J.; Isenberg, D.A. Elevated levels of beta 2 glycoprotein-I (beta 2 GPI) in antiphospholipid antibody syndrome are due to increased amounts of beta 2 GPI in association with other plasma constituents. Blood Coagul. Fibrinolysis 1995, 6, 411–416. [Google Scholar] [CrossRef]
- Zucker, M.B.; Katz, I.R. Platelet factor 4: Production, structure, and physiologic and immunologic action. Proc. Soc. Exp. Biol. Med. 1991, 198, 693–702. [Google Scholar] [CrossRef]
- Peters, T. All about Albumin: Biochemistry, Genetics and Medical Applications; Academic Press Limited: San Diego, CA, USA, 1996. [Google Scholar]
- Smiles, A.M.; Macy, E.M.; Sakkinen, P.A.; Bovill, E.G.; Mann, K.G.; Tracy, R.P. Variability in plasma prothrombin concentration: Implications for use in epidemiology. Blood Coagul. Fibrinolysis 1998, 9, 525–531. [Google Scholar] [CrossRef]
- Butenas, S.; Parhami-Seren, B.; Undas, A.; Fass, D.N.; Mann, K.G. The “normal” factor VIII concentration in plasma. Thromb. Res. 2010, 126, 119–123. [Google Scholar] [CrossRef]
- Schmidt, A.E.; Bajaj, S.P. Structure–function relationships in factor IX and factor IXa. Trends Cardiovasc. Med. 2003, 13, 39–45. [Google Scholar] [CrossRef]
- Sakai, H.; Goto, S.; Kim, J.Y.; Aoki, N.; Abe, S.; Ichikawa, N.; Yoshida, M.; Nagaoka, Y.; Handa, S. Plasma concentration of von Willebrand factor in acute myocardial infarction. Thromb. Haemost. 2000, 84, 204–209. [Google Scholar] [PubMed]
- Eigenthaler, M.; Nolte, C.; Halbrügge, M.; Walter, U. Concentration and regulation of cyclic nucleotides, cyclic-nucleotide-dependent protein kinases and one of their major substrates in human platelets. Estimating the rate of cAMP-regulated and cGMP-regulated protein phosphorylation in intact cells. Eur. J. Biochem. 1992, 205, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, K.; Naik, D.; Ray, S.; Vadiraja, B.M.; Kamath, A. Serum phosphodiesterase levels in oral cancer. J. Cancer Res. Ther. 2011, 7, 180–182. [Google Scholar] [CrossRef]
- Manon-Jensen, T.; Kjeld, N.G.; Karsdal, M.A. Collagen-mediated hemostasis. J. Thromb. Haemost. 2016, 14, 438–448. [Google Scholar] [CrossRef]
- Okhota, S.; Melnikov, I.; Avtaeva, Y.; Kozlov, S.; Gabbasov, Z. Shear stress-induced activation of von Willebrand Factor and cardiovascular pathology. Int. J. Mol. Sci. 2020, 21, 7804. [Google Scholar] [CrossRef]
- Childers, K.C.; Peters, S.C.; Clint Spiegel, P., Jr. Structural insights into blood coagulation factor VIII: Procoagulant complexes, membrane binding, and antibody inhibition. J. Thromb. Haemost. 2022, 20, 1957–1970. [Google Scholar] [CrossRef] [PubMed]
- Franchini, M.; Lippi, G. Von Willebrand factor-containing factor VIII concentrates and inhibitors in haemophilia A. A critical literature review. Thromb. Haemost. 2010, 104, 931–940. [Google Scholar]
- Shannon, O.; Herwald, H.; Oehmcke, S. Modulation of the coagulation system during severe streptococcal disease. Curr. Top. Microbiol. Immunol. 2013, 368, 189–205. [Google Scholar] [CrossRef]
- de Groot, P.G.; Meijers, J.C. β(2) -Glycoprotein I: Evolution, structure and function. J. Thromb. Haemost. 2011, 9, 1275–1284. [Google Scholar] [CrossRef]
- Cole, R.; Proulx, P. Further studies on the cardiolipin phosphodiesterase of Escherichia coli. Can. J. Biochem. 1977, 55, 1228–1232. [Google Scholar] [CrossRef]
- Pichard, A.L.; Cheung, W.Y. Cyclic 3′:5′-nucleotide phosphodiesterase. Stimulation of bovine brain cytoplasmic enzyme by lysophosphatidylcholine. J. Biol. Chem. 1977, 252, 4872–4875. [Google Scholar] [CrossRef]
- Rossen, R.D.; Michael, L.H.; Hawkins, H.K.; Youker, K.; Dreyer, W.J.; Baughn, R.E.; Entman, M.L. Cardiolipin-protein complexes and initiation of complement activation after coronary artery occlusion. Circ. Res. 1994, 75, 546–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, A.T.; Boyd, R.J.; Sarkar, D.; Teijeira-Crespo, A.; Chan, C.K.; Bates, E.; Waraich, K.; Vant, J.; Wilson, E.; Truong, C.D.; et al. ChAdOx1 interacts with CAR and PF4 with implications for thrombosis with thrombocytopenia syndrome. Sci. Adv. 2021, 7, eabl8213. [Google Scholar] [CrossRef] [PubMed]
- Steinert, M.; Ramming, I.; Bergmann, S. Impact of Von Willebrand Factor on bacterial pathogenesis. Front. Med. 2020, 7, 543. [Google Scholar] [CrossRef] [PubMed]
- Lukomski, S.; Bachert, B.A.; Squeglia, F.; Berisio, R. Collagen-like proteins of pathogenic streptococci. Mol. Microbiol. 2017, 103, 919–930. [Google Scholar] [CrossRef]
- Thomas, S.; Arora, S.; Liu, W.; Churion, K.; Wu, Y.; Höök, M. vhp is a fibrinogen-binding protein related to vWbp in Staphylococcus aureus. mBio 2021, 12, e0116721. [Google Scholar] [CrossRef]
- Vojdani, A.; Kharrazian, D. Potential antigenic cross-reactivity between SARS-CoV-2 and human tissue with a possible link to an increase in autoimmune diseases. Clin. Immun. 2020, 217, 108480. [Google Scholar] [CrossRef]
- Vojdani, A.; Vojdani, E.; Kharrazian, D. Reaction of human monoclonal antibodies to SARS-CoV-2 proteins with tissue antigens: Implications for autoimmune diseases. Front. Immunol. 2021, 11, 617089. [Google Scholar] [CrossRef]
- Passariello, M.; Vetrei, C.; Amato, F.; De Lorenzo, C. Interactions of spike-RBD of SARS-CoV-2 and Platelet Factor 4: New insights in the etiopathogenesis of thrombosis. Int. J. Mol. Sci. 2021, 22, 8562. [Google Scholar] [CrossRef]
- Pai, M. Epidemiology of VITT. Semin. Hematol. 2022, 59, 72–75. [Google Scholar] [CrossRef]
- Qiu, S.; Zeng, G.; Li, P.; Li, X.; Liu, H.; Du, X.; Zhang, H.; Xiang, X.; Wang, H.; Chen, X.; et al. pneumonia patients caused by co-infection with SARS-CoV-2 and human adenovirus in China. Front. Med. 2021, 8, 735779. [Google Scholar] [CrossRef] [PubMed]
- Swets, M.C.; Russell, C.D.; Harrison, E.M.; Docherty, A.B.; Lone, N.; Girvan, M.; Hardwick, H.E.; ISARIC4C Investigators; Visser, L.G.; Openshaw, P.; et al. SARS-CoV-2 co-infection with influenza viruses, respiratory syncytial virus, or adenoviruses. Lancet 2022, 399, 1463–1464. [Google Scholar] [CrossRef]
- Sreenath, K.; Batra, P.; Vinayaraj, E.V.; Bhatia, R.; SaiKiran, K.V.; Singh, V.; Singh, S.; Verma, N.; Singh, U.B.; Mohan, A.; et al. Coinfections with other respiratory pathogens among patients with COVID-19. Microbiol. Spectr. 2021, 9, e0016321. [Google Scholar] [CrossRef]
- Alwen, J.; Emmerson, A.M. Antibodies against adeno-, cytomegalo and rubella viruses in Australia-antigen-negative sera from patients with infectious hepatitis. J. Hyg. 1974, 72, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Krashias, G.; Pafiti, A.; Deeba, E.; Pantzaris, M.; Lambrianides, A. SARS CoV- 2 vaccination 1 induces antibodies against cardiolipin? BMC Res. Notes 2022, in press. [CrossRef]
- Thurm, C.; Reinhold, A.; Borucki, K.; Kahlfuss, S.; Feist, E.; Schreiber, J.; Reinhold, D.; Schraven, B. Homologous and Heterologous Anti-COVID-19 Vaccination Does Not Induce New-Onset Formation of Autoantibodies Typically Accompanying Lupus Erythematodes, Rheumatoid Arthritis, Celiac Disease and Antiphospholipid Syndrome. Vaccines 2022, 10, 333. [Google Scholar] [CrossRef]
- Borghi, M.O.; Bombaci, M.; Bodio, C.; Lonati, P.A.; Gobbini, A.; Lorenzo, M.; Torresani, E.; Dubini, A.; Bulgarelli, I.; Solari, F.; et al. Anti-Phospholipid Antibodies and Coronavirus Disease 2019: Vaccination Does Not Trigger Early Autoantibody Production in Healthcare Workers. Front. Immunol. 2022, 15, 930074. [Google Scholar] [CrossRef]
- Liu, T.; Dai, J.; Yang, Z.; Yu, X.; Xu, Y.; Shi, X.; Wei, D.; Tang, Z.; Xu, G.; Xu, W.; et al. Inactivated SARS-CoV-2 vaccine does not influence the profile of prothrombotic antibody nor increase the risk of thrombosis in a prospective Chinese cohort. Sci. Bull. 2021, 66, 2312–2319. [Google Scholar] [CrossRef] [PubMed]
- Foret, T.; Dufrost, V.; Salomon Du Mont, L.; Costa, P.; Lefevre, B.; Lacolley, P.; Regnault, V.; Zuily, S.; Wahl, D. Systematic review of antiphospholipid antibodies in COVID-19 patients: Culprits or bystanders? Curr. Rheumatol. Rep. 2021, 23, 65. [Google Scholar] [CrossRef]
- Gasparini, G.; Canepa, P.; Verdiani, S.; Carmisciano, L.; Cozzani, E.; De Grazia, D.; Andrea, O.; Icardi, G.; Parodi, A. A retrospective study on the prevalence of anti-phospholipid antibodies, thrombotic events and cutaneous signs of vasculopathy in 173 hospitalized COVID-19 patients. Int. J. Immunopathol. Pharmacol. 2021, 35, 20587384211042115. [Google Scholar] [CrossRef]
- Hörkkö, S.; Miller, E.; Dudl, E.; Reaven, P.; Curtiss, L.K.; Zvaifler, N.J.; Terkeltaub, R.; Pierangeli, S.S.; Branch, D.W.; Palinski, W.; et al. Antiphospholipid antibodies are directed against epitopes of oxidized phospholipids. Recognition of cardiolipin by monoclonal antibodies to epitopes of oxidized low density lipoprotein. J. Clin. Invest. 1996, 98, 815–825. [Google Scholar] [CrossRef]
- Chayoua, W.; Kelchtermans, H.; Gris, J.C.; Moore, G.W.; Musiał, J.; Wahl, D.; de Groot, P.G.; de Laat, B.; Devreese, K.M.J. The (non-)sense of detecting anti-cardiolipin and anti-β2glycoprotein I IgM antibodies in the antiphospholipid syndrome. J. Thromb. Haemost. 2020, 18, 169–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinuzzo, M.E.; Forastiero, R.R.; Adamczuk, Y.; Pombo, G.; Carreras, L.O. Antiplatelet factor 4--heparin antibodies in patients with antiphospholipid antibodies. Thromb. Res. 1999, 95, 271–279. [Google Scholar] [CrossRef]
- Hörkkö, S.; Miller, E.; Branch, D.W.; Palinski, W.; Witztum, J.L. Annexin A2 mediates endothelial cell activation by antiphospholipid/anti-β2 glycoprotein I antibodies. Proc. Natl. Acad. Sci. USA 1997, 94, 10356–10361. [Google Scholar] [CrossRef]
- Larsson, A.; Egberg, N.; Lindahl, T.L. Platelet activation and binding of complement components to platelets induced by immune complexes. Platelets 1994, 5, 149–155. [Google Scholar] [CrossRef]
- Lindahl, T.L.; Larsson, A. C1q binding to platelets induced by monoclonal antibodies and immune complexes-a flow cytometric analysis. Platelets 1993, 4, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Peerschke, E.I.; Ghebrehiwet, B. C1q augments platelet activation in response to aggregated Ig. J. Immunol. 1997, 159, 5594–5598. [Google Scholar]
- Nazy, I.; Jevtic, S.D.; Moore, J.C.; Huynh, A.; Smith, J.W.; Kelton, J.G.; Arnold, D.M. Platelet-activating immune complexes identified in critically ill COVID-19 patients suspected of heparin-induced thrombocytopenia. J. Thromb. Haemost. 2021, 19, 1342–1347. [Google Scholar] [CrossRef]
- Jevtic, S.D.; Nazy, I. The COVID complex: A review of platelet activation and immune complexes in COVID-19. Front. Immunol. 2022, 13, 807934. [Google Scholar] [CrossRef]
- Uzun, G.; Pelzl, L.; Singh, A.; Bakchoul, T. Immune-mediated platelet activation in COVID-19 and vaccine-induced immune thrombotic thrombocytopenia. Front. Immunol. 2022, 13, 837629. [Google Scholar] [CrossRef]
- Nevzorova, T.A.; Mordakhanova, E.R.; Daminova, A.G.; Ponomareva, A.A.; Andrianova, I.A.; Le Minh, G.; Rauova, L.; Litvinov, R.I.; Weisel, J.W. Platelet factor 4-containing immune complexes induce platelet activation followed by calpain-dependent platelet death. Cell Death Discov. 2019, 5, 106. [Google Scholar] [CrossRef] [PubMed]
- Haile, L.A.; Rao, R.; Polumuri, S.K.; Arepally, G.M.; Keire, D.A.; Verthelyi, D.; Sommers, C.D. PF4-HIT antibody (KKO) complexes activate broad innate immune and inflammatory responses. Thromb. Res. 2017, 159, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Althaus, K.; Möller, P.; Uzun, G.; Singh, A.; Beck, A.; Bettag, M.; Bösmüller, H.; Guthoff, M.; Dorn, F.; Petzold, G.C.; et al. Antibody-mediated procoagulant platelets in SARS-CoV-2-vaccination associated immune thrombotic thrombocytopenia. Haematologica 2021, 106, 2170–2179. [Google Scholar] [CrossRef] [PubMed]
- Holm, S.; Kared, H.; Michelsen, A.E.; Kong, X.Y.; Dahl, T.B.; Schultz, N.H.; Nyman, T.A.; Fladeby, C.; Seljeflot, I.; Ueland, T.; et al. Immune complexes, innate immunity, and NETosis in ChAdOx1 vaccine-induced thrombocytopenia. Eur. Heart J. 2021, 42, 4064–4072. [Google Scholar] [CrossRef]
- Perdomo, J.; Leung, H.H.L.; Ahmadi, Z.; Yan, F.; Chong, J.J.H.; Passam, F.H.; Chong, B.H. Neutrophil activation and NETosis are the major drivers of thrombosis in heparin-induced thrombocytopenia. Nat. Commun. 2019, 10, 1322. [Google Scholar] [CrossRef]
- Root-Bernstein, R. Age and location in severity of COVID-19 pathology: Do lactoferrin and pneumococcal vaccination explain low infant mortality and regional differences? BioEssays 2020, 42, 2000076. [Google Scholar] [CrossRef]
