Dysregulation of the Nitric Oxide/Dimethylarginine Pathway in Hypoxic Pulmonary Vasoconstriction—Molecular Mechanisms and Clinical Significance
 Juliane Hannemann1,2†
Juliane Hannemann1,2†  Rainer Böger1,2*†
Rainer Böger1,2*†- 1Institute of Clinical Pharmacology and Toxicology, University Medical Center Hamburg-Eppendorf, Hamburg, Germany
- 2Institute DECIPHER, German-Chilean Institute for Research on Pulmonary Hypoxia and its Health Sequelae, Hamburg, Germany
The pulmonary circulation responds to hypoxia with vasoconstriction, a mechanism that helps to adapt to short-lived hypoxic episodes. When sustained, hypoxic pulmonary vasoconstriction (HPV) may become deleterious, causing right ventricular hypertrophy and failure, and contributing to morbidity and mortality in the late stages of several chronic pulmonary diseases. Nitric oxide (NO) is an important endothelial vasodilator. Its release is regulated, amongst other mechanisms, by the presence of endogenous inhibitors like asymmetric dimethylarginine (ADMA). Evidence has accumulated in recent years that elevated ADMA may be implicated in the pathogenesis of HPV and in its clinical sequelae, like pulmonary arterial hypertension (PAH). PAH is one phenotypic trait in experimental models with disrupted ADMA metabolism. In high altitude, elevation of ADMA occurs during long-term exposure to chronic or chronic intermittent hypobaric hypoxia; ADMA is significantly associated with high altitude pulmonary hypertension. High ADMA concentration was also reported in patients with chronic obstructive lung disease, obstructive sleep apnoea syndrome, and overlap syndrome, suggesting a pathophysiological role for ADMA-mediated impairment of endothelium-dependent, NO-mediated pulmonary vasodilation in these clinically relevant conditions. Improved understanding of the molecular (dys-)regulation of pathways controlling ADMA concentration may help to dissect the pathophysiology and find novel therapeutic options for these diseases.
Introduction
Hypoxia is a deadly threat to every cell and to the organism as a whole. It is therefore not surprising that complex molecular mechanisms have evolved that help the cell to maintain its integrity during short-lived periods of hypoxia, as well as physiological mechanisms that help the organism to adapt to conditions of low oxygen supply.
In most organs, the response to a mismatch between oxygen demand and supply is an increase in blood flow. This has been demonstrated for the coronary, cerebral, renal, and other vascular beds (1–3). Hypoxia in the systemic circulation may result from local vascular occlusion (either by vasospasm or thromboembolism), low oxygen delivery with the blood stream (either because of anemia or reduced arterial hemoglobin oxygen content), or reduced perfusion volume (e.g., in chronic heart failure). In each case, compensatory mechanisms aiming at increasing local blood flow are activated to minimize ischemic tissue damage. Recurrent brief periods of ischemia in the systemic circulation activate mechanisms leading to improved protection of tissues from ischemic cell death. This interesting phenomenon called ischemic pre-conditioning has been extensively investigated and reviewed (4–6); further detailed description is beyond the scope of this review.
By contrast, the vast majority of tissue oxygen tension in the lung results from oxygen diffusing from the alveoli rather than being delivered with the blood stream of the bronchial arteries. Hypoxia in the lung is therefore most frequently a result of blocked airflow through the bronchial tree into the alveoli. In the lung, the vascular system responds to hypoxia with vasoconstriction rather than vasodilation. This obvious difference between hypoxic systemic vasodilation and hypoxic pulmonary vasoconstriction has aroused intense research interest for many decades ever since it was first described in the early 20th century (7, 8). However, its molecular mechanisms have remained elusive to this date.
