 Ying Zhang
Ying Zhang Wei Guo
Wei Guo Zhumei Zhan
Zhumei Zhan Ou Bai
Ou Bai- Department of Hematology, The First Hospital of Jilin University, Changchun, Jilin, China
The development of lymphoma is a complex multistep process that integrates numerous experimental findings and clinical data that have not yet yielded a definitive explanation. Studies of oncogenic viruses can help to deepen insight into the pathogenesis of lymphoma, and identifying associations between lymphoma and viruses that are established and unidentified should lead to cellular and pharmacologically targeted antiviral strategies for treating malignant lymphoma. This review focuses on the pathogenesis of lymphomas associated with hepatitis B and C, Epstein-Barr, and human immunodeficiency viruses as well as Kaposi sarcoma-associated herpesvirus to clarify the current status of basic information and recent advances in the development of virus-associated lymphomas.
1 Introduction
The most consistent risk factors for malignant lymphoma comprise immune dysfunction and infectious agents that are primarily viruses. The concept of virus-induced lymphoma is not new, because viruses are associated with ~ 15% of all types of cancer (1). The pathogenesis of virus-associated lymphoma is complex and involves viral infection, immune disorders or deprivation of immunity, the tumor microenvironment (TME), and several viral co-infections. The complex biological properties of the virus itself, a delicate balance between viral and host immunity, and difficulties with establishing animal models have hindered research and understanding of the pathogenesis of virus-associated lymphoma. Lymphoma-associated viruses are very diverse (Table 1). Examples are large double-stranded DNA genomes (Epstein-Barr virus, EBV; Kaposi sarcoma-associated herpesvirus, KSHV), small double-stranded DNA genomes (hepatitis B virus; HBV), and positive-sense single-stranded RNA genomes (hepatitis C virus; HCV). Sufficient evidence indicates that human immunodeficiency virus (HIV), EBV and KSHV are pathogenic factors in lymphoma. However, other evidence indicates a possible relationship between HIV and viruses that cause hepatitis (HBV and HCV) and might be more limited and indirect than EBV and KSHV (2–4). Overall, general pathogenic mechanisms for the development of virus-associated lymphoma have been identified. Viruses can directly infect and transform lymphocytes, and viral antigen products or soluble factors induce chronic B-cell activation and promote transformation. Long-term immunodeficiency, such as that caused by HIV, facilitates viral evasion of the immune response and leads to tumor cloning. Current options for treating virus-associated lymphoma include radiotherapy, chemotherapy, immunotherapy, as well as antiretroviral, antiviral, and targeted therapy. Nevertheless, most virus-associated lymphomas are typically more chemoresistant and have a poorer prognosis than solid tumors. Therefore, a deeper understanding of the molecular mechanisms of virus-associated lymphoma will provide directions to develop targeted therapies.
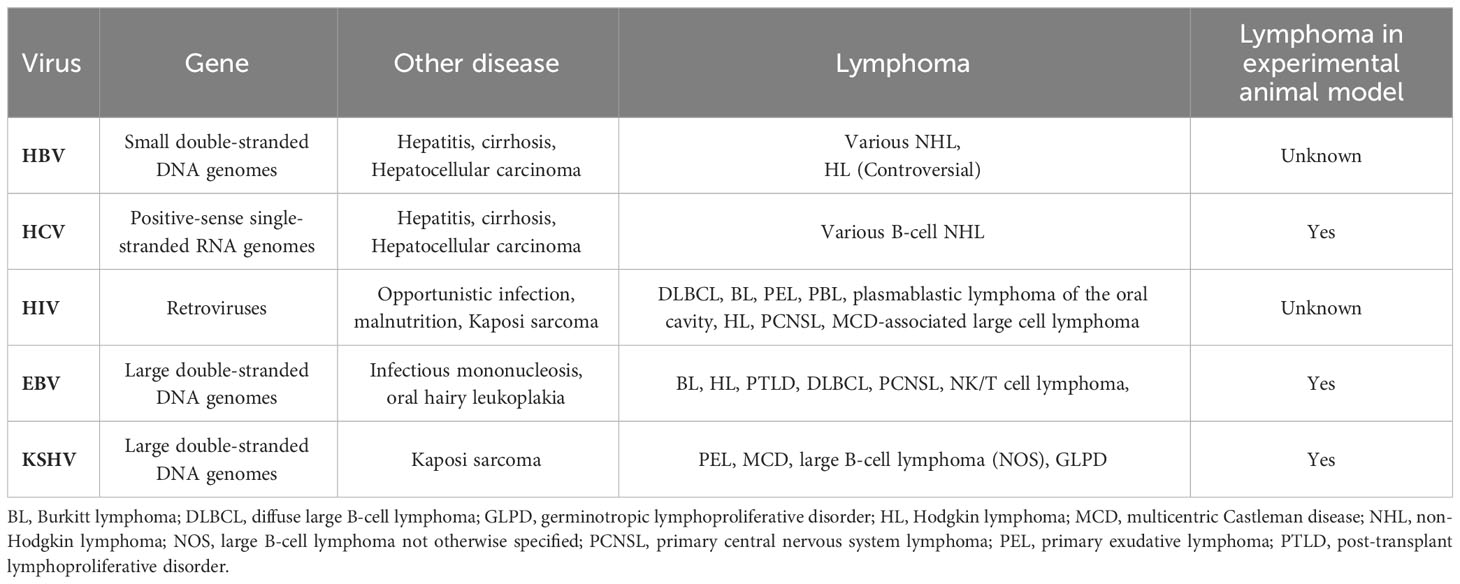
Table 1 Human viruses that are associated with lymphoma and other diseases.
2 Epstein-Barr virus
Epstein-Barr virus (EBV) is the most prevalent human oncovirus (5), and > 90% of adults are infected during their lifetime (6). The main mode of transmission of EBV is through oral transmission via saliva, and the current study confirms that the main tropism of EBV is for B cells and epithelial cells, and the presence of EBV has been demonstrated in tumor cells derived from NK/T cells and leiomyosarcoma (7). When EBV was first isolated from a Burkitt lymphoma (BL) cell line in 1964 (8), its association with cancer was widely studied. According to the 2016 WHO classification, EBV is associated with lymphomas, including mature B-cell tumors, mature T-cell and Natural killer (NK)-cell tumors, Hodgkin lymphoma (HL), and post-transplant lymphoproliferative disorders (9). The prognosis is worse for patients with HL and diffuse large B-cell lymphoma (DLBCL) who are EBV+ than EBV-. (10) NK/T-cell lymphoma (NKTCL), a rare subtype of EBV-associated non-Hodgkin lymphoma (NHL), has similarly shown poorer outcomes (11).
2.1 Epstein-Barr virus structure
Epstein-Barr virus (also known as human herpesvirus 4; HHV-4), belongs to the gamma herpesvirus family. The EBV virion has a diameter of 150-170 nm and consists of a lipoprotein capsule and an icosahedral nucleocapsid, including 162 capsid particles. The viral genome comprises double-stranded DNA of ~ 170 kb. This virus is permanently latent in lymphocytes, free in the cytoplasm as circular DNA and can integrate into cellular chromosomes (12). The life cycle of EBV is biphasic, with lytic replication and a latent phase, and the usual progression of EBV latency in B cells from type III to types II to I has been detailed in a review (13). After infecting resting naïve B cells, EBV enters type III latency, when all latency genes are expressed. The production of highly immunogenic viral proteins triggers a powerful cytotoxic T cell response. Subsequently, the virus restricts gene expression and enters type II latency by expressing Epstein-Barr nuclear antigen (EBNA)-1, latent membrane protein (LMP)-1, and LMP-2. B cells differentiate into memory B cells during this phase. Finally, EBV restricts gene expression to latency type I, where only EBNA-1 and EBV-encoded small RNAs (EBERs) are expressed (14). Table 2 shows EBV gene expression during various latent infections.
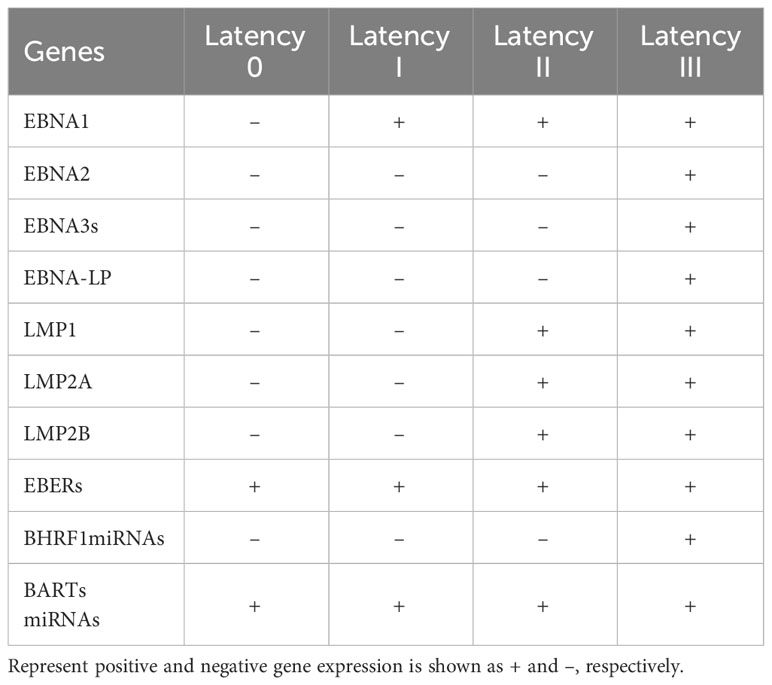
Table 2 EBV viral gene expression during different types of latent infection.
2.2 Carcinogenic mechanisms of EBV
The range of EBV-associated lymphomas is extraordinarily broad, and each has unique developmental pathways. Differences in EBV gene expression among them reflect the different pathogenic roles of EBV. Despite the current scale of research into the relationship between EBV and lymphoma, the etiological role of EBV is difficult to explain. This is partly because the virus acts differently on various tumors and partly because current disease models do not adequately replicate subtle changes in the virus-host balance among EBV-associated cancers. Moreover, although 95% of adults are persistently infected with EBV, most do not develop EBV-associated lymphomas. Therefore, the virus does not act alone, which warrants further exploration. Therefore, we would like to further summarize the mechanism of EBV-associated lymphoma from the perspective of the virus itself.
2.2.1 Expression of viral protein
Latent proteins are essential for the transformation of normal B lymphocytes into lymphoblastoid cell lines (LCLs), and they are involved not only in driving the overexpression of oncogenes, the silencing of tumor suppressors, the cell cycle, migration, but also in the regulation of adhesion.
2.2.1.1 EBNA1
EBNA1 is the only consistently expressed viral protein during the latent phase of EBV, and it is indispensable for the propagation and propagation of the latent viral genome. The current study finds that EBNA1 has significant pleiotropic effects, (15) including disruption of p53 stability (16–18) and promyelocytic leukemia (PML) nuclear bodies (19), and EBNA1 also affects several currently known signaling pathways involved in cell proliferation and apoptosis, known to include interference of EBNA1 with TGF-β signaling (20, 21) and inhibition of NF-κB activity. (22) Moreover, previous studies have also found that stable or transient infection with EBNA1 leads to oxidative stress, allowing reactive oxygen species accumulation and has a variety of effects on cell growth and survival, involving the induction of apoptosis as well as DNA damage. (23, 24) In particular, EBNA1 can co-immunoprecipitate with Nm23-H1 in lymphocytes, which may contribute to the spread of EBV-associated tumors (25, 26). In fact, EBNA1 is actually highly antigenic, and T cells targeting ENBA1 are present in infected individuals (27). Therefore clarifying the immunomodulatory role of EBNA1 for the host has long been a focus of attention for researchers, which has been comprehensively summarized in a recently published review (28). Most published studies have now been limited to immune evasion or immunosuppression (28), include that EBNA1 can specifically bind to viral and cellular DNA for sequences (29–31) and can also enhance and inhibit the transcription of viral and cellular genes (32, 33), and mediate the maintenance of the EBV genome (34). Recent studies have confirmed the trans-immune evasion ability of EBNA1. EBNA1 can inhibit the expression of these genes and enhance the survival and proliferation of infected cells by binding to DNA near the transcriptional start site of NKG2D ligand and c-Myc gene (35). In another study (36), EBNA1 was found to target c-Myc by chromatin immunoprecipitation (ChIP) sequencing of endogenous bromodomain-containing protein 7 (BRD7) in Burkitt lymphoma(BL), thereby regulating the viral infection status by coordinating with host BRD7. In addition, other studies have found that the expression of Galectin-9 (Gal-9) is positively regulated by EBNA1 at both the mRNA and protein levels (37), and Gal-9 has been shown to be a ligand for immune proteins on immune cell subpopulations and is also involved in cell proliferation and differentiation (38).
2.2.1.2 EBNA2
Many of the virus’ latent genes are expressed in currently established EBV-infected cell lines. Of high interest, Pich et al. (39) explored in depth the first 8 days of infection by using EBV derivatives with a single mutation in EBV and found that EBNA2 played and its important role in activating naïve human B lymphocytes, inducing growth, and facilitating division, and in particular EBNA2 prevented the death of a subpopulation of infected cells. However, EBNA-LP, LMP2A, and miRNAs only have supportive and auxiliary functions. Even EBNA1, which has been in the spotlight, seems to be nonessential for cell activation in early viral infection. Previously known studies have extensively explored the mechanism of action of EBNA2, which is not only a potent activator of transcription of genes such as CD23 (40) and C-myc (41), but also negatively regulates genes such as BCL6 and lg (42). Of interest is the previous finding that restricted expression of EBV latent genes contributes to viral persistence by down-regulating the plasma cell master regulator Blimp1, which induces and maintains the mature B-cell phenotype (43). EBNA2 is also a functional homologue of activated Notch (44), while both C-myc and activated Notch have oncogenic properties. In a recent study by Zhang et al. (45) it was demonstrated that LMP1 and EBNA2 constitute the minimum EBV proteins required for B-cell transformation, emphasizing the important role of EBNA2 in B-cell transformation, even though the study did not provide an in-depth investigation of the mechanism. EBNA2 is involved in host immunomodulation through its regulation of miRNAs. In B-cell lymphoma, EBNA2 positively regulates miRNA-21 and negatively regulates the expression of miRNA-146a, which affects the antiviral response of the innate immune system and is involved in EBV-induced B-cell transformation. The detailed mechanism has not been published up to now. The study by Anastasiadou et al. (46) found that EBNA2 down-regulated miRNA-34 by recruiting early B-cell factor 1 (EBF1) to the promoter and increased PD-L1 expression in BL and DLBCL. Other research found that EBNA2 also reduces ICOSL expression by inducing miRNA-24 while maintaining pro-proliferative C-myc levels to evade host immune responses (47).
2.2.1.3 EBNA-LP
Current studies on EBNA-LP are limited. Like EBNA2, EBNA-LP is also expressed early in infection, and EBNA-LP acts mainly as a co-activator of EBNA2 and participates in B-cell transformation by activating viral and cellular transcription (48). In addition, some studies have demonstrated other effects of EBNA-LP. These include regulation of specific alternative splicing (49), promotion of transcription factor recruitment, and involvement in cell growth and survival (50).
2.2.1.4 EBNA3
The EBNA3 family, consisting of the EBNA3A, EBNA3B, and EBNA3C genes, is thought to be a nonredundant family of EBV genes that likely arose from gene duplication during the evolution of primate gamma herpesviruses (51). The production of EBNA3 proteins is thought to be tightly regulated and, because of their low protein levels and turnover efficiency, these proteins are very stable (52). Interestingly, the EBNA3 family has conflicting roles in carcinogenesis. EBNA3A and EBNA3C promote carcinogenesis, whereas EBNA3B inhibits carcinogenesis (53). EBNA3A stimulates cell proliferation by inhibiting p21WAF/CIPI, targeting tumor suppressor pathways and altering cell cycle regulation (54). The mechanisms by which EBNA3C promotes lymphoma development are more diverse, including regulation of cyclin D2 (55) and targeting of tumor suppressor pathways (53). The role of EBNA3 family proteins in EBV-associated B-cell lymphomagenesis has been systematically described (51). Numerous synergistic collaborations between the EBNA3 protein families have been recognized, mostly involving cooperation between EBNA3C and EBNA3A or EBNA3B. Only in the absence of EBNA3C is there moderate cooperation between EBNA3A and 3B. The cooperation between the EBNA3 protein families has been described in detail in the review by Styles et al. (56).
2.2.1.5 LMP1
Among the proteins expressed during EBV viral latency, LMP1 has been of great interest, which is expressed in HL, DLBCL, and post-transplant lymphoproliferative disorder(PTLD) (57, 58), and is essential for the transformation of viral B cells into lymphoblastoid cell lineages, which has been meticulously reviewed in many previous studies (59) (60, 61) (62). The oncogenic mechanism of LMP1 in EBV-associated lymphomas is very complex. EBV not only promotes oncogenic pathways such as Janus kinase/signal transducer, nuclear factor-κB (NF-κB), phosphatidylinositol-3-kinase/protein kinase B (PI3K/AKT), mitogen-activated protein kinase (MAPK), and transcriptional activator of transcription (JAK/STAT) (63), but also, because of its own weaker immunogenicity, it can bypass the targeting effect of CD8+ T-cells and fail to elicit an appreciable immune response in EBV-positive healthy people (62). More importantly, LMP1 was associated with increased expression of PD-L1 in a variety of lymphomas (64), which provided new clues to further explore the immunomodulatory role of LMP1. The latest study by Giehler et al. (65) demonstrates a direct protein-protein interaction between LMP1 and TNF receptor-associated factor 6 (TRAF6), which underlies C-terminal activation region 2 (CTAR2) signaling and the survival of LMP1-transformed B-cells, resolving what we have always wondered.
2.2.1.6 LMP2A and LMP2B
LMP2A is expressed in various B-cell malignancies, including HL, PTLD, and BL, but our current studies on the mechanism by which LMP2A promotes lymphomagenesis are not in-depth. Using transgenic mice, Fish et al. (66, 67) demonstrated that LMP2A accelerated lymphoma development in vivo by exploiting the role of MYC in the cell cycle, particularly during p27kip1 degradation. The latest study utilized phosphoproteomics and transcriptomics to further explore the molecular mechanisms by which LMP2A affects B-cell biology, and found that LMP2A down-regulates cyclic checkpoint genes, including CDKN1B(p27) and CHEK1, as well as the tumor suppressor RB1 (68).
The function of LMP2B is largely unknown. Earlier studies demonstrated that LMP2B negatively regulates the function of LMP2A to prevent the transition from latent to lytic EBV replication (69). In addition, LMP2B affects epithelial cell behavior, such as cell adhesion and motility (70).