- Root-Bernstein, R. Pneumococcal and influenza vaccination rates and pneumococcal invasive disease rates set geographical and ethnic population susceptibility to serious COVID-19 cases and deaths. Vaccines 2021, 9, 474. [Google Scholar] [CrossRef]
- Root-Bernstein, R. Possible cross-reactivity between SARS-CoV-2 proteins.; CRM197 and proteins in pneumococcal vaccines may protect against symptomatic SARS-CoV-2 disease and death. Vaccines 2020, 8, 559. [Google Scholar] [CrossRef]
- Nunes, M.C.; Cutland, C.L.; Klugman, K.P.; Madhi, S.A. Pneumococcal conjugate vaccine protection against coronavirus-associated pneumonia hospitalization in children living with and without HIV. mBio 2021, 12, e02347-20. [Google Scholar] [CrossRef]
- Jehi, L.; Ji, X.; Milinovich, A.; Erzurum, S.; Rubin, B.P.; Gordon, S.; Young, J.B.; Kattan, M.W. Individualizing risk prediction for positive coronavirus disease 2019 testing. Chest 2020, 158, 1364–1375. [Google Scholar] [CrossRef]
- Pawlowski, C.; Puranik, A.; Bandi, H.; Venkatakrishnan, A.J.; Agarwal, V.; Kennedy, R.; O’Horo, J.C.; Gores, G.J.; Williams, A.W.; Halamka, J.; et al. Exploratory analysis of immunization records highlights decreased SARS-CoV-2 rates in individuals with recent non-COVID-19 vaccinations. Sci. Rep. 2021, 11, 1–20. [Google Scholar] [CrossRef]
- Noale, M.; Trevisan, C.; Maggi, S.; Incalzi, R.A.; Pedone, C.; Di Bari, M.; Adorni, F.; Jesuthasan, N.; Sojic, A.; Galli, M.; et al. the association between influenza and pneumococcal vaccinations and SARS-Cov-2 infection: Data from the EPICOVID19 web-based survey. Vaccines 2020, 8, 471. [Google Scholar] [CrossRef] [PubMed]
- Lewnard, A.J.; Bruxvoort, K.J.; Fischer, H.; Hong, V.X.; Grant, L.R.; Jódar, L.; Gessner, B.D.; Tartof, S.Y. Prevention of COVID-19 among older adults receiving pneumococcal conjugate vaccine suggests interactions between Streptococcus pneumoniae and SARS-CoV-2 in the respiratory tract. J. Infect. Dis. 2021, 225, 1710–1720. [Google Scholar] [CrossRef] [PubMed]
- Thindwa, D.; Garcia Quesada, M.; Liu, Y.; Bennett, J.; Cohen, C.; Knoll, M.D.; von Gottberg, A.; Hayford, K.; Flasche, S. Use of seasonal influenza and pneumococcal polysaccharide vaccines in older adults to reduce COVID-19 mortality. Vaccine 2020, 38, 5398–5401. [Google Scholar] [CrossRef] [PubMed]
- Amin-Chowdhury, Z.; Aiano, F.; Mensah, A.; Sheppard, C.L.; Litt, D.; Fry, N.K.; Andrews, N.; Ramsay, M.E.; Ladhani, S.N. Impact of the Coronavirus Disease 2019 (COVID-19) Pandemic on Invasive Pneumococcal Disease and Risk of Pneumococcal coinfection with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2): Prospective national cohort study, England. Clin. Infect. Dis. 2021, 72, e65–e75. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.J.; Lai, C.C.; Chao, C.M. Changing epidemiology of respiratory tract infection during COVID-19 pandemic. Antibiotics 2022, 11, 315. [Google Scholar] [CrossRef]
- Root-Bernstein, R. Synergistic activation of Toll-Like and NOD receptors by complementary antigens as facilitators of autoimmune disease: Review, model and novel predictions. Int. J. Mol. Sci. 2020, 21, 4645. [Google Scholar] [CrossRef]
