4. Experimental Section
The 1H (400 or 500 MHz) and 13C (100 or 125 MHz) NMR spectra were determined on Bruker Biospin spectrometers (Bruker, Billerica, MA, USA) with solutions in CDCl3 unless otherwise noted. HRMS were obtained in TOF-ESI mode on Bruker Solarix FT-ICR instrument (Bruker, Billerica, MA, USA) unless otherwise noted. TLC was performed with Merck (Darmstadt, Germany) kieselgel 60-F254 sheets and products were detected with 254 nm light or by visualization with Ce(SO4)2/(NH4)6Mo7O24∙4H2O/H2SO4/H2O reagent. Merck kieselgel 60 (230–400 mesh) was used for column chromatography. Reagent grade chemicals were used, and solvents were dried by reflux over and distillation from CaH2 (except for THF/potassium) under argon or by passing the solvents through activated alumina cartridges using a solvent purification system.
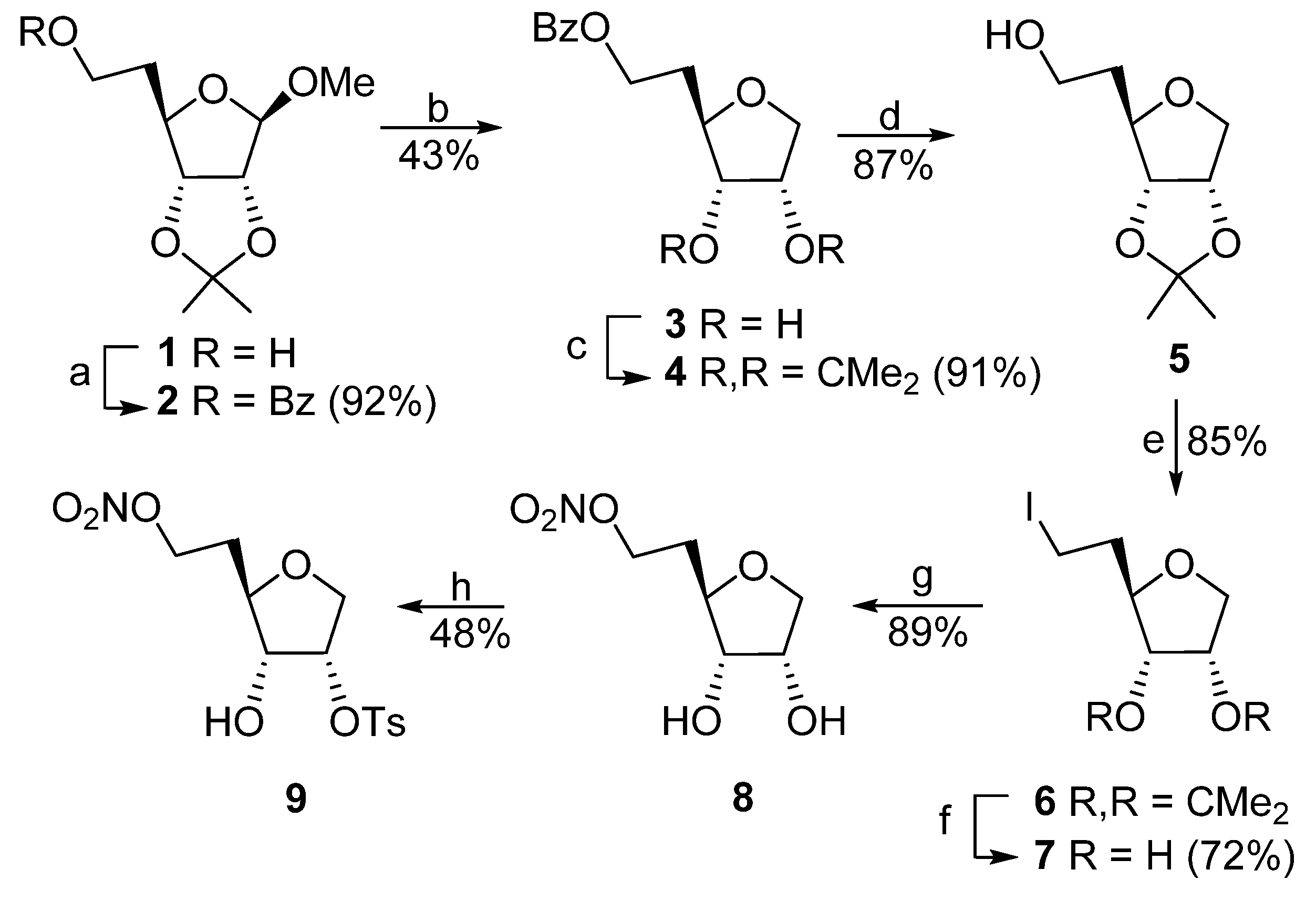
Methyl 6-O-Benzoyl-1,5-dideoxy-2,3-O-isopropylidene-β-D-ribo-hexofuranoside (2). To a solution of methyl 1,5-dideoxy-2,3-isopropylidene-β-
D-
ribo-hexofuranoside [
29] (
1; 13.0 g, 58.8 mmol) and Et
3N (8.9 g, 12.3 mL, 88.2 mol) in CH
2Cl
2 (50 mL) was added dropwise BzCl (12.4 g, 10.2 mL, 88.2 mmol) at 0 °C. The cooling bath was removed and after 1 h the reaction mixture was quenched by addition of MeOH (2 mL). After additional 30 min the volatiles were evaporated and the residue was column chromatographed (hexanes → hexanes/EtOAc, 6:1) to give
2 as pale yellow oil (17.3 g, 92%):
1H NMR δ 1.32, 1.49 (2 × s, 2 × 3H), 2.01–2.04 (m, 2H), 3.38 (s, 3H), 4.39–4.52 (m, 3H), 4.62–4.65 (m, 2H), 4.98 (s, 1H), 7.42–7.57 (m, 3H), 8.04–8.06 (m, 2H);
13C NMR δ 24.6, 26.2, 33.9, 54.8, 61.6, 83.6, 84.0, 85.2, 109.6, 112.0, 128.1, 129.3, 129.9, 132.6, 166.0; MS FAB
m/
z 345 (3, [M + Na]
+), 291 (100, [M − OMe]
+); HRMS ESI
m/
z calcd for C
17H
22O
6Na [M + Na]
+ 345.1314, found 345.1317.
6-O-Benzoyl-1,5-dideoxy-D-ribo-hexofuranose (3) and 6-O-Benzoyl-1,5-dideoxy-2,3-O- isopropylidene-D-ribo-hexofuranose (4). To a stirred solution of 2 (18.0 g, 55.9 mmol) and Et3SiH (19.4 g, 26.7 mL, 167.7 mmol) in CH2Cl2 (10 mL) was added BF3•OEt2 (23.8 g, 21.3 mL, 167.7 mmol). Mildly exothermic reaction ensued after approx. 15 min and the reaction mixture was allowed to stir for an additional 3 h. The reaction flask was placed in an ice slush and a saturated NaHCO3/H2O solution (200 mL) was added slowly with vigorous stirring. CH2Cl2 was added (50 mL), aqueous phase was separated and washed with CH2Cl2 (50 mL). Organic fractions were combined and evaporated to dryness. Column chromatography (hexanes/EtOAc, 6:1 → EtOAc) gave contaminated (~10%) 4 (1.5 g, 9%) and 3 (6.1 g, 43%) as syrup. Compound 3 had: 1H NMR (CD3OD) δ 1.87–1.94 (m, 1H), 2.12–2.19 (m, 1H), 3.70 (dd, J = 2.9, 9.8 Hz, 1H), 3.79 (dd, J = 5.4, 7.3 Hz, 1H), 3.85–3.89 (m, 1H), 4.07 (dd, J = 4.9, 9.8 Hz, 1H), 4.16 (td, J = 2.9, 4.9 Hz, 1H), 4.38–4.50 (m, 2H), 7.45–7.61 (m, 3H), 8.02–8.04 (m, 2H); 13C NMR (CD3OD) δ 32.4, 61.9, 70.8, 72.2, 75.9, 78.9, 128.3, 129.5, 129.9, 133.0, 166.8; MS FAB m/z 275 (100, [M + Na]+); HRMS ESI m/z calcd for C13H16O5Na [M + Na]+ 275.1531, found 275.0902.
Diol 3 (6.1 g, 24.2 mmol) and TsOH hydrate (0.5 g, 2.6 mmol) were dissolved in a mixture of acetone (40 mL) and 2,2-dimethoxypropane (10 mL) and left to stir at room temperature for 30 min. Neutralization with sat. NaHCO3 (100 mL) followed by EtOAc extraction (2 × 50 mL) and evaporation to dryness provided oil that was filtered through silica to give 4 (6.4 g, 91%): 1H NMR δ 1.33, 1.52 (2 × s, 2 × 3H), 1.86–1.92 (m, 2H), 3.86 (dd, J = 4.4, 10.7 Hz, 1H), 3.97 (dd, J = 1.5, 10.7 Hz, 1H), 4.23–4.26 (m, 1H), 4.36–4.49 (m, 2H), 4.52 (dd, J = 1.5, 6.3 Hz, 1H), 4.80–4.83 (m, 1H), 7.42–7.57 (m, 3H), 8.03–8.05 (m, 2H); 13C NMR δ 24.9, 26.5, 29.7, 61.5, 71.6, 80.8, 81.2, 85.0, 112.8, 128.3, 129.5, 130.1, 132.9, 166.4. MS FAB m/z 293 (100, [M + H]+), 315 (15, [M + Na]+); HRMS ESI m/z calcd for C16H20O5Na [M + Na]+ 315.1208, found 315.1209.
1,5-Dideoxy-2,3-O-isopropylidene-D-ribo-hexofuranose (5). To a stirred solution of
4 (14.0 g, 47.9 mmol) in MeOH (50 mL) was added a solution of KOH (2.0 g, 35.7 mol) in MeOH (50 mL). After 1 h the volatiles were evaporated and the residue was column chromatographed (hexanes/EtOAc, 2:1 → EtOAc) to give
5 [
45] (8.0 g, 87%):
1H NMR δ 1.33, 1.51 (2 × s, 2 × 3H), 1.63–1.76 (m, 2H), 3.74–3.82 (m, 2H), 3.89 (dd,
J = 4.4, 10.4 Hz, 1H), 3.94 (d,
J = 10.3 Hz, 1H), 4.19–4.22 (m, 2H), 4.48 (dd,
J = 1.5, 5.8 Hz, 1H), 4.81 (t,
J = 5.1 Hz, 1H);
13C NMR δ 24.5, 26.2, 32.4, 59.2, 71.1, 80.4, 82.2, 84.6, 112.3. MS FAB
m/
z 189 (100, [M + H]
+); HRMS ESI
m/z calcd for C
9H
17O
4 [M + H]
+ 189.1127, found 189.1123.