2.2.2 Genetic instability
Genetic instability is one of the major common features of cancer and can be observed at the chromosomal or genetic level in malignant cells (71). Integration of EBV into the host genome may be a common occurrence in lymphomas, but our understanding of this is limited. On the one hand, the large size of the EBV genome itself makes it difficult to determine the integration site with the host genome and to analyze it further, on the other hand, the highly methylated DNA hinders the mapping of the EBV genome, and not only that, multiple copies of the viral exons can generate interference noise at the integration site (72), which makes it more difficult to study it in depth. Previous studies have demonstrated the integration of EBV in the chromosomal genome of BL (73) and other B-cell lymphomas (74, 75). Takakuwa et al. (71) demonstrated in Raji that integration of EBV into 6q15 resulted in loss of expression of the human Bach2 gene (BACH2) at the mRNA and protein levels. BACH2 has been shown to have a significant inhibitory effect on cellular proliferation, and deletion of BACH2 expression may contribute to the development of B-cell lymphomas, including BL. Related studies have previously analyzed copy number alterations (CNAs) and gene expression profiles of EBV+ and EBV-DLBCL samples confirming that EBV+ DLBCL has fewer genomic alterations (76). In a recent whole-exome sequencing of EBV+DLBCL, it was shown that a heterogeneous mutational landscape is associated with DNA double-strand break-homologous recombination repair failure, and genes found to have a high number and frequency of mutations include serine protease 3 (PRSS3), MUC3A and MUC16 (77). A recent study by Zhou et al. (78) demonstrated an elevated frequency of mutations in MYC and RHOA in patients with EBV+DLBCL. An updated mutational map of EBV+DLBCL has been comprehensively characterized, complementing previous studies with recurrent alterations in CCR6, CCR7, DAPK1, TNFRSF21, and YY1 (79), further elucidating the mechanism by which EBV leads to B-cell transformation.
2.2.3 MicroRNAs
EBV was the first virus to detect viral miRNAs (80). The EBV genome encodes 44 mature miRNAs belonging to two distinct classes, BamHI-A region rightward transcript (BART) and Bam HI fragment H rightward open reading frame 1 (BHRF1), which have different expression levels in different EBV latency types (81). Among them, BART transcripts encode 22 miRNA precursors and 40 mature miRNAs, while BHRF1 transcripts express three miRNA precursors to produce four mature miRNAs. Current published literature has demonstrated that EBV-encoded miRNAs play an important role in the development and progression of EBV-associated malignancies, including cell proliferation, apoptosis, invasion, and transformation (82, 83).Moreover, EBV miRNAs can even directly target immune-related genes, allowing infected cells to evade surveillance and destruction of the immune system (84), (85). However, EBV miRNAs have different expression profiles in different cancer types. In EBV-infected DLBCL, all EBV-miRNAs except BHRF1 cluster and EBV-miR-BART15 and -20 could be detected, as demonstrated in Imig et al. And in NK/T-cell lymphomas, the most highly expressed viral miRNAs were miR-BART1-5p, miR-BART5, miR-BART7, miR-BART11-5p, and miR-BART19-3p, accounting for 50% of viral miRNAs and approximately 1% of total miRNAs (86). Studies have described the presence and expression levels of EBV miRNAs and host miRNAs in different lymphomas, with some focusing on patient samples and others on different cell line models for in vitro experiments. EBV microRNA profiles and human microRNA profiles for EBV-associated lymphomas are detailed in a recent study by Soltani et al. (87) What’s more, published studies have confirmed that EBV-encoded miRNAs may interfere with host miRNAs, which actually leads to even more complications (87).
EBV miRNAs are essential for regulating the viral life cycle. It was demonstrated as early as lizasa et al. (88) that EBV-miRNA-BART6-5p targets four sites within the 3’-UTR of human Dicer mRNA and comprehensively affects the maturation of the miRNAs, resulting in the total repression of these molecules, which helps to maintain latent infection. Of particular note, in addition to EBV miRNAs, EBV-associated products also contribute to the downregulation of Dicer, such as the EBNA1 protein, which has been described in detail in Mansouri et al. (89) EBV miRNA biogenesis and action are also affected by adenosine to inosine (A-to-I) RNA editing. A-to-I editing of pri-miR-BART6-5p was found in EBV-infected BL to activate Zta and Rta viral proteins encoding EBNA2 viral oncogenes and essential for lysis and replication, leading to the transition of the viral cycle to type III latency (88). Of interest, EBV-encoded miRNAs are also involved in host cell growth, cell cycle, and apoptosis. PRDM1/Blimp1 is a major regulator of terminal B-cell differentiation and is well known as an oncogene in aggressive lymphomas. Nie et al. (90) have demonstrated that the cellular target of the EBV-miRNA-BHRF1-2 is PRDM1, and that by inhibiting the PRDM1-mediated function and conferred a growth advantage to EBV-infected B cells, promoting lymphoma development. Another study confirmed that the EBV-miRNAs-BART9 were involved in the proliferation of Nasal NK/T cell lymphomas (NKTCL) by regulating the level of LMP-1 (91).
The success and persistence of any viral infection depends on a complex balance with the host immune system, and EBV miRNAs are also involved in the regulation of the host immune system. (Figure 1) EBV-miRNAs-BART6-3p was found to mediate down-regulation of the interleukin-6 receptor (IL-6R) in BL (92), which is involved in regulating key cellular processes, including cell proliferation, survival, and response to host pathogens after dimerization receptor binding to interferon-α, IL-12, or IL-27 (93). In addition, the EBV-miRNAs-BART20-5p were shown to inhibit T-bet translation through secondary inhibition of p53 (94). The role of EBV-encoded miRNAs in immunomodulation has been well and exhaustively described (84, 95–97). The latest research has confirmed that in DLBCL, EBV-miRNA-BHRF1-2-5p targets LMP1 to drive the expression of PD-L1 and PD-L2, exerting context-dependent immune counter-regulation, leading to immune escape and contributing to persistent viral infection (98). In another study, Murer et al. (99) used NOD-SCID γc null (NSG) and HLA-A2 transgenic NSG mice to construct a mice model infected with an EBV variant infection lacking viral miRNAs and a mice model infected with wild-type EBV, which found that the viral load in mice infected with EBV variants lacking viral miRNAs was significantly reduced, and the proliferation frequency of EBV-infected B cells was also decreased. What’s more, the depletion of T CD8+ cells led to the formation of lymphomas n the mouse model infected with the viral miRNA-deficient variant, which supports the notion that EBV miRNAs play a major role in immune evasion in vivo and support tumor development. The role of EBV virus-encoded microRNAs in human lymphomas can be found in the review by Navari et al. (82).
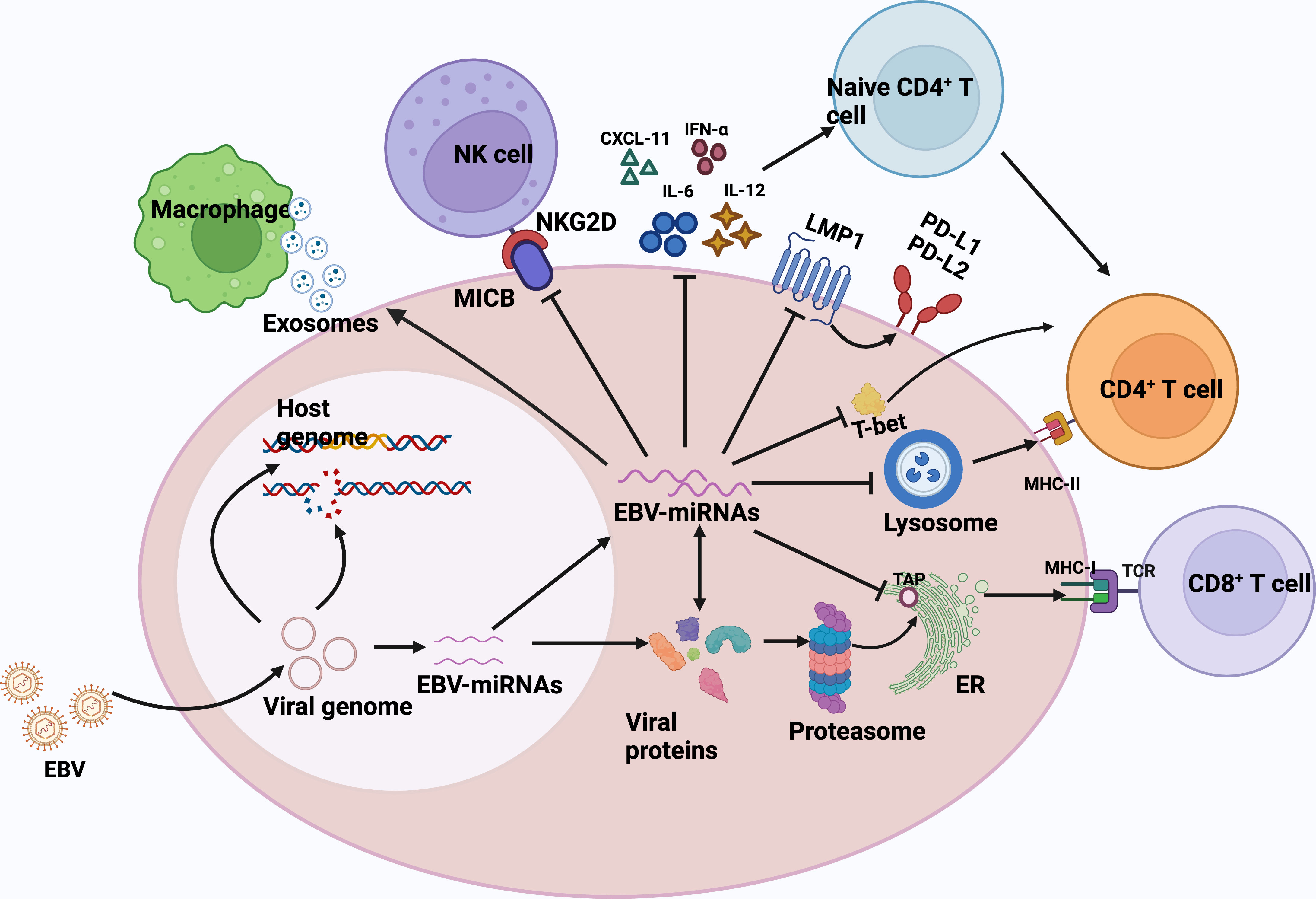
Figure 1 EBV miRNAs are involved in regulating the host immune response. Biogenesis of EBV-encoded miRNAs is dependent on host mechanisms and comprehensively controls the antiviral adaptive immune response of infected B cells. Immediately after infection, the viral DNA genome is circularized and virally encoded and noncoded RNAs are expressed. EBV miRNAs support immune evasion at multiple levels.1) EBV miRNA-BHRF1-2-5p targets the viral antigen LMP1, driving the expression of PD-L1 and PD-L2, and facilitating viral persistence in host cells.2) EBV miRNAs also effectively interfere with MHC class I-mediated antigen presentation by targeting the antigen transporter protein, TAP2. TAP2 is a target of miRNA-BHRF -13 and -BART17.3)EBV miRNA inhibits the expression of lysosomal enzymes (IFI30, LGMN, and CTSB), of which IFI30 and LGMN are under the control of miR-BART1 and -BART2, respectively, and CTSB is controlled by miRNA-BART2 and -BHRF1-2, inhibiting the antigen presentation ability to CD4+ T cells via MHC class II.4) EBV miRNA-BART20-5p inhibits T-bet translation by secondary inhibition of p53 and thus inhibits T-bet translation.5) EBV miRNAs also control the expression of inflammatory cytokines (IL-12, IL-6, and IFN-α), thus inhibiting cytokine-mediated immune response.6) miRNA -BHRF1-3 reduces the secretion of the NK cell ligand CXCL-11, allowing infected B cells to evade immunization by NK cells and T cells.7) EBV acts in trans on uninfected macrophages in tumors by secreting exosomes and promotes lymphoma development. CXCL-11,C-X-C motif chemokine ligand 11; ER, endoplasmic reticulum; TCR, T-cell receptor; MHC, major histocompatibility complex; NKG2D, natural killer group 2D; MICB,MHC class I chain-related molecule B.
3 Hepatitis B virus
According to the World Health Organization (WHO), 257 million people worldwide have chronic HBV infection defined as hepatitis B surface antigen (HbSAg) positivity. The geographic epidemiological profile of HBV is clear according to the WHO; the prevalence is 6.1% in Africa, the Western Pacific, and Southeast Asia, and 1.6% in Europe and North America. (100) Worldwide, the most common route of transmission of HBV is perinatal, but it can also be transmitted percutaneously and via mucous membranes, as well as through sexual intercourse. (101) When infection occurs, the host may experience acute infection with full recovery, or chronic infection or an acute course leading to hepatic failure. (102) The relationship between HBV infection and NHL has been explored (103–105). However, HBsAg+ is not associated with elevated risk of HL, multiple myeloma (MM), or various types of leukemia (106). Compared with HBsAg- DLBCL, the median age of HBsAg+ DLBCL onset is younger, with more frequent splenic or retroperitoneal lymph node involvement, more advanced disease, and significantly worse outcomes (107). The results of other studies are similar (106, 108–110). A meta-analysis of 58 studies revealed that HBV infection leads to a 2.5-fold increased risk of NHL, and data from a stratified analysis suggest a closer association between HBV infection and B-cell, than T-cell NHL (111). Why HBV infection is more closely associated with B- than T-cell lymphoma requires elucidation in functional studies.
3.1 Hepatitis B virus structure
The hepatitis B virus (HBV) is a prototype that belongs to a family of small, enveloped, hepatotropic DNA viruses that infect a narrow host range of mammals and birds and preferentially orientate towards hepatocytes (112). After HBV infection of hepatocytes, the genome of HBV is delivered into the nucleus and repaired in the nucleus to form covalently closed circular DNA (cccDNA), which is then used as a template to guide the transcription of viral RNA. cccDNA is highly stable in the nucleus of infected hepatocytes, which is why chronic hepatitis B is difficult to treat thoroughly (113). The HBV genome contains four overlapping open reading frames (ORFs), four promoters, two enhancer elements (EN1 and EN2), a polyadenylation site for viral RNA transcription and several cis-acting signals for DNA replication. The ORFs P, S, C, and X in the negative strand respectively encode DNA polymerase, HBsAg protein, core and pre-core proteins, and X protein (HBx). (114).
Such DNA viruses have unusual replication features through RNA intermediates and can integrate into the host genome.
3.2 Carcinogenic mechanisms
The biological mechanisms through which HBV infection causes lymphoma are unclear. Those specific to HBV-associated lymphoma have been inferred primarily from studies of HBV-associated hepatocellular carcinoma (HCC) and HCV-associated lymphoma. We emphasize the importance of the humoral and cellular immune systems are important for viral clearance (115), as both are activated by HBV infection and exert antiviral effects. The two immune system might destroy host cells that are already infected with HBV. Therefore, the potential role of HBV in the development of lymphoid disease might be very complex. Various hypotheses have been proposed to explain the mechanisms through which HBV causes lymphoma, and these are summarized below (Figure 2).
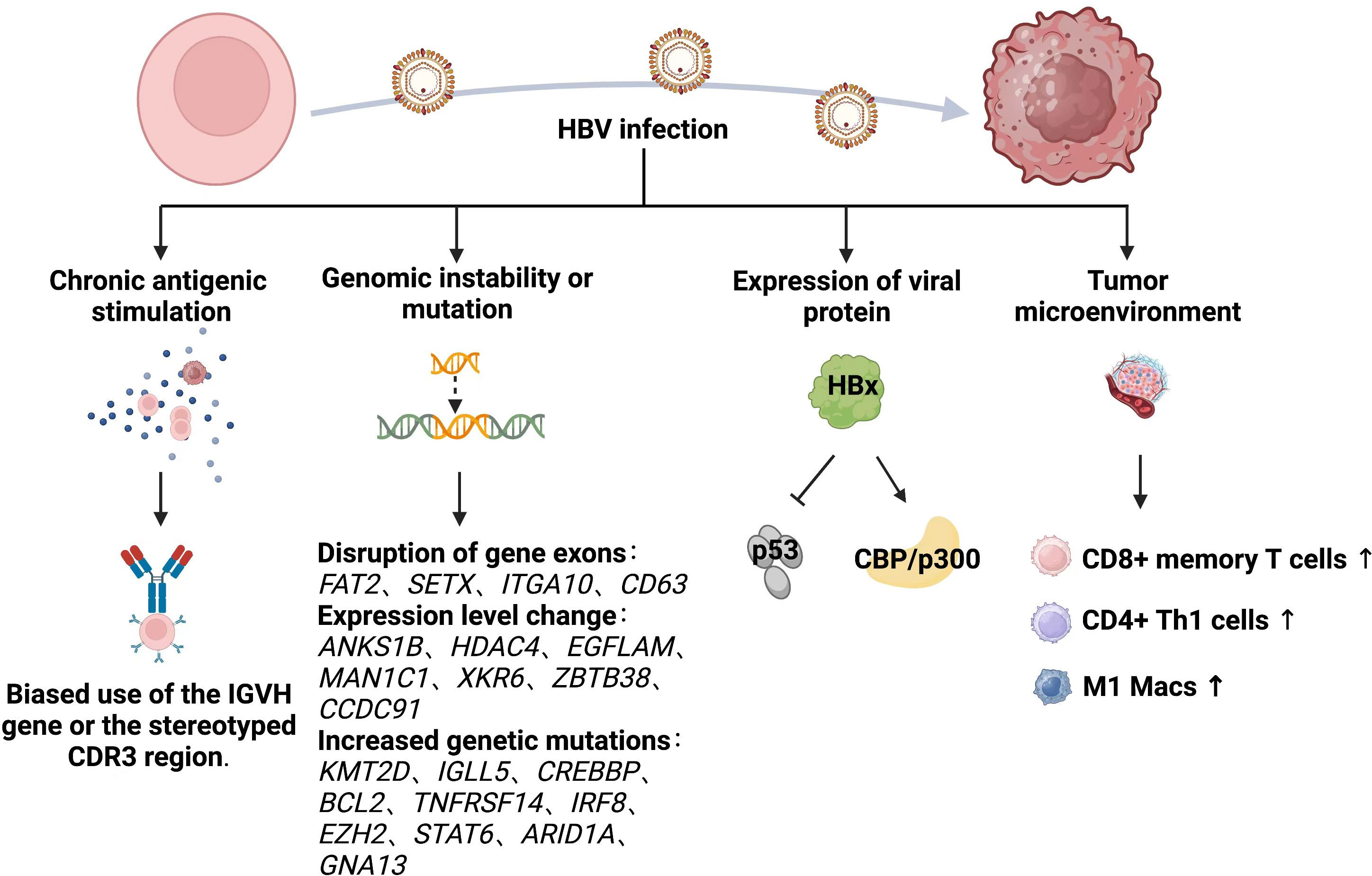
Figure 2 Mechanism of HBV causing lymphoma development.
3.2.1 Chronic antigenic stimulation
The hypothesis that chronic antigenic stimulation causes lymphomas remains controversial. Chronic local antigen-stimulated immune responses caused by HBV infection might be associated with the development of lymphoma (116). A large 14-year follow-up cohort study in Korea (106) consistently associated HBsAg+ with elevated risk of NHL, suggesting that chronic infection promotes the development of lymphoma. Risk of B-NHL is not increased in individuals previously infected with HBV or vaccinated against HBV (117, 118). Nucleic acid sequences specific to HBV have been detected in peripheral blood nuclei and hematopoietic tumor cells of patients with HBsAg+ (3, 119, 120), which might result in chronically stimulated B cells that transform into B-cell NHL. Peripheral blood mononuclear cells (PBMCs) derived from patients with chronic HBV infection have immortalization potential when cultured in vitro (121). New cells identified in the peripheral blood of some patients with non-lymphoid chronical HBV infection were later confirmed as being of B-cell origin. Moreover, the immunophenotype of these cells was similar to that of most HBsAg+ B-cell NHL. This supported the relevance of HBV-induced B-cell NHL, although none of the patients developed lymphoma during > 1 year of follow-up. Furthermore, a 42.1% and 65.5% bias towards Immunoglobulin Heavy Variable 4-34 (IGHV4-34) heavy, and Immunoglobulin Kappa Variable 4-1 (IGKV4-1), light-chain genes respectively in HBsAg+ DLBCL, exceeded that in normal peripheral blood B cells and B-cell NHLs (107). However, these results were contradicted by a study that found no evidence of biased IGVH gene usage or the stereotyped third complementarity determining region (CDR3) (122). Unlike classical antigen-driven hepatitis C virus-associated lymphoma, the chronic antigen stimulation model seems less applicable to HBV-associated DLBCL.
3.2.2 Genomic instability or mutation
Hepatitis B viral DNA is integrated into the chromosomal DNA of lymph node cells (123). A genome-wide investigation of HBV integration in HCC found that HBV integration alters chromosomal stability and gene expression, and shortens the overall survival of infected individuals (124). Approximately 50% of woodchuck hepatitis virus (WHV) is integrated into the myelocytomatosis oncogene (MYC) family of genes and it affects the proto-oncogene in woodchuck models of HCC with chronic WHV infection (125). In fact, HBV integration is common, occurring in 80%-90% of HBV-associated HCC (126, 127).
Hepatitis B viral DNA can be integrated into the genome of NHL cells, and like HCC, it has preferential targets in NHL, since exons of the protein-coding genes FAT Atypical Cadherin 1 (FAT2), Senataxin (SETX), Integrin Subunit Alpha 10 (ITGA10) and Granulophysin (CD63) are disrupted by HBV DNA and the expression of seven HBV preferential target genes, Ankyrin Repeat and Sterile Alpha Motif Domain Containing 1B (ANKS1B), Histone Deacetylase 4 (HDAC4), EGF Like, Fibronectin Type III And Laminin G Domains (EGFLAM), Mannosidase Alpha Class 1C Member 1 (MAN1C1), XK-Related 6 (XKR6), Zinc Finger And BTB Domain Containing 38 (ZBTB38), and Coiled-Coil Domain Containing 91 (CCDC91) is significantly altered in NHL (128). The expression of six of these genes is increased in NHL whereas that of HDAC4 is not, suggesting that HBV integration leads to the cis-activation of primary oncogenes rather than the inactivation of tumor suppressor genes. However, no evidence of HBV DNA integration into the tumor genome has been found in either HBV-associated FL (129, 130) or DLBCL (122). A trend towards an increased genome-wide mutational load has been identified by whole genome, or whole exon sequencing in the coding regions of HBsAg+ follicular lymphoma (FL) with significantly more non-silent mutations per tumor (129). The most significantly mutated genes were Histone-Lysine N-methyltransferase 2D (KMT2D), Immunoglobulin Lambda‐Like Polypeptide 5 (IGLL5), CREB-binding protein (CREBBP), B Cell Lymphoma 2 (BCL2), Tumor Necrosis Factor Receptor Superfamily 14 (TNFRSF14), Interferon Regulatory Factor 8 (IRF8), Enhancer Of Zeste 2 Polycomb Repressive Complex 2 (EZH2), Signal Transducer And Activator Of Transcription 6 (STAT6), AT-Rich Interaction Domain 1A (ARID1A), and Guanine Nucleotide-Binding Protein Subunit Alpha-13 (GNA13). Furthermore, the most obvious mutational pathways were HBV infection-associated, followed by the Forkhead Box O (FoxO), Wingless/Integrated (Wnt), Janus Kinase/STAT (JAK-STAT), B-Cell Receptor (BCR), Phosphatidylinositol-3 Kinase (PI3K), and Nuclear Factor Kappa B (NF-κB) signaling pathways.
3.2.3 Expression of viral protein
The HBx protein encoded by the X gene was once named “viral oncoprotein.” This protein is involved in hepatocyte transformation through regulation of the cell cycle and the pleiotropic activity of DNA repair and signaling pathways (131–133). The expression of HBV antigens, especially HBx protein, is abundant in HBV+ DLBCL sera (103). These findings were consistent with the significantly elevated HBx levels in HCC due to stable HBV integration (124, 134). The HBx protein inhibits p53 in hepatocytes, which leads to abnormal hepatocyte division and HCC (135, 136). A similar B cell mechanism might contribute to the malignant transformation and development of B cell NHL (3). Among the various activities of HBx, its transactivation might play a crucial role in carcinogenesis. Interaction between HBx and the acetyltransferase CREBBP/p300 facilitates the recruitment of these cofactors to the CREB-responsive promoter, which leads to the activation of gene expression (112). A Chinese study of HBV-associated FL found significantly upregulated CREBBP-binding genes in HBsAg+, compared with HBsAg- FL (129). This could explain the low dependence of HBsAg+ FL on CREBBP mutations in that study, as interaction between HBx and CREBBP/p300 might mimic the role of mutant CREBBP during the early stages of lymphoma. The contribution of HBx to the pathogenesis of lymphoma remains obscure, and further investigation is needed to verify its mechanism of action.
3.2.4 Tumor microenvironment
The tumor microenvironment is a complex system of cellular and subcellular components with reciprocal signaling pathways that play key roles in carcinogenesis (137). Tumorigenesis is dependent on the TME, and stroma is uniformly and inappropriately activated in cancer, thus contributing to the malignant features of tumors (138). Chronic and persistent HBV infection induces immune cell dysfunction, T-cell failure, as well as the extensive activation and production of numerous cytokines, chemokines and growth factors that constitute a sophisticated TME that might affect cancer development (139, 140). Hepatitis B surface antigen-positive FLs might have an altered TME with increased infiltration of cluster of differentiation (CD)8+ memory T cells, CD4+ Th1 cells, M1-macrophages and increased T cell failure (129). This was consistent with similar findings in HCC associated with HBV.
The unique biological characteristics of HBV complicates exploring curative mechanisms, and animal models have various strengths and weaknesses. This might explain to some degree, the limited progress of investigations into HBV-related lymphoma.
4 Hepatitis C virus
An estimated 71.1 million people worldwide are infected with HCV, with an annual incidence of 1.75 million (141). The most common routes of HCV transmission are blood transfusions, health care-related injections and injecting drug use (142). Most people (75-80%) will develop chronic infection after exposure to HCV, and the clinical cases of acute hepatitis C are less than 25% (142). In addition to infecting hepatocytes, HCV can infect other cells, such as lymphocytes (143). A possible association between HCV infection and NHL was first described in 1994 (144). A study of 150,000 patients with HCV in the USA found that HCV infection increased risk of lymphoma by 20%–30% (145). Epidemiological data show no, or only a slight increase in the risk of T-cell NHL and HL (146, 147), while the strongest evidence is for B-cell NHL (148). A meta-analysis found that the prevalence of HCV infection in patients with B-cell NHL is ~ 15% (149), and others have reached similar conclusions (150–152). We found that the histological subtypes of NHL most closely associated with HCV infection were marginal zone lymphoma (MZL), lymphoplasmacytic lymphoma, and DLBCL (153–156). Clinical HCV+ NHL usually occurs after infection for >15 years (157) and patients with HCV+ DLBCL usually have higher International Prognostic Index (IPI) scores and LDH levels (158, 159).
4.1 Hepatitis C virus structure
The life cycle of HCV begins with the binding of HCV to specific entry factors on hepatocytes, after which the virus is internalized into the cytoplasm. Subsequently, its genomic RNA is released and used for multiprotein translation and viral replication (143). The small, enveloped, positive-sense, single-stranded RNA HCV belongs to the Flaviviridae family of the genus Hepatophilus. The icosahedral diameter of the envelope particles is 56-65 nm (160), whereas that of the viral core is ~ 45 nm (161). The HCV genome is a positive single-stranded RNA comprising ~ 9,600 nucleotides. It encodes a single open reading frame (ORF) flanked by five and three untranslated regions (UTRs). The HCV polyprotein encoded by a single ORF is ~ 3,000 amino acids long and undergoes co-translational and post-translational processing by cellular and viral proteases to form three structural proteins (core, E1, and E2), an ion channel protein (p7), and the nonstructural (NS) proteins, NS2, NS3A, NS4A, NS4B, NS5A, and NS5B. The structural and NS proteins are located at the N-terminus, whereas other proteins are located at the C-terminal end (162).
4.2 Carcinogenic mechanisms
The integration of single-stranded RNA into HCV nucleic acid sequences of the host genome appears to be impossible owing to the absence of a reverse transcriptase. Therefore, it indirectly exerts oncogenic effects by modulating the host immune system (163). Liver cells and lymphocytes share the HCV receptor, CD81 (164, 165). Activation-mediated CD81 differs from other B cell stimuli because it induces the preferential proliferation of naïve B cells. Expression of the C-X-C Motif chemokine receptor 3(CXCR3) is upregulated in CD81-activated B lymphocytes, but decreased when stimulated with different substances (166). This interaction between HCV and the immune system might underlie the immune and lymphoid tissue proliferative disorders that frequently accompany chronic HCV infections. Three theories might explain HCV transformation (Figure 3).
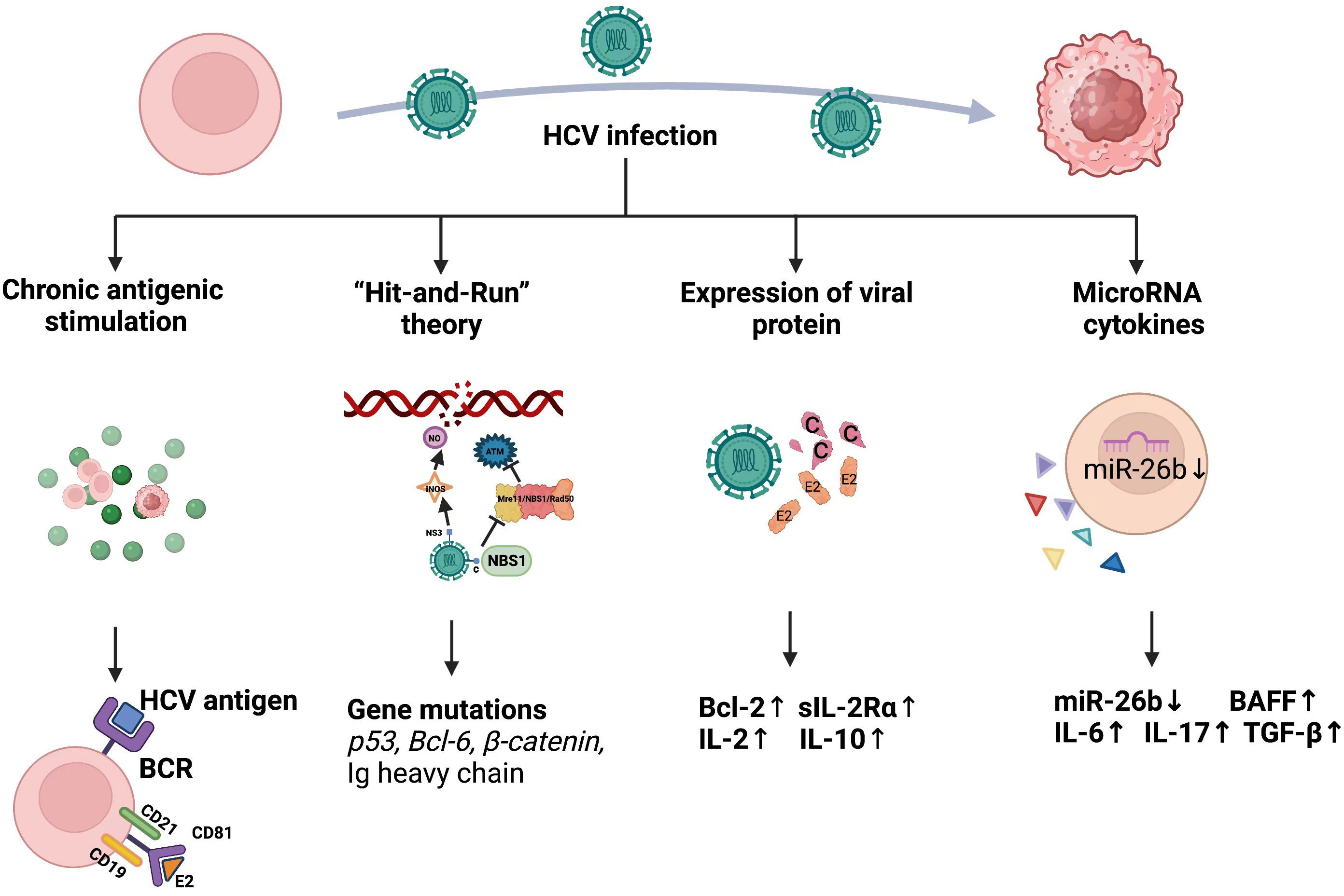
Figure 3 Mechanism of HCV causing lymphoma development.
4.2.1 Chronic antigenic stimulation
The defined pathogenic link between chronic Helicobacter pylori infection and the development of mucosa-associated lymphoid tissue (MALT) gastric lymphoma suggests that chronic antigenic stimulation can determine the likelihood of NHL (167). Notably, the regression of MALT lymphoma after HP eradication makes this possibility more plausible (168). Splenic lymphoma regression after antiviral therapy similarly eradicates HCV (169). About 10% of patients with type II mixed cryoglobulinemia (MC) develop overt B-NHL after 5-7 years of follow-up (170), and HCV is a major etiological factor in MC and might also be the cause of its evolution to overt NHL (171–173). HCV-associated type II MC expresses immunoglobulins encoded mainly by germline VH1-69 and VκA27 genes. A preference for the VH1-69/VκA27 combination in HCV-associated lymphomas is consistent with the possible role of antigen selection in the expansion of B cell clones (174). In addition, B-cell receptors expressed by lymphomas in patients infected with HCV rarely react with viral proteins (175). Notably, the highly biased stereotyped BCR sequence of HCV+ B-NHL has also been found in other HCV-B-cell malignancies (176). This confirmed that HCV-associated lymphoma cells originate from precursors with autoimmune properties rather than from B cells that express antiviral BCR.
The HCV envelope protein E2 can bind to CD81 expressed on B cells (164). This receptor is upregulated in HCV infection and MC and positively correlates with viral load (177). Moreover, CD81 forms a conjugate complex with CD19 and CD21 in human B cells (178, 179), and the attachment of the B cell antigen receptor (BCR) to any component of this complex decreases the threshold required for BCR-mediated B-cell proliferation (180). Bound E2-CD81 is also involved in activating the transcription factor NF-κB, which subsequently increases the expression of Bcl-2 protein, thus enhancing B cell survival and protecting human B lymphocytes from Fas-mediated apoptosis (181). In addition, HCV E2 binds to CD81 antibodies on neonatal human B cells, which leads to the activation and sequential proliferation of the C-JUN N-terminal kinase pathway (166). Furthermore, HCV-E2 binding to CD81 directly prevents the functional activation of NK cells, providing an effective immune escape strategy for the virus (182). Overall, the interaction between HCV and CD81 promotes chronic infection and facilitates the development of HCV-associated B-cell lymphoma.
4.2.2 Hit-and-run theory
Some evidence indicates that intracellular viral replication is not required for tumor transformation (183). The hit-and-run theory suggests that viruses play a predisposing role in cancer formation and that the viral genome can be completely lost after the host cell has accumulated numerous mutations (184). This mechanism was suggested for HCV (185). Infection with HCV results in a 5-10-fold increase in the frequency of mutations in the Ig heavy chain, B cell Lymphoma 6 (BCL-6), Protein 53 (p53) and Catenin genes in HCV-infected B-cell lines and HCV-associated peripheral blood mononuclear cells, lymphomas, and HCC in vitro. The authors concluded that HCV induces a mutator phenotype by causing changes in proto-oncogenes and oncogenes that successively lead to oncogenic B cell transformation, even when the virus might have already left the cells. The same group also conducted RNA interference experiments and found that HCV induced error-prone DNA polymerases ζ, ι, and activation-induced cytidine deaminase. All these together contribute to increased mutation frequency, complementing the oncogenic mechanism of HCV causing lymphoma. Some controversy remains regarding the clinical applicability of these findings, as they have not been confirmed in vivo (186, 187).
Infection with HCV stimulates nitric oxide (NO) production by activating the inducible NOS (iNOS) gene through the viral core and NS3 protein (188). Nitric oxide causes DNA breaks and enhances DNA mutations. The HCV core protein binds to NBS1 and inhibits formation of the Mre11/NBS1/Rad50 complex, thus affecting Ataxia Telangiectasia-Mutated (ATM) activation and inhibiting DNA binding by repair enzymes (189). Infection with HCV inhibits multiple DNA repair processes and leads to chromosomal instability, which explains its oncogenicity from a different perspective.
4.2.3 Expression of viral protein
Hepatitis C viral RNA and protein were detected in an established HCV-infected B-NHL cell line in vitro using RNase protection assays and immunoblotting (190). That study confirmed that HCV can infect primary human hepatocytes, PBMCs and established Raji B cell lines in vitro, indicating that HCV can replicate in B cells. Ample evidence supports the notion that intracellular viral proteins contribute to oncogenic transformation. Interferon regulatory factor-1-null (irf-1(-/-)) mice with inducible and persistent expression of HCV structural proteins (irf-1/CN2 mice) have been established (191). These mice have a high incidence of lymphoma and lymphoproliferative disorders. The HCV core and E2 proteins are responsible for the expression of interleukin (IL)-2, -10, and -12, as well as the induction of Bcl-2 in the presence of nucleocapsid proteins in the context of complex signaling networks in these mice (191). Another transgenic mouse model expressing HCV core protein frequently developed follicular center cell-type lymphoma, and HCV core mRNA was detected in lymphoma tissues (192). Transgenic RzCD19Cre mice express the complete HCV genome in B cells (193). However, the incidence of DLBCL in RzCD19Cre mice was only 25%. The incidence of B-cell lymphoma correlated significantly with serum levels of soluble interleukin-2 receptor α subunit (sIL-2Rα) only in the RzCD19Cre mice.
4.2.4 MicroRNA and cytokines
Small non-coding MicroRNAs (miRNAs) sequence-specifically regulate gene expression at the post-transcriptional level (194). They play roles in controlling various biological functions such as developmental patterns, cell differentiation, proliferation, genomic rearrangement and transcriptional regulation (195). MicroRNA-26b is significantly downregulated (P = 0.0016) in HCV+ splenic marginal zone lymphoma (SMZL) and this might cause miR-26b to stop inhibiting never in mitosis gene A (NIMA)-related Kinase 6 (NEK6) and have oncogenic potential in HCV-associated SMZL (196). MicroRNA-26b functions not only in the specific area of HCV-associated SMZL, but also in HCV-associated NHL, including MZL and DLBCL (197). Overall, these findings suggest that miRNA network dysregulation is involved in the development of HCV-associated lymphomas.
Cytokines are small glycoproteins and peptides that usually have relatively short half-lives and act via autocrine and paracrine signaling. Cytokines mediate interactions between immune and non-immune cells in tumors and can promote or inhibit cancer cell growth (198). B-cell activating factor (BAFF) is a key survival factor for B cells that is upregulated during HCV infection (199). An excess of BAFF in the absence of protective tumor necrosis factor (TNF) leads to a high incidence of lymphoma in BAFF transgenic mice, suggesting that BAFF functions in promoting B-cell malignancy (200). Notably, other cytokines and growth factors, including IL-6, -17, -10 and TGF-ß, also contribute to B-cell proliferation in HCV infection (201–203).
However, the molecular mechanisms underlying the development of HCV-associated lymphomas remain poorly understood. The prevailing views are not mutually exclusive and might involve parallel pathways leading to HCV-associated lymphoma, as it is likely that a combination of translational conditions is required to eventually lead to the development of lymphoma. Additional bridging studies combining in vivo and ex vivo investigations are required to further explore this topic.
5 Human immunodeficiency virus
It is estimated that 38.6 million people are currently infected with HIV-1 worldwide, that some 25 million people have died, and that heterosexual transmission remains the dominant mode of transmission, accounting for about 85 per cent of all HIV infections (204). HIV infection carries multiple immune cell types for CD4 and CXCR4/CCR5 co-receptors. This includes helper T cells, macrophages. If untreated, it may also infect microglia and astrocytes of the nervous system (205).An association between HIV and aggressive lymphoma was initially reported in 1982 (206). As the most prevalent malignancies among patients infected with HIV, the relative risks of NHL and HL are 60-200- and 8-10-fold higher than patients with lymphoma without HIV infection, respectively (207, 208). The WHO classification system recognizes subtypes of HIV-NHL (9b). Over 95% of malignancies are of B-cell origin, including DLBCL and BL, whereas plasmablastic, T-cell, and primary effusion lymphomas, are rare, and primary central nervous system (CNS) lymphoma is a very rare B-cell subtype that was more prevalent during the early stages of the AIDS epidemic. These lymphomas have high-grade features such as typically late presentation, extra-nodal involvement, and a marked tendency to involve the gastrointestinal tract, CNS, liver, bone marrow and perinodal soft tissues (209). Despite the introduction of highly active antiretroviral therapy (HAART) and the improved survival rates of patients infected with HIV during the past 20 years, malignant lymphoma remains the leading cause of morbidity and mortality (210).
5.1 Human immunodeficiency virus structure
The two types of HIV isolates comprise types 1 (HIV-1) and 2 (HIV-2). The globally predominant pathogen of AIDS is HIV-1, whereas HIV-2 is restricted to certain areas of West and Central Africa (211).Human immunodeficiency virus forms spherical, membrane-enveloped, pleomorphic virions, 1,000–1,500 Å in diameter. that contain two copies of a single-stranded, positive-sense RNA genome (212) This virus is characterized by the structural genes gag, pol, env (211). Like other retroviruses, gag genes encode the structural proteins of the core (p24, p7, and p6) and matrix (p17), and env genes encode the viral envelope glycoproteins gp120 and gp41. The pol encodes enzymes that are essential for viral replication.
5.2 Carcinogenic mechanisms
That HIV causes chronic antigenic stimulation, immune dysregulation, is generally accepted. However, the high incidence of lymphoma in patients who are HIV+ despite the introduction of HAART suggests that incomplete immune reconstitution or factors unrelated to immune dysfunction also play causative roles. Although HIV-1 infects a subpopulation of human cells, namely CD4+ cells, soluble HIV-1 proteins that are detectable in serum from infected individuals invade and/or bind to receptors in uninfected cells, including B lymphocytes and endothelial cells. These proteins interfere with host gene expression and other cellular processes, ultimately leading to cellular transformation and the development of HIV-associated lymphomas. This section summarizes current mainstream views (Figure 4).
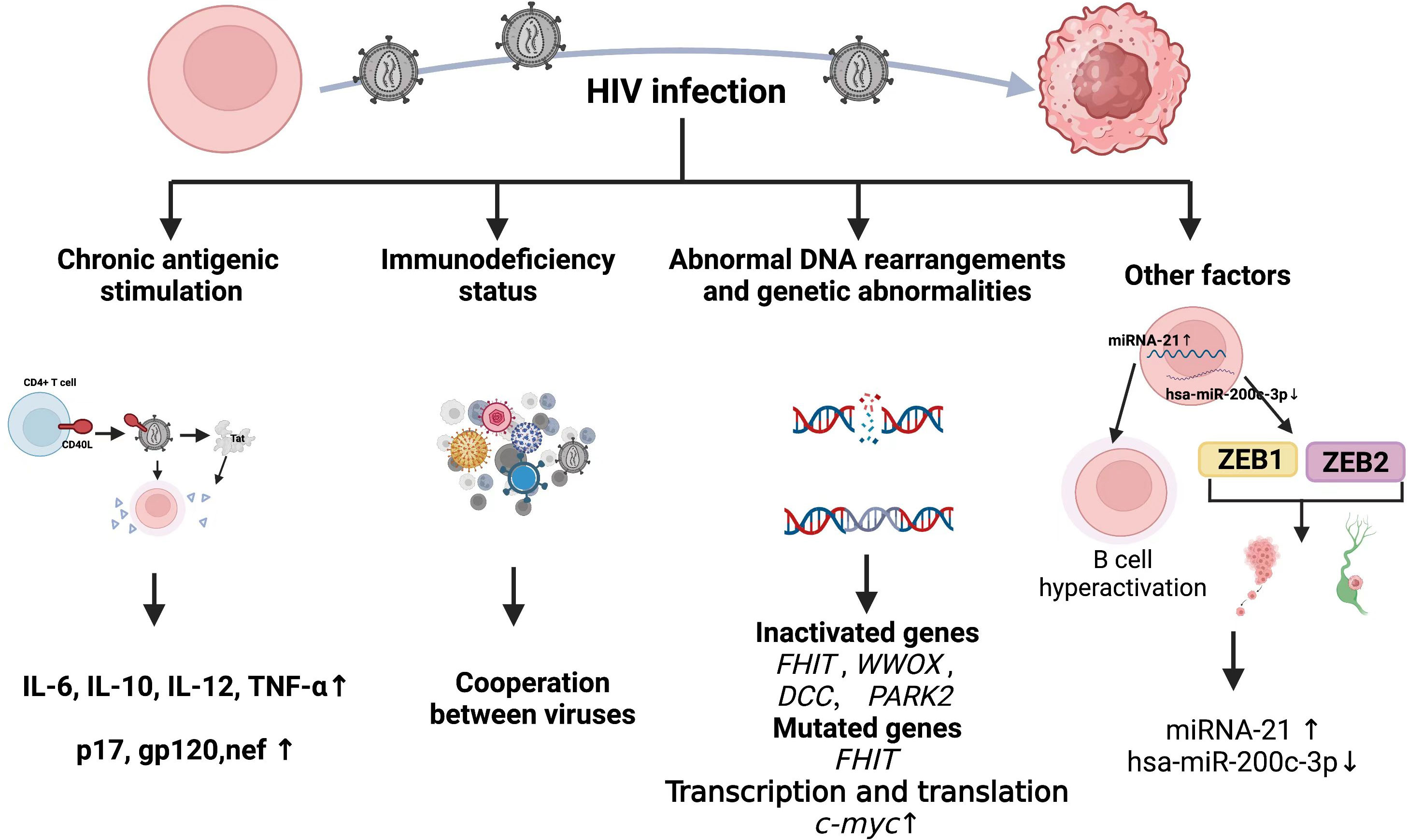
Figure 4 Mechanism of HIV causing lymphoma development.
5.2.1 Chronic antigen stimulation and cytokines
Although HIV infection is characterized by a reduction in the function or number of CD4+ T cells (213), the obviously increased B cell activation in HIV infection is primarily driven by the abnormal production of B cell-stimulating cytokines such as IL-6 and chronic antigenic stimulation. Elevated levels of circulating free immunoglobulin light chains in patients at increased risk of HIV-associated lymphoma might represent a marker for polyclonal B-cell activation (214). In addition, evidence indicates a skewed IGHV repertoire in specific HIV-NHL categories. Heterogeneous expression of IGHV genes in HIV-NHL might be related to specific pathways of antigenic stimulation (215).
Serum levels of IL6, IL10, C-reactive protein (CRP), soluble (s)CD23, sCD27, and sCD30 are significantly higher in patients with HIV-NHL compared with HIV+ or AIDS controls after adjusting for numbers of CD4+ T-cells (216). The CD40 ligand (CD40L) can insert itself into the surface of HIV-1 particles when budding from activated CD4+ T cells (217), and HIV containing CD40 ligand (CD40L) activates B cells, which leads to secretion of the cytokines, IL-6, IL-10, IL-12 and TNF-α (218), in a way that mimics physiological stimulation. The role of CD40L in cancer has been detailed in a review (219). The HIV-1 trans-activator of transcription (Tat) induces the expression of IL-6 and IL-10 at the cellular level. Findings were similar at the individual level by in transgenic mice (220), and numerous spleens from Tat-transgenic mice had malignant lymphomas of B-cell origin. The HIV Tat also enhances the intrinsic antibody diversification mechanism by increasing activation-induced deaminase (AID)-induced somatic mutations in the variable heavy chain (VH) region of human B cells (221), which might lead to genome-wide mutations in malignant B cells among patients with HIV.
Mice transgenic for a defective HIV-1 provirus lacking part of the gag-pol region overexpress the HIV proteins p17, gp120, and negative regulatory factor (nef), then develop B-cell lymphoma (222). This supports the pathogenic role of aberrant HIV protein and B-cell-stimulating cytokine expression during lymphoma formation. Indeed, the HIV-1 matrix protein p17 persists in the germinal center after HIV-1 drug inhibition, and its variants (vp17s) activate Akt signaling and promote the growth of transformed B cells. This protein might also upregulate LMP-1 in B lymphocytes infected with EBV, leading to lymphoma development (223). Infection HIV can directly induce lymphoma formation. The oncogenic effects of HIV-1 proteins have been reviewed in detail elsewhere (224) and are not discussed herein.
5.2.2 Immunodeficiency status
For immunity, although multiple mechanisms may contribute to the development of lymphoma in HIV-infected individuals, two mechanisms appear to be involved: (1) loss of immunoregulatory control of EBV and/or KSHV; (2) chronic B-cell activation due to immune dysfunction caused by HIV infection. The cooperation of HIV, EBV, and KSHV in the pathogenesis of lymphoma and resulting microenvironmental abnormalities have been reviewed in detail elsewhere (225, 226). Table 3 shows associations between HIV-associated lymphoma and EBV and KSHV infections. It has long been shown that B-cell activation and immature phenotypic changes in vivo are accompanied by polyclonal Ig production in HIV-infected individuals (228). Notably, recent studies suggest that HIV may contribute to lymphomagenesis by acting directly on B lymphocytes as a key microenvironmental factor. It is worth noting that recent studies have shown that HIV may lead to lymphomagenesis by acting directly on B lymphocytes as a key microenvironmental factor. Various HIV-encoded proteins, including gp120, may trigger and maintain abnormal activation of B cells, abnormal secretion of cytokines IL6 and IL10, and so on, which have been stated in other subsections of HIV-associated lymphomas in this paper. Perhaps it is time to revisit the second immune-related mechanism.
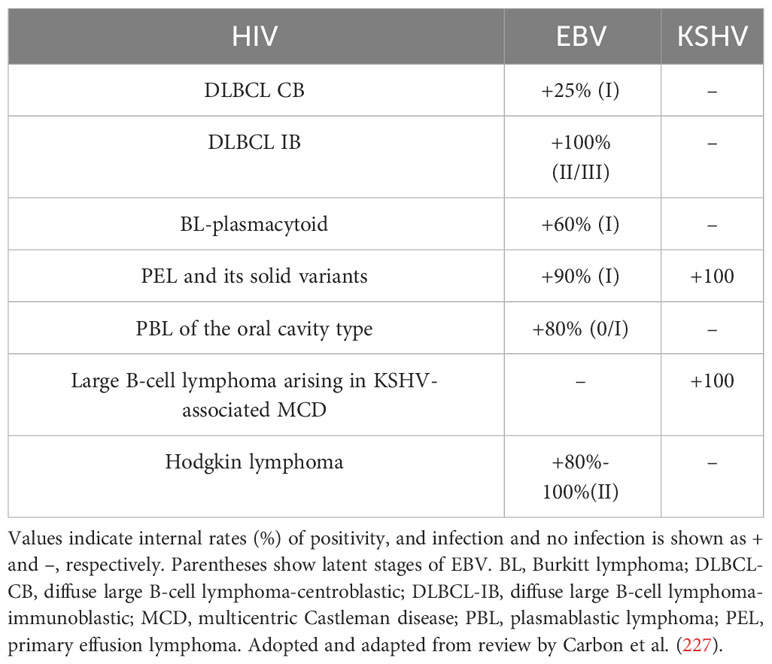
Table 3 Lymphomas in patients infected with HIV include pathological subtypes with different virus-specific associations.
5.2.3 Abnormal DNA rearrangements and genetic abnormalities
Retroviruses damage DNA via various mechanisms such as genome integration, replication, inflammation, and direct interaction of viral proteins with DNA and HIV might be randomly integrated into the human genome. However, a pattern of integrated duplicated Alu elements and introns of Breast Cancer Gene 1 (BRCA1) has been identified (229) that supports the tendency of HIV-1 to integrate near the Alu class of human repetitive elements (230).
A genome-wide analysis of 57 HIV lymphomas found that genes associated with fragile sites such as Fragile Histidine Triad (FHIT; FRA3B), WW domain-containing oxidoreductase (WWOX; FRA16D), Deleted in Colon Cancer (DCC; FRA18B), and Parkinson Protein 2 (PARK2; FRA6E), are frequently inactivated by mesenchymal deletions in HIV-NHL, and that the prevalence of FHIT alterations is significantly higher in HIV-DLBCL (231). Among these, FHIT, WWOX and DCC are tumor suppressor genes that are frequently inactivated in various human malignancies (232–234). Thus, HIV might act directly at the genomic level to promote the pathogenesis of HIV-NHL, and this translational effect is partially independent of the expression of viral oncogenes. Human immunodeficiency virus induces c-myc dysregulation in B cells, and levels of viral RNA and myc expression correlate (235). Expression of the highly oncogenic transcription factor c-myc is enhanced at the transcriptional and translational levels in the presence of HIV-1 Tat protein (236).
5.2.4 Other factors
Viruses and their components manipulate the expression of host miRNAs and play important roles in cancer pathogenesis. Hsa-miR-200c-3p is significantly downregulated in HIV-associated BL, and the zinc finger E-box binding homeobox epithelial-mesenchymal transition (EMT) transcription factors ZEB1 and ZEB2 are upregulated and actively help to promote tumor metastasis and invasion (237). Moreover, miRNA-21 is significantly elevated in peripheral B cells of patients infected with HIV, suggesting that it might contribute to the maintenance of B cell hyperactivation (238). A proteomic analysis of plasma proteins from AIDS-NHL recently identified 20 host proteins and a set of protein combinations that might serve as biomarkers for the pathogenesis of AIDS-NHL (239). This indicates a new direction towards a better understanding of the pathogenesis of HIV lymphoma.
6 Kaposi sarcoma-associated herpes virus
This virus (human herpesvirus-8, HHV-8) is the causative agent of Kaposi sarcoma (KS) and is associated with the lymphoproliferative primary exudative lymphoma (PEL) and the plasmablastic form of MCD (240, 241). The other types of lymphoma associated with KSHV are KSHV-positive large B-cell lymphoma not otherwise specified (NOS) and GLPD. The geographic distribution of KSHV is variable, with the prevalence of infections being highest in sub-Saharan Africa (seropositivity > 50%), intermediate in Mediterranean, Middle Eastern, and Caribbean countries (seropositivity 20%-30%), and lowest in Asia, Europe, and the USA (seropositivity < 9%) (242). At present, the transmission route of KSHV is not completely clear, but it is believed that the infection mainly occurs through salivary transmission (243). Several studies have shown that KSHV can infect almost any type of cells, including epithelial cells, monocytes, macrophages, dendritic cells, T cells and fibroblasts. (243) Lymphoproliferative primary exudative lymphoma is a rare HIV-associated non-Hodgkin lymphoma (NHL) that accounts for ~ 4% of all HIV-associated NHL. This type of lymphoma tends to locate in the pleural space, pericardium, and peritoneum. It is morphologically variable with an empty lymphocyte immunophenotype and evidence of KSHV infection (244). It is aggressive, rapidly progressive, and is associated with high mortality rate; the average survival of patients with PEL is 2- 6 months (245).
6.1 Kaposi sarcoma-associated herpes virus structure
The KSHV genome consists of linear double-stranded DNA that is cyclized during latent infection. It contains a unique coding sequence of ~ 140 kb flanked by 25-30 kb repetitive terminal repeats (246). The life cycle of KSHV is biphasic, with consecutive latent and lytic replication phases (247, 248), each of which has a unique gene expression profile like EBV (249). The viral oncoproteins, KSHV latency-associated nuclear antigen (LANA; ORF73), vCyclin, and latent viral FADD-like interleukin-1-converting enzyme (FLICE) inhibitory protein (vFLIP) are encoded by KSHV during the latent phase, whereas KSHV G protein-coupled receptor (vGPCR), viral B cell lymphoma 2 (vBcl-2), vIL-6, viral IFN regulatory factor 1(vIRF) 1/vIRF 3, K1, K15, and viral protein kinase (vPK) (250) are encoded during the lytic phase.
6.2 Carcinogenic mechanisms
KSHV has evolved to produce a large number of viral gene products that intricately subvert normal cellular pathways. The proteins encoded by KSHV that are thought to have transformative and oncogenic properties include latent proteins, which increase the survival and proliferation of infected cells, and lytic proteins, which are thought to mediate tumor growth. Due to space constraints, this section only summarizes the main mechanisms.
6.2.1 viral proteins
6.2.1.1 LANA
The mechanisms underlying KSHV carcinogenesis remain unclear. Analysis of infected cells by immunofluorescence and immunohistochemistry confirmed that LANA is one of the latent proteins consistently present in all KSHV-infected tumor cells of Kaposi’s sarcoma, PEL and MCD. (251) As a multifunctional protein, LANA is involved in the regulation of transcription, chromatin remodeling, exome maintenance, DNA replication, and the control of latency and lytic phase reactivation. In addition, LANA is also involved in cell cycle regulation, which has been described in the review by Wei et al. (251) LANA binds to and inactivates the tumor suppressor proteins TP53 and retinoblastoma (RB1), thereby regulating cell growth. (252) LANA expression also affects MYC levels by binding to the negative regulator GSK-3β and thus promotes lymphomagenesis. (253) Based on current knowledge, LANA appears to provide the basis for at least the formation of KSHV-associated lymphomas.
6.2.1.2 Viral cyclin
Viral cyclin (ORF72) is a viral homolog of cell cycle protein D (254) which plays an important role in lymphangiogenesis via several functions. Physiologically, cyclin D forms a complex with cyclin-dependent kinase (CDK) and CDK4 that phosphorylates retinoblastoma protein (Rb) and leads to the release of E2F transcription factors (255). The KSHV vCyclin interacts with CDK6 to promote cell cycle progression (256, 257). Moreover, the vCyclin/CDK6 complex can phosphorylate nuclear phospholipid histone chaperones, leading to genomic instability (258).
6.2.1.3 vFLIP
vFLIP is the viral homologue of cellular FLIP. Transgenic mice expressing vFLIP exhibit B cell transdifferentiation and acquire the ability to express histiocyte/dendritic cell markers (259). These mice have hematological properties typical of PEL and MCD. Previously, it has been found that vFLIP prevents apoptosis by up-regulating NF-κB. (260) In addition, the study of Lee et al. demonstrated that vFLIP can protect cells by preventing autophagy to further maintain latency. (261).
6.2.1.4 vIL-6
vIL-6 is the viral homologue of hulL-6, and immunohistochemistry has shown that it is expressed in variable proportions in KSHV+ lymphoproliferative lesions. (251) One characteristic of KSHV-driven PEL is elevated serum human IL-6 (hIL-6) levels. Notably, v-IL6 can replace hIL-6, activating it constitutively via the rat sarcoma/mitogen activated protein kinase (Ras/MAPK) and JAK/STAT pathways (262).
6.2.2 miRNAs
KSHV miRNAs are generated from 12 pre-miRNA transcripts in the latency region, ultimately producing at least 17 mature miRNAs. (263) The biogenesis of KSHV miRNAs and their role in the development of KSHV-associated malignant tumors has recently been described in detail. (242, 264) Among the large number of miRNAs encoded by KSHV, KSHV-miRNA-K11 compares particularly because it shows significant homology to cellular miRNA-155. (265) MiRNA-155/bic overexpression can be observed in many human B-cell lymphomas, (266) and B-cell lymphomas can be induced in mice. (267).
7 Conclusions
The main aspect of virus-driven lymphangiogenesis initially focuses on the direct transforming activity of a single viral oncogenic product. However, cooperation among different viruses also plays crucial roles in the development, survival, and dissemination of lymphoid malignancies. Therefore, many studies have targeted relationships among the microenvironment, oncogenesis, tumor growth, and dissemination. How EBV and KSHV support each other in terms of persistence and lymphangiogenesis has been explained in recent reviews (268), (269). A relationship between EBV and HCV replication markers has not been identified in patients with AIDS (270), which is in contrast to other known coinfections. Indeed, HCV and HBV co-infection inhibits HCV replication, whereas HCV and HIV co-infection stimulates HCV replication and exacerbates HIV-associated immunosuppression, and EBV and HIV co-infection stimulates HIV replication in CD4T cells (271, 272). All of these complicate understanding the mechanisms through which co-infection causes carcinogenesis. To further elucidate and characterize the mechanisms of viral induction of lymphoma is a considerable challenge that will require an integrated multidisciplinary approach involving epidemiologists, molecular biologists, and immunopathologists.
Author contributions
YZ: Writing – original draft, Writing – review and editing, Validation. WG: Investigation, Writing – review and editing. ZZ: Investigation, Writing – review and editing. OB: Supervision, Writing – review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by Department of Science and Technology of Jilin Province (20220402064GH) (20200201591JC).
Acknowledgments
We would like to thank Editage (www.editage.cn) for English language editing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Talbot SJ, Crawford DH. Viruses and tumours - an update. Eur J Cancer. (2004) 40:1998–2005. doi: 10.1016/j.ejca.2003.11.039
2. Bouvard V, Baan R, Straif K, Grosse Y, Secretan B, El Ghissassi F, et al. A review of human carcinogens-Part B: biological agents. Lancet Oncol. (2009) 10:321–2. doi: 10.1016/S1470-2045(09)70096-8
3. Marcucci F, Mele A. Hepatitis viruses and non-Hodgkin lymphoma: epidemiology, mechanisms of tumorigenesis, and therapeutic opportunities. Blood. (2011) 117:1792–8. doi: 10.1182/blood-2010-06-275818
4. De Paoli P, Carbone A. Carcinogenic viruses and solid cancers without sufficient evidence of causal association. Int J Cancer. (2013) 133:1517–29. doi: 10.1002/ijc.27995
5. Munz C. Latency and lytic replication in Epstein-Barr virus-associated oncogenesis. Nat Rev Microbiol. (2019) 17:691–700. doi: 10.1038/s41579-019-0249-7
6. Wong Y, Meehan MT, Burrows SR, Doolan DL, Miles JJ. Estimating the global burden of Epstein-Barr virus-related cancers. J Cancer Res Clin Oncol. (2022) 148:31–46. doi: 10.1007/s00432-021-03824-y
7. Bu GL, Xie C, Kang YF, Zeng MS, Sun C. How EBV infects: the tropism and underlying molecular mechanism for viral infection. Viruses. (2022) 14(11):2372. doi: 10.3390/v14112372
8. Epstein MA, Achong BG, Barr YM. Virus particles in cultured lymphoblasts from burkitt’s lymphoma. Lancet. (1964) 1:702–3. doi: 10.1016/S0140-6736(64)91524-7
9. Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. (2016) 127:2375–90. doi: 10.1182/blood-2016-01-643569
10. Oyama T, Ichimura K, Suzuki R, Suzumiya J, Ohshima K, Yatabe Y, et al. Senile EBV plus B-cell Lymphoproliferative disorders - A clinicopathologic study of 22 patients. Am J Surg Pathol. (2003) 27:16–26. doi: 10.1097/00000478-200301000-00003
11. He L, Chen N, Dai L, Peng X. Advances and challenges of immunotherapies in NK/T cell lymphomas. iScience. (2023) 26:108192. doi: 10.1016/j.isci.2023.108192
12. Yin H, Qu J, Peng Q, Gan R. Molecular mechanisms of EBV-driven cell cycle progression and oncogenesis. Med Microbiol Immunol. (2019) 208:573–83. doi: 10.1007/s00430-018-0570-1
13. Taylor GS, Long HM, Brooks JM, Rickinson AB, Hislop AD. The immunology of Epstein-Barr virus-induced disease. Annu Rev Immunol. (2015) 33:787–821. doi: 10.1146/annurev-immunol-032414-112326
14. Yu H, Robertson ES. Epstein-barr virus history and pathogenesis. Viruses. (2023) 15(3):714. doi: 10.3390/v15030714
15. Frappier L. The epstein-barr virus EBNA1 protein. Scientifica (Cairo). (2012) 2012:438204. doi: 10.6064/2012/438204
16. Saridakis V, Sheng Y, Sarkari F, Holowaty MN, Shire K, Nguyen T, et al. Structure of the p53 binding domain of HAUSP/USP7 bound to Epstein-Barr nuclear antigen 1 implications for EBV-mediated immortalization. Mol Cell. (2005) 18:25–36. doi: 10.1016/j.molcel.2005.02.029
17. Sheng Y, Saridakis V, Sarkari F, Duan S, Wu T, Arrowsmith CH, et al. Molecular recognition of p53 and MDM2 by USP7/HAUSP. Nat Struct Mol Biol. (2006) 13:285–91. doi: 10.1038/nsmb1067
18. Chatterjee K, Das P, Chattopadhyay NR, Mal S, Choudhuri T. The interplay between Epstein-Bar virus (EBV) with the p53 and its homologs during EBV associated Malignancies. Heliyon. (2019) 5:e02624. doi: 10.1016/j.heliyon.2019.e02624
19. Sivachandran N, Sarkari F, Frappier L. Epstein-Barr nuclear antigen 1 contributes to nasopharyngeal carcinoma through disruption of PML nuclear bodies. PloS Pathog. (2008) 4:e1000170. doi: 10.1371/journal.ppat.1000170
20. Wood VH, O’neil JD, Wei W, Stewart SE, Dawson CW, Young LS. Epstein-Barr virus-encoded EBNA1 regulates cellular gene transcription and modulates the STAT1 and TGFbeta signaling pathways. Oncogene. (2007) 26:4135–47. doi: 10.1038/sj.onc.1210496
21. Flavell JR, Baumforth KR, Wood VH, Davies GL, Wei W, Reynolds GM, et al. Down-regulation of the TGF-beta target gene, PTPRK, by the Epstein-Barr virus encoded EBNA1 contributes to the growth and survival of Hodgkin lymphoma cells. Blood. (2008) 111:292–301. doi: 10.1182/blood-2006-11-059881
22. Valentine R, Dawson CW, Hu C, Shah KM, Owen TJ, Date KL, et al. Epstein-Barr virus-encoded EBNA1 inhibits the canonical NF-kappaB pathway in carcinoma cells by inhibiting IKK phosphorylation. Mol Cancer. (2010) 9:1. doi: 10.1186/1476-4598-9-1
23. Gruhne B, Sompallae R, Marescotti D, Kamranvar SA, Gastaldello S, Masucci MG. The Epstein-Barr virus nuclear antigen-1 promotes genomic instability via induction of reactive oxygen species. Proc Natl Acad Sci U.S.A. (2009) 106:2313–8. doi: 10.1073/pnas.0810619106
24. Kamranvar SA, Masucci MG. The Epstein-Barr virus nuclear antigen-1 promotes telomere dysfunction via induction of oxidative stress. Leukemia. (2011) 25:1017–25. doi: 10.1038/leu.2011.35
25. Murakami M, Lan K, Subramanian C, Robertson ES. Epstein-Barr virus nuclear antigen 1 interacts with Nm23-H1 in lymphoblastoid cell lines and inhibits its ability to suppress cell migration. J Virol. (2005) 79:1559–68. doi: 10.1128/JVI.79.3.1559-1568.2005
26. Kaul R, Murakami M, Choudhuri T, Robertson ES. Epstein-Barr virus latent nuclear antigens can induce metastasis in a nude mouse model. J Virol. (2007) 81:10352–61. doi: 10.1128/JVI.00886-07
27. Leen A, Meij P, Redchenko I, Middeldorp J, Bloemena E, Rickinson A, et al. Differential immunogenicity of Epstein-Barr virus latent-cycle proteins for human CD4(+) T-helper 1 responses. J Virol. (2001) 75:8649–59. doi: 10.1128/JVI.75.18.8649-8659.2001
28. Dinh VT, Loaec N, Quillevere A, Le Senechal R, Keruzore M, Martins RP, et al. The hide-and-seek game of the oncogenic Epstein-Barr virus-encoded EBNA1 protein with the immune system: An RNA G-quadruplex tale. Biochimie. (2023) 214:57–68. doi: 10.1016/j.biochi.2023.07.010
29. Dresang LR, Vereide DT, Sugden B. Identifying sites bound by Epstein-Barr virus nuclear antigen 1 (EBNA1) in the human genome: defining a position-weighted matrix to predict sites bound by EBNA1 in viral genomes. J Virol. (2009) 83:2930–40. doi: 10.1128/JVI.01974-08
30. Lu F, Wikramasinghe P, Norseen J, Tsai K, Wang P, Showe L, et al. Genome-wide analysis of host-chromosome binding sites for Epstein-Barr Virus Nuclear Antigen 1 (EBNA1). Virol J. (2010) 7:262. doi: 10.1186/1743-422X-7-262
31. Kim KD, Tanizawa H, De Leo A, Vladimirova O, Kossenkov A, Lu F, et al. Epigenetic specifications of host chromosome docking sites for latent Epstein-Barr virus. Nat Commun. (2020) 11:877. doi: 10.1038/s41467-019-14152-8
32. Gahn TA, Sugden B. An EBNA-1-dependent enhancer acts from a distance of 10 kilobase pairs to increase expression of the Epstein-Barr virus LMP gene. J Virol. (1995) 69:2633–6. doi: 10.1128/jvi.69.4.2633-2636.1995
33. Altmann M, Pich D, Ruiss R, Wang J, Sugden B, Hammerschmidt W. Transcriptional activation by EBV nuclear antigen 1 is essential for the expression of EBV’s transforming genes. Proc Natl Acad Sci U.S.A. (2006) 103:14188–93. doi: 10.1073/pnas.0605985103
34. Lupton S, Levine AJ. Mapping genetic elements of Epstein-Barr virus that facilitate extrachromosomal persistence of Epstein-Barr virus-derived plasmids in human cells. Mol Cell Biol. (1985) 5:2533–42. doi: 10.1128/MCB.5.10.2533
35. Westhoff Smith D, Chakravorty A, Hayes M, Hammerschmidt W, Sugden B. The epstein-barr virus oncogene EBNA1 suppresses natural killer cell responses and apoptosis early after infection of peripheral B cells. mBio. (2021) 12:e0224321. doi: 10.1128/mBio.02243-21
36. Li S, Yang L, Li YL, Yue WX, Xin SY, Li J, et al. Epstein-Barr Virus Synergizes with BRD7 to Conquer c-Myc-Mediated Viral Latency Maintenance via Chromatin Remodeling. Microbiol Spectrum. (2023) 11(2):e01237-22. doi: 10.1128/spectrum.01237-22
37. Xu JX, Zhang R, Huang DJ, Tang Y, Ping LQ, Huang BJ, et al. Galectin-9 facilitates epstein-barr virus latent infection and lymphomagenesis in human B cells. Microbiol Spectr. (2023) 11:e0493222. doi: 10.1128/spectrum.04932-22
38. Hirashima M, Kashio Y, Nishi N, Yamauchi A, Imaizumi TA, Kageshita T, et al. Galectin-9 in physiological and pathological conditions. Glycoconj J. (2002) 19:593–600. doi: 10.1023/B:GLYC.0000014090.63206.2f
39. Pich D, Mrozek-Gorska P, Bouvet M, Sugimoto A, Akidil E, Grundhoff A, et al. First days in the life of naive human B lymphocytes infected with epstein-barr virus. mBio. (2019) 10(5):10.1128. doi: 10.1128/mBio.01723-19
40. Wang F, Kikutani H, Tsang SF, Kishimoto T, Kieff E. Epstein-Barr virus nuclear protein 2 transactivates a cis-acting CD23 DNA element. J Virol. (1991) 65:4101–6. doi: 10.1128/jvi.65.8.4101-4106.1991
41. Kaiser C, Laux G, Eick D, Jochner N, Bornkamm GW, Kempkes B. The proto-oncogene c-myc is a direct target gene of Epstein-Barr virus nuclear antigen 2. J Virol. (1999) 73:4481–4. doi: 10.1128/JVI.73.5.4481-4484.1999
42. Boccellato F, Anastasiadou E, Rosato P, Kempkes B, Frati L, Faggioni A, et al. EBNA2 interferes with the germinal center phenotype by downregulating BCL6 and TCL1 in non-Hodgkin’s lymphoma cells. J Virol. (2007) 81:2274–82. doi: 10.1128/JVI.01822-06
43. Anastasiadou E, Vaeth S, Cuomo L, Boccellato F, Vincenti S, Cirone M, et al. Epstein-Barr virus infection leads to partial phenotypic reversion of terminally differentiated Malignant B cells. Cancer Lett. (2009) 284:165–74. doi: 10.1016/j.canlet.2009.04.025
44. Hayward SD. Viral interactions with the Notch pathway. Semin Cancer Biol. (2004) 14:387–96. doi: 10.1016/j.semcancer.2004.04.018
45. Zhang J, Sommermann T, Li X, Gieselmann L, de la Rosa K, Stecklum M, et al. LMP1 and EBNA2 constitute a minimal set of EBV genes for transformation of human B cells. Front Immunol. (2023) 14:1331730. doi: 10.3389/fimmu.2023.1331730
46. Anastasiadou E, Stroopinsky D, Alimperti S, Jiao AL, Pyzer AR, Cippitelli C, et al. Epstein-Barr virus-encoded EBNA2 alters immune checkpoint PD-L1 expression by downregulating miR-34a in B-cell lymphomas. Leukemia. (2019) 33:132–47. doi: 10.1038/s41375-018-0178-x
47. Leopizzi M, Mundo L, Messina E, Campolo F, Lazzi S, Angeloni A, et al. Epstein-Barr virus encoded EBNA2 downregulates ICOSL by inducing miR-24 in B-cell lymphoma. Blood. (2023). doi: 10.1182/blood.2023021346
48. Kempkes B, Ling PD. EBNA2 and its coactivator EBNA-LP. Curr Top Microbiol Immunol. (2015) 391:35–59. doi: 10.1007/978-3-319-22834-1_2
49. Manet E, Polveche H, Mure F, Mrozek-Gorska P, Roisne-Hamelin F, Hammerschmidt W, et al. Modulation of alternative splicing during early infection of human primary B lymphocytes with Epstein-Barr virus (EBV): a novel function for the viral EBNA-LP protein. Nucleic Acids Res. (2021) 49:10657–76. doi: 10.1093/nar/gkab787
50. Szymula A, Palermo RD, Bayoumy A, Groves IJ, Ba Abdullah M, Holder B, et al. Epstein-Barr virus nuclear antigen EBNA-LP is essential for transforming naive B cells, and facilitates recruitment of transcription factors to the viral genome. PloS Pathog. (2018) 14:e1006890. doi: 10.1371/journal.ppat.1006890
51. Bhattacharjee S, Ghosh Roy S, Bose P, Saha A. Role of EBNA-3 family proteins in EBV associated B-cell lymphomagenesis. Front Microbiol. (2016) 7:457. doi: 10.3389/fmicb.2016.00457
52. Touitou R, O’nions J, Heaney J, Allday MJ. Epstein-Barr virus EBNA3 proteins bind to the C8/alpha7 subunit of the 20S proteasome and are degraded by 20S proteasomes in vitro, but are very stable in latently infected B cells. J Gen Virol. (2005) 86:1269–77. doi: 10.1099/vir.0.80763-0
53. Allday MJ, Bazot Q, White RE. The EBNA3 family: two oncoproteins and a tumour suppressor that are central to the biology of EBV in B cells. Curr Top Microbiol Immunol. (2015) 391:61–117. doi: 10.1007/978-3-319-22834-1_3
54. Tursiella ML, Bowman ER, Wanzeck KC, Throm RE, Liao J, Zhu J, et al. Epstein-Barr virus nuclear antigen 3A promotes cellular proliferation by repression of the cyclin-dependent kinase inhibitor p21WAF1/CIP1. PloS Pathog. (2014) 10:e1004415. doi: 10.1371/journal.ppat.1004415
55. Pei Y, Singh RK, Shukla SK, Lang F, Zhang S, Robertson ES. Epstein-barr virus nuclear antigen 3C facilitates cell proliferation by regulating cyclin D2. J Virol. (2018) 92(18):10.1128. doi: 10.1128/JVI.00663-18
56. Styles CT, Paschos K, White RE, Farrell PJ. The cooperative functions of the EBNA3 proteins are central to EBV persistence and latency. Pathogens. (2018) 7(1):31. doi: 10.3390/pathogens7010031
57. Young LS, Rickinson AB. Epstein-Barr virus: 40 years on. Nat Rev Cancer. (2004) 4:757–68. doi: 10.1038/nrc1452
58. Chen YP, Zhang WN, Chen L, Tang LL, Mao YP, Li WF, et al. Effect of latent membrane protein 1 expression on overall survival in Epstein-Barr virus-associated cancers: a literature-based meta-analysis. Oncotarget. (2015) 6:29311–23. doi: 10.18632/oncotarget.v6i30
59. Kaye KM, Izumi KM, Kieff E. Epstein-Barr virus latent membrane protein 1 is essential for B-lymphocyte growth transformation. Proc Natl Acad Sci U.S.A. (1993) 90:9150–4. doi: 10.1073/pnas.90.19.9150
60. Dirmeier U, Neuhierl B, Kilger E, Reisbach G, Sandberg ML, Hammerschmidt W. Latent membrane protein 1 is critical for efficient growth transformation of human B cells by epstein-barr virus. Cancer Res. (2003) 63:2982–9.