- Smith, A.P.; Williams, E.P.; Plunkett, T.R.; Selvaraj, M.; Lane, L.C.; Zalduondo, L.; Xue, Y.; Vogel, P.; Channappanavar, R.; Jonsson, C.B.; et al. Time-dependent increase in susceptibility and severity of secondary bacterial infections during SARS-CoV-2. Front. Immunol. 2022, 13, 894534. [Google Scholar] [CrossRef]
- Barman, T.K.; Singh, A.K.; Bonin, J.L.; Nafiz, T.N.; Salmon, S.L.; Metzger, D.W. Lethal synergy between SARS-CoV-2 and Streptococcus pneumoniae in hACE2 mice and protective efficacy of vaccination. JCI Insight 2022, 7, e159422. [Google Scholar] [CrossRef]
- Moreno-García, E.; Puerta-Alcalde, P.; Letona, L.; Meira, F.; Dueñas, G.; Chumbita, M.; Garcia-Pouton, N.; Monzó, P.; Lopera, C.; Serra, L.; et al. Bacterial co-infection at hospital admission in patients with COVID-19. Int. J. Infect. Dis. 2022, 118, 197–202. [Google Scholar] [CrossRef]
- Garcia-Vidal, C.; Sanjuan, G.; Moreno-García, E.; Puerta-Alcalde, P.; Garcia-Pouton, N.; Chumbita, M.; Fernandez-Pittol, M.; Pitart, C.; Inciarte, A.; Bodro, M.; et al. Incidence of co-infections and superinfections in hospitalized patients with COVID-19: A retrospective cohort study. Clin. Microbiol. Infect. 2021, 27, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Said, K.B.; Alsolami, A.; Moussa, S.; Alfouzan, F.; Bashir, A.I.; Rashidi, M.; Aborans, R.; Taha, T.E.; Almansour, H.; Alazmi, M.; et al. COVID-19 clinical profiles and fatality rates in hospitalized patients reveal case aggravation and selective co-infection by limited gram-negative bacteria. Int. J. Environ. Res. Public Health 2022, 19, 5270. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.C.; Wang, C.Y.; Hsueh, P.R. Co-infections among patients with COVID-19: The need for combination therapy with non-anti-SARS-CoV-2 agents? J. Microbiol. Immunol. Infect. 2020, 53, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Root-Bernstein, R.; Dobbelstein, C. Insulin binds to glucagon forming a complex that is hyper-antigenic and inducing complementary antibodies having an idiotype-antiidiotype relationship. Autoimmunity 2001, 33, 153–169. [Google Scholar] [CrossRef]
- Root-Bernstein, R. Antigenic complementarity among AIDS-associated infectious agents and molecular mimicry of lympohocyte proteins as inducers of lymphocytotoxic antibodies and circulating immune complexes. J. Clin. Virol. 2004, 31S, S16–S25. [Google Scholar] [CrossRef]
- Root-Bernstein, R. Antigenic complementarity between HIV and other AIDS-associated infections results in idiotype-antiidiotype antibody complexes that cross-react with lymphocyte proteins. Vaccine 2005, 23, 2160–2163. [Google Scholar] [CrossRef]
- Svačina, M.K.R.; Röth, P.; Bobylev, I.; Sprenger, A.; Zhang, G.; Sheikh, K.A.; Lehmann, H.C. Changes of serum IgG dimer levels after treatment with IVIg in Guillain-Barré Syndrome. J. Neuroimmune Pharmacol. 2019, 14, 642–648. [Google Scholar] [CrossRef]





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Function | Interactions |
|---|---|---|
| Coagulation Proteins | ||
| Factor 2 (F2) (Phosphatidylserine/prothrombin) | When cleaved to thrombin, converts fibrinogen to fibrin (i.e., clot formation) | Activated by FX |