6-Iodo-1,5,6-trideoxy-2,3-O-isopropylidene-D-ribo-hexofuranose (6). To a stirred solution of 5 (9.0 g, 47.9 mmol), Ph3P (15.1 g, 57.6 mmol) and imidazole (7.9 g, 116.2 mmol) in toluene (225 mL) was added iodine (14.6 g, 57.5 mmol) at 70 °C. The suspension was vigorously stirred another 2 h, then allowed to cool to ambient temperature and the solution decanted to another flask. Volatiles were evaporated and the residue was column chromatographed (hexanes → hexanes/EtOAc, 6:1) to give 6 (13.03 g, 85%): 1H NMR δ 1.33, 1.52 (2 × s, 2 × 3H), 1.88–1.97 (m, 2H), 3.14–3.27 (m, 2H), 3.78 (dd, J = 4.4, 10.7 Hz, 1H), 3.95 (dd, J = 1.5, 10.7 Hz, 1H), 4.10 (ddd, J = 1.5, 4.9, 9.2 Hz, 1H), 4.23 (dd, J = 2.0, 6.3 Hz, 1H), 4.78 (m, 1H); 13C NMR δ 1.0, 24.7, 26.2, 34.0, 71.2, 80.4, 83.6, 84.1, 112.3; MS FAB m/z 321 (12, [M + Na]+), 319 (100); HRMS ESI m/z calcd for C9H15IO3Na [M + Na]+ 320.9964, found 320.9970.
6-Iodo-1,5,6-trideoxy-D-ribo-hexofuranose (7). A solution of 6 (14.0 g, 47.0 mmol) in a mixture of MeOH (100 mL), H2O (25 mL) and conc. HCl (25 mL) was stirred at room temperature for 3 h. Volatiles were evaporated and the residue was column chromatographed (hexanes/EtOAc, 2:1 → EtOAc) to give 7 (8.73 g, 72%): 1H NMR δ 2.00–2.07 (m, 1H), 2.16–2.25 (m, 1H), 3.22–3.45 (m, 4H), 3.74–3.77 (m, 2H), 3.84 (br s, 1H), 4.10 (dd, J = 4.9, 10.3 Hz, 1H), 4.27 (br s, 1H); 13C NMR δ 1.7, 37.3, 70.9, 72.8, 75.6, 81.6; MS FAB m/z 258 (10, [M]+), 74 (100); HRMS ESI m/z calcd for C6H11IO3, 257.9753, found 257.9740.
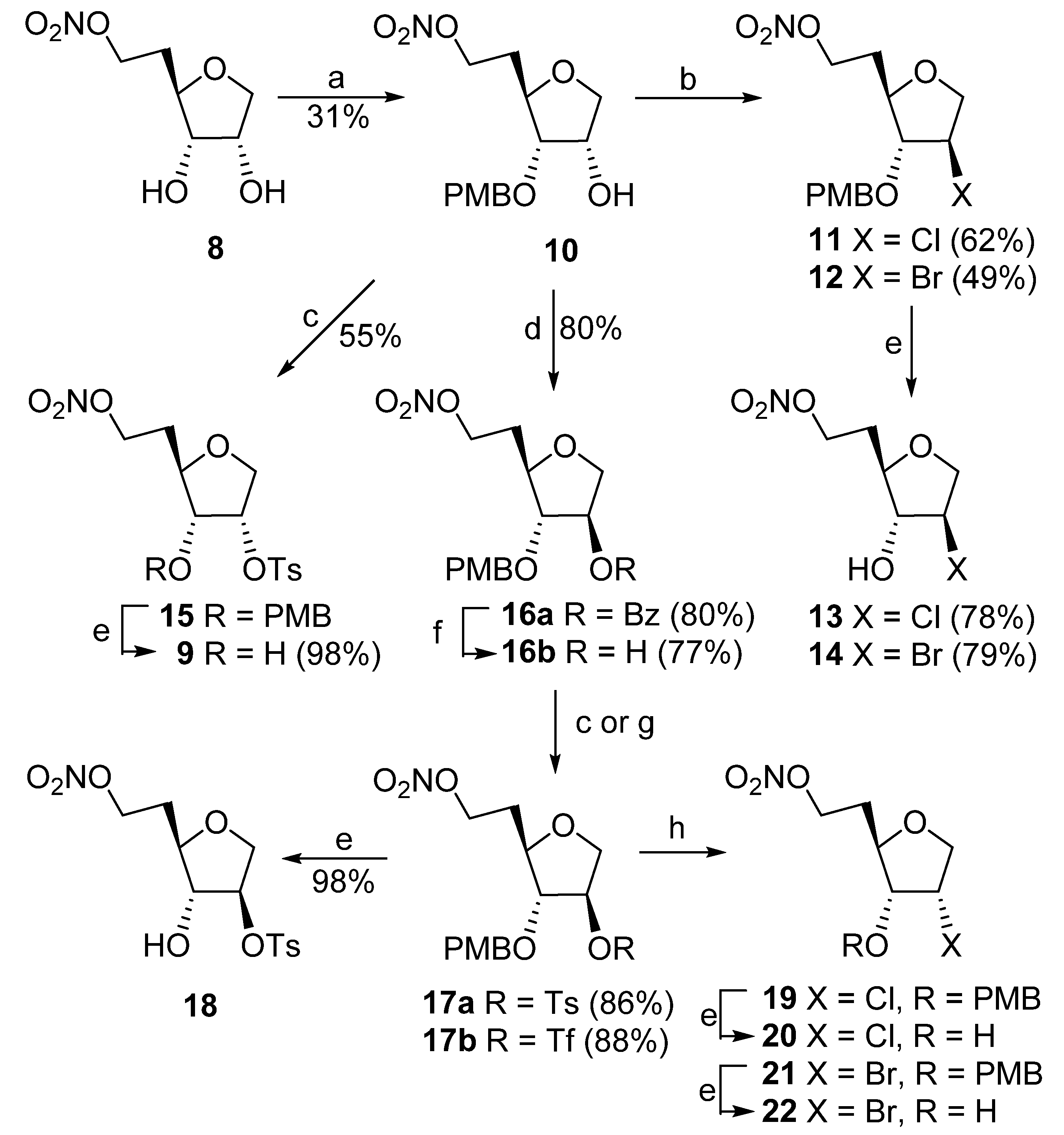
1,5-Dideoxy-6-O-nitro-D-ribo-hexofuranose (8). A suspension of 7 (12.0 g, 46.5 mmol) and AgNO3 (15.8 g, 93.0 mmol) in CH3CN (150 mL) was stirred at room temperature for 2 days. The yellow precipitate was filtered off, washed with EtOAc and the volatiles were evaporated. Column chromatography (hexanes/EtOAc, 1:1 → EtOAc) gave 8 (7.99 g, 89%): 1H NMR δ 1.88–1.96 (m, 1H), 2.08–2.15 (m, 1H), 3.43 (br s, 2H), 3.73–3.83 (m, 3H), 4.12 (dd, J = 4.9, 10.3 Hz, 1H), 4.23–4.26 (m, 1H), 4.56–4.66 (m, 2H); 13C NMR δ 30.2, 70.1, 70.8, 72.6, 75.9, 77.8; MS FAB m/z 216 (100, [M + Na]+); HRMS ESI m/z calcd for C6H11NO6Na [M + Na]+, 216.0484 found 216.0497.
1,5-Dideoxy-6-O-nitro-2-O-tosyl-D-ribo-hexofuranose (9). Method A. A solution of diol 8 (370 mg, 1.92 mmol) and Bu2SnO (477 mg, 1.92 mmol) in anhydrous MeOH (40 mL) was heated in a sealed flask at 75 °C for 1 h. After the flask was cooled to 0 °C, Et3N (1.15 g, 1.6 mL, 11.4 mmol) was added with stirring followed by TsCl (2.17 g, 11.4 mmol) dissolved in a minimum volume of acetone. The volatiles were evaporated, and the residue was suspended in acetone and deposited on silica. Column chromatography (hexanes/EtOAc, 6:1 → 3:1) gave a 5:2 mixture of two isomers (670 mg) from which the main product 9 (320 mg, 48%) was isolated after second column chromatography: 1H NMR δ 1.85–1.92 (m, 1H), 2.09–2.17 (m, 1H), 2.46 (s, 3H), 2.76 (d, J = 7.8 Hz, 1H), 3.75 (dt, J = 3.9, 8.3 Hz, 1H), 3.80 (dd, J = 3.1, 11.2 Hz, 1H), 3.89 (dt, J = 5.4, 7.8 Hz, 1H), 4.06 (dd, J = 4.9, 11.2 Hz, 1H), 4.52–4.62 (m, 2H), 4.93 (dt, J = 3.0, 5.3 Hz, 1H), 7.38, 7.82 (2 × d, J = 8.3 Hz, 2 × 2H); 13C NMR δ 21.6, 30.2, 69.8, 70.1, 74.9, 77.8, 79.4, 127.8, 130.1, 132.8, 145.6; MS FAB m/z 370 (100, [M + Na]+); HRMS ESI m/z calcd for C13H17NO8SNa [M + Na]+ 370.0573, found 370.0587.
Method B. Step a: TsCl (66 mg, 0.35 mmol) was added to a stirred solution of 10 (100 mg, 0.32 mmol) in anhydrous pyridine (1 mL) at ambient temperature. After 16 h, the volatiles were evaporated and residue was partitioned between ice-cold AcOH/H2O (1:99, 30 mL) and CHCl3 (30 mL). The aqueous layer was extracted with CHCl3 and the combined organic phase was washed with ice-cold saturated NaHCO3/H2O (30 mL), brine (30 mL) and dried (Na2SO4). Column chromatography (EtOAc/hexane, 5:95 → 35:65) gave 1,5-dideoxy-3-O-(p-methoxybenzyl)- 6-O-nitro-2-O-tosyl-D-ribo-hexofuranose (15; 82 mg, 55%): 1H NMR δ 1.78–1.81 (m, 1H), 2.00–2.02 (m, 1H), 2.47 (s, 3H), 3.59 (dd, J = 5.0, 8.6 Hz, 1H), 3.83 (s, 3H), 3.86 (dt, J = 2.2, 9.6 Hz, 2H), 4.04 (dd, J = 4.5, 11.2 Hz, 2H), 4.26 (d, J = 11.2 Hz, 1H), 4.47 (m, 2H), 4.58 (d, J = 11.2Hz, 1H), 5.13 (dt, J = 4.7, 7.0 Hz, 1H), 6.88 and 7.21 (2 × d, J = 9.5 Hz, 2 × 2H), 7.34 and 7.84 (2 × d, J = 8.3 Hz, 2 × 2H). In step b, A solution of CAN (176 mg, 0.32 mmol) and 15 (75 mg, 0.16 mmol) in MeCN (1.5 mL) and water (0.15 mL) was stirred at ambient temperature for 22 h. Volatiles were evaporated and the residue was column chromatographed (EtOAc/hexane, 3:97 → 30:70) to give 9 (54 mg, 98%) with data as above.