61. El-Sharkawy A, Al Zaidan L, Malki A. Epstein-barr virus-associated Malignancies: roles of viral oncoproteins in carcinogenesis. Front Oncol. (2018) 8:265. doi: 10.3389/fonc.2018.00265
62. Awasthi P, Dwivedi M, Kumar D, Hasan S. Insights into intricacies of the Latent Membrane Protein-1 (LMP-1) in EBV-associated cancers. Life Sci. (2023) 313:121261. doi: 10.1016/j.lfs.2022.121261
63. Luo Y, Liu YT, Wang CK, Gan RL. Signaling pathways of EBV-induced oncogenesis. Cancer Cell Int. (2021) 21:1–11. doi: 10.1186/s12935-021-01793-3
64. Kume A, Shinozaki-Ushiku A, Kunita A, Kondo A, Ushiku T. Enhanced PD-L1 expression in LMP1-positive cells of epstein-barr virus-associated Malignant lymphomas and lymphoproliferative disorders: A single-cell resolution analysis with multiplex fluorescence immunohistochemistry and in situ hybridization. Am J Surg Pathol. (2022) 46:1386–96. doi: 10.1097/PAS.0000000000001919
65. Giehler F, Ostertag MS, Sommermann T, Weidl D, Sterz KR, Kutz H, et al. Epstein-Barr virus-driven B cell lymphoma mediated by a direct LMP1-TRAF6 complex. Nat Commun. (2024) 15:414. doi: 10.1038/s41467-023-44455-w
66. Fish K, Chen J, Longnecker R. Epstein-Barr virus latent membrane protein 2A enhances MYC-driven cell cycle progression in a mouse model of B lymphoma. Blood. (2014) 123:530–40. doi: 10.1182/blood-2013-07-517649
67. Fish K, Sora RP, Schaller SJ, Longnecker R, Ikeda M. EBV latent membrane protein 2A orchestrates p27(kip1) degradation via Cks1 to accelerate MYC-driven lymphoma in mice. Blood. (2017) 130:2516–26. doi: 10.1182/blood-2017-07-796821
68. Fish K, Comoglio F, Shaffer AL 3rd, Ji Y, Pan KT, Scheich S, et al. Rewiring of B cell receptor signaling by Epstein-Barr virus LMP2A. Proc Natl Acad Sci U.S.A. (2020) 117:26318–27. doi: 10.1073/pnas.2007946117
69. Rechsteiner MP, Berger C, Zauner L, Sigrist JA, Weber M, Longnecker R, et al. Latent membrane protein 2B regulates susceptibility to induction of lytic Epstein-Barr virus infection. J Virol. (2008) 82:1739–47. doi: 10.1128/JVI.01723-07
70. Allen MD, Young LS, Dawson CW. The Epstein-Barr virus-encoded LMP2A and LMP2B proteins promote epithelial cell spreading and motility. J Virol. (2005) 79:1789–802. doi: 10.1128/JVI.79.3.1789-1802.2005
71. Takakuwa T, Luo WJ, Ham MF, Sakane-Ishikawa F, Wada N, Aozasa K. Integration of Epstein-Barr virus into chromosome 6q15 of Burkitt lymphoma cell line (Raji) induces loss of BACH2 expression. Am J Pathol. (2004) 164:967–74. doi: 10.1016/S0002-9440(10)63184-7
72. Anvret M, Karlsson A, Bjursell G. Evidence for integrated EBV genomes in Raji cellular DNA. Nucleic Acids Res. (1984) 12:1149–61. doi: 10.1093/nar/12.2.1149
73. Wolf J, Pawlita M, Jox A, Kohls S, Bartnitzke S, Diehl V, et al. Integration of Epstein Barr virus near the breakpoint of a translocation 11;19 in a Burkitt’s lymphoma cell line. Cancer Genet Cytogenet. (1993) 67:90–4. doi: 10.1016/0165-4608(93)90158-I
74. Gulley ML, Raphael M, Lutz CT, Ross DW, Raab-Traub N. Epstein-Barr virus integration in human lymphomas and lymphoid cell lines. Cancer. (1992) 70:185–91. doi: 10.1002/(ISSN)1097-0142
75. Daibata M, Taguchi T, Nemoto Y, Saito T, Machida H, Imai S, et al. Epstein-Barr virus (EBV)-positive pyothorax-associated lymphoma (PAL): chromosomal integration of EBV in a novel CD2-positive PAL B-cell line. Br J Haematol. (2002) 117:546–57. doi: 10.1046/j.1365-2141.2002.03466.x
76. Yoon H, Park S, Ju H, Ha SY, Sohn I, Jo J, et al. Integrated copy number and gene expression profiling analysis of Epstein-Barr virus-positive diffuse large B-cell lymphoma. Genes Chromosomes Cancer. (2015) 54:383–96. doi: 10.1002/gcc.22249
77. Liu F, Wang Z, Zhou X, Liu Q, Chen G, Xiao H, et al. Genetic heterogeneity and mutational signature in Chinese Epstein-Barr virus-positive diffuse large B-cell lymphoma. PloS One. (2018) 13:e0201546. doi: 10.1371/journal.pone.0201546
78. Zhou Y, Xu Z, Lin W, Duan Y, Lu C, Liu W, et al. Comprehensive genomic profiling of EBV-positive diffuse large B-cell lymphoma and the expression and clinicopathological correlations of some related genes. Front Oncol. (2019) 9:683. doi: 10.3389/fonc.2019.00683
79. Gebauer N, Kunstner A, Ketzer J, Witte HM, Rausch T, Benes V, et al. Genomic insights into the pathogenesis of Epstein-Barr virus-associated diffuse large B-cell lymphoma by whole-genome and targeted amplicon sequencing. Blood Cancer J. (2021) 11:102. doi: 10.1038/s41408-021-00493-5
80. Pfeffer S, Zavolan M, Grasser FA, Chien M, Russo JJ, Ju J, et al. Identification of virus-encoded microRNAs. Science. (2004) 304:734–6. doi: 10.1126/science.1096781
81. Klinke O, Feederle R, Delecluse HJ. Genetics of Epstein-Barr virus microRNAs. Semin Cancer Biol. (2014) 26:52–9. doi: 10.1016/j.semcancer.2014.02.002
82. Navari M, Etebari M, Ibrahimi M, Leoncini L, Piccaluga PP. Pathobiologic roles of epstein-barr virus-encoded microRNAs in human lymphomas. Int J Mol Sci. (2018) 19(4):1168. doi: 10.3390/ijms19041168
83. Caetano BFR, Jorge BAS, Muller-Coan BG, Elgui De Oliveira D. Epstein-Barr virus microRNAs in the pathogenesis of human cancers. Cancer Lett. (2021) 499:14–23. doi: 10.1016/j.canlet.2020.11.019
84. Zidovec Lepej S, Matulic M, Grskovic P, Pavlica M, Radmanic L, Korac P. miRNAs: EBV mechanism for escaping host’s immune response and supporting tumorigenesis. Pathogens. (2020) 9(5):353. doi: 10.3390/pathogens9050353
85. Li W, He C, Wu J, Yang D, Yi W. Epstein barr virus encodes miRNAs to assist host immune escape. J Cancer. (2020) 11:2091–100. doi: 10.7150/jca.42498
86. Motsch N, Alles J, Imig J, Zhu J, Barth S, Reineke T, et al. MicroRNA profiling of Epstein-Barr virus-associated NK/T-cell lymphomas by deep sequencing. PloS One. (2012) 7:e42193. doi: 10.1371/journal.pone.0042193
87. Soltani S, Zakeri A, Tabibzadeh A, Zakeri AM, Zandi M, Siavoshi S, et al. A review on EBV encoded and EBV-induced host microRNAs expression profile in different lymphoma types. Mol Biol Rep. (2021) 48:1801–17. doi: 10.1007/s11033-021-06152-z
88. Iizasa H, Wulff BE, Alla NR, Maragkakis M, Megraw M, Hatzigeorgiou A, et al. Editing of Epstein-Barr virus-encoded BART6 microRNAs controls their dicer targeting and consequently affects viral latency. J Biol Chem. (2010) 285:33358–70. doi: 10.1074/jbc.M110.138362
89. Mansouri S, Pan Q, Blencowe BJ, Claycomb JM, Frappier L. Epstein-Barr virus EBNA1 protein regulates viral latency through effects on let-7 microRNA and dicer. J Virol. (2014) 88:11166–77. doi: 10.1128/JVI.01785-14
90. Ma J, Nie K, Redmond D, Liu Y, Elemento O, Knowles DM, et al. EBV-miR-BHRF1-2 targets PRDM1/Blimp1: potential role in EBV lymphomagenesis. Leukemia. (2016) 30:594–604. doi: 10.1038/leu.2015.285
91. Ramakrishnan R, Donahue H, Garcia D, Tan J, Shimizu N, Rice AP, et al. Epstein-Barr virus BART9 miRNA modulates LMP1 levels and affects growth rate of nasal NK T cell lymphomas. PloS One. (2011) 6:e27271. doi: 10.1371/journal.pone.0027271
92. Zhang YM, Yu Y, Zhao HP. EBV−BART−6−3p and cellular microRNA−197 compromise the immune defense of host cells in EBV−positive Burkitt lymphoma. Mol Med Rep. (2017) 15:1877–83. doi: 10.3892/mmr.2017.6173
93. Peters M, Jacobs S, Ehlers M, Vollmer P, Mullberg J, Wolf E, et al. The function of the soluble interleukin 6 (IL-6) receptor in vivo: sensitization of human soluble IL-6 receptor transgenic mice towards IL-6 and prolongation of the plasma half-life of IL-6. J Exp Med. (1996) 183:1399–406. doi: 10.1084/jem.183.4.1399
94. Lin TC, Liu TY, Hsu SM, Lin CW. Epstein-Barr virus-encoded miR-BART20-5p inhibits T-bet translation with secondary suppression of p53 in invasive nasal NK/T-cell lymphoma. Am J Pathol. (2013) 182:1865–75. doi: 10.1016/j.ajpath.2013.01.025
95. Wang M, Yu F, Wu W, Wang Y, Ding H, Qian L. Epstein-Barr virus-encoded microRNAs as regulators in host immune responses. Int J Biol Sci. (2018) 14:565–76. doi: 10.7150/ijbs.24562
96. Ungerleider N, Bullard W, Kara M, Wang X, Roberts C, Renne R, et al. EBV miRNAs are potent effectors of tumor cell transcriptome remodeling in promoting immune escape. PloS Pathog. (2021) 17:e1009217. doi: 10.1371/journal.ppat.1009217
97. Albanese M, Tagawa T, Hammerschmidt W. Strategies of Epstein-Barr virus to evade innate antiviral immunity of its human host. Front Microbiol. (2022) 13:955603. doi: 10.3389/fmicb.2022.955603
98. Cristino AS, Nourse J, West RA, Sabdia MB, Law SC, Gunawardana J, et al. EBV microRNA-BHRF1-2-5p targets the 3’UTR of immune checkpoint ligands PD-L1 and PD-L2. Blood. (2019) 134:2261–70. doi: 10.1182/blood.2019000889
99. Murer A, Ruhl J, Zbinden A, Capaul R, Hammerschmidt W, Chijioke O, et al. MicroRNAs of epstein-barr virus attenuate T-cell-mediated immune control in vivo. mBio. (2019) 10(1):10-1128. doi: 10.1128/mBio.01941-18
100. Lemaitre M, Brice P, Frigeni M, Hermine O, Arcaini L, Thieblemont C, et al. Hepatitis B virus-associated B-cell non-Hodgkin lymphoma in non-endemic areas in Western Europe: Clinical characteristics and prognosis. J Infect. (2020) 80:219–24. doi: 10.1016/j.jinf.2019.12.005
101. Chang MH. Hepatitis B virus infection. Semin Fetal Neonatal Med. (2007) 12:160–7. doi: 10.1016/j.siny.2007.01.013
102. Rosenberg M, Poluch M, Thomas C, Sindaco P, Khoo A, Porcu P. Hepatitis B Virus and B-cell lymphoma: evidence, unmet need, clinical impact, and opportunities. Front Oncol. (2023) 13:1275800. doi: 10.3389/fonc.2023.1275800
103. Wang YC, Wang HJ, Pan SK, Hu T, Shen JB, Zheng H, et al. Capable infection of hepatitis B virus in diffuse large B-cell lymphoma. J Cancer. (2018) 9:1575–81. doi: 10.7150/jca.24384
104. Zhou X, Pan HX, Yang P, Ye P, Cao HY, Zhou H. Both chronic HBV infection and naturally acquired HBV immunity confer increased risks of B-cell non-Hodgkin lymphoma. BMC Cancer. (2019) 19. doi: 10.1186/s12885-019-5718-x
105. Kang XD, Bai L, Han C, Qi XG. Clinical analysis and prognostic significance of hepatitis B virus infections with diffuse large B-cell lymphoma. Cancer Manage Res. (2020) 12:2839–51. doi: 10.2147/CMAR.S244381
106. Engels EA, Cho ER, Jee SH. Hepatitis B virus infection and risk of non-Hodgkin lymphoma in South Korea: a cohort study. Lancet Oncol. (2010) 11:827–34. doi: 10.1016/S1470-2045(10)70167-4
107. Deng LJ, Song YQ, Young KH, Hu SM, Ding N, Song WW, et al. Hepatitis B virus-associated diffuse large B-cell lymphoma: unique clinical features, poor outcome, and hepatitis B surface antigen-driven origin. Oncotarget. (2015) 6:25061–73. doi: 10.18632/oncotarget.v6i28
108. Wei Z, Zou SH, Li F, Cheng ZX, Li JM, Wang JM, et al. HBsAg is an independent prognostic factor in diffuse large B cell lymphoma patients in rituximab era: result from a multicenter retrospective analysis in China. Med Oncol. (2014) 31:1–9. doi: 10.1007/s12032-014-0845-3
109. Yan X, Zhou M, Lou ZZ, Mu QT, Sheng LX, Zhang P, et al. Diffuse large B-cell lymphoma with concurrent hepatitis B virus infection in the MabThera era: Unique clinical features and worse outcomes. J Cancer Res Ther. (2018) 14:S248–53. doi: 10.4103/0973-1482.187285
110. Cheng CL, Huang SC, Chen JH, Wei CH, Fang WQ, Su TH, et al. Hepatitis B surface antigen positivity is an independent unfavorable prognostic factor in diffuse large B-cell lymphoma in the rituximab era. Oncologist. (2020) 25:793–802. doi: 10.1634/theoncologist.2019-0756
111. Li M, Gan Y, Fan C, Yuan H, Zhang X, Shen Y, et al. Hepatitis B virus and risk of non-Hodgkin lymphoma: An updated meta-analysis of 58 studies. J Viral Hepatitis. (2018) 25:894–903. doi: 10.1111/jvh.12892
112. Neuveut C, Wei Y, Buendia MA. Mechanisms of HBV-related hepatocarcinogenesis. J Hepatol. (2010) 52:594–604. doi: 10.1016/j.jhep.2009.10.033
113. Guidotti LG, Chisari FV. Immunobiology and pathogenesis of viral hepatitis. Annu Rev Pathol. (2006) 1:23–61. doi: 10.1146/annurev.pathol.1.110304.100230
114. Wan X, Young KH, Bai O. HBV-associated DLBCL of poor prognosis: advance in pathogenesis, immunity and therapy. Front Immunol. (2023) 14:1216610. doi: 10.3389/fimmu.2023.1216610
115. Huang CF, Lin SS, Ho YC, Chen FL, Yang CC. The immune response induced by hepatitis B virus principal antigens. Cell Mol Immunol. (2006) 3:97–106.
116. Talamo TS, Dekker A, Gurecki J, Singh G. Primary hepatic Malignant lymphoma: its occurrence in a patient with chronic active hepatitis, cirrhosis, and hepatocellular carcinoma associated with hepatitis B viral infection. Cancer. (1980) 46:336–9. doi: 10.1002/(ISSN)1097-0142
117. Taborelli M, Polesel J, Montella M, Libra M, Tedeschi R, Battiston M, et al. Hepatitis B and C viruses and risk of non-Hodgkin lymphoma: a case-control study in Italy. Infect Agents Cancer. (2016) 11:1–6. doi: 10.1186/s13027-016-0073-x
118. Huang CE, Yang YH, Chen YY, Chang JJ, Chen KJ, Lu CH, et al. The impact of hepatitis B virus infection and vaccination on the development of non-Hodgkin lymphoma. J Viral Hepatitis. (2017) 24:885–94. doi: 10.1111/jvh.12713
119. Yoffe B, Noonan CA, Melnick JL, Hollinger FB. Hepatitis B virus DNA in mononuclear cells and analysis of cell subsets for the presence of replicative intermediates of viral DNA. J Infect Dis. (1986) 153:471–7. doi: 10.1093/infdis/153.3.471
120. Galun E, Ilan Y, Livni N, Ketzinel M, Nahor O, Pizov G, et al. Hepatitis B virus infection associated with hematopoietic tumors. Am J Pathol. (1994) 145:1001–7.
121. Qi XY, Gui X, Zhuang K. Establishment and characterization of HBV-associated B lymphocytes with an immortalization potential. PloS One. (2019) 14(5):e0217161. doi: 10.1371/journal.pone.0217161
122. Ren WC, Ye XF, Su H, Li W, Liu DB, Pirmoradian M, et al. Genetic landscape of hepatitis B virus-associated diffuse large B-cell lymphoma. Blood. (2018) 131:2670–81. doi: 10.1182/blood-2017-11-817601
123. Umeda M, Marusawa H, Seno H, Katsurada A, Nabeshima M, Egawa H, et al. Hepatitis B virus infection in lymphatic tissues in inactive hepatitis B carriers. J Hepatol. (2005) 42:806–12. doi: 10.1016/j.jhep.2005.01.016
124. Sung WK, Zheng HC, Li SY, Chen RH, Liu X, Li YR, et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat Genet. (2012) 44:765–U188. doi: 10.1038/ng.2295
125. Hsu T, Moroy T, Etiemble J, Louise A, Trepo C, Tiollais P, et al. Activation of c-myc by woodchuck hepatitis virus insertion in hepatocellular carcinoma. Cell. (1988) 55:627–35. doi: 10.1016/0092-8674(88)90221-8
126. Li XJ, Zhang JB, Yang ZW, Kang JT, Jiang SZ, Zhang T, et al. The function of targeted host genes determines the oncogenicity of HBV integration in hepatocellular carcinoma. J Hepatol. (2014) 60:975–84. doi: 10.1016/j.jhep.2013.12.014
127. Zhao LH, Liu X, Yan HX, Li WY, Zeng X, Yang Y, et al. Genomic and oncogenic preference of HBV integration in hepatocellular carcinoma. Nat Commun. (2016) 7(1):12992. doi: 10.1038/ncomms13591
128. Li MG, Shen YL, Chen YM, Gao HF, Zhou JQ, Wang Q, et al. Characterization of hepatitis B virus infection and viral DNA integration in non-Hodgkin lymphoma. Int J Cancer. (2020) 147:2199–209. doi: 10.1002/ijc.33027
129. Ren WC, Wang XH, Yang MY, Wan H, Li XB, Ye XF, et al. Distinct clinical and genetic features of hepatitis B virus-associated follicular lymphoma in Chinese patients. Blood Adv. (2022) 6:2731–44. doi: 10.1182/bloodadvances.2021006410
130. Wang N, Qin W, Zheng Z, Zhao M, Xiong J, Fang H, et al. Hepatitis B virus-associated follicular lymphoma presents T-cell inflamed phenotype and response to lenalidomide. Cancer Commun. (2022) 42:170–4. doi: 10.1002/cac2.12241
131. Andrisani OM, Barnabas S. The transcriptional function of the hepatitis B virus X protein and its role in hepatocarcinogenesis (Review). Int J Oncol. (1999) 15:373–9. doi: 10.3892/ijo
132. Bouchard MJ, Schneider RJ. The enigmatic X gene of hepatitis B virus. J Virol. (2004) 78:12725–34. doi: 10.1128/JVI.78.23.12725-12734.2004
133. Tang H, Oishi N, Kaneko S, Murakami S. Molecular functions and biological roles of hepatitis B virus x protein. Cancer Sci. (2006) 97:977–83. doi: 10.1111/j.1349-7006.2006.00299.x
134. Tarocchi M, Polvani S, Marroncini G, Galli A. Molecular mechanism of hepatitis B virus-induced hepatocarcinogenesis. World J Gastroenterol. (2014) 20:11630–40. doi: 10.3748/wjg.v20.i33.11630
135. Wang XW, Forrester K, Yeh H, Feitelson MA, Gu JR, Harris CC. Hepatitis B virus X protein inhibits p53 sequence-specific DNA binding, transcriptional activity, and association with transcription factor ERCC3. Proc Natl Acad Sci U.S.A. (1994) 91:2230–4. doi: 10.1073/pnas.91.6.2230
136. Dewantoro O, Gani RA, Akbar N. Hepatocarcinogenesis in viral Hepatitis B infection: the role of HBx and p53. Acta Med Indones. (2006) 38:154–9.
137. Xiao Y, Yu DH. Tumor microenvironment as a therapeutic target in cancer. Pharmacol Ther. (2021) 221:107753. doi: 10.1016/j.pharmthera.2020.107753
138. Cheng CL, Fang WQ, Lin YJ, Yuan CT, Ko BS, Tang JL, et al. Hepatitis B surface antigen positivity is associated with progression of disease within 24 months in follicular lymphoma. J Cancer Res Clin Oncol. (2022) 148:1211–22. doi: 10.1007/s00432-021-03719-y
139. Yang PY, Markowitz GJ, Wang XF. The hepatitis B virus-associated tumor microenvironment in hepatocellular carcinoma. Natl Sci Rev. (2014) 1:396–412. doi: 10.1093/nsr/nwu038
140. Li TY, Yang Y, Zhou G, Tu ZK. Immune suppression in chronic hepatitis B infection associated liver disease: A review. World J Gastroenterol. (2019) 25:3527–37. doi: 10.3748/wjg.v25.i27.3527
141. Polaris Observatory, H. C. V. C. Global prevalence and genotype distribution of hepatitis C virus infection in 2015: a modelling study. Lancet Gastroenterol Hepatol. (2017) 2:161–76. doi: 10.1016/S2468-1253(16)30181-9
142. Spearman CW, Dusheiko GM, Hellard M, Sonderup M. Hepatitis C. Lancet. (2019) 394:1451–66. doi: 10.1016/S0140-6736(19)32320-7
143. Li HC, Yang CH, Lo SY. Cellular factors involved in the hepatitis C virus life cycle. World J Gastroenterol. (2021) 27:4555–81. doi: 10.3748/wjg.v27.i28.4555
144. Ferri C, Caracciolo F, La Civita L, Monti M, Longombardo G, Greco F, et al. Hepatitis C virus infection and B-cell lymphomas. Eur J Cancer. (1994) 30A:1591–2. doi: 10.1016/0959-8049(94)90066-3
145. Giordano TP, Henderson L, Landgren O, Chiao EY, Kramer JR, El-Serag H, et al. Risk of non-Hodgkin lymphoma and lymphoproliferative precursor diseases in US veterans with hepatitis C virus. Jama-J Am Med Assoc. (2007) 297:2010–7. doi: 10.1001/jama.297.18.2010
146. Nieters A, Kallinowski B, Brennan P, Ott M, Maynadie M, Benavente Y, et al. Hepatitis C and risk of lymphoma: Results of the European Multicenter Case-Control Study EPILYMPH. Gastroenterology. (2006) 131:1879–86. doi: 10.1053/j.gastro.2006.09.019
147. De Sanjose S, Benavente Y, Vajdic CM, Engels EA, Morton LM, Bracci PM, et al. Hepatitis C and non-Hodgkin lymphoma among 4784 cases and 6269 controls from the international lymphoma epidemiology consortium. Clin Gastroenterol Hepatol. (2008) 6:451–8. doi: 10.1016/j.cgh.2008.02.011
148. Vannata B, Arcaini L, Zucca E. Hepatitis C virus-associated B-cell non-Hodgkin’s lymphomas: what do we know? Ther Adv Hematol. (2016) 7:94–107. doi: 10.1177/2040620715623924
149. Gisbert JP, Garcia-Buey L, Pajares JM, Moreno-Otero R. Prevalence of hepatitis C virus infection in B-cell non-Hodgkin’s lymphoma: Systematic review and meta-analysis. Gastroenterology. (2003) 125:1723–32. doi: 10.1053/j.gastro.2003.09.025
150. Engels EA, Chatterjee N, Cerhan JR, Davis S, Cozen W, Severson RK, et al. Hepatitis C virus infection and non-Hodgkin lymphoma: Results of the NCI-SEER multi-center case-control study. Int J Cancer. (2004) 111:76–80. doi: 10.1002/ijc.20021
151. Schollkopf C, Smedby KE, Hjalgrim H, Rostgaard K, Panum I, Vinner L, et al. Hepatitis C infection and risk of Malignant lymphoma. Int J Cancer. (2008) 122:1885–90. doi: 10.1002/ijc.23416
152. Kim M, Lee YK, Park B, Oh DJ, Choi HG. Hepatitis virus B and C infections are associated with an increased risk of non-Hodgkin lymphoma: A nested case-control study using a national sample cohort. J Med Virol. (2020) 92:1214–20. doi: 10.1002/jmv.25653
153. Peveling-Oberhag J, Arcaini L, Hansmann ML, Zeuzem S. Hepatitis C-associated B-cell non-Hodgkin lymphomas. Epidemiology, molecular signature and clinical management. J Hepatol. (2013) 59:169–77. doi: 10.1016/j.jhep.2013.03.018
154. Visco C, Finotto S. Hepatitis C virus and diffuse large B-cell lymphoma: Pathogenesis, behavior and treatment. World J Gastroenterol. (2014) 20:11054–61. doi: 10.3748/wjg.v20.i32.11054
155. Sulyok M, Makara M, Ujhelyi E, Valyi-Nagy I. Non-hodgkin lymphoma and hepatitis C: where we are and what next? Pathol Oncol Res. (2015) 21:1–7. doi: 10.1007/s12253-014-9845-z
156. Frigeni M, Besson C, Visco C, Fontaine H, Goldaniga M, Visentini M, et al. Interferon-free compared to interferon-based antiviral regimens as first-line therapy for B-cell lymphoproliferative disorders associated with hepatitis C virus infection. Leukemia. (2020) 34:1462–6. doi: 10.1038/s41375-019-0687-2
157. Bachy E, Besson C, Suarez F, Hermine O. Hepatitis C virus infection and lymphoma. Mediterr J Hematol Infect Dis. (2010) 2:e2010004. doi: 10.4084/mjhid.2010.004
158. Besson C, Canioni D, Lepage E, Pol S, Morel P, Lederlin P, et al. Characteristics and outcome of diffuse large B-cell lymphoma in hepatitis C virus-positive patients in LNH 93 and LNH 98 Groupe d’Etude des Lymphomes de l’Adulte programs. J Clin Oncol. (2006) 24:953–60. doi: 10.1200/JCO.2005.01.5016
159. Merli M, Visco C, Spina M, Luminari S, Ferretti VV, Gotti M, et al. Outcome prediction of diffuse large B-cell lymphomas associated with hepatitis C virus infection: a study on behalf of the Fondazione Italiana Linfomi. Haematologica. (2014) 99:489–96. doi: 10.3324/haematol.2013.094318
160. Kaito M, Watanabe S, Tsukiyama-Kohara K, Yamaguchi K, Kobayashi Y, Konishi M, et al. Hepatitis C virus particle detected by immunoelectron microscopic study. J Gen Virol. (1994) 75:1755–60. doi: 10.1099/0022-1317-75-7-1755
161. Gastaminza P, Dryden KA, Boyd B, Wood MR, Law M, Yeager M, et al. Ultrastructural and biophysical characterization of hepatitis C virus particles produced in cell culture. J Virol. (2010) 84:10999–1009. doi: 10.1128/JVI.00526-10
162. Tang H, Grise H. Cellular and molecular biology of HCV infection and hepatitis. Clin Sci (Lond). (2009) 117:49–65. doi: 10.1042/CS20080631
163. Ferri C, La Civita L, Zignego AL, Pasero G. Viruses and cancers: possible role of hepatitis C virus. Eur J Clin Invest. (1997) 27:711–8. doi: 10.1046/j.1365-2362.1997.1790728.x
164. Pileri P, Uematsu Y, Campagnoli S, Galli G, Falugi F, Petracca R, et al. Binding of hepatitis C virus to CD81. Science. (1998) 282:938–41. doi: 10.1126/science.282.5390.938
165. Petracca R, Falugi F, Galli G, Norais N, Rosa D, Campagnoli S, et al. Structure-function analysis of hepatitis C virus envelope-CD81 binding. J Virol. (2000) 74:4824–30. doi: 10.1128/jvi.74.10.4824-4830.2000
166. Rosa D, Saletti G, De Gregorio E, Zorat F, Comar C, D’oro U, et al. Activation of naive B lymphocytes via CD81, a pathogenetic mechanism for hepatitis C virus-associated B lymphocyte disorders. Proc Natl Acad Sci United States America. (2005) 102:18544–9. doi: 10.1073/pnas.0509402102
167. Wotherspoon AC, Doglioni C, Isaacson PG. Low-grade gastric B-cell lymphoma of mucosa-associated lymphoid tissue (MALT): a multifocal disease. Histopathology. (1992) 20:29–34. doi: 10.1111/j.1365-2559.1992.tb00912.x
168. Roggero E, Zucca E, Pinotti G, Pascarella A, Capella C, Savio A, et al. Eradication of Helicobacter pylori infection in primary low-grade gastric lymphoma of mucosa-associated lymphoid tissue. Ann Intern Med. (1995) 122:767–9. doi: 10.7326/0003-4819-122-10-199505150-00006
169. Emens LA, Sulkowski MS. Regression of splenic lymphoma after treatment of hepatitis C virus infection. New Engl J Med. (2002) 347:2168–9. doi: 10.1056/NEJM200212263472614
170. Ferri C, Greco F, Longombardo G, Palla P, Moretti A, Marzo E, et al. Antibodies to hepatitis C virus in patients with mixed cryoglobulinemia. Arthritis Rheum. (1991) 34:1606–10. doi: 10.1002/art.1780341221
171. Dammacco F, Sansonno D, Cornacchiulo V, Mennuni C, Carbone R, Lauletta G, et al. Hepatitis C virus infection and mixed cryoglobulinemia: a striking association. Int J Clin Lab Res. (1993) 23:45–9. doi: 10.1007/BF02592281
172. Agnello V. The aetiology of mixed cryoglobulinaemia associated with hepatitis C virus infection. Scand J Immunol. (1995) 42:179–84. doi: 10.1111/j.1365-3083.1995.tb03643.x
173. Dammacco F, Gatti P, Sansonno D. Hepatitis C virus infection, mixed cryoglobulinemia, and non-Hodgkin’s lymphoma: an emerging picture. Leuk Lymphoma. (1998) 31:463–76. doi: 10.3109/10428199809057606
174. Chan CH, Hadlock KG, Foung SKH, Levy S. V(H)1-69 gene is preferentially used by hepatitis C virus-associated B cell lymphomas and by normal B cells responding to the E2 viral antigen. Blood. (2001) 97:1023–6. doi: 10.1182/blood.V97.4.1023
175. Ng PP, Kuo CC, Wang S, Einav S, Arcaini L, Paulli M, et al. B-cell receptors expressed by lymphomas of hepatitis C virus (HCV)-infected patients rarely react with the viral proteins. Blood. (2014) 123:1512–5. doi: 10.1182/blood-2013-10-532895
176. Armand M, Boudjoghra M, Xochelli A, Canioni D, Tavernier ML, Colombo M, et al. Auto-immune origin of B cells from HCV-associated lymphoma. Blood. (2015) 126(23):1464. doi: 10.1182/blood.V126.23.1464.1464
177. Zuckerman E, Slobodin G, Kessel A, Sabo E, Yeshurun D, Halas K, et al. Peripheral B-cell CD5 expansion and CD81 overexpression and their association with disease severity and autoimmune markers in chronic hepatitis C virus infection. Clin Exp Immunol. (2002) 128:353–8. doi: 10.1046/j.1365-2249.2002.01844.x
178. Bradbury LE, Kansas GS, Levy S, Evans RL, Tedder TF. The CD19/CD21 signal transducing complex of human B lymphocytes includes the target of antiproliferative antibody-1 and Leu-13 molecules. J Immunol. (1992) 149:2841–50. doi: 10.4049/jimmunol.149.9.2841
179. Fearon DT, Carter RH. The CD19/CR2/TAPA-1 complex of B lymphocytes: linking natural to acquired immunity. Annu Rev Immunol. (1995) 13:127–49. doi: 10.1146/annurev.iy.13.040195.001015
180. Carter RH, Fearon DT. CD19: lowering the threshold for antigen receptor stimulation of B lymphocytes. Science. (1992) 256:105–7. doi: 10.1126/science.1373518
181. Chen ZH, Zhu YZ, Ren YL, Tong YM, Hua X, Zhu FH, et al. Hepatitis C virus protects human B lymphocytes from fas-mediated apoptosis via E2-CD81 engagement. PloS One. (2011) 6(4):e18933. doi: 10.1371/journal.pone.0018933
182. Crotta S, Stilla A, Wack A, D’andrea A, Nuti S, D’oro U, et al. Inhibition of natural killer cells through engagement of CD81 by the major hepatitis C virus envelope protein. J Exp Med. (2002) 195:35–41. doi: 10.1084/jem.20011124
183. Ambinder RF. Gammaherpesviruses and “hit-and-run” oncogenesis. Am J Pathol. (2000) 156:1–3. doi: 10.1016/S0002-9440(10)64697-4
184. Ferreira DA, Tayyar Y, Idris A, Mcmillan NAJ. A “hit-and-run” affair - A possible link for cancer progression in virally driven cancers. Biochim Biophys Acta Rev Cancer. (2021) 1875:188476. doi: 10.1016/j.bbcan.2020.188476
185. Machida K, Cheng KTN, Sung VMH, Shimodaira S, Lindsay KL, Levine AM, et al. Hepatitis C virus induces a mutator phenotype: Enhanced mutations of immunoglobulin and protooncogenes. Proc Natl Acad Sci United States America. (2004) 101:4262–7. doi: 10.1073/pnas.0303971101
186. Hofmann WP, Fernandez B, Herrmann E, Welsch C, Mihm U, Kronenberger B, et al. Somatic hypermutation and mRNA expression levels of the BCL-6 gene in patients with hepatitis C virus-associated lymphoproliferative diseases. J Viral Hepatitis. (2007) 14:484–91. doi: 10.1111/j.1365-2893.2006.00833.x
187. Tucci FA, Broering R, Johansson P, Schlaak JF, Kuppers R. B cells in chronically hepatitis C virus-infected individuals lack a virus-induced mutation signature in the TP53, CTNNB1, and BCL6 genes. J Virol. (2013) 87:2956–62. doi: 10.1128/JVI.03081-12
188. Machida K, Cheng KTH, Sung VMH, Lee KJ, Levine AM, Lai MMC. Hepatitis C virus infection activates the immunologic (type II) isoform of nitric oxide synthase and thereby enhances DNA damage and mutations of cellular genes. J Virol. (2004) 78:8835–43. doi: 10.1128/JVI.78.16.8835-8843.2004
189. Machida K, Mcnamara G, Cheng KTH, Huang J, Wang CH, Comai L, et al. Hepatitis C virus inhibits DNA damage repair through reactive oxygen and nitrogen species and by interfering with the ATM-NBS1/mre11/rad50 DNA repair pathway in monocytes and hepatocytes. J Immunol. (2010) 185:6985–98. doi: 10.4049/jimmunol.1000618
190. Sung VM, Shimodaira S, Doughty AL, Picchio GR, Can H, Yen TS, et al. Establishment of B-cell lymphoma cell lines persistently infected with hepatitis C virus in vivo and in vitro: the apoptotic effects of virus infection. J Virol. (2003) 77:2134–46. doi: 10.1128/JVI.77.3.2134-2146.2003
191. Machida K, Tsukiyama-Kohara K, Sekiguch S, Seike E, Tone S, Hayashi Y, et al. Hepatitis C virus and disrupted interferon signaling promote lymphoproliferation via type II CD95 and interleukins. Gastroenterology. (2009) 137:285–96. doi: 10.1053/j.gastro.2009.03.061
192. Ishikawa T, Shibuya K, Yasui K, Mitamura K, Ueda S. Expression of hepatitis C virus core protein associated with Malignant lymphoma in transgenic mice. Comp Immunol Microbiol Infect Dis. (2003) 26:115–24. doi: 10.1016/S0147-9571(02)00038-3
193. Kasama Y, Sekiguchi S, Saito M, Tanaka K, Satoh M, Kuwahara K, et al. Persistent expression of the full genome of hepatitis C virus in B cells induces spontaneous development of B-cell lymphomas in vivo. Blood. (2010) 116:4926–33. doi: 10.1182/blood-2010-05-283358
194. Ali Syeda Z, Langden SSS, Munkhzul C, Lee M, Song SJ. Regulatory mechanism of microRNA expression in cancer. Int J Mol Sci. (2020) 21(5):1723. doi: 10.3390/ijms21051723
195. He L, Hannon GJ. Micrornas: Small RNAs with a big role in gene regulation. Nat Rev Genet. (2004) 5:522–31. doi: 10.1038/nrg1379
196. Jee HJ, Kim AJ, Song N, Kim HJ, Kim M, Koh H, et al. Nek6 overexpression antagonizes p53-induced senescence in human cancer cells. Cell Cycle. (2010) 9:4703–10. doi: 10.4161/cc.9.23.14059
197. Fognani E, Giannini C, Piluso A, Gragnani L, Monti M, Caini P, et al. Role of microRNA profile modifications in hepatitis C virus-related mixed cryoglobulinemia. PloS One. (2013) 8(5):e62965. doi: 10.1371/journal.pone.0062965
198. Atallah-Yunes SA, Robertson MJ. Cytokine based immunotherapy for cancer and lymphoma: biology, challenges and future perspectives. Front Immunol. (2022) 13. doi: 10.3389/fimmu.2022.872010
199. Sene D, Limal N, Ghillani-Dalbin P, Saadoun D, Piette JC, Cacoub P. Hepatitis C virus-associated B-cell proliferation - the role of serum B lymphocyte stimulator (BLyS/BAFF). Rheumatology. (2007) 46:65–9. doi: 10.1093/rheumatology/kel177
200. Batten M, Fletcher C, Ng LG, Groom J, Wheway J, Laabi Y, et al. TNF deficiency fails to protect BAFF transgenic mice against autoimmunity and reveals a predisposition to B cell lymphoma. J Immunol. (2004) 172:812–22. doi: 10.4049/jimmunol.172.2.812
201. Feldmann G, Nischalke HD, Nattermann J, Banas B, Berg T, Teschendorf C, et al. Induction of interleukin-6 by hepatitis C virus core protein in hepatitis C-associated mixed cryoglobulinemia and B-cell non-Hodgkin’s lymphoma. Clin Cancer Res. (2006) 12:4491–8. doi: 10.1158/1078-0432.CCR-06-0154
202. Machida K, Cheng KTH, Sung VMH, Levine AM, Foung S, Lai MMC. Hepatitis C virus induces toll-like receptor 4 expression, leading to enhanced production of beta interferon and interleukin-6. J Virol. (2006) 80:866–74. doi: 10.1128/JVI.80.2.866-874.2006