Nitric oxide (NO) is a critically important mediator of vasodilation under a variety of physiological and pathophysiological conditions. The generation of NO, which occurs mainly in the vascular endothelium, is regulated (a) by transcriptional and posttranscriptional mechanisms affecting the NO-producing enzyme, endothelial nitric oxide synthase (eNOS), (b) by factors regulating the enzymatic activity of eNOS, and (c) by reactive oxygen species that rapidly react—and thereby inactivate—NO once released from the endothelium. The enzymatic activity of eNOS is also regulated by the presence of methylarginines (9). Asymmetric dimethylarginine (ADMA) is a competitive inhibitor of eNOS; elevated ADMA concentration has been shown to lead to impaired NO generation and endothelial dysfunction which is reversible by L-arginine (10). Individuals with elevated circulating ADMA concentration are at increased risk of cardiovascular events and mortality (11, 12). ADMA levels are regulated through its biosynthesis, which occurs during arginine methylation of proteins by protein arginine N-methyltransferases (PRMTs) (13, 14), and through its metabolism, which is facilitated by dimethylarginine dimethylaminohydrolases (DDAH) 1 and 2 (15, 16). An alternative metabolic pathway is mediated by alanine glyoxylate aminotransferase-2 (AGXT-2) (17, 18). Dysregulation of the activity or expression of enzymes regulating ADMA concentration may thus contribute to impaired NO generation, endothelial dysfunction, vasospasm, and elevated vascular resistance, both in the systemic and pulmonary circulation (19). Figure 1 depicts the enzymatic pathways involved in the biosynthesis and degradation of ADMA.
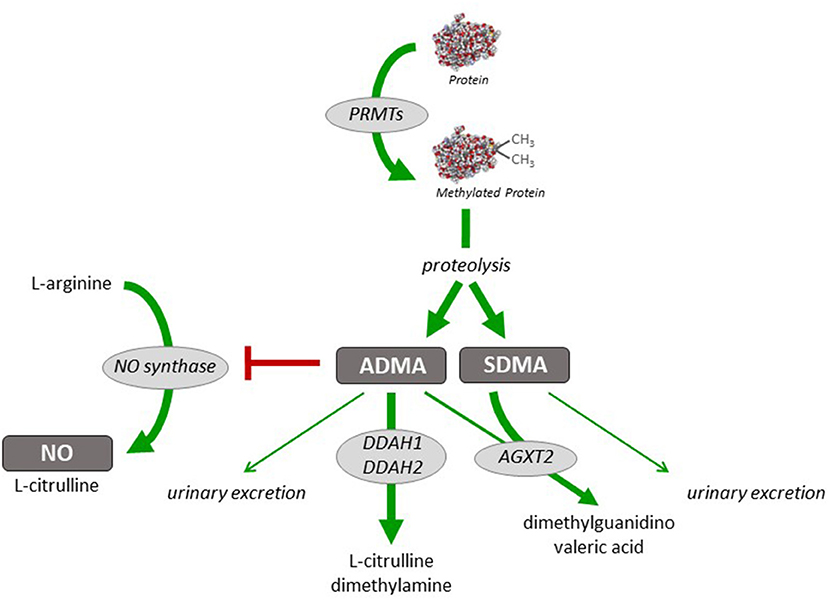
Figure 1. Schematic representation of pathways of dimethylarginine biosynthesis and metabolism. Dimethylarginines are formed during (di-)methylation of protein-bound L-arginine residues by a family of protein arginine N-methyltransferases (PRMTs). Free ADMA and SDMA are released during physiological hydrolytic protein turnover. Asymmetric dimethylarginine (ADMA) inhibits nitric oxide (NO) synthesis from L-arginine, whilst symmetric dimethylarginine (SDMA) does not directly interfere with NO synthase activity. ADMA is metabolically degraded to L-citrulline and dimethylamine by either of two isoforms of dimethylarginine dimethylaminohydrolase (DDAH). Both ADMA and SDMA can be cleaved by alanine glyoxylate aminotransferase-2 (AGXT2); this enzyme is the major pathway of SDMA clearance. Minor amounts of both ADMA and SDMA can also be excreted into the urine.