| Factor VIII (FVIII) | Cofactor for FX activation by Factor Xa | Forms complex with FX |
| Factor IX (FIX) | Activated by tissue factor (TF) and Factor VIIa to activate FVIII | Forms complex with FVIII; complex forms on phospholipid scaffolds on platelets |
| Factor X (FX) | Cleaves prothrombin (F2) to thrombin | Forms complex with F2 |
| Factor Xa (FXa) | Activated by tissue factor (TF) and Factor VIIa; in concert with FXIII, activates FX | Complex forms on phospholipid scaffolds on platelets |
| Von Willebrand Factor (vWF) | Facilitates platelet aggregation to form temporary clots | Binds to FIX and GP1a on activated platelets and collagens exposed on damaged cells |
| Platelet Proteins | ||
| Glycoprotein 1b (GP1b) | Activates platelet aggregation | Binds Factor V and FIX to the surfaces of platelets to activate them |
| Lupus anticoagulant (LA) | Antibodies against PL, PLBP, and PDE, inactivating them | Bind to PL and PLBP |
| Phosphodiesterases (PDE) | Regulate platelet activation by regulating phospholipid-binding proteins | Modify phospholipid-binding proteins such as β2GPI |
| Beta-2 Glycoprotein I (β2GPI) | Phospholipid binding protein that acts as an anti-coagulant | Bind to phospholipids such as CL to inactivate them |
| Cardiolipin (CL)(a phospholipid) | Promote coagulation by acting as scaffolds for blood factor binding | Necessary for FVIII and FXa activity; bind to phospholipid binding proteins such as β2GPI |
| Platelet Factor 4 (PF4) | Released by activated platelets to neutralize heparin permitting coagulation | Binds to and inactivates heparin-like molecules |
| Tissue Proteins | ||
| Collagens | Extracellular cell matrix proteins involved in cell adhesion and integrity | vWF binds to exposed collagens to initiate FIX and GP1a platelet binding |
| Protein | Significant Kd Found Here | Conc. in Serum | Conc. in Platelets | Source |
|---|---|---|---|---|
| Cardiolipin | 400 nM–2 µM | 10 µM | [65] | |
| β2 Glycoprotein I | 2–200 nM | 3 µM | [66] | |
| Platelet Factor 4 | 5–100 nM | 0.4 nM | 400 nM | [67] |
| Serum Albumin | NONE | 500 µM | [68] | |
| Factor II (Prothrombin) | 2–200 nM | 2 µM | [69] | |
| Factor VIII | 0.4–0.5 nM | 1 nM | [70] | |
| Factor IX | 0.3–0.9 nM | 90 nM | [71] | |
| von Willebrand Factor | 10–30 nM | 2 µM | [72] | |
| Phosphodiesterases | 8–40 nM | 12 µM | 7 µM | [73,74] |
| Collagen 1 | 4–8 nM | n/r | n/r |
| Antibody Target | Species | Supplier | Product # |
|---|---|---|---|
| Adenovirus | Goat | Millipore (Burlington, MA, USA) | AB1056 |
| Clostridia | Rabbit | Invitrogen (Waltham, MA, USA) | PA1-7210 |
| Clostridia | Rabbit | Invitrogen | PA1-7210 |
| Clostridium sp. HRP | Rabbit | US Biological (Swampscott, MA, USA) | C5853-25C |
| Coxsackie Virus B-Blend | Mouse | Millipore | MAB9410 |
| Escherichia coli | Goat | Abcam (Cambridge, England) | AB13627 |
| Goat Anti-Mouse IgG HRP | Goat | Sigma-Aldrich (Burlington, MA, USA) | A9917 |
| Goat Anti-Rabbit IgG HRP | Goat | Invitrogen | 65-6120 |
| Herpes Simplex Virus Type 1 | Goat | Invitrogen | PA1-7493 |
| Influenza A HRP | Goat | Biodesign International (Palo Alto, CA, USA) | B65243G |