203. Kondo Y, Ninomiya M, Kimura O, Machida K, Funayama R, Nagashima T, et al. HCV infection enhances th17 commitment, which could affect the pathogenesis of autoimmune diseases. PloS One. (2014) 9(6):e98521. doi: 10.1371/journal.pone.0098521
204. Simon V, Ho DD, Abdool Karim Q. HIV/AIDS epidemiology, pathogenesis, prevention, and treatment. Lancet. (2006) 368:489–504. doi: 10.1016/S0140-6736(06)69157-5
205. Sadowski I, Hashemi FB. Strategies to eradicate HIV from infected patients: elimination of latent provirus reservoirs. Cell Mol Life Sci. (2019) 76:3583–600. doi: 10.1007/s00018-019-03156-8
206. Pongas GN, Ramos JC. HIV-associated lymphomas: progress and new challenges. J Clin Med. (2022) 11(5):1447. doi: 10.3390/jcm11051447
207. Grogg KL, Miller RF, Dogan A. HIV infection and lymphoma. J Clin Pathol. (2007) 60:1365–72. doi: 10.1136/jcp.2007.051953
208. Grulich AE, Van Leeuwen MT, Falster M, Vajdic CM. Incidence of cancers in people with HIV/AIDS compared with immunosuppressed transplant recipients: a meta-analysis. Lancet. (2007) 370:59–67. doi: 10.1016/S0140-6736(07)61050-2
209. Dolcetti R, Gloghini A, Caruso A, Carbone A. A lymphomagenic role for HIV beyond immune suppression? Blood. (2016) 127:1403–9. doi: 10.1182/blood-2015-11-681411
210. Gloghini A, Dolcetti R, Carbone A. Lymphomas occurring specifically in HIV-infected patients: From pathogenesis to pathology. Semin Cancer Biol. (2013) 23:457–67. doi: 10.1016/j.semcancer.2013.08.004
211. Fanales-Belasio E, Raimondo M, Suligoi B, Butto S. HIV virology and pathogenetic mechanisms of infection: a brief overview. Annali Dell Istituto Superiore Di Sanita. (2010) 46:5–14. doi: 10.1590/S0021-25712010000100002
212. Ganser-Pornillos BK, Yeager M, Pornillos O. Assembly and architecture of HIV. Viral Mol Machines. (2012) 726:441–65. doi: 10.1007/978-1-4614-0980-9_20
213. Martinez-Maza O, Widney D, van der Meijden M, Knox R, Echeverri A, Breen EC, et al. Immune dysfunction and the pathogenesis of AIDS-associated non-Hodgkin’s lymphoma. Mem Inst Oswaldo Cruz. (1998) 93:373–81. doi: 10.1590/s0074-02761998000300019
214. Landgren O, Goedert JJ, Rabkin CS, Wilson WH, Dunleavy K, Kyle RA, et al. Circulating serum free light chains as predictive markers of AIDS-related lymphoma. J Clin Oncol. (2010) 28:773–9. doi: 10.1200/JCO.2009.25.1322
215. Capello D, Martini M, Gloghini A, Cerri M, Rasi S, Deambrogi C, et al. Molecular analysis of immunoglobulin variable genes in human immunodeficiency virus-related non-Hodgkin’s lymphoma reveals implications for disease pathogenesis and histogenesis. Haematologica-the Hematol J. (2008) 93:1178–85. doi: 10.3324/haematol.12705
216. Breen EC, Hussain SK, Magpantay L, Jacobson LP, Detels R, Rabkin CS, et al. B-cell stimulatory cytokines and markers of immune activation are elevated several years prior to the diagnosis of systemic AIDS-associated non-hodgkin B-cell lymphoma. Cancer Epidemiol Biomarkers Prev. (2011) 20:1303–14. doi: 10.1158/1055-9965.EPI-11-0037
217. Martin G, Roy J, Barat C, Ouellet M, Gilbert C, Tremblay MJ. Human immunodeficiency virus type 1-associated CD40 ligand transactivates B lymphocytes and promotes infection of CD4(+) T cells. J Virol. (2007) 81:5872–81. doi: 10.1128/JVI.02542-06
218. Bishop GA, Hostager BS. The CD40-CD154 interaction in B cell-T cell liaisons. Cytokine Growth Factor Rev. (2003) 14:297–309. doi: 10.1016/S1359-6101(03)00024-8
219. Graham JP, Arcipowski KM, Bishop GA. Differential B-lymphocyte regulation by CD40 and its viral mimic, latent membrane protein 1. Immunol Rev. (2010) 237:226–48. doi: 10.1111/j.1600-065X.2010.00932.x
220. Kundu RK, Sangiorgi F, Wu LY, Pattengale PK, Hinton DR, Gill PS, et al. Expression of the human immunodeficiency virus-Tat gene in lymphoid tissues of transgenic mice is associated with B-cell lymphoma. Blood. (1999) 94:275–82. doi: 10.1182/blood.V94.1.275.413a30_275_282
221. Wang XH, Duan Z, Yu GJ, Fan MX, Scharff MD. Human immunodeficiency virus tat protein aids V region somatic hypermutation in human B cells. Mbio. (2018) 9(2):10-1128. doi: 10.1128/mBio.02315-17
222. Curreli S, Krishnan S, Reitz M, Lunardi-Iskandar Y, Lafferty MK, Garzino-Demo A, et al. B cell lymphoma in hiv transgenic mice. Retrovirology. (2013) 10:1–13. doi: 10.1186/1742-4690-10-92
223. Martorelli D, Muraro E, Mastorci K, Dal Col J, Fae DA, Furlan C, et al. A natural HIV p17 protein variant up-regulates the LMP-1 EBV oncoprotein and promotes the growth of EBV-infected B-lymphocytes: Implications for EBV-driven lymphomagenesis in the HIV setting. Int J Cancer. (2015) 137:1374–85. doi: 10.1002/ijc.29494
224. Isaguliants M, Bayurova E, Avdoshina D, Kondrashova A, Chiodi F, Palefsky JM. Oncogenic effects of HIV-1 proteins, mechanisms behind. Cancers. (2021) 13(2):305. doi: 10.3390/cancers13020305
225. Da Silva SR, De Oliveira DE. HIV, EBV and KSHV: Viral cooperation in the pathogenesis of human Malignancies. Cancer Lett. (2011) 305:175–85. doi: 10.1016/j.canlet.2011.02.007
226. De Paoli P, Carbone A. Microenvironmental abnormalities induced by viral cooperation: Impact on lymphomagenesis. Semin Cancer Biol. (2015) 34:70–80. doi: 10.1016/j.semcancer.2015.03.009
227. Carbone A, Cesarman E, Spina M, Gloghini A, Schulz TF. HIV-associated lymphomas and gamma-herpesviruses. Blood. (2009) 113:1213–24. doi: 10.1182/blood-2008-09-180315
228. Martinez-Maza O, Crabb E, Mitsuyasu RT, Fahey JL, Giorgi JV. Infection with the human immunodeficiency virus (HIV) is associated with an in vivo increase in B lymphocyte activation and immaturity. J Immunol. (1987) 138:3720–4. doi: 10.4049/jimmunol.138.11.3720
229. Lyn D, Bennett NA, Shiramizu BT, Herndier BG, Igietseme JU. Sequence analysis of HIV-1 insertion sites in peripheral blood lymphocytes. Cell Mol Biol. (2001) 47:981–6.
230. Stevens SW, Griffith JD. Sequence analysis of the human DNA flanking sites of human immunodeficiency virus type 1 integration. J Virol. (1996) 70:6459–62. doi: 10.1128/jvi.70.9.6459-6462.1996
231. Capello D, Scandurra M, Poretti G, Rancoita PMV, Mian M, Gloghini A, et al. Genome wide DNA-profiling of HIV-related B-cell lymphomas. Br J Haematol. (2010) 148:245–55. doi: 10.1111/j.1365-2141.2009.07943.x
232. Teyssier JR, Rousset F, Garcia E, Cornillet P, Laubriet A. Upregulation of the netrin receptor (DCC) gene during activation of B lymphocytes and modulation by interleukins. Biochem Biophys Res Commun. (2001) 283:1031–6. doi: 10.1006/bbrc.2001.4902
233. Ishii H, Vecchione A, Furukawa Y, Sutheesophon K, Han SY, Druck T, et al. Expression of FRA16D/WWOX and FRA3B/FHIT genes in hematopoietic Malignancies. Mol Cancer Res. (2003) 1:940–7.
234. Kameoka Y, Tagawa H, Tsuzuki S, Karnan S, Ota A, Suguro M, et al. Contig array CGH at 3p14.2 points to the FRA3B/FHIT common fragile region as the target gene in diffuse large B-cell lymphoma. Oncogene. (2004) 23:9148–54. doi: 10.1038/sj.onc.1208136
235. Laurence J, Astrin SM. Human immunodeficiency virus induction of Malignant transformation in human B lymphocytes. Proc Natl Acad Sci U.S.A. (1991) 88:7635–9. doi: 10.1073/pnas.88.17.7635
236. Rios LAD, Mapekula L, Mdletshe N, Chetty D, Mowla S. HIV-1 transactivator of transcription (Tat) co-operates with AP-1 factors to enhance c-MYC transcription. Front Cell Dev Biol. (2021) 9. doi: 10.3389/fcell.2021.693706
237. Ramorola BR, Goolam-Hoosen T, Rios LAD, Mowla S. Modulation of cellular microRNA by HIV-1 in burkitt lymphoma cells-A pathway to promoting oncogenesis. Genes. (2021) 12(9):1302. doi: 10.3390/genes12091302
238. Sekar D, Islam VIH, Thirugnanasambantham K, Saravanan S. Relevance of miR-21 in HIV and non-HIV-related lymphomas. Tumor Biol. (2014) 35:8387–93. doi: 10.1007/s13277-014-2068-9
239. Zhuang K, Zhang YX, Mo PZ, Deng LP, Jiang Y, Yu L, et al. Plasma proteomic analysis reveals altered protein abundances in HIV-infected patients with or without non-Hodgkin lymphoma. J Med Virol. (2022) 94:3876–89. doi: 10.1002/jmv.27775
240. Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, Babinet P, et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood. (1995) 86:1276–80. doi: 10.1182/blood.V86.4.1276.bloodjournal8641276
241. Dupin N, Fisher C, Kellam P, Ariad S, Tulliez M, Franck N, et al. Distribution of human herpesvirus-8 latently infected cells in Kaposi’s sarcoma, multicentric Castleman’s disease, and primary effusion lymphoma. Proc Natl Acad Sci United States America. (1999) 96:4546–51. doi: 10.1073/pnas.96.8.4546
242. Hussein HAM, Alfhili MA, Pakala P, Simon S, Hussain J, Mccubrey JA, et al. miRNAs and their roles in KSHV pathogenesis. Virus Res. (2019) 266:15–24. doi: 10.1016/j.virusres.2019.03.024
243. Dollery SJ. Towards understanding KSHV fusion and entry. Viruses. (2019) 11(11):1073. doi: 10.3390/v11111073
244. Chen YB, Rahemtullah A, Hochberg E. Primary effusion lymphoma. Oncologist. (2007) 12:569–76. doi: 10.1634/theoncologist.12-5-569
245. Komanduri KV, Luce JA, Mcgrath MS, Herndier BG, Ng VL. The natural history and molecular heterogeneity of HIV-associated primary Malignant lymphomatous effusions. J Acquir Immune Defic Syndr Hum Retrovirol. (1996) 13:215–26. doi: 10.1097/00042560-199611010-00003
246. Renne R, Lagunoff M, Zhong W, Ganem D. The size and conformation of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) DNA in infected cells and virions. J Virol. (1996) 70:8151–4. doi: 10.1128/jvi.70.11.8151-8154.1996
247. Zhang F, Liang DG, Lin XX, Zou Z, Sun R, Wang X, et al. NDRG1 facilitates the replication and persistence of Kaposi’s sarcoma-associated herpesvirus by interacting with the DNA polymerase clamp PCNA. PloS Pathog. (2019) 15(2):e1007628. doi: 10.1371/journal.ppat.1007628
248. Li XJ, Huang L, Xiao YJ, Yao XY, Long XB, Zhu FX, et al. Development of an ORF45-derived peptide to inhibit the sustained RSK activation and lytic replication of kaposi’s sarcoma-associated herpesvirus. J Virol. (2019) 93(10):10-1128. doi: 10.1128/JVI.02154-18
249. Uppal T, Jha HC, Verma SC, Robertson ES. Chromatinization of the KSHV genome during the KSHV life cycle. Cancers. (2015) 7:112–42. doi: 10.3390/cancers7010112
250. Lopes AD, Marinho PD, Medeiros LDD, De Paula VS. Human gammaherpesvirus 8 oncogenes associated with kaposi’s sarcoma. Int J Mol Sci. (2022) 23(13):7203. doi: 10.3390/ijms23137203
251. Wei F, Gan J, Wang C, Zhu C, Cai Q. Cell cycle regulatory functions of the KSHV oncoprotein LANA. Front Microbiol. (2016) 7:334. doi: 10.3389/fmicb.2016.00334
252. Friborg J Jr., Kong W, Hottiger MO, Nabel GJ. p53 inhibition by the LANA protein of KSHV protects against cell death. Nature. (1999) 402:889–94. doi: 10.1038/47266
253. Liu J, Martin HJ, Liao G, Hayward SD. The Kaposi’s sarcoma-associated herpesvirus LANA protein stabilizes and activates c-Myc. J Virol. (2007) 81:10451–9. doi: 10.1128/JVI.00804-07
254. Li M, Lee H, Yoon DW, Albrecht JC, Fleckenstein B, Neipel F, et al. Kaposi’s sarcoma-associated herpesvirus encodes a functional cyclin. J Virol. (1997) 71:1984–91. doi: 10.1128/jvi.71.3.1984-1991.1997
255. Swanton C, Mann DJ, Fleckenstein B, Neipel F, Peters G, Jones N. Herpes viral cyclin/Cdk6 complexes evade inhibition by CDK inhibitor proteins. Nature. (1997) 390:184–7. doi: 10.1038/36606
256. Godden-Kent D, Talbot SJ, Boshoff C, Chang Y, Moore P, Weiss RA, et al. The cyclin encoded by Kaposi’s sarcoma-associated herpesvirus stimulates cdk6 to phosphorylate the retinoblastoma protein and histone H1. J Virol. (1997) 71:4193–8. doi: 10.1128/jvi.71.6.4193-4198.1997
257. Ojala PM, Tiainen M, Salven P, Veikkola T, Castanos-Velez E, Sarid R, et al. Kaposi’s sarcoma-associated herpesvirus-encoded v-cyclin triggers apoptosis in cells with high levels of cyclin-dependent kinase 6. Cancer Res. (1999) 59:4984–9.
258. Sarek G, Jarviluoma A, Moore HM, Tojkander S, Vartia S, Biberfeld P, et al. Nucleophosmin phosphorylation by v-cyclin-CDK6 controls KSHV latency. PloS Pathog. (2010) 6(3):e1000818. doi: 10.1371/journal.ppat.1000818
259. Ballon G, Chen K, Perez R, Tam W, Cesarman E. Kaposi sarcoma herpesvirus (KSHV) vFLIP oncoprotein induces B cell transdifferentiation and tumorigenesis in mice. J Clin Invest. (2011) 121:1141–53. doi: 10.1172/JCI44417
260. Chaudhary PM, Jasmin A, Eby MT, Hood L. Modulation of the NF-kappa B pathway by virally encoded death effector domains-containing proteins. Oncogene. (1999) 18:5738–46. doi: 10.1038/sj.onc.1202976
261. Lee JS, Li Q, Lee JY, Lee SH, Jeong JH, Lee HR, et al. FLIP-mediated autophagy regulation in cell death control. Nat Cell Biol. (2009) 11:1355–62. doi: 10.1038/ncb1980
262. Direkze S, Laman H. Regulation of growth signalling and cell cycle by Kaposi’s sarcoma-associated herpesvirus genes. Int J Exp Pathol. (2004) 85:305–19. doi: 10.1111/j.0959-9673.2004.00407.x
263. Wu YH, Hu TF, Chen YC, Tsai YN, Tsai YH, Cheng CC, et al. The manipulation of miRNA-gene regulatory networks by KSHV induces endothelial cell motility. Blood. (2011) 118:2896–905. doi: 10.1182/blood-2011-01-330589
264. Qin J, Li W, Gao SJ, Lu C. KSHV microRNAs: tricks of the devil. Trends Microbiol. (2017) 25:648–61. doi: 10.1016/j.tim.2017.02.002
265. Gottwein E, Mukherjee N, Sachse C, Frenzel C, Majoros WH, Chi JT, et al. A viral microRNA functions as an orthologue of cellular miR-155. Nature. (2007) 450:1096–9. doi: 10.1038/nature05992
266. Eis PS, Tam W, Sun L, Chadburn A, Li Z, Gomez MF, et al. Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc Natl Acad Sci U.S.A. (2005) 102:3627–32. doi: 10.1073/pnas.0500613102
267. Costinean S, Zanesi N, Pekarsky Y, Tili E, Volinia S, Heerema N, et al. Pre-B cell proliferation and lymphoblastic leukemia/high-grade lymphoma in E(mu)-miR155 transgenic mice. Proc Natl Acad Sci U.S.A. (2006) 103:7024–9. doi: 10.1073/pnas.0602266103
268. Boni M, Rieble L, Munz C. Co-infection of the epstein-barr virus and the kaposi sarcoma-associated herpesvirus. Viruses-Basel. (2022) 14(12):2709. doi: 10.3390/v14122709
269. Sugden AU, Hayes M, Sugden B. How epstein-barr virus and kaposi’s sarcoma-associated herpesvirus are maintained together to transform the same B-cell. Viruses-Basel. (2021) 13(8):1478. doi: 10.3390/v13081478
270. Challine D, Buisson M, Cadilhac M, Germanidis G, Joab I, Eliaszewicz M, et al. Hepatitis C virus-Epstein-Barr virus interaction in patients with AIDS. J Med Virol. (2002) 67:510–5. doi: 10.1002/jmv.10130
271. Guan M, Zhang RD, Wu B, Henderson EE. Infection of primary CD4+ and CD8+ T lymphocytes by Epstein-Barr virus enhances human immunodeficiency virus expression. J Virol. (1996) 70:7341–6. doi: 10.1128/jvi.70.10.7341-7346.1996
Keywords: virus, lymphoma, pathogenesis, EBV, HBV, HCV, HIV
Citation: Zhang Y, Guo W, Zhan Z and Bai O (2024) Carcinogenic mechanisms of virus-associated lymphoma. Front. Immunol. 15:1361009. doi: 10.3389/fimmu.2024.1361009
Received: 24 December 2023; Accepted: 12 February 2024;
Published: 28 February 2024.
Edited by:
Luwen Zhang, University of Nebraska-Lincoln, United StatesReviewed by:
Pankaj Trivedi, Sapienza University of Rome, ItalyChristopher Jondle, Western Michigan University, United States
Copyright © 2024 Zhang, Guo, Zhan and Bai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ou Bai, baiou@jlu.edu.cn