This review aims to summarize our current understanding of the molecular mechanisms and clinical significance of hypoxic pulmonary vasoconstriction, and addresses the possible role of dysregulation of the L-arginine - dimethylarginine - NO pathway in this condition, based on recent experimental and clinical studies.
The Physiology of Hypoxic Pulmonary Vasoconstriction
Obviously, the lung's physiological function is to deliver fully oxygenated blood into the systemic circulation. Any regional reduction in lung ventilation—as it may occur by blocked airflow through the bronchial tree—threatens to result in suboptimal oxygenation of the blood delivered from the lung into the systemic circulation. Therefore, pulmonary vasoconstriction in a region of hypoventilation is a mechanism to redirect blood flow to better ventilated areas of the lungs, ensuring optimal oxygen supply to all tissues (Figures 2A,B).
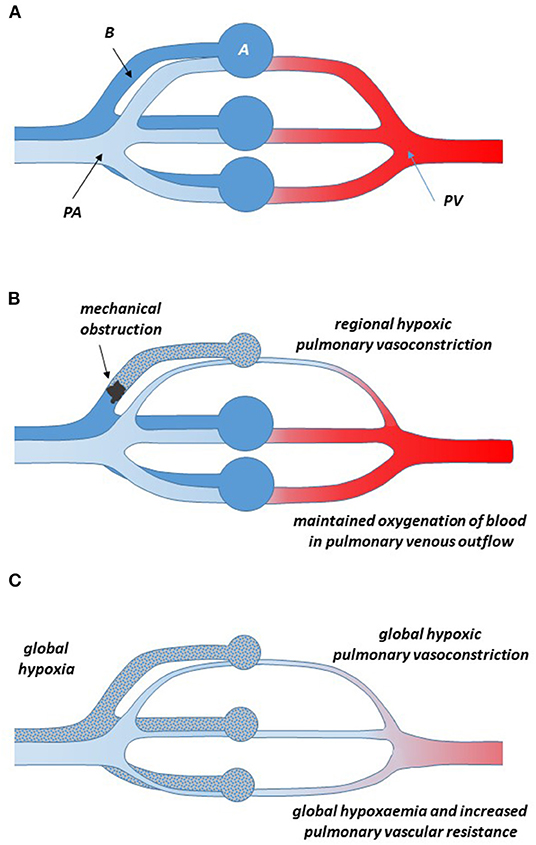
Figure 2. Schematic representation of the pulmonary circulation in normoxia (A) and when one bronchus is obstructed and the respective alveoli are hypoventilated (B). During normoxia in the healthy state, deoxygenated blood from the pulmonary artery flows through the capillary bed surrounding the alveoli, where it takes up oxygen and, fully oxygenated, returns through the pulmonary vein to the left atrium of the heart. Local hypoventilation of an area of the lungs causes vasoconstriction of the pulmonary arteries in the same area; thus, less blood flows through the hypoventilated area and relatively more through other, better ventilated areas, resulting in a minimal reduction of the oxygenation status of the blood returning into the systemic circulation through the pulmonary vein (Euler-Liljestrand mechanism). (C) In global hypoxia, hypoxic pulmonary vasoconstriction occurs throughout the lung. This obviously does not improve the oxygenation status of the blood, but it causes a major increase in total pulmonary vascular resistance. When this situation is maintained for longer time periods, pulmonary hypertension may occur, resulting in right ventricular hypertrophy and failure.
It was the seminal work of Euler and Liljestrand in pulmonary arteries of the cat who first linked pulmonary vasoconstriction to the maintenance of full oxygenation of the blood (20). They concluded that “[…] oxygen want and carbon dioxide accumulation have exactly the reverse local effects on the vessels of the systemic and pulmonary circulations, respectively […]. They cause a dilatation of the vessels of the working organs which need a greater blood supply than during rest, but they call forth a contraction of the lung vessels, thereby increasing the blood flow to better aerated lung areas, which leads to improