 Xin Wu
Xin Wu Sidhant Singla
Sidhant Singla Jianhua J. Liu2
Jianhua J. Liu2 Liang Hong
Liang Hong- 1Department of Medicine, University of Illinois at Chicago, Chicago, IL, United States
- 2Department of Pathology, University of Illinois at Chicago, Chicago, IL, United States
- 3Department of Physiology and Biophysics, University of Illinois at Chicago, Chicago, IL, United States
Atherosclerosis is a complex inflammatory disease that affects the arteries and can lead to severe complications such as heart attack and stroke. Macrophages, a type of immune cell, play a crucial role in atherosclerosis initiation and progression. Emerging studies revealed that ion channels regulate macrophage activation, polarization, phagocytosis, and cytokine secretion. Moreover, macrophage ion channel dysfunction is implicated in macrophage-derived foam cell formation and atherogenesis. In this context, exploring the regulatory role of ion channels in macrophage function and their impacts on the progression of atherosclerosis emerges as a promising avenue for research. Studies in the field will provide insights into novel therapeutic targets for the treatment of atherosclerosis.
1 Introduction
Atherosclerosis is a complex process that involves the interaction between oxidized low-density lipoproteins (oxLDLs), macrophages, endothelial cells, and vascular smooth muscle cells (1). It is an inflammatory disease of the large artery driven by macrophage activation and infiltration. Through scavenger receptors, macrophages uptake oxLDLs and other lipids, leading to foam cell formation and fatty streak lesions characteristic of early atherosclerosis. The macrophage-derived foam cells frequently undergo apoptosis to give rise to cholesterol-rich necrotic cores to advance atherosclerotic lesions (2).
Ion channels, transmembrane proteins that allow the flow of ions across cell membranes, are emerging as important regulators of macrophage function (3, 4). Recent studies have revealed diverse roles of ion channels in macrophages and their implications in immune responses and inflammatory diseases, including atherosclerosis (5). In the present review, we discuss ion channel regulation of macrophage function and summarize studies in macrophage ion channel dysfunctions associated with pathogenesis of atherosclerosis.
2 Macrophage in the progression of atherosclerosis
Macrophages play a crucial role in the initiation, progression, and complications of atherosclerosis (2, 6, 7). In the presence of risk factors such as high blood pressure, dyslipidemia, diabetes, and smoking, plasma lipoproteins enter the intima by permeating through the vessel wall, where they are modified into oxidized lipoproteins including oxLDLs (8–11). This leads to the activation of endothelial cells, triggering the expression of adhesion and chemotactic factors that attract monocytes. Consequently, monocytes differentiate into macrophages and transform into foam cells in the intima.
The foam cells are formed by the uptake and accumulation of oxLDLs through scavenger receptors in macrophages, leading to the development of fatty streak lesions and atherosclerotic plaques (7). Macrophages express several scavenger receptors including SR-A1 (Scavenger Receptor - Class A1) and CD36 (i.e., SR-B2, Scavenger Receptor - Class B2). The SR-A1 binds to the modified LDL such as oxLDL and is involved in the subendothelial translocation of LDL. SR-A1 function is associated with JNK2 signal pathway, it was reported that JNK2-dependent phosphorylation of SR-A1 promotes the uptake of LDL in macrophage and enhances the foam cell formation (12). The CD36 is the predominant scavenger receptor for oxLDL, and the binding between CD36 and oxLDL triggers TLRs signaling pathway to promote pro-inflammatory responses to mediate the progression of foam cell formation and atherosclerosis (13).
Meanwhile, macrophages in atherosclerotic lesions produce various inflammatory cytokines such as IL-1β, IL-6, IL-12, IL-18, and TNF to promote smooth muscle cell proliferation and recruit other inflammatory cells (14). For example, IL-1β induces angiogenesis through the recruitment of myeloid and endothelial cells. IL-6 promotes activation of endothelial cells and induces smooth muscle cells proliferation. IL-12 stimulates the differentiation of T cells and recruits T cells into the atherosclerotic plaques. IL-18 enhances the expression of scavenger receptor CD36 to accelerate atherosclerosis. And TNF induces expression of adhesion molecules and enhances the recruitment of other inflammatory cytokines into the plaques. Taken together, these sustained inflammatory responses create an atherogenic environment, further regulating plaque progression (2).
Within the microenvironment of atherosclerotic plaques, macrophages receive various stimulation and polarize into distinct subtypes. The macrophage subtypes, including M1 and M2, play diverse roles in the advancement of atherosclerosis. The M1 macrophages secrete pro-inflammatory cytokines, contribute to plaque instability, and promote thrombosis. In contrast, M2 macrophages exhibit anti-inflammatory properties, promote tissue repair, and aid in the resolution of inflammation. Imbalances in macrophage polarization affect plaque stabilization in the development of atherosclerosis (6).
3 Ion channels in macrophages
Ion channels are transmembrane proteins that enable the passage of ions across cell membranes and regulate physiological processes (15–23). Recent studies have shown ion channels regulate macrophage function (3, 4). Several ion channels in macrophages are implicated in immune responses and inflammatory diseases. The macrophage ion channels include potassium channel, transient receptor potential (TRP) channel, calcium channel, mechanosensitive Piezo channel, chloride channel, and proton channel (3–5).
Potassium channels are crucial regulators of macrophage membrane potential and ion homeostasis. Macrophages express a variety of potassium channels, including inward-rectifying potassium channels (Kir), voltage-gated potassium channels (Kv), and Ca2+-activated potassium channels (KCa) (5, 24). These channels modulate macrophage membrane potential, calcium signaling, cytokine release, and phagocytosis. Dysregulation of potassium channels can lead to abnormal macrophage activation and cause chronic inflammation (25, 26).
TRP channels are a diverse family of cation channels that play important roles in various physiological and pathological processes (27). Several TRP channels, such as TRPA1, TRPC3, TRPM2, and TRPV4, are expressed in macrophages (5). Emerging evidence suggests their involvement in macrophage function and inflammatory responses. TRP channels have been shown to modulate macrophage M1/M2 polarization and play roles in calcium homeostasis and reactive oxygen species (ROS) production. Some TRP channels have been implicated in the formation of macrophage-derived foam cells and the development of atherosclerosis (28–34).