1,5-Dideoxy-3-O-(p-methoxybenzyl)-6-O-nitro-D-ribo-hexofuranose (10). A suspension of diol 8 (1.20 g, 6.22 mmol) and Bu2SnO (1.70 g, 6.84 mmol) in anhydrous MeOH (10 mL) was heated in a sealed flask at 75 °C for 1 h. After the flask was cooled to ambient temperature, the volatiles were evaporated. DMF (5 mL) and PMBCl (1.95 g, 1.7 mL, 12.44) were added, and the reaction mixture was heated at 90 °C for 18 h. Volatiles were evaporated, and the residue was chromatographed (hexanes/EtOAc, 6:1 → 1:1) to give 1,5-dideoxy-2,3-di-O-(p-methoxybenzyl)-6-O-nitro-D-ribo- hexofuranose (1.55 g; 20% contaminated) followed by 1,5-dideoxy-2-O-(p-methoxybenzyl)-6-O-nitro-D-ribo-hexofuranose (0.47 g, 24%) and 10 (0.60 g; 31%) of sufficient purity for use in subsequent reaction. Sample of 10 was repurified on column chromatography for spectroscopic characterization: 1H NMR δ 1.77–1.85 (m, 1H), 1.93–2.00 (m, 1H), 3.20 (br s, 1H), 3.60 (dd, J = 5.4, 7.3 Hz, 1H), 3.76 (dd, J = 2.9, 10.3 Hz, 1H), 3.81 (s, 3H), 3.85 (dt, J = 3.9, 7.3 Hz, 1H), 4.01 (dd, J = 4.9, 10.3 Hz, 1H), 4.18 (m, 1H), 4.47–4.61 (m, 4H), 6.88–6.92 (m, 2H), 7.24–7.30 (m, 2H); 13C NMR δ 30.5, 55.2, 69.1, 70.0, 72.6, 73.3, 76.1, 82.4, 114.0, 128.8, 129.7, 159.7; MS FAB m/z 336 (100, [M + Na]+); HRMS ESI m/z calcd for C14H19NO7Na [M + Na]+ 336.1059, found 336.1072.
2-Chloro-3-O-(p-methoxybenzyl)-6-O-nitro-1,2,5-trideoxy-D-arabino-hexofuranose (11). To a solution of Ph3P (2.18 g, 8.31 mmol) and DIAD (1.26 g, 1.22 mL, 6.23 mmol) in THF (14 mL) was added a solution of 10 (0.65 g, 2.08 mmol) in THF (6 mL) followed by freshly prepared HCl•pyridine (0.72 g, 6.23 mmol; prepared by slow addition of TMSCl (0.68 g, 0.8 mL, 6.31 mmol) to a solution of pyridine (0.98 g, 1.0 mL, 12.4 mmol) in MeOH (0.32 g, 0.4 mL, 9.9 mmol) and CH2Cl2 (10 mL) at 0 °C. Volatiles were evaporated and the crystalline residue was dried at 80 °C under vacuum overnight). The suspension was stirred overnight at room temperature. Volatiles were evaporated, and the residue was column chromatographed (hexanes/EtOAc, 10:1 → 6:1) to give 11 (0.43 g, 62%): 1H NMR δ 2.05–2.10 (m, 2H), 3.82 (br s, 4H), 3.81–3.86 (m, 1H), 3.90 (br d, J = 3.4 Hz, 1H), 4.02 (d, J = 10.3 Hz, 1H), 4.13 (dd, J = 4.4, 10.7 Hz, 1H), 4.47–4.65 (m, 4H), 6.90–6.93 (m, 2H), 7.25–7.28 (m, 2H); 13C NMR δ 30.9, 55.2, 60.1, 70.0, 72.1, 74.2, 80.9, 89.8, 113.9, 128.8, 129.6, 159.5; HRMS ESI m/z Calcd for C14H18ClNO6Na [M + Na]+ 354.0720, found 354.0706.
2-Chloro-1,2,5-trideoxy-6-O-nitro-D-arabino-hexofuranose (13). A solution of 11 (0.43 g, 1.30 mmol) and CAN (1.42 g, 2.59 mmol) in CH3CN (7 mL) and H2O (0.7 mL) was stirred for 1.5 h. The reaction mixture was concentrated, and the residue was deposited on silica gel and column chromatographed (hexanes/EtOAc, 10:1 → 3:1) to give 13 (0.21 g, 78%): 1H NMR δ 2.10–2.22 (m, 2H), 2.69 (br s, 1H), 3.80–3.84 (m, 1H), 4.02 (br d, J = 10.8 Hz, 1H), 4.14–4.25 (m, 3H), 4.58–4.68 (m, 2H); 13C NMR δ 30.9, 62.3, 70.0, 73.3, 81.7, 83.2; MS FAB m/z 212 (5, [M + H]+), 120 (100); HRMS ESI m/z calcd for C6H11ClNO5 [M + H]+ 212.0326, found 212.0327.
2-Bromo-1,2,5-trideoxy-6-O-nitro-D-arabino-hexofuranose (14). Step a: To a solution of Ph3P (2.85 g, 10.09 mmol) and DIAD (1.65 g, 8.15 mmol) in THF (30 mL) was added a stirred solution of 10 (1.75 g, 5.43 mmol) in THF (10 mL) followed by freshly prepared HBr•pyridine (1.30 g, 8.15 mmol; prepared by slow addition of TMSBr (0.93 g, 0.8 mL, 6.06 mmol) to a solution of pyridine (0.98 g, 1.0 mL, 12.4 mmol) in MeOH (0.32 g, 0.4 mL, 9.9 mmol) and CH2Cl2 (10 mL) at 0 °C. Volatiles were evaporated and the crystalline residue was dried at 80 °C under vacuum overnight). The suspension was stirred at room temperature for 22 h. Volatiles were evaporated, and the residue was column chromatographed (hexanes/EtOAc, 10:1 → 6:1) to give crude 2-bromo-3-O-(p-methoxybenzyl)-6-O-nitro-1,2,5-trideoxy-D-arabino-hexofuranose (12; 1.0 g, 49%) which was directly used in next step. Step b. A solution of crude 12 (1.0 g, 2.66 mmol) and CAN (2.91 g, 5.32 mmol) in a mixture of CH3CN (14 mL) and H2O (1.4 mL) was stirred for 1.5 h. The resulting mixture was concentrated and the residue was deposited on silica and column chromatographed (hexanes/EtOAc, 10:1 → 4:1) to give 14 (0.54 g, 79%): 1H NMR δ 2.14–2.26 (m, 2H), 3.80–3.86 (dt, J = 5.2, 8.8 Hz, 1H), 4.10 (dd, J = 4.0, 10.6 Hz, 1H), 4.16–4.22 (m, 1H), 4.26–4.33 (m, 2H), 4.58–4.72 (m, 2H); 13C NMR δ 31.2, 51.5, 69.9, 73.6, 81.5, 83.5. HRMS ESI/DART m/z calcd for C6H1479BrN2O5 [M + NH4]+ 273.0081, found 273.0084; calcd for C6H1481BrN2O5 [M + NH4]+ 275.0061, found 275.0061.
1,5-Dideoxy-3-O-(p-methoxybenzyl)-6-O-nitro-D-arabino-hexofuranose (16b). Step a: A solution of compound 10 (2.70 g, 8.63 mmol) in THF (8 mL) followed by a solution of DIAD (2.09 g, 2.03 mL, 10.35 mmol) in THF (3 mL) were slowly (12 min) added to a stirred solution of Ph3P (2.71 g, 10.35 mmol) and PhCO2H (1.26 g, 10.35 mmol) in THF (40 mL) at –50 °C under nitrogen atmosphere. The resulting mixture was allowed to warm to ambient temperature within 1 h (it became colorless at –20 °C). Volatiles were evaporated, and the residue was column chromatographed (hexanes → hexanes/EtOAc, 3:1) to give 2-O-benzoyl-1,5-dideoxy-3-O-(p-methoxybenzyl)-6-O-nitro-D-arabino-hexofuranose (16a; 2.87 g, 80%): 1H NMR δ 2.0–2.07 (m, 2H), 3.79 (s, 3H), 3.85 (td, J = 1.0, 4.6 Hz, 1H), 3.90 (td, J = 5.0, 7.6 Hz, 1H), 4.08–4.12 (m, 2H), 4.51–4.59 (m, 3H), 4.75 (d, J = 11.7 Hz, 1H), 5.43 (td, J = 1.4, 2.7 Hz, 1H), 6.86 (d, J = 8.7 Hz, 2H), 7.27 (d, J = 8.7, 2H), 7.47 (t, J = 7.7 Hz, 2H), 7.61 (tt, J = 1.5, 7.5 Hz, 1H), 8.03 (d, J = 7.6 Hz, 2H). Step b: A suspension of KOH (0.40 g, 7.14 mol) in MeOH (20 mL) was added to a stirred solution of 16a (2.82 g, 6.76 mmol) in MeOH (20 mL) at ambient temperature. After 1 h, the solution was neutralized with AcOH to pH 7. The volatiles were evaporated and the residue was column chromatographed (hexanes/EtOAc, 3:1 → 2:1) to give 16b (1.64 g, 77%): 1H NMR δ 1.26 (d, J = 2.9 Hz, 1H), 2.00–2.04 (m, 2H), 3.64 (d, J = 2.9 Hz, 1H), 3.80 (br s, 4H), 3.83 (d, J = 10.3 Hz, 1H), 3.91 (dd, J = 3.9, 10.3 Hz, 1H), 4.27–4.29 (m, 1H), 4.48–4.58 (m, 4H), 6.88–6.91 (m, 2H), 7.24–7.27 (m, 2H); 13C NMR δ 30.9, 55.2, 70.2, 71.8, 74.1, 76.1 80.3, 89.2, 113.9, 128.4, 129.5, 159.4; MS FAB m/z 336 (100, [M + Na]+); HRMS ESI m/z calcd for C14H19NO7Na [M + Na]+ 336.1059, found 336.1047.
1,5-Dideoxy-6-O-nitro-2-O-tosyl-D-arabino-hexofuranose (18). Step a: TsCl (134 mg, 0.70 mmol) was added to a stirred solution of 16b (100 mg, 0.32 mmol) in anhydrous pyridine (1mL) at ambient temperature. After 16 h, the volatiles were evaporated and residue was partitioned between ice-cold AcOH/H2O (1:99, 30 mL) and CHCl3 (30 mL). The organic layer was separated, and the aqueous layer was extracted with CHCl3 (30 mL). Combined organic phase was washed with ice-cold saturated NaHCO3/H2O (30 mL), brine (30 mL) and dried (Na2SO4), concentrated in vacuo and column chromatographed (EtOAc/hexane, 5:95 → 35:65) to give 1,5-dideoxy-3-O-(p-methoxybenzyl)-6-O-nitro-2-O-tosyl-D-arabino-hexofuranose (17a; 128 mg, 86%) as a colorless oil: 1H NMR δ 1.90–1.96 (m, 2H), 2.46 (s, 3H), 3.72–3.78 (m, 1H), 3.82 (s, 3H), 3.84 (dt, J = 1.2, 4.6 Hz, 1H), 3.87 (d, J = 4.0 Hz, 1H), 3.92 (d, J = 11.3 Hz, 1H), 4.33 (d, J = 11.3 Hz, 1H), 4.42–4.49 (m, 3H), 4.93 (dt, J = 1.2, 4.0 Hz, 1H), 6.87 (d, J = 8.7 Hz, 2H), 7.16 (d, J = 8.7 Hz, 2H), 7.38 (d, J = 8.2 Hz, 2H), 7.81 (d, J = 8.2 Hz, 2H); 13C NMR δ 21.6, 30.3, 55.3, 69.8, 71.4, 71.8, 80.2, 83.9, 86.3, 114.0, 127.8, 128.8, 129.5, 130.1, 133.4, 145.4, 159.6. Step b: A solution of CAN (352 mg, 0.64 mmol) and 17a (100 mg, 0.21 mmol) in MeCN (1 mL) and H2O (0.1 mL) was stirred at ambient temperature for 22 h. Volatiles were evaporated and the residue was column chromatographed (EtOAc/hexane, 3:97 → 30:70) to give 18 (73 mg, 98%) as a colorless oil: 1H NMR δ 1.97–2.07 (m, 1H), 2.07–2.17 (m, 1H), 2.47 (s, 3H), 3.69–3.76 (m, 1H), 3.85–3.90 (m, 1H), 3.95 (dd, J = 5.5, 11.3 Hz, 1H), 4.17 (dd, J = 2.5, 6.0 Hz, 1H), 4.51–4.63 (m, 2H), 4.76 (dt, J = 2.5, 5.5 Hz, 1H), 7.38 (d, J = 8.5 Hz, 2H), 7.80 (d, J = 8.5 Hz, 2H); 13C NMR δ 21.6, 30.2, 69.7, 70.3, 80.5, 80.9, 86.5, 127.9, 130.1, 132.8, 145.6. HRMS ESI/DART m/z calcd for C13H21N2O8S [M + NH4]+ 365.1013, found 365.1031.
2-Chloro-1,2,5-trideoxy-3-O-(p-methoxybenzyl)-6-O-nitro-D-ribo-hexofuranose (19). Step a: TfCl (160 mg, 0.95 mmol) was added to a stirred solution of 16b (250 mg, 0.80 mmol) and DMAP (295 mg, 2.4 mmol) in anhydrous CH2Cl2 (2 mL) at 0 °C (ice-bath). After 1 h, the reaction mixture was partitioned between ice-cold AcOH/H2O (1:99, 30 mL) and CH2Cl2 (30 mL). The aqueous layer was extracted with CH2Cl2 (30 mL) and the combined organic phase was washed with ice-cold saturated NaHCO3/H2O (30 mL), brine (30 mL) and dried (Na2SO4) to give 1,5-dideoxy-3-O-(p-methoxybenzyl)-2-O-(trifluoromethanesulfonyl)-6-O-nitro-D-arabino-hexofuraose as a colorless oil (17b; 313 mg, 88%) of sufficient purity to be used in next step. Column chromatography (EtOAc/hexane, 5:95 → 30:70) gave pure sample of 17b: 1H NMR δ 1.93–2.02 (m, 2H), 3.80–3.84 (m, 1H), 3.82 (s, 3H), 3.92 (dt, J = 1.2, 4.6 Hz, 1H), 4.02 (dd, J = 3.5, 12.0 Hz, 1H), 4.18 (br d, J = 12.1 Hz, 1H), 4.47–4.52 (m, 3H), 4.66 (d, J = 11.5 Hz, 1H), 5.32 (d, J = 3.5 Hz, 1H), 6.91 (d, J = 8.8 Hz, 2H), 7.24 (d, J = 8.8 Hz, 2H). Step b: A solution of crude 17b (50 mg, 0.11 mmol; from step a) and dried LiCl (23 mg, 0.54 mmol) in DMF (1 mL) was stirred for 5 h at ambient temperature under N2. Volatiles were evaporated and residue was partitioned between ice-cold saturated NaHCO3/H2O (15 mL) and EtOAc (15 mL). The separated organic phase was washed with brine (30 mL), dried (Na2SO4) and column chromatographed (EtOAc/hexane, 5:95 → 15:85) to give 19 (30 mg, 80.6%) as a colorless oil: 1H NMR δ 1.82–1.91 (m, 1H), 2.01–2.1 (m, 1H), 3.72 (dd, J = 5.2, 8.2 Hz, 1H), 3.82 (s, 3H), 3.99 (dt, J = 4.0, 8.2 Hz, 1H), 4.06 (dd, J = 2.8, 10.7 Hz, 1H), 4.30 (dd, J = 5.07,10.7 Hz, 1H), 4.40 (d, J = 11.2 Hz, 1H), 4.44 (dt, J = 2.2, 5.2 Hz, 2H), 4.48–4.59 (m, 2H), 4.69 (d, 11.3 Hz, 1H), 6.90 (d, J = 8.7 Hz, 2H), 7.30 (d, J = 8.7 Hz, 2H); 13C NMR δ 30.4, 55.2, 57.8, 69.9, 72.1, 74.0, 76.3, 81.5, 114.0, 128.8, 129.8, 159.7. HRMS ESI m/z calcd for C14H1835ClNO6Na [M + Na]+ 354.0715, found 354.0718; calcd for C14H1837ClNO6Na [M + Na]+ 356.0691, found 356.0694.
2-Chloro-1,2,5-trideoxy-6-O-nitro-D-ribo-hexofuranose (20). A solution of CAN (74 mg, 0.13 mmol) and 19 (15 mg, 0.04 mmol) in MeCN (1mL) and H2O (0.1 mL) was stirred at ambient temperature for 22 h. Volatiles were evaporated and the residue was column chromatographed (EtOAc/hexane, 3:97 → 30:70) to give 20 (6.2 mg, 66%) as a colorless oil: 1H NMR δ 1.87–1.98 (m, 1H), 2.11–2.20 (m, 1H), 2.25 (d, J = 8.7 Hz, 1H), 3.82–3.88 (m, 1H), 3.93–4.00 (m, 1H), 4.02 (dd, J = 4.0, 10.7 Hz, 1H), 4.33 (dd, J = 5.4, 10.5 Hz, 1H), 4.44–4.48 (m, J = 4.2, 5.3 Hz, 1H), 4.55–4.67 (m, 2H); 13C NMR δ 30.5, 61.1, 69.8, 72.9, 75.7, 78.3; HRMS ESI/DART m/z calcd for C6H1435ClN2O5 [M + NH4]+ 229.0586, found 229.0588; calcd for C6H1437ClN2O5 [M + NH4]+ 231.0562, found 231.0561.
2-Bromo-1,2,5-trideoxy-3-O-(p-methoxybenzyl)-6-O-nitro-D-ribo-hexofuranose (21). A solution of 17b (30 mg, 0.06 mmol; prepared as described for 19, step a) and dried LiBr (29 mg, 0.33 mmol) in DMF (1 mL) was stirred for 7 h at ambient temperature under N2. Volatiles were evaporated and the resulting residue was partitioned between ice-cold saturated NaHCO3/H2O (15 mL) and EtOAc (15 mL). The separated organic phase was washed with brine (30 mL), dried (Na2SO4) and column chromatographed (EtOAc/hexane, 5:95 → 15:85) to give 21 (18 mg, 71%) as a colorless oil: 1H NMR δ 1.83–1.94 (m, 1H), 2.02–2.12 (m, 1H), 3.6 (dd, J = 5.0, 7.6 Hz, 1H), 3.84 (s, 3H), 4.03 (dt, J = 4.0, 8.0 Hz, 1H), 4.22 (dd, J = 2.8, 10.6 Hz, 1H), 4.44 (d, J = 11.1 Hz, 1H), 4.43–4.51 (m, 2 H), 4.51–4.61 (m, 2H), 4.71 (d, J = 11.1 Hz, 1H) 6.92 (d, J = 8.6 Hz, 2H), 7.33 (d, J = 8.6 Hz, 2H); 13C NMR δ 30.4, 49.7, 55.3, 69.9, 72.1, 74.1, 76.6, 81.2, 114.0, 128.81, 129.8, 159.71. HRMS ESI m/z calcd for C14H1879BrNO6Na [M + Na]+ 398.0210, found 398.0203; calcd for C14H1881BrNO6Na [M + Na]+ 400.0191, found 400.0183.
2-Bromo-1,2,5-trideoxy-6-O-nitro-D-ribo-hexofuranose (22). A solution of CAN (78 mg, 0.14 mmol) and 21 (18 mg, 0.05 mmol) in MeCN (1 mL) and water (0.1 mL) was stirred at ambient temperature for 22 h. Volatiles were evaporated and the residue was column chromatographed (EtOAc/hexane, 3:97 → 30:70) to give 22 (12 mg, 96%) as a colorless oil: 1H NMR δ 1.89–1.98 (m, 1H), 2.11–2.20 (m, 2H), 3.78–3.84 (m, 1H), 3.85–3.92 (m, 1H), 4.13 (dd, J = 4.6 Hz, 1H), 4.43 (dd, J = 5.4 Hz, 1H), 4.48 (dd, J = 5.0 Hz, 1H), 4.55–4.70 (m, 2H); 13C NMR δ 31.6, 53.7, 69.8, 72.9, 75.3, 78.8. HRMS ESI/DART m/z calcd for C6H1479BrN2O5 [M + NH4]+ 273.0081, found 273.0077; calcd for C6H1481BrN2O5 [M + NH4]+ 275.0066, found 275.0058.
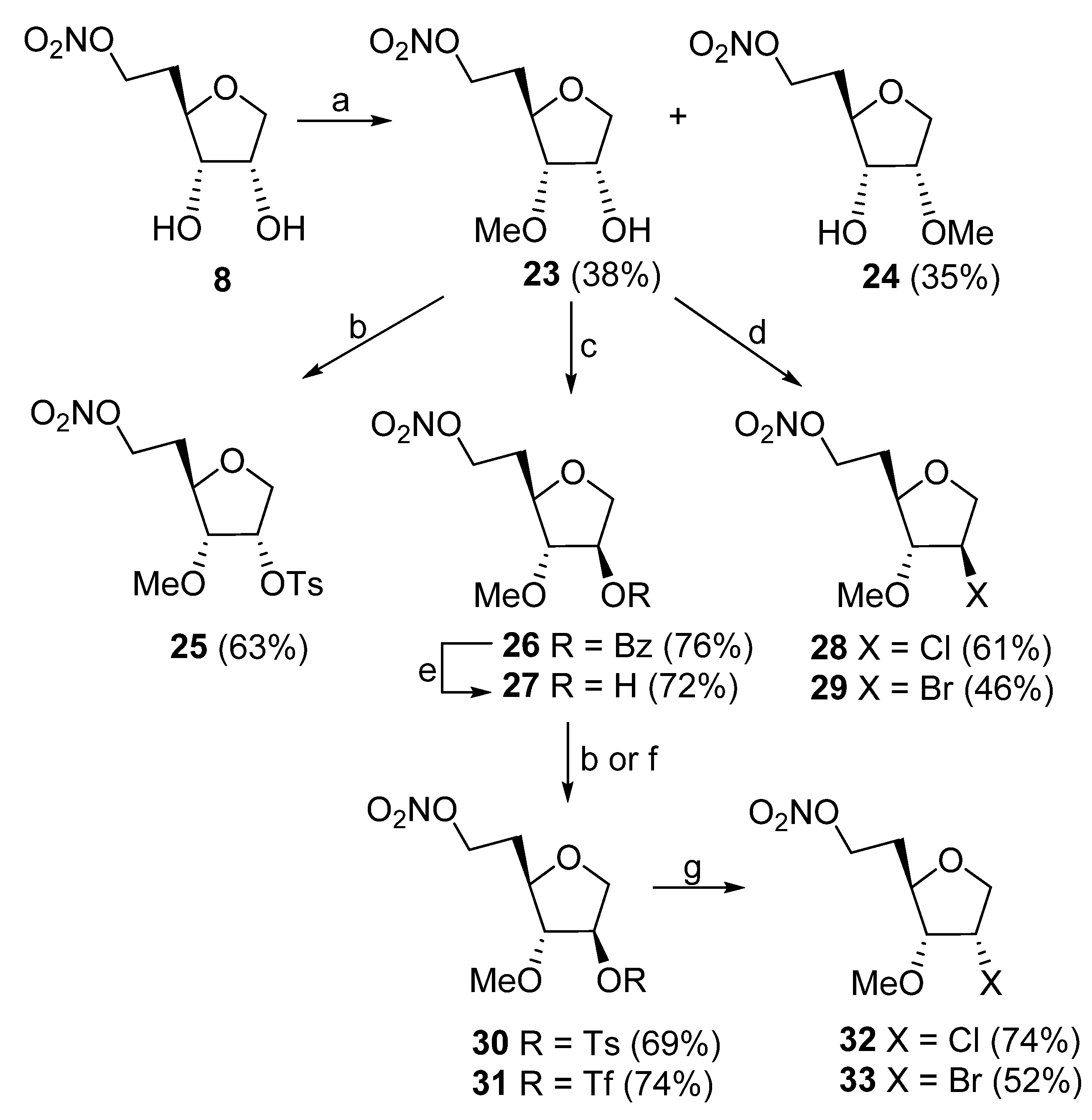
1,5-Dideoxy-3-O-methyl-6-O-nitro-D-ribo-hexofuranose (23) and 1,5-Dideoxy-2-O-methyl- 6-O-nitro-D-ribo-hexofuranose (24) and: A suspension of 8 (0.34 g, 1.76 mmol) and Bu2SnO (0.44 g, 1.76 mmol) in anhydrous MeOH (8 mL) was refluxed for 30 min. Volatiles were evaporated after the flask was cooled to ambient temperature. DMF (1 mL) and MeI (1.14 g, 0.5 mL, 8.03 mmol) were added, the flask was sealed and the solution was stirred at 40 °C for 12 h. Volatiles were evaporated and the residue was column chromatographed (hexanes/EtOAc, 4:1 → 1:1) to give 23 (145 mg, 38%) and 24 (134 mg, 35%). Compound 23 had: 1H NMR δ 1.85–1.92 (m, 1H), 2.09–2.16 (m, 1H), 2.79 (d, J = 8.8 Hz, 1H), 3.44 (s, 3H), 3.67 (ddd, J = 3.9, 7.8, 8.8 Hz, 1H), 3.74–3.84 (m, 3H), 4.06 (dd, J = 4.4, 5.4 Hz, 1H), 4.56–4.65 (m, 2H); 13C NMR δ 30.5, 57.7, 70.0, 70.1, 75.4, 78.6, 79.4; MS FAB m/z 230 (100, [M + Na]+); HRMS ESI m/z calcd for C7H13NO6Na [M + Na]+ 230.0641, found 230.0651. Compound 24 had: 1H NMR δ 1.89–1.96 (m, 1H), 2.04–2.11 (m, 1H), 2.70 (d, J = 3.9 Hz, 1H), 3.45 (dd, J = 4.9, 6.8 Hz, 1H), 3.47 (s, 3H), 3.79 (dd, J = 2.9, 9.8 Hz, 1H), 3.86 (ddd, J = 3.9, 6.8, 8.3 Hz, 1H), 3.79 (dd, J = 4.4, 10.2 Hz, 1H), 4.31 (m, 1H), 4.55–4.65 (m, 2H); 13C NMR δ 30.8, 58.3, 68.9, 70.0, 73.2, 75.9, 85.2; MS FAB m/z 208 (100, [M + H]+); HRMS ESI m/z calcd for C7H14NO6 [M + H]+ 208.0821, found 208.0819.
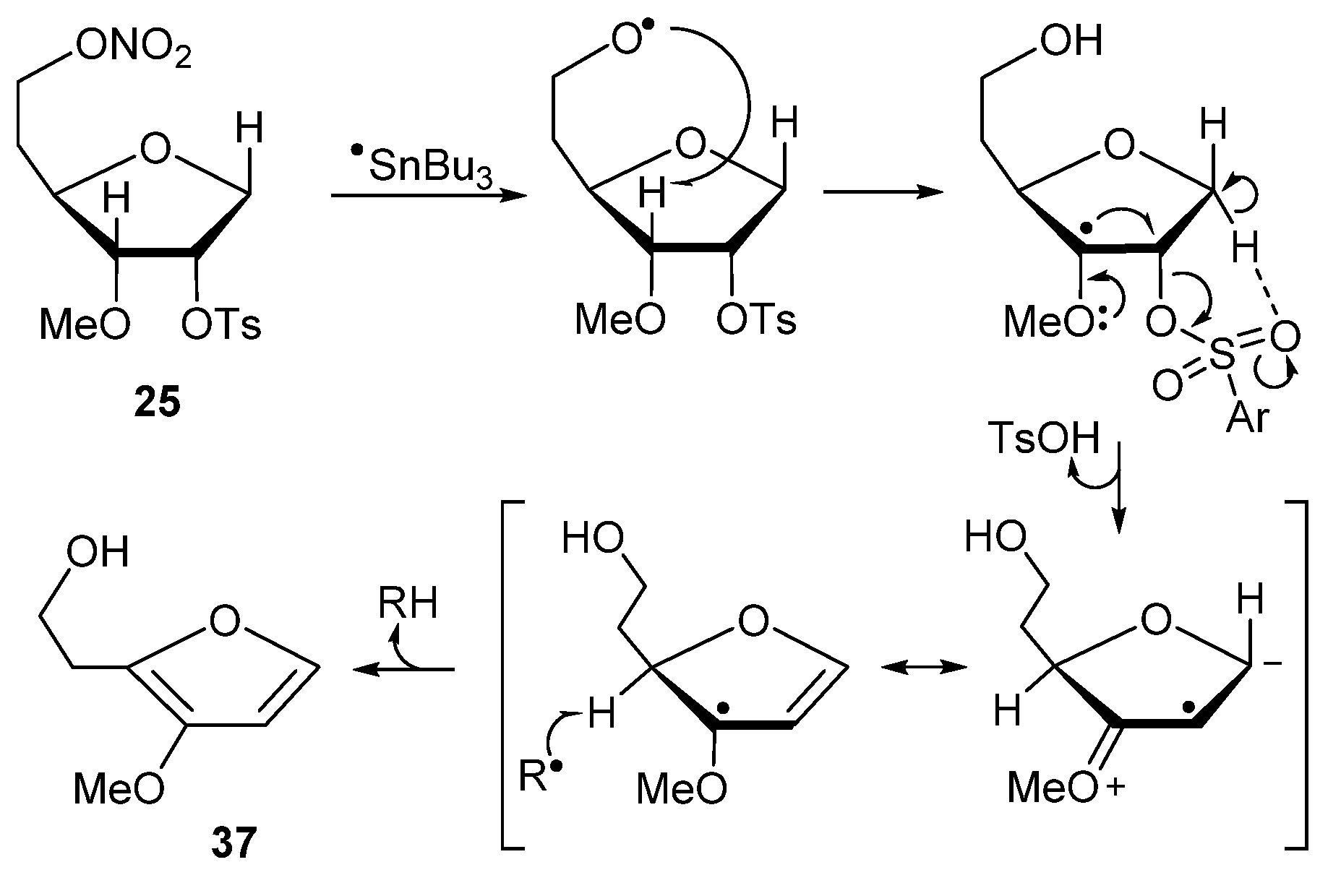
1,5-Dideoxy-2-O-tosyl-3-O-methyl-6-O-nitro-D-ribo-hexofuranose (25). TsCl (101 mg, 0.53 mmol) was added to a stirred solution of 23 (100 mg, 0.48 mmol) in anhydrous pyridine (1 mL) at ambient temperature. After 16 h, the volatiles were evaporated and residue was partitioned between ice-cold AcOH/H2O (1:99, 30 mL) and CHCl3 (30 mL). The organic layer was separated, and the aqueous layer was extracted with CHCl3 (30 mL). Combined organic phase was washed with ice-cold NaHCO3/H2O (30 mL), brine (30 mL), dried (Na2SO4), concentrated in vacuo and column chromatographed (EtOAc/hexane, 5:95 → 35:65) to give 25 (110 mg, 63%) as a colorless oil: 1H NMR δ 1.86–1.96 (m, 1H), 2.05–2.15 (m, 1H), 2.48 (s, 3H), 3.31 (s, 3H), 3.47 (dd, J = 5.0, 8.6 Hz, 1H), 3.85 (dd, J = 4.2, 8.6 Hz, 1H), 3.89 (dd, J = 2.44, 11.5 Hz, 1H), 4.07 (dd, J = 4.44, 11.5 Hz, 1H), 4.51–4.63 (m, 2H), 5.12 (dt, J = 2.2, 4.7 Hz, 1H), 7.39 (d, J = 8.15 Hz, 2H), 7.85 (d, J = 8.15 Hz, 2H); 13C NMR δ 21.6, 30.5, 58.3, 69.8, 70.9, 75.9, 76.7, 83.7, 127.8, 129.9, 133.7, 145.2; HRMS ESI/DART m/z calcd for C14H23N2O8S [M + NH4]+ 379.1170, found 379.1172.
2-O-Benzoyl-1,5-dideoxy-3-O-methyl-6-O-nitro-D-arabino-hexofuranose (26). Compound 23 (0.20 g, 0.97 mmol) in THF (6 mL) was added to a stirred solution of Ph3P (0.31 g, 1.16 mmol) and PhCO2H (0.14 g, 1.16 mmol) in THF (5 mL) at −50 °C. After 5 min. DIAD (0.23 g, 0.22 mL, 1.16 mmol) in THF (2 mL) was added slowly over 12 min. The reaction mixture was allowed to warm to room temperature within 1 h (it became colorless at −20 °C). Volatiles were evaporated, and the residue was column chromatographed (hexanes → hexanes/EtOAc, 10:1) to give 26 (0.23 g, 76%): 1H NMR δ 2.07–2.17 (m, 2H), 3.50 (s, 3H), 3.71 (d, J = 3.4 Hz, 1H), 3.86–3.90 (m, 1H), 4.03–4.07 (m, 2H), 4.57–4.66 (m, 2H), 5.38–5.40 (m, 1H), 7.44–7.62 (m, 3H), 8.02–8.14 (m, 2H); 13C NMR δ 30.8, 58.0, 70.0, 71.7, 77.9, 80.4, 89.6, 128.4, 129.4, 130.1, 133.4, 165.7; MS FAB m/z 334 (15, [M + Na]+), 312 (100, [M + H]+); HRMS ESI m/z calcd for C14H17O7NNa [M + Na]+ 334.0903, found 334.0904.
1,5-Dideoxy-3-O-methyl-6-O-nitro-D-arabino-hexofuranose (27). KOH (1.62 g, 28.9 mmol) in MeOH (80 mL) was added to a stirred solution of 26 (1.82 g, 5.85 mmol) in MeOH (80 mL). The reaction mixture was left to stir at room temperature for 1 h, then was neutralized with 5% HCl/H2O. Volatiles were evaporated, and the residue was column chromatographed (hexanes/EtOAc, 3:1 → 1:1) to give 27 (0.87 g, 72%): 1H NMR (300 MHz) δ 2.07–2.14 (m, 2H), 2.80 (br s, 1H), 3.42 (s, 3H), 3.48 (td, J = 1.1, 3.9 Hz, 1H), 3.79 (ddd, J = 3.9, 6.1, 7.3 Hz, 1H), 3.85 (br d, J = 10.3 Hz, 1H), 3.90 (dd, J = 3.7, 10.3 Hz, 1H), 4.27–4.29 (m, 1H), 4.54–4.67 (m, 2H); 13C NMR δ 30.9, 57.5, 70.2, 73.9, 75.2, 80.2, 91.6; MS FAB m/z 208 (5, [M + H]+), 71 (100); HRMS ESI m/z calcd for C7H14NO6 [M + H]+ 208.0821, found 208.0807.
2-Chloro-3-O-methyl-6-O-nitro-1,2,5-trideoxy-D-arabino-hexofuranose (28). Solution of 23 (0.60 g, 2.90 mmol) in THF (15 mL) was added to a stirred solution of Ph3P (1.52 g, 5.78 mmol) and DIAD (0.89 g, 0.86 mL, 4.38 mmol) in THF (15 mL) followed by addition of freshly prepared HCl•pyridine (0.50 g, 4.33 mmol). The suspension was stirred overnight at room temperature. Volatiles were evaporated, and the residue was chromatographed (hexanes → hexanes/EtOAc, 6:1) to give 28 (0.38 g, 61%): 1H NMR δ 2.14–2.18 (m, 2H), 3.44 (s, 3H), 3.73 (d, J = 3.9 Hz, 1H), 3.78–3.82 (m, 1H), 4.02 (d, J = 10.7 Hz, 1H), 4.10 (dd, J = 4.4, 10.7 Hz, 1H), 4.24–4.27 (m, 1H), 4.56–4.67 (m, 2H); 13C NMR δ 31.3, 58.1, 59.4, 70.0, 74.2, 80.9, 92.8; HRMS ESI/DART m/z calcd for C7H1635ClN2O5 [M + NH4]+ 243.0742, found 243.0752; calcd for C7H1637ClN2O5 [M + NH4]+ 245.0716, found 245.0715.
2-Bromo-3-O-methyl-6-O-nitro-1,2,5-trideoxy-D-arabino-hexofuranose (29). Solution of 23 (0.65 g, 3.13 mmol) in THF (15 mL) was added to a stirred solution of Ph3P (1.64 g, 6.25 mmol) and DIAD (0.95 g, 0.92 mL, 4.69 mmol) in THF (15 mL) followed by addition of freshly prepared HBr•pyridine (0.75 g, 4.69 mmol). The suspension was stirred overnight at room temperature. Volatiles were evaporated, and the residue was column chromatographed (hexanes/EtOAc, 10:1 → 6:1) to give 29 (0.39 g, 46%): 1H NMR δ 2.19–2.27 (m, 2H), 3.49 (s, 3H), 3.82 (dt, J = 4.2, 6.8 Hz, 1H), 3.92 (d, J = 3.8 Hz, 1H), 4.14 (dd, J = 1.86, 11.0 Hz, 1H), 4.21 (dd, J = 4.47, 11.0 Hz, 1H), 4.27–4.31 (m, 1H), 4.58–4.70 (m, 2H); 13C NMR δ 31.5, 48.5, 58.0, 70.0, 74.6, 81.2, 93.0; HRMS ESI/DART m/z calcd for C7H1679BrN2O5 [M + NH4]+ 287.0237, found 287.0252; calcd for C7H1681BrN2O5 [M + NH4]+ 289.0218, found 289.0233.
1,5-Dideoxy-2-O-tosyl-3-O-methyl-6-O-nitro-D-arabino-hexofuranose (30). TsCl (116 mg, 1.057 mmol) was added to a stirred solution of 27 (200 mg, 0.965 mmol) in anhydrous pyridine (1 mL) at ambient temperature. After 16 h, the volatiles were evaporated and residue was partitioned between ice-cold AcOH/H2O (1:99, 30 mL) and CHCl3 (30 mL). The organic layer was separated, and the aqueous layer was extracted with CHCl3 (30 mL). Combined organic phase was washed with ice-cold saturated NaHCO3/H2O (30 mL), brine (30 mL) and dried (Na2SO4), concentrated in vacuo and column chromatographed (EtOAc/hexane, 5:95 → 35:65) to give 30 (240 mg, 69%) as a colorless oil: 1H NMR δ 1.96–2.11 (m, 2H), 2.47 (s, 3H), 3.29 (s, 3H), 3.65 (td, J = 1.2, 4.4 Hz, 1H), 3.69–3.75 (m, 1H), 3.81 (dd, J = 4.1, 11.4 Hz, 1H), 3.92 (d, J = 11.4 Hz, 1H), 4.48–4.60 (m, 2H), 4.86 (td, J = 1.2, 4.1 Hz, 1H), 7.38 (d, J = 8.4 Hz, 2H), 7.81 (d, J = 8.4 Hz, 2H). 13C NMR δ 21.6, 30.5, 58.0, 69.8, 71.3, 80.1, 83.1, 89.4, 127.8, 130.0, 133.4, 145.4; HRMS ESI/DART m/z calcd for C14H23N2O8S [M + NH4]+ 379.1170, found 379.1169.
2-Chloro-1,2,5-trideoxy-3-O-methyl-6-O-nitro-D-ribo-hexofuranose (32). Step a: TfCl (123 μL, 195 mg, 1.16 mmol) was added to a stirred solution of 27 (200 mg, 0.96 mmol) and DMAP (354 mg, 2.9 mmol) in anhydrous CH2Cl2 (2 mL) at 0 °C (ice-bath). After 1 h, the reaction mixture was partitioned between ice-cold AcOH/H2O (1:99, 30 mL) and CH2Cl2 (30 mL). The aqueous layer was extracted with CH2Cl2 (30 mL) and the combined organic phase was washed with ice-cold saturated NaHCO3/H2O (30 mL), brine (30 mL) and dried (Na2SO4) to give 31 as a colorless oil (243 mg, 74%) of sufficient purity to be used in next step. Column chromatography (EtOAc/hexane, 5:95 → 30:70) gave pure sample of 31: 1H NMR δ 2.01–2.09 (m, 1H), 2.09–2.19 (m, 1H), 3.46 (s, 3H), 3.75–3.82 (m, 2H), 3.98 (dd, J = 3.6, 12.1 Hz, 1H), 4.17 (d, J = 12.1 Hz, 1H), 4.53–4.65 (m, 2H), 5.26 (d, J = 3.5 Hz, 1H); Step b. A solution of crude 31 (100 mg, 0.29 mmol; from step a) and dried LiCl (62.5 mg, 1.47 mmol) in DMF (1 mL) was stirred for 5 h at ambient temperature under N2. Volatiles were evaporated and residue was partitioned between ice-cold saturated NaHCO3/H2O (15 mL) and EtOAc (15 mL). The separated organic phase was washed with brine (15 mL), dried (Na2SO4) and column chromatographed (EtOAc/hexane, 5:95 → 15:85) to give 32 (49 mg, 74%) as a colorless oil: 1H NMR δ 1.89–2.02 (m, 1H), 2.06–2.18 (m, 1H), 3.44 (s, 3H), 3.58 (dd, J = 5.1, 8.0 Hz, 1H), 3.95 (td, J = 4.1, 8.3 Hz, 1H), 4.05 (dd, J = 2.7, 10.8 Hz, 1H), 4.32 (dd, J = 4.8, 10.8 Hz, 1H), 4.49 (td, J = 2.8, 5.0 Hz, 1H), 4.53–4.64 (m, 2H); 13C NMR δ 30.7, 57.6, 58.1, 69.9, 73.9, 76.1, 84.7; HRMS ESI/DART m/z calcd for C7H1635ClN2O5 [M + NH4]+ 243.0742, found 243.0747; calcd for C7H1637ClN2O5 [M + NH4]+ 245.0718, found 245.0716.
2-Bromo-1,2,5-trideoxy-3-O-methyl-6-O-nitro-D-ribo-hexofuranose (33). A solution of crude 31 (100 mg, 0.29 mmol; prepared as described for 32, step a) and dried LiBr (77 mg, 0.88 mmol) in DMF (1 mL) was stirred for 5 h at ambient temperature under N2. Volatiles were evaporated and residue was partitioned between ice-cold saturated NaHCO3/H2O (15 mL) and EtOAc (15 mL). The separated organic phase was washed with brine (15 mL), dried (Na2SO4) and column chromatographed (EtOAc/hexane, 5:95 → 15:85) to give 33 (41 mg, 52%) as a colorless oil: 1H NMR δ 1.90–2.0 (m, 1H), 2.06–2.16 (m, 1H), 3.39–3.45 (m, 4H), 3.97 (td, J = 4.2, 8.0 Hz, 1H), 4.20 (dd, J = 3.0, 11 Hz, 1H), 4.45 (dd, J = 4.8, 11.0 Hz, 1H), 4.50 (td, J = 2.9, 5.0 Hz, 1H), 4.53–4.64 (m, 2H); 13C NMR δ 30.7, 49.5, 58.2, 69.9, 74.0, 76.5, 84.4; HRMS ESI/DART m/z calcd for C7H1679BrN2O5 [M + NH4]+ 287.0237, found 287.0239; calcd for C7H1681BrN2O5 [M + NH4]+ 289.0222, found 289.0218.
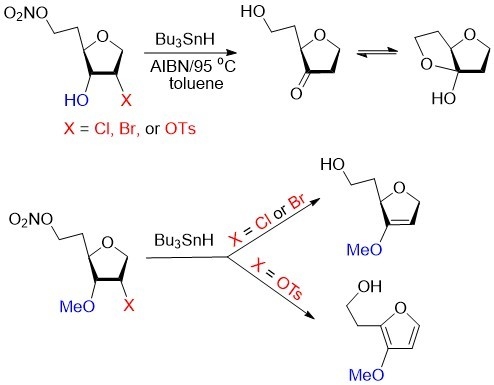
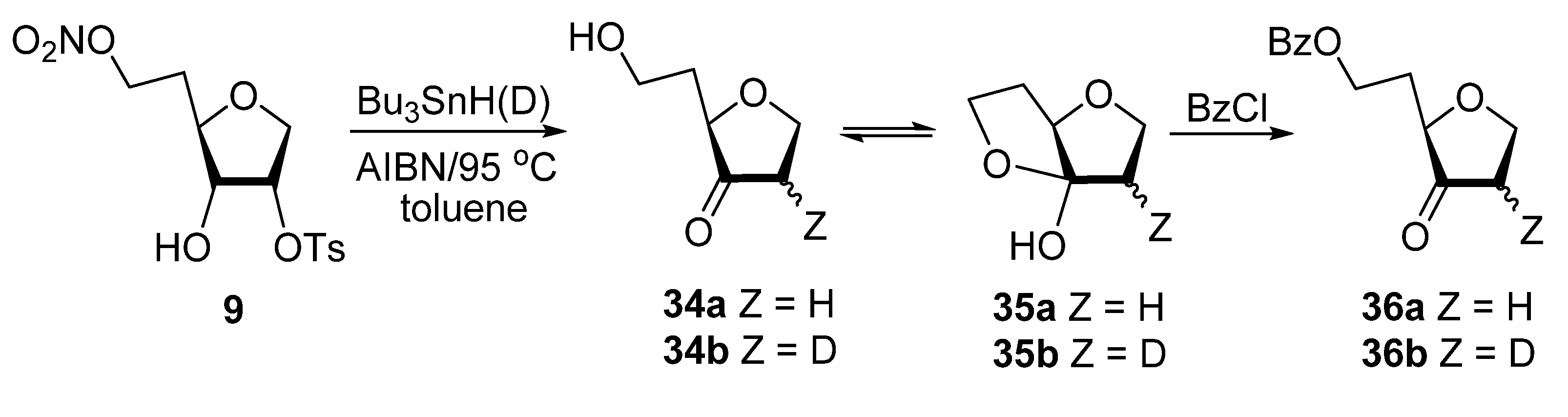
Biomimetic studies with 6-O-nitro-1,4-anhydrohexitols. Typical Procedure. A solution of 9 (20 mg, 0.06 mmol), Bu3SnH (77 μL, 83 mg, 0.28 mmol), and AIBN (18 mg, 0.12 mmol) in dried toluene (2 mL) was deoxygenated (Ar) for 20 min and then heated at 95 °C for 1 h. Volatiles were evaporated carefully (at 25 °C and diminished pressure ~40 mmHg) and the residue was purified by column chromatography (EtOAc/hexane, 10:90 → 70:30) to give 1,2,5-trideoxy-D-glycero-hexofuranose-3-ulose 34a in equilibrium mixture (~1:1) with cyclic hemiacetal 35a (5 mg, 67%) as a colorless oil: HRMS ESI/DART m/z calcd for C6H11O3 [M + H]+ 131.0703, found 131.0707. Ketone 34a had: 1H NMR δ 1.84–1.93 (m, 1H), 1.95–2.06 (m, 2H), 2.15–2.27 (m, 2H), 3.74–3.85 (m, 2H), 3.88 (dd, J = 4.9, 7.4 Hz, 1H), 4.04–4.16 (m, 2H); 13C NMR δ 32.9, 38.9, 59.8, 68.9, 78.6, 215.9. Hemiacetal 35a had: 1.95–2.06 (m, 1H), 2.15–2.27 (m, 1H), 2.46–2.64 (m, 3H), 3.93–4.03 (m, 2H), 4.04–4.16 (m, 1H) 4.28 (dd, J = 2.27, 5.6 Hz, 1H), 4.37 (dt, J = 4.1, 9.2 Hz, 1H); 13C NMR δ 32.1, 36.7, 64.6, 68.3, 85.7, 114.9.