| Klebsiella pneumoniae HRP | Rabbit | Invitrogen | PA1-73176 |
| Mycobacterium tuberculosis | Rabbit | ABD Serotec (Kidlington, UK) | OBT0947 |
| Mycobacterium tuberculosis | Guinea Pig | MyBioSource (San Diego, CA, USA) | MBS315001 |
| Rabbit Anti-Goat IgG HRP | Rabbit | Millipore | AP106P |
| Rabbit Anti-Guinea Pig HRP | Rabbit | abcam | AB6771 |
| SARS-CoV-2 Envelope protein | Rabbit | Invitrogen | PA1-41158 |
| SARS-CoV-2 Matrix protein | Rabbit | Invitrogen | PA1-41160 |
| SARS-CoV-2 Nucleocapsid | Rabbit | Invitrogen | PA5-116894 |
| SARS-CoV-2 Spike Protein RBD | Rabbit | Millipore | ABF1064 |
| SARS-CoV-2 Spike Protein S1 | Rabbit | Millipore | ABF1065 |
| SARS-CoV-2 Spike Protein S1 | Rabbit | Invitrogen | PA5-116916 |
| SARS-CoV-2 Spike Protein S2 | Rabbit | Millipore | ABF1063 |
| Staphylococcus aureus | Rabbit | Invitrogen | PA1-7246 |
| Staphylococcus aureus HRP | Rabbit | Invitrogen | PA1-73173 |
| Streptococcus Group A | Goat | Invitrogen | PA1-7249 |
| Streptococcus Group A HRP | Rabbit | Acris Antibodies (Herford, Germany) | BP2026HRP |
| Streptococcus pneumoniae | Rabbit | Biodesign International | B65831R |
| Streptococcus pneumoniae | Rabbit | Invitrogen | PA1-7259 |
| Protein | Species | Supplier | Product # | Purity |
|---|---|---|---|---|
| Beta 2 glycoprotein I | Human | Prolytix | B2G1-0001 | >95% by SDS-PAGE |
| Cardiolipin sodium salt | Bovine | Sigma-Aldrich | C0563 | ≥97% (TLC) |
| Coagulation Factor VIII (rDNA) | Human | Sigma-Aldrich | H0920000 | >99%, European Pharmacopoeia Reference Standard |
| Coagulation Factor IX (rDNA) | Human | Sigma-Aldrich | Y0001659 | >99%, European Pharmacopoeia Reference Standard |
| Collagen 1 | Human | Sigma-Aldrich | C7774 | >95% SDS Electrophoresis |
| Platelet Factor 4 | Murine | Sigma-Aldrich | SRP3231 | ≥98% (SDS-PAGE), ≥98% (HPLC) |
| Prothrombin | Human | Sigma-Aldrich | 539515 | >95% (SDS-PAGE) |
| Phosphodiesterase II | Bovine | Sigma-Aldrich | P9041 | ≥5.0 units/mg protein |
| Serum Albumin | Human | Sigma-Aldrich | A1653 | ≥96% (agarose gel electrophoresis) |
| von Willebrand Factor | Human | Sigma-Aldrich | 681300 | ≥95% (SDS-PAGE) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Root-Bernstein, R.; Huber, J.; Ziehl, A. Complementary Sets of Autoantibodies Induced by SARS-CoV-2, Adenovirus and Bacterial Antigens Cross-React with Human Blood Protein Antigens in COVID-19 Coagulopathies. Int. J. Mol. Sci. 2022, 23, 11500. https://doi.org/10.3390/ijms231911500
Root-Bernstein R, Huber J, Ziehl A. Complementary Sets of Autoantibodies Induced by SARS-CoV-2, Adenovirus and Bacterial Antigens Cross-React with Human Blood Protein Antigens in COVID-19 Coagulopathies. International Journal of Molecular Sciences. 2022; 23(19):11500. https://doi.org/10.3390/ijms231911500
Chicago/Turabian StyleRoot-Bernstein, Robert, Jack Huber, and Alison Ziehl. 2022. "Complementary Sets of Autoantibodies Induced by SARS-CoV-2, Adenovirus and Bacterial Antigens Cross-React with Human Blood Protein Antigens in COVID-19 Coagulopathies" International Journal of Molecular Sciences 23, no. 19: 11500. https://doi.org/10.3390/ijms231911500