Macrophages also express multiple types of calcium channels, including store-operated calcium channels (SOCCs) and voltage-gated calcium channels (Cav) (3, 5). Calcium ions (Ca2+) serve as universal second messengers and play a central role in macrophage signaling. Calcium influx through the calcium channels triggers intracellular signaling cascades, leading to macrophage activation, phagocytosis, and cytokine secretion. Abnormal calcium signaling in macrophages has been associated with various inflammatory diseases (35, 36).
In addition, macrophages express chloride channels involved in cellular volume regulation and mechanosensitive Piezo1 that mediated cellular mechano-signaling. Macrophages also express proton channels regulating the activation of phagocyte NADPH oxidase to regulate the process of phagocytosis (3, 5).
4 Macrophage ion channels associated with atherosclerosis
In the development of atherosclerosis, macrophage ion channels play important roles in several key cellular events, such as macrophage polarization and infiltration, cell proliferation and migration, and foam cell apoptosis. Recent studies indicated that macrophage ion channels are associated with the pathogenesis of atherosclerosis (Table 1).
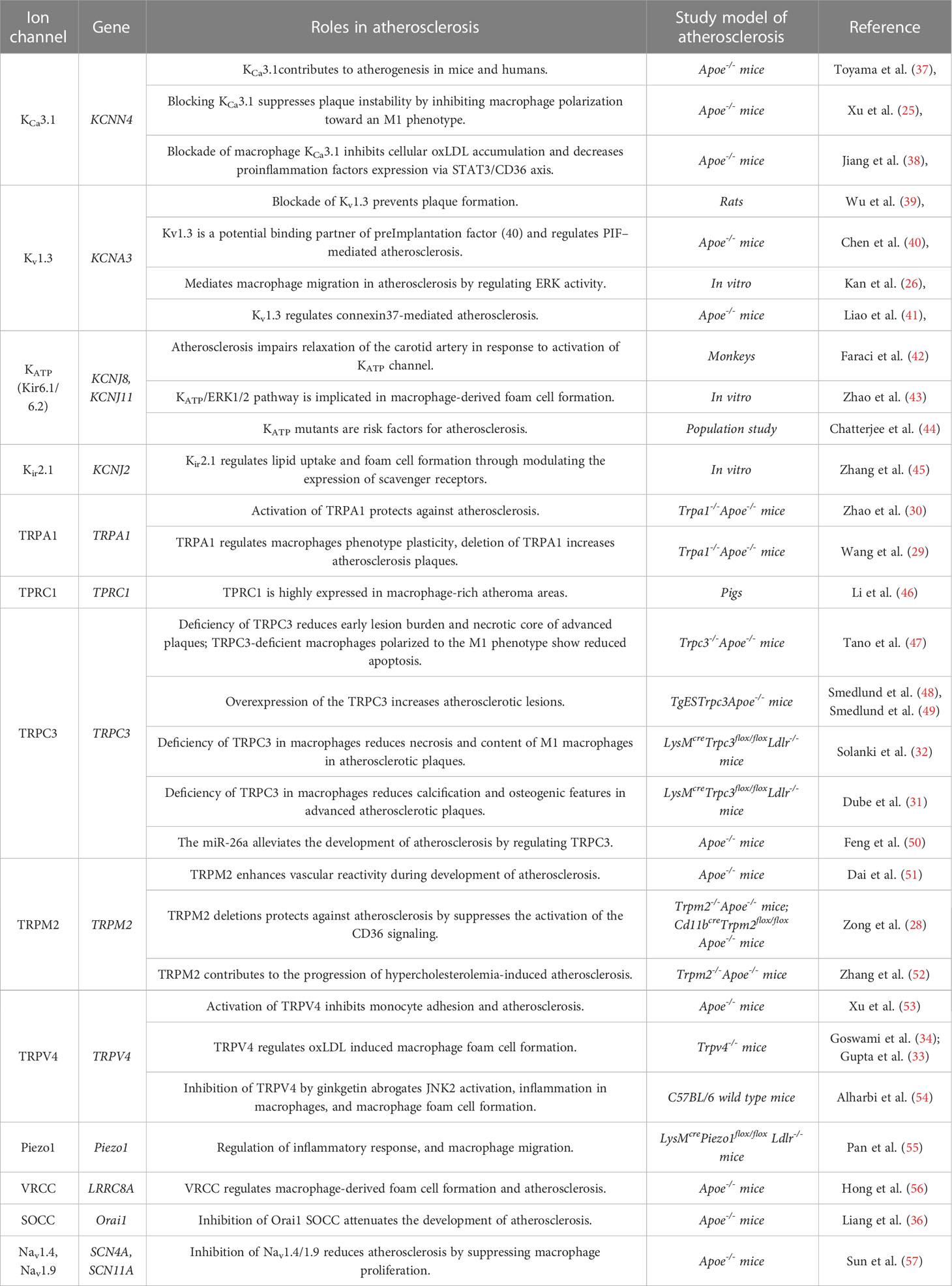
Table 1 The role of macrophage ion channels in atherosclerosis.
4.1 Calcium-activated potassium channel (KCa)
The KCa3.1 is the predominant subtype of calcium-activated potassium channels in macrophages. KCa3.1 regulates macrophage activity and plays an essential role in the progression of atherosclerosis.
One study found that KCa3.1 expression is upregulated in macrophages within atherosclerotic plaques in Apoe-/- mouse model of atherosclerosis and human patients. Inhibition of KCa3.1 with the treatment of TRAM-34 and clotrimazole prevents macrophage activation (37). Moreover, the migratory response of KCa3.1-/- macrophages was significantly reduced than in KCa3.1+/+ macrophages, indicating a role of KCa3.1 in the activation of macrophages during the atherosclerosis progression. Xu et al. showed that KCa3.1 regulates macrophage polarization. Blocking KCa3.1 suppresses macrophage polarization towards the M1 phenotype, reducing atherosclerotic plaque instability (25). Moreover, a recent study discovered that KCa3.1 modifies the development of atherosclerosis via the STAT3/CD36 signaling axis (38).
4.2 Voltage dependent potassium channel (Kv)
Kv1.3 is one of the voltage-gated potassium channels predominantly expressed in macrophages. Kv1.3 regulates the membrane potential of immune cells and is an important modulator of calcium signaling and cytokine production. In macrophages, Kv1.3 regulates the activation and proliferation of the cells, as well as the production of pro-inflammatory cytokines such as tumor necrosis factor-alpha (TNF-α) and interleukin-1 beta (IL-1β).
Previous studies showed that pharmacological inhibition of Kv1.3 channels reduces atherosclerotic lesion area in mouse models (39), suggesting a pro-atherosclerotic role of Kv1.3. The mechanisms by which Kv1.3 underlies the development of atherosclerosis are associated with preimplantation factor (58) and extracellular signal-regulated kinase (ERK) signaling pathway. Kv1.3 is proposed to be a binding partner of PIF and regulates PIF-mediated atherosclerosis (40). Kv1.3 has also been shown to modify ERK activity to promote macrophage migration during the progression of atherosclerosis. Inhibition of Kv1.3 channel attenuates macrophage migration and reduces the phosphorylation level of ERK1/2 (26). Additionally, other studies reported that connexin is involved in Kv1.3-mediated atherosclerosis (41).
4.3 Inward-rectifying potassium channel (Kir)
The primary macrophage Kir involved in atherosclerosis is ATP-sensitive potassium channels (Kir6.1/6.2, or KATP). In macrophages, the KATP channel is essential for the regulation of inflammation and the immune response.
The first study showing the relationship between KATP channel and atherosclerosis is from a monkey model of atherosclerosis, and it was shown that atherosclerosis impairs the relaxation of the carotid artery in response to activation of the KATP channel (42). Further studies revealed that KATP participates in macrophage-derived foam cell formation. Zhao et al. found that the downregulations of total cholesterol and esterified cholesterol concentrations induced by hydrogen sulfide (H2S), were reversed by KATP blocker glibenclamide, suggesting that KATP channel promotes the formation of macrophage-derived foam cells (43). Moreover, a population study indicated that KATP mutants are risk factors for atherosclerosis (44).
In addition to KATP, another inward-rectifying potassium channel Kir2.1 also participates in the development of atherosclerosis. Kir2.1 regulates macrophage maturation and differentiation and plays a crucial role in lipid uptake and foam cell formation by modulating the expression of scavenger receptors (45).
4.4 Transient receptor potential ankyrin channel (TRPA)
The transient receptor potential ankyrin 1 (TRPA1) channel is a non-selective cation channel widely expressed in immune cells, including macrophages. TRPA1 channels play a key role in regulating inflammation. In recent years, several studies have explored the potential role of TRPA1 channels in atherosclerosis.
TRPA1 has been shown to regulate the cholesterol metabolism of macrophage-derived foam cells. OxLDL-induced lipid accumulation of macrophages is exacerbated by either inhibition or loss of function of TRPA1, leading to the progression of atherosclerotic plaques. On the other hand, treatment with TRPA1 agonists alleviates the development of atherosclerosis in Apoe-/- mice, indicating that TRPA1 protects against atherosclerosis (30). A recent study found that TRPA1 modifies macrophage phenotype plasticity. Inhibition of TRPA1 enhances M1 marker genes expression whereas downregulates M2 genes expression (29).
4.5 Transient receptor potential canonical channel (TRPC)
TPRC3 is one of the primary TRPC channels expressed in macrophages. In atherosclerosis, macrophage TRPC3 channel activation enhances inflammation and the development of atherosclerotic plaques. TRPC3 channel activation in macrophages can increase the expression of inflammatory cytokines and chemokines, promote the recruitment of additional immune cells to the site of inflammation, and contribute to the formation of atherosclerotic lesions.
A bone marrow transplantation study revealed that macrophage deficiency of TRPC3 reduces early lesion burden and necrotic core of advanced plaques in Apoe-/- mice (47). Moreover, macrophage-specific deletion of TRPC3 was reported to decrease necrosis and content of apoptotic M1 macrophages in advanced atherosclerotic plaques of mice (31, 32). The miR-26a was shown to alleviate the development of atherosclerosis by regulating TRPC3 (50). Additionally, studies in endothelial cells discovered that endothelial overexpression of the human TRPC3 channel increased the size and cellularity of advanced atherosclerotic lesions in mice model of atherosclerosis (48, 49).
In addition to TRPC3, another TRPC channel TRPC1 has been shown to be predominantly expressed in macrophage-rich atheroma areas, indicating macrophage TRPC1 plays a role in atherogenesis (46). The mechanisms by which TRPC1 channels regulate macrophage function in atherosclerosis are not fully understood. Further research is required to elucidate the signaling pathways and molecular mechanisms involved in TRPC1-mediated atherosclerosis.
4.6 Transient receptor potential melastatin channel (TRPM)
Transient receptor potential melastatin channel member 2 (TRPM2) is highly expressed in macrophages and promotes atherosclerotic progression (28, 51, 52). It was shown that both global and macrophage-specific TRPM2 deletions could protect Apoe-/- mice against atherosclerosis (28, 52). Inhibition of TRPM2 channel activity in macrophages decreases the production of ROS and pro-inflammatory cytokines, and reduces the size of atherosclerotic lesions in multiple mice models of atherosclerosis (28, 52). TRPM2 deficiency in macrophages decreases the uptake of oxLDL, and reduces macrophage infiltration, foam cell formation, and inflammatory responses. Further studies showed that TRPM2 activation is required for CD36-induced oxLDL uptake and macrophage inflammatory responses. Deletion of the TRPM2 gene or inhibiting TRPM2 channel activity suppresses the activation of the CD36 signaling, suggesting that the TRPM2–CD36 axis plays a vital role in atherogenesis (28).
4.7 Transient receptor potential vanilloid channel (TRPV)
Transient receptor potential vanilloid channel member 4 (TRPV4) has been implicated in the formation of macrophage-derived foam cells and the development of atherosclerosis.
TRPV4 is expressed and functional in mouse macrophages. It is required for oxLDL-induced macrophage foam cell formation and regulates the uptake of oxLDL (33, 34). Inhibition of TRPV4 by ginkgetin abrogates JNK2 activation, inflammation in macrophages, and macrophage foam cell formation (54). These results indicate that TRPV4 activity is essential for macrophage foam cell formation and atherosclerosis progression. In addition to its direct effects on macrophages, TRPV4 contributes to atherosclerosis by regulating endothelial cell function. In a study using human monocytes, inhibition of TRPV4 reduces monocyte/macrophage adhesion to endothelial cells to regulate the progression of atherosclerosis (53).
4.8 Other ion channels
In addition to potassium channels and TRP channels, several other macrophage ion channels were implicated in atherosclerosis.
Recent studies have revealed that Piezo type mechanosensitive ion channel 1 (Piezo1) in macrophage has been implicated in atherosclerosis (55, 59, 60). This channel appears to play a pro-atherosclerotic role in atherogenesis. It was shown that macrophage specific deletion of Piezo1 gene significantly reduced atherosclerotic plaques in Ldlr-/- mice model of atherosclerosis (55). Piezo1 was proposed to participate in macrophage inflammatory activation, proliferation, and migration/infiltration to mediate the progression of atherosclerosis (59, 60).
Additionally, macrophage chloride channels have been linked with atherosclerosis (56). It was reported that volume-regulated chloride channel (VRCC) plays an essential role in macrophage foam cell formation (56), and the activity of VRCC was enhanced in Apoe-/- mice model of atherosclerosis. The activation of VRCC accelerated the formation of macrophage foam cells, whereas the chloride blockers inhibition of VRCC impaired the foam cell formation (56).
Moreover, macrophage calcium and sodium channels are also linked with atherogenesis. It was shown that Orai1 store-operated calcium channel (SOCC) is required for oxLDL-induced Ca2+ influx in macrophages. And in vivo studies revealed that inhibition of Orai1 SOCC attenuates the development of atherosclerosis (36). In addition, inhibition of voltage-gated sodium channel (Nav) suppressed macrophage proliferation and reduced atherosclerotic lesions in the Apoe-/- mouse model, shedding light on the role of Nav sodium channels in atherogenesis (57).
5 Conclusions and perspectives
We discuss ion channels regulation of macrophage function during the progression of atherosclerosis, as well as summarize recent studies in macrophage ion channel families associated with atherosclerosis. Macrophages are critical players in the pathogenesis of atherosclerosis, influencing multiple stages of plaque development and plaque stability. Ion channels play critical roles in macrophage biology, regulating diverse cellular processes that impact macrophage activation, polarization, phagocytosis, and cytokine secretion. Dysregulation of ion channels is implicated in macrophage-mediated atherogenesis, making them attractive targets for therapeutic intervention.
Although significant progress was made in the functional characterization of the K+, TRP, Ca2+, and Cl- channels in the progression of atherosclerosis (Figure 1), emerging evidence indicated other macrophage ion channels, such as intracellular channels and H+ channels, are potentially novel targets against atherosclerosis.
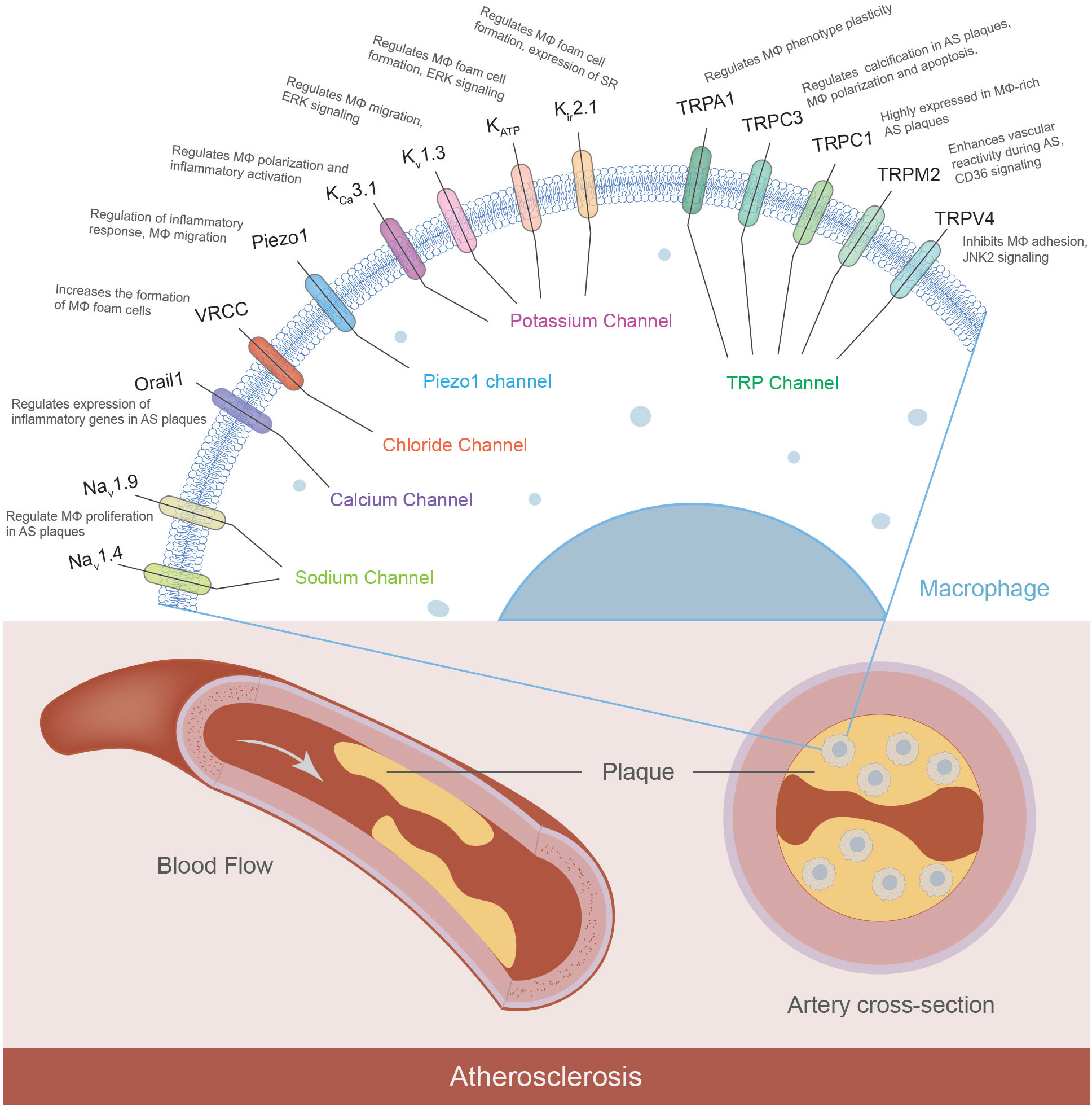
Figure 1 Macrophage ion channels in atherosclerosis. Macrophages express a variety of ion channels, and some of them have been characterized to contribute to the pathophysiology of atherosclerosis. The macrophage ion channels implicated in atherogenesis include K+ channels (KCa3.1, Kv1.3, KATP, Kir2.1), TRP channels (TRPA1, TRPC1, TRPC3, TRPM2, TRPV4), Orai1 Ca2+ channel, volume-regulated Cl- channel VRCC, mechanosensitive Piezo1, and voltage-gated Na+ channels Nav. (MΦ: macrophages; AS: atherosclerosis; SR: scavenger receptor).
For example, studies have shown that ryanodine receptor 3 (RyR3) channel mutations are associated with atherosclerosis in populations (61). RyR3 is one of the ryanodine receptor channel isoforms expressed on the endoplasmic reticulum of immune cells, including macrophages (62, 63), RyR3 mutations may cause the dysfunction of the channel, which in turn, lead to abnormal calcium signaling linked with the development of atherosclerosis.
Meanwhile, accumulated evidence suggests that Hv1 proton channel is associated with atherosclerosis. Hv1 channel controls acid extrusion from cells and regulates cellular pH homeostasis (64). It is highly expressed in macrophages, and its activity promotes macrophage migration and inflammatory cytokines secretion (65, 66). The microarray data has revealed that Hv1 was remarkably upregulated during atherogenesis and downregulated along with the atherosclerotic lesion regression (67), indicating that Hv1 is linked with atherogenesis and involved in the pathological process of this disease.
Moreover, acid-sensing ion channel member 1 (ASIC1) was recently proposed to play a role in atherosclerotic development. ASIC1 channel in macrophages decreases ATP-binding cassette transporter A1 (ABCA1)-mediated cholesterol efflux, indicating a role of macrophage ASIC1 in lipid metabolism and atherosclerosis progression (68).
Future studies are required to characterize new roles of these channels in the pathogenesis of atherosclerosis. Meanwhile, studies have shown that ion channels regulate genetic expression in various cells (69–72), it remains to determine if ion channels regulate the gene transcriptional networks controlling macrophage activation linked with atherogenesis. The investigation in this field will extend our understanding of the function of macrophage ion channels in human diseases and discover novel targets against atherosclerosis.
Author contributions
LH conceptualized the review. XW created the Figure and Table. XW, SS, JL, and LH wrote and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported in part by the National Institute of Health Grant R01GM139991 (LH), American Heart Association Grant 19CDA34630041 (LH), and University of Illinois Chicago Center for Clinical and Translational Science Grant UL1TR002003 (LH).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bjorkegren JLM, Lusis AJ. Atherosclerosis: Recent developments. Cell (2022) 185(10):1630–45. doi: 10.1016/j.cell.2022.04.004
2. Tabas I, Bornfeldt KE. Intracellular and Intercellular Aspects of Macrophage Immunometabolism in Atherosclerosis. Circ Res (2020) 126(9):1209–27. doi: 10.1161/CIRCRESAHA.119.315939
3. Selezneva A, Gibb AJ, Willis D. The contribution of ion channels to shaping macrophage behaviour. Front Pharmacol (2022) 13:970234. doi: 10.3389/fphar.2022.970234
4. Eder C. Ion channels in microglia (brain macrophages). Am J Physiol (1998) 275(2):C327–42. doi: 10.1152/ajpcell.1998.275.2.C327
5. Feske S, Wulff H, Skolnik EY. Ion channels in innate and adaptive immunity. Annu Rev Immunol (2015) 33:291–353. doi: 10.1146/annurev-immunol-032414-112212
6. Eshghjoo S, Kim DM, Jayaraman A, Sun Y, Alaniz RC. Macrophage Polarization in Atherosclerosis. Genes (Basel) (2022) 13(5):756. doi: 10.3390/genes13050756
7. Choudhury RP, Lee JM, Greaves DR. Mechanisms of disease: macrophage-derived foam cells emerging as therapeutic targets in atherosclerosis. Nat Clin Pract Cardiovasc Med (2005) 2(6):309–15. doi: 10.1038/ncpcardio0195
8. Boullier A, Bird DA, Chang MK, Dennis EA, Friedman P, Gillotre-Taylor K, et al. Scavenger receptors, oxidized LDL, and atherosclerosis. Ann N Y Acad Sci (2001) 947:214–22; discussion 22-3. doi: 10.1111/j.1749-6632.2001.tb03943.x
9. Chen K, Febbraio M, Li W, Silverstein RL. A specific CD36-dependent signaling pathway is required for platelet activation by oxidized low-density lipoprotein. Circ Res (2008) 102(12):1512–9. doi: 10.1161/CIRCRESAHA.108.172064
10. Ganesan R, Henkels KM, Wrenshall LE, Kanaho Y, Di Paolo G, Frohman MA, et al. Oxidized LDL phagocytosis during foam cell formation in atherosclerotic plaques relies on a PLD2-CD36 functional interdependence. J Leukoc Biol (2018) 103(5):867–83. doi: 10.1002/JLB.2A1017-407RR
11. Tang M, Hong L, Li H, Chen W, Tai L, Minshall R, et al. Stiffness of aortic arch and carotid arteries increases in ApoE-knockout mice with high-fat diet: evidence from echocardiography. Am J Transl Res (2021) 13(3):1352–64.
12. Ricci R, Sumara G, Sumara I, Rozenberg I, Kurrer M, Akhmedov A, et al. Requirement of JNK2 for scavenger receptor A-mediated foam cell formation in atherogenesis. Science (2004) 306(5701):1558–61. doi: 10.1126/science.1101909
13. Hoebe K, Georgel P, Rutschmann S, Du X, Mudd S, Crozat K, et al. CD36 is a sensor of diacylglycerides. Nature (2005) 433(7025):523–7. doi: 10.1038/nature03253
14. Farahi L, Sinha SK, Lusis AJ. Roles of Macrophages in Atherogenesis. Front Pharmacol (2021) 12:785220. doi: 10.3389/fphar.2021.785220
15. Alexander SPH, Mathie A, Peters JA, Veale EL, Striessnig J, Kelly E, et al. THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Ion channels. Br J Pharmacol (2019) 176 Suppl 1:S142–228. doi: 10.1111/bph.14749
16. Wu X, Hong L. Calmodulin Interactions with Voltage-Gated Sodium Channels. Int J Mol Sci (2021) 22(18):9798. doi: 10.3390/ijms22189798
17. Hong L, Zhang M, Sridhar A, Darbar D. Pathogenic mutations perturb calmodulin regulation of Nav1.8 channel. Biochem Biophys Res Commun (2020) 533(1):168-74. doi: 10.1016/j.bbrc.2020.08.010
18. Ly OT, Chen H, Brown GE, Hong L, Wang X, Han YD, et al. Mutant ANP induces mitochondrial and ion channel remodeling in a human iPSC-derived atrial fibrillation model. JCI Insight (2022) 7(7):e155640. doi: 10.1172/jci.insight.155640
19. Wu X, Li Y, Hong L. Effects of Mexiletine on a Race-specific Mutation in Nav1.5 Associated With Long QT Syndrome. Front Physiol (2022) 13:904664. doi: 10.3389/fphys.2022.904664
20. Wu X, Zhang L, Hong L. The role of Phe150 in human voltage-gated proton channel. iScience (2022) 25(11):105420. doi: 10.1016/j.isci.2022.105420
21. Zhao C, Hong L, Riahi S, Lim VT, Tobias DJ, Tombola F. A novel Hv1 inhibitor reveals a new mechanism of inhibition of a voltage-sensing domain. J Gen Physiol (2021) 153(9)::e202012833. doi: 10.1085/jgp.202012833
22. Zhao C, Hong L, Galpin JD, Riahi S, Lim VT, Webster PD, et al. HIFs: New arginine mimic inhibitors of the Hv1 channel with improved VSD-ligand interactions. J Gen Physiol (2021) 153(9):e202012832. doi: 10.1085/jgp.202012832
23. Alkhalil A, Hong L, Nguitragool W, Desai SA. Voltage-dependent inactivation of the plasmodial surface anion channel via a cleavable cytoplasmic component. Biochim Biophys Acta (2012) 1818(3):367–74. doi: 10.1016/j.bbamem.2011.11.010
24. Man Q, Gao Z, Chen K. Functional Potassium Channels in Macrophages. J Membr Biol (2023) 256(2):175–87. doi: 10.1007/s00232-022-00276-4
25. Xu R, Li C, Wu Y, Shen L, Ma J, Qian J, et al. Role of KCa3.1 Channels in Macrophage Polarization and Its Relevance in Atherosclerotic Plaque Instability. Arterioscler Thromb Vasc Biol (2017) 37(2):226–36. doi: 10.1161/ATVBAHA.116.308461
26. Kan XH, Gao HQ, Ma ZY, Liu L, Ling MY, Wang YY. Kv1.3 potassium channel mediates macrophage migration in atherosclerosis by regulating ERK activity. Arch Biochem Biophys (2016) 591:150–6. doi: 10.1016/j.abb.2015.12.013
27. Santoni G, Morelli MB, Amantini C, Santoni M, Nabissi M, Marinelli O, et al. "Immuno-Transient Receptor Potential Ion Channels": The Role in Monocyte- and Macrophage-Mediated Inflammatory Responses. Front Immunol (2018) 9:1273. doi: 10.3389/fimmu.2018.01273
28. Zong P, Feng J, Yue Z, Yu AS, Vacher J, Jellison ER, et al. TRPM2 deficiency in mice protects against atherosclerosis by inhibiting TRPM2-CD36 inflammatory axis in macrophages. Nat Cardiovasc Res (2022) 1(4):344–60. doi: 10.1038/s44161-022-00027-7
29. Wang Q, Chen K, Zhang F, Peng K, Wang Z, Yang D, et al. TRPA1 regulates macrophages phenotype plasticity and atherosclerosis progression. Atherosclerosis (2020) 301:44–53. doi: 10.1016/j.atherosclerosis.2020.04.004
30. Zhao JF, Shyue SK, Kou YR, Lu TM, Lee TS. Transient Receptor Potential Ankyrin 1 Channel Involved in Atherosclerosis and Macrophage-Foam Cell Formation. Int J Biol Sci (2016) 12(7):812–23. doi: 10.7150/ijbs.15229
31. Dube PR, Chikkamenahalli LL, Birnbaumer L, Vazquez G. Reduced calcification and osteogenic features in advanced atherosclerotic plaques of mice with macrophage-specific loss of TRPC3. Atherosclerosis (2018) 270:199–204. doi: 10.1016/j.atherosclerosis.2017.12.025
32. Solanki S, Dube PR, Birnbaumer L, Vazquez G. Reduced Necrosis and Content of Apoptotic M1 Macrophages in Advanced Atherosclerotic Plaques of Mice With Macrophage-Specific Loss of Trpc3. Sci Rep (2017) 7:42526. doi: 10.1038/srep42526
33. Gupta N, Goswami R, Alharbi MO, Biswas D, Rahaman SO. TRPV4 is a regulator in P. gingivalis lipopolysaccharide-induced exacerbation of macrophage foam cell formation. Physiol Rep (2019) 7(7):e14069. doi: 10.14814/phy2.14069
34. Goswami R, Merth M, Sharma S, Alharbi MO, Aranda-Espinoza H, Zhu X, et al. TRPV4 calcium-permeable channel is a novel regulator of oxidized LDL-induced macrophage foam cell formation. Free Radic Biol Med (2017) 110:142–50. doi: 10.1016/j.freeradbiomed.2017.06.004
35. Soldatov NM. Cav1.2, cell proliferation, and new target in atherosclerosis. ISRN Biochem (2013) 2013:463527. doi: 10.1155/2013/463527
36. Liang SJ, Zeng DY, Mai XY, Shang JY, Wu QQ, Yuan JN, et al. Inhibition of Orai1 Store-Operated Calcium Channel Prevents Foam Cell Formation and Atherosclerosis. Arterioscler Thromb Vasc Biol (2016) 36(4):618–28. doi: 10.1161/ATVBAHA.116.307344
37. Toyama K, Wulff H, Chandy KG, Azam P, Raman G, Saito T, et al. The intermediate-conductance calcium-activated potassium channel KCa3.1 contributes to atherogenesis in mice and humans. J Clin Invest (2008) 118(9):3025–37. doi: 10.1172/JCI30836
38. Jiang XX, Bian W, Zhu YR, Wang Z, Ye P, Gu Y, et al. Targeting the KCa3.1 channel suppresses diabetes-associated atherosclerosis via the STAT3/CD36 axis. Diabetes Res Clin Pract (2022) 185:109776. doi: 10.1016/j.diabres.2022.109776
39. Wu X, Xu R, Cao M, Ruan L, Wang X, Zhang C. Effect of the Kv1.3 voltage-gated potassium channel blocker PAP-1 on the initiation and progress of atherosclerosis in a rat model. Heart Vessels (2015) 30(1):108–14. doi: 10.1007/s00380-013-0462-7
40. Chen YC, Rivera J, Fitzgerald M, Hausding C, Ying YL, Wang X, et al. PreImplantation factor prevents atherosclerosis via its immunomodulatory effects without affecting serum lipids. Thromb Haemost (2016) 115(5):1010–24. doi: 10.1160/TH15-08-0640
41. Liao M, Chen L, Lu J, Liang G, Yao Y, Ouyang S, et al. Connexin 37 Regulates the Kv1.3 Pathway and Promotes the Development of Atherosclerosis. Mediators Inflammation (2022) 2022:2689918. doi: 10.1155/2022/2689918
42. Faraci FM, Orgren K, Heistad DD. Impaired relaxation of the carotid artery during activation of ATP-sensitive potassium channels in atherosclerotic monkeys. Stroke (1994) 25(1):178–82. doi: 10.1161/01.STR.25.1.178
43. Zhao ZZ, Wang Z, Li GH, Wang R, Tan JM, Cao X, et al. Hydrogen sulfide inhibits macrophage-derived foam cell formation. Exp Biol Med (Maywood) (2011) 236(2):169–76. doi: 10.1258/ebm.2010.010308
44. Chatterjee R, Davenport CA, Raffield LM, Maruthur N, Lange L, Selvin E, et al. KCNJ11 variants and their effect on the association between serum potassium and diabetes risk in the Atherosclerosis Risk in Communities (ARIC) Study and Jackson Heart Study (JHS) cohorts. PloS One (2018) 13(8):e0203213. doi: 10.1371/journal.pone.0203213
45. Zhang W, Lei XJ, Wang YF, Wang DQ, Yuan ZY. Role of Kir2.1 in human monocyte-derived foam cell maturation. J Cell Mol Med (2016) 20(3):403–12. doi: 10.1111/jcmm.12705
46. Li W, Chen X, Riley AM, Hiett SC, Temm CJ, Beli E, et al. Long-term spironolactone treatment reduces coronary TRPC expression, vasoconstriction, and atherosclerosis in metabolic syndrome pigs. Basic Res Cardiol (2017) 112(5):54. doi: 10.1007/s00395-017-0643-0
47. Tano JY, Solanki S, Lee RH, Smedlund K, Birnbaumer L, Vazquez G. Bone marrow deficiency of TRPC3 channel reduces early lesion burden and necrotic core of advanced plaques in a mouse model of atherosclerosis. Cardiovasc Res (2014) 101(1):138–44. doi: 10.1093/cvr/cvt231
48. Smedlund KB, Birnbaumer L, Vazquez G. Increased size and cellularity of advanced atherosclerotic lesions in mice with endothelial overexpression of the human TRPC3 channel. Proc Natl Acad Sci U.S.A. (2015) 112(17):E2201–6. doi: 10.1073/pnas.1505410112
49. Smedlund K, Dube P, Vazquez G. Early steatohepatitis in hyperlipidemic mice with endothelial-specific gain of TRPC3 function precedes changes in aortic atherosclerosis. Physiol Genomics (2016) 48(8):644–9. doi: 10.1152/physiolgenomics.00067.2016
50. Feng M, Xu D, Wang L. miR-26a inhibits atherosclerosis progression by targeting TRPC3. Cell Biosci (2018) 8:4. doi: 10.1186/s13578-018-0203-9
51. Dai YQ, Yan XX, Lin YC, Chen HY, Liu XR. Transient Receptor Potential Melastatin 2 Enhances Vascular Reactivity During Development of Atherosclerosis in Mice. Am J Hypertens (2021) 34(1):121–2. doi: 10.1093/ajh/hpaa154
52. Zhang Y, Ying F, Tian X, Lei Z, Li X, Lo CY, et al. TRPM2 Promotes Atherosclerotic Progression in a Mouse Model of Atherosclerosis. Cells (2022) 11(9):1423. doi: 10.3390/cells11091423
53. Xu S, Liu B, Yin M, Koroleva M, Mastrangelo M, Ture S, et al. A novel TRPV4-specific agonist inhibits monocyte adhesion and atherosclerosis. Oncotarget (2016) 7(25):37622–35. doi: 10.18632/oncotarget.9376
54. Alharbi MO, Dutta B, Goswami R, Sharma S, Lei KY, Rahaman SO. Identification and functional analysis of a biflavone as a novel inhibitor of transient receptor potential vanilloid 4-dependent atherogenic processes. Sci Rep (2021) 11(1):8173. doi: 10.1038/s41598-021-87696-9
55. Pan X, Wan R, Wang Y, Liu S, He Y, Deng B, et al. Inhibition of chemically and mechanically activated Piezo1 channels as a mechanism for ameliorating atherosclerosis with salvianolic acid B. Br J Pharmacol (2022) 179(14):3778–814. doi: 10.1111/bph.15826
56. Hong L, Xie ZZ, Du YH, Tang YB, Tao J, Lv XF, et al. Alteration of volume-regulated chloride channel during macrophage-derived foam cell formation in atherosclerosis. Atherosclerosis (2011) 216(1):59–66. doi: 10.1016/j.atherosclerosis.2011.01.035
57. Sun H, Jiang J, Gong L, Li X, Yang Y, Luo Y, et al. Voltage-gated sodium channel inhibitor reduces atherosclerosis by modulating monocyte/macrophage subsets and suppressing macrophage proliferation. BioMed Pharmacother (2019) 120:109352. doi: 10.1016/j.biopha.2019.109352
58. Zare F, Seifati SM, Dehghan-Manshadi M, Fesahat F. Preimplantation Factor (PIF): a peptide with various functions. JBRA Assist Reprod (2020) 24(2):214–18. doi: 10.5935/1518-0557.20190082
59. Shinge SAU, Zhang D, Achu Muluh T, Nie Y, Yu F. Mechanosensitive Piezo1 Channel Evoked-Mechanical Signals in Atherosclerosis. J Inflammation Res (2021) 14:3621–36. doi: 10.2147/JIR.S319789
60. Shinge SAU, Zhang D, Din AU, Yu F, Nie Y. Emerging Piezo1 signaling in inflammation and atherosclerosis; a potential therapeutic target. Int J Biol Sci (2022) 18(3):923–41. doi: 10.7150/ijbs.63819
61. Zhao C, Ikeda S, Arai T, Naka-Mieno M, Sato N, Muramatsu M, et al. Association of the RYR3 gene polymorphisms with atherosclerosis in elderly Japanese population. BMC Cardiovasc Disord (2014) 14:6. doi: 10.1186/1471-2261-14-6
62. Hosoi E, Nishizaki C, Gallagher KL, Wyre HW, Matsuo Y, Sei Y. Expression of the ryanodine receptor isoforms in immune cells. J Immunol (2001) 167(9):4887–94. doi: 10.4049/jimmunol.167.9.4887
63. Guidarelli A, Cerioni L, Fiorani M, Cantoni O. Differentiation-associated loss of ryanodine receptors: a strategy adopted by monocytes/macrophages to prevent the DNA single-strand breakage induced by peroxynitrite. J Immunol (2009) 183(7):4449–57. doi: 10.4049/jimmunol.0901260
64. DeCoursey TE. Voltage-gated proton channels: molecular biology, physiology, and pathophysiology of the H(V) family. Physiol Rev (2013) 93(2):599–652. doi: 10.1152/physrev.00011.2012
65. Canton J, Khezri R, Glogauer M, Grinstein S. Contrasting phagosome pH regulation and maturation in human M1 and M2 macrophages. Mol Biol Cell (2014) 25(21):3330–41. doi: 10.1091/mbc.e14-05-0967
66. Yu Y, Yu Z, Xie M, Wang W, Luo X. Hv1 proton channel facilitates production of ROS and pro-inflammatory cytokines in microglia and enhances oligodendrocyte progenitor cells damage from oxygen-glucose deprivation in vitro. Biochem Biophys Res Commun (2018) 498(1):1–8. doi: 10.1016/j.bbrc.2017.06.197
67. Li R, Paul A, Ko KW, Sheldon M, Rich BE, Terashima T, et al. Interleukin-7 induces recruitment of monocytes/macrophages to endothelium. Eur Heart J (2012) 33(24):3114–23. doi: 10.1093/eurheartj/ehr245
68. Zhang RJ, Yin YF, Xie XJ, Gu HF. Acid-sensing ion channels: Linking extracellular acidification with atherosclerosis. Clin Chim Acta (2020) 502:183–90. doi: 10.1016/j.cca.2019.12.027
69. House CD, Wang BD, Ceniccola K, Williams R, Simaan M, Olender J, et al. Voltage-gated Na+ Channel Activity Increases Colon Cancer Transcriptional Activity and Invasion Via Persistent MAPK Signaling. Sci Rep (2015) 5:11541. doi: 10.1038/srep11541
70. Wu X, Li Y, Maienschein-Cline M, Feferman L, Wu L, Hong L. RNA-Seq Analyses Reveal Roles of the HVCN1 Proton Channel in Cardiac pH Homeostasis. Front Cell Dev Biol (2022) 10:860502. doi: 10.3389/fcell.2022.860502
71. Yawei L, Yuan L. STdGCN: accurate cell-type deconvolution using graph convolutional networks in spatial transcriptomic data. bioRxiv (2023) 2023:03. doi: 10.1101/2023.03.10.532112
Keywords: macrophage, atherosclerosis, ion channel, foam cell formation, inflammatory disease
Citation: Wu X, Singla S, Liu JJ and Hong L (2023) The role of macrophage ion channels in the progression of atherosclerosis. Front. Immunol. 14:1225178. doi: 10.3389/fimmu.2023.1225178
Received: 18 May 2023; Accepted: 10 July 2023;
Published: 31 July 2023.
Edited by:
Yogesh Bhaskar Narkhede, University of Notre Dame, United StatesReviewed by:
Jun Chen, Hubei University of Medicine, ChinaYuanli Chen, Hefei University of Technology, China
Hong-Feng Gu, University of South China, China
Copyright © 2023 Wu, Singla, Liu and Hong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liang Hong, hong2004@uic.edu