Also isolated from the reaction mixture was 1,5-dideoxy-2-O-tosyl-D-ribo-hexofuranose (3.3 mg, 19%): 1H NMR δ 1.81–1.94 (m, 2H), 2.46 (s, 3H), 3.74–3.85 (m, 4H), 3.86–3.93 (m, 1H), 4.12 (dd, J = 4.9, 11.2 Hz, 1H), 4.95 (dt, J = 3.0, 5.3 Hz, 1H), 7.38 & 7.82 (2 × d, J = 8.3 Hz, 2 × 2H).
Analogous treatment of 9 (20 mg, 0.06 mmol) with Bu3SnD (77 μL, 83 mg, 0.28 mmol), instead of Bu3SnH gave 2-deuterio epimers (2R/S, ~1:1) of 34b in equilibrium mixture (~1:1) with 35b (5.2 mg, 71%) as a colorless oil: 1H NMR spectrum of 34b/35b corresponded to this of the above 34a/35a with reduction of the integrated intensity for the H2/2′ signal at δ 2.15–2.27 and 2.46–2.64 to approximately half and simplification of the H1/1′ signals at δ 4.04–4.16 (m, 1H) and 4.33–4.40 (m, 1H). 13C NMR spectrum of 34b/35b showed triplets at δ 36.7 and 38.9 (J = 20.1 Hz) for C2 carbons because of splitting to deuterium and two close peaks of equal intensity for each hemiacetal carbons. HRMS ESI/DART m/z calcd for C6H10DO3 [M + H]+ 132.0765, found 132.0768. Isotopic incorporation (MS) was calculated to be 85–95% for [2H] isotopomers of 34b/35b depends on the experiments. The 13C NMR spectrum for the sample of 34b/35b (2R/S, ~1:1, [2H] incorporation ~85%) showed residual peaks at 38.9 ppm and 36.7 for the unlabeled 34a and 35a, respectively and isotopically upfield shifted carbon signals for 34b (two sets of triplets of equal intensity at 38.60 and 38.63 ppm with JC2–D = 20.1 Hz) and 35b two sets of triplets of equal intensity at 36.35 and 3.36 with JC2–D = 20.1 Hz), respectively.
6-O-Benzoyl-1,2,5-trideoxy-D-glycero-hexofuranose-3-ulose (36a). BzCl (23 µL, 28 mg, 0.2 mmol), pyridine (44 µL, 43 mg, 0.54 mmol), and DMAP (4 mg, 0.032 mmol) were added to a stirred solution of 34a/35a (30 mg, 0.23 mmol), in CH2Cl2 (2 mL). Stirring was continued at ambient temperature for 3 h and MeOH (0.3 mL) was added. Volatiles were evaporated and the residue was chromatographed (EtOAc/hexane, 5:95 → 15:85) to give 36a (22 mg, 81%) as an colorless oil: 1H NMR δ 2.06–2.16 (m, 1H), 2.18–2.28 (m, 1H), 2.51 (dd, J = 6.50, 8.26 Hz, 2H), 3.90 (dd, J = 4.71, 7.0 Hz, 1H), 4.05–4.13 (m, 1H), 4.28–4.36 (m, 1H), 4.39–4.46 (m, 1H), 4.47–4.54 (m, 1H), 7.41–7.47(m, 2H), 7.56 (tt, J = 1.5, 7.4 Hz, 1H), 8.0 (d, J = 8.57 Hz, 2H); 13C NMR δ 29.9, 36.8, 60.9, 64.7, 76.8, 128.5, 129.7, 130.2, 133.1, 166.5, 215.5; HRMS TOF/DART m/z calcd for C13H18NO4 [M + NH4]+ 252.1230, found 252.1234.
6-O-Benzoyl-2-deuterio-1,2,5-trideoxy-D-glycero-hexofuranose-3-ulose (36b). Treatment of 34b/35b (30 mg, 0.23 mmol) with BzCl, as described for 36a, gave 36b (12 mg, 67%) as a colorless oil: 1H NMR spectrum of 36b corresponded to this of the above 36a with reduction of the integrated intensity for the H2/2′ signal at δ 2.51 to half and simplification of the H1/1′ signals at 4.05–4.13 and 4.28–4.36. 13C NMR spectrum showed triplet at δ 36.4 (J = 20.1 Hz) for C2 because of splitting to deuterium. HRMS ESI/DART m/z calcd for C13H13DNaO4 [M + Na]+ 258.0847, found 258.0836.
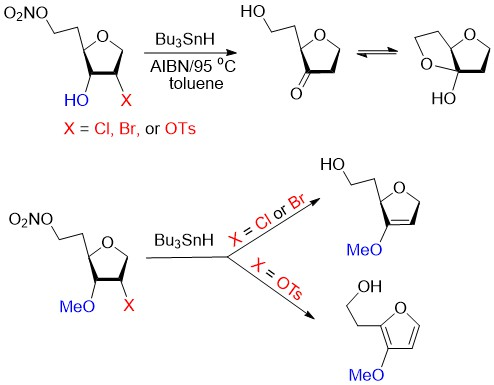
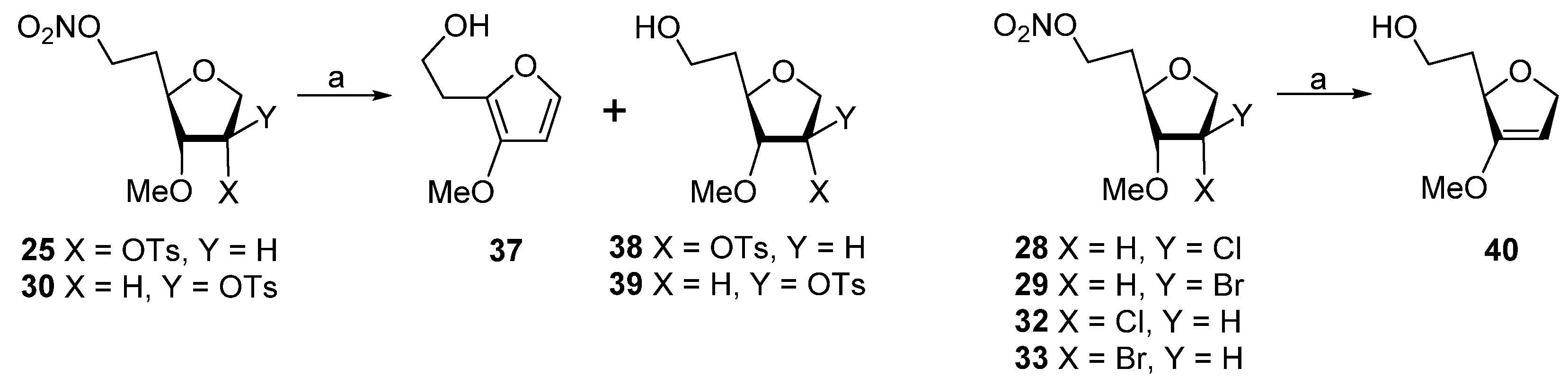
Biomimetic studies with 3-O-methyl-6-O-nitro-1,4-anhydrohexitols. 2-(2-Hydroxyethyl)-3-methoxyfuran (37). A solution of 25 (25 mg, 0.069 mmol), Bu3SnH (92 μL, 100 mg, 0.34 mmol), and AIBN (22.7 mg, 0.14 mmol) in dried toluene (2 mL), was deoxygenated (Ar) for 20 min and then heated at 95 °C for 1.5 h. Volatiles were evaporated and the residue was purified by column chromatography (EtOAc/hexane, 10:90 → 75:25) to give 37 (6.2 mg, 63%) followed by 1,5-dideoxy-2-O-tosyl-3-O-methyl-D-ribo-hexofuranose (38, 4.8 mg, 22%) as a colorless oils. Compound 37 had: 1H NMR δ 2.89 (t, J = 6.0 Hz, 2H), 3.76 (s, 3H), 3.86 (t, J = 6.0 Hz, 2H), 6.31 (d, J = 2.1 Hz, 1H), 7.16 (d, J = 2.1 Hz, 1H); 13C NMR δ 29.3, 59.4, 60.9, 102.9, 136.7, 139.7, 144.3; HRMS ESI/FT-ICR m/z calcd for C7H11O3 [M + H]+ 143.0702, found 143.0701. Compound 38 had: 1H NMR δ 1.74–1.84 (m, 1H), 1.85–1.94 (m, 1H), 2.29 (t, J = 5.9 Hz, 1H), 2.46 (s, 3H), 3.32 (s, 3H), 3.49 (dd, J = 4.9, 8.3 Hz, 1H), 3.72–3.79 (m, 2H), 3.85–3.94 (m, 2H), 4.07 (dd, J = 4.7, 11.3 Hz, 1H), 5.11 (dt, J = 2.4, 4.8 Hz, 1H), 7.36 (d, J = 8.1 Hz, 2H), 7.83 (d, J = 8.1 Hz, 2H); 13C NMR δ 21.6, 35.6, 58.3, 60.6, 70.9, 76.5, 79.1, 83.8, 127.8, 129.9, 133.8, 145.1; HRMS ESI/DART m/z calcd for C14H21O6S [M + H]+ 317.1053, found 317.1055.
Analogous treatment of 25 with Bu3SnD gave 37 (6.0 mg, 61%) and (38, 4.8 mg, 22%) with spectroscopic data as above.
Treatment of 30 (25 mg, 0.069 mmol) with Bu3SnH, as described for 37, gave 37 (5.0 mg, 51%) with data as above and 1,5-dideoxy-2-O-tosyl-3-O-methyl-D-arabino-hexofuranose 39 (8.3 mg, 38%) as a colorless oil: 1H NMR δ 1.86–1.94 (m, 2H), 2.04–2.11 (m, 1H), 2.47 (s, 3H), 3.31 (s, 3H), 3.70 (dt, J = 1.2, 5.0 Hz, 1H), 3.73–3.82 (m, 3H), 3.83 (d, J = 4.3 Hz, 1H), 3.93 (d, J = 11.7 Hz, 1H), 4.88 (dt, J = 1.3, 4.2 Hz, 1H), 7.37 (d, J = 8.4 Hz, 2H), 7.81 (d, J = 8.4 Hz, 2H); 13C NMR δ 21.6, 35.3, 58.0, 60.5, 71.3, 82.7, 83.4, 89.7, 127.8, 130.0, 133.5, 145.3; HRMS ESI/DART m/z calcd for C14H21O6S [M + H]+ 317.1053, found 317.1050.
Analogous treatment of 30 with Bu3SnD gave 37 (4.9 mg, 50%) and 39 (8.2 mg, 38%) with spectroscopic data as above.
1,2,5-Trideoxy-3-O-methyl-D-glycero-hex-2-enofuranose (40). Typical Procedure. A solution of
29 (20 mg, 0.074 mmol), Bu
3SnD (99 μL, 108 mg, 0.369 mmol), and AIBN (24 mg, 0.146 mmol) in dried toluene (2 mL), was deoxygenated (Ar) for 20 min and then heated at 95 °C for 2 h. Volatiles were evaporated and the residue was purified by column chromatography (EtOAc/hexane, 10:90 → 70:30) to give
40 [
22] (5.0 mg, 47%) as a colorless oil:
1H NMR δ 1.73–1.87 (m, 1H), 1.94–2.09 (m, 1H), 2.58 (t,
J = 5.6 Hz, 1H), 3.68 (s, 3H), 3.76–3.82 (m, 2H), 4.60–4.70 (m, 3H), 4.73–4.79 (m, 1H);
13C NMR δ 35.6, 57.6, 60.6, 72.8, 81.2, 90.2, 157.7; HRMS ESI/DART
m/
z calcd for C
7H
16NO
3 [M + NH
4]
+ 162.1125, found 162.1131.
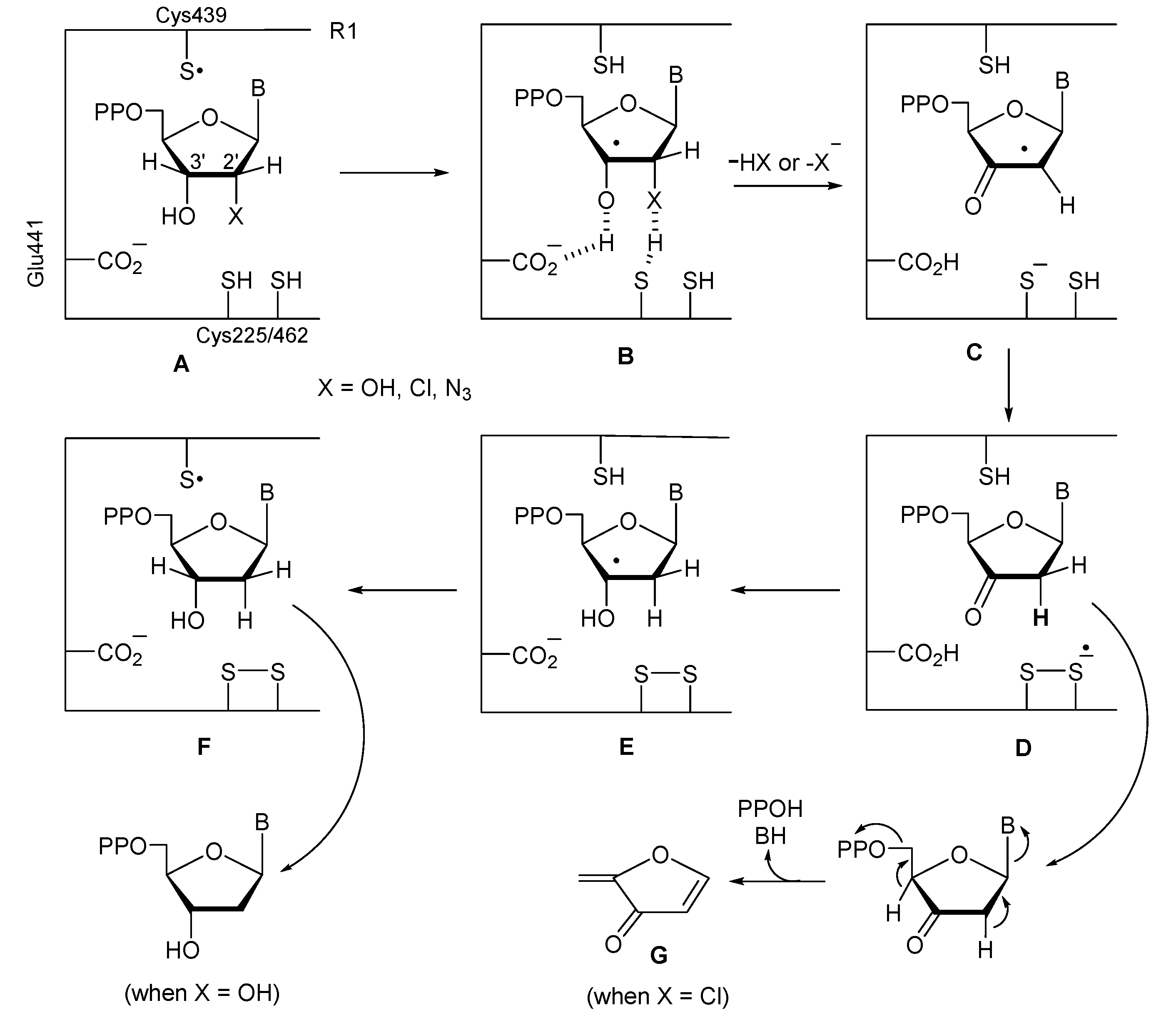
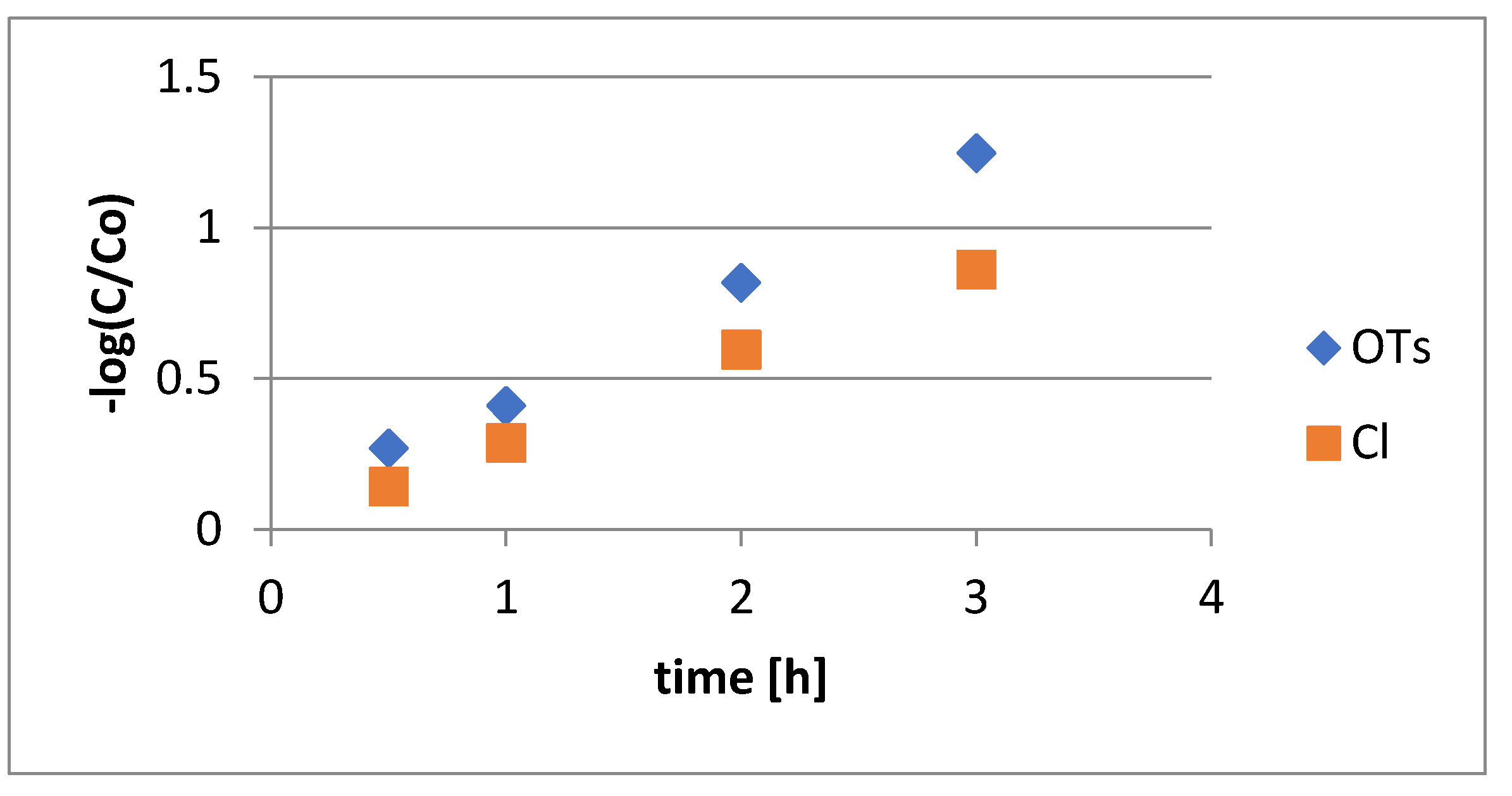
Comparison of Reaction Rates of 9, 13 or 18 with Bu3SnH. Independent solutions of 0.057 mmol samples of
9,
13 and
18 in toluene–
d8 (2.0 mL) were treated with 5 molar equiv of Bu
3SnH and 2 molar equiv of AIBN at 75 °C. Aliquots of the individual reaction mixtures (0.3 mL) were diluted in toluene–
d8 (0.2 mL) and directly analyzed by
1H NMR. The
34a/35a (1:1)
/starting material ratios were obtained by integrating disappearance of the peak at 4.55 ppm for H2 of
9 or
18 or at 3.96 ppm for H6 of
13 and appearance of the peak at 4.10 ppm for the H4 of
34a. The determinations were conducted under the pseudo-first-order conditions:
where
C/Co is the ratio of the concentration of starting material
9,
18, or
13 in the mixture at time
t to the initial concentration of starting material. Values of the term [−log(
C/Co)] were plotted against [
t(min)
k(s
−1) =
k1 (min
−1)/3600].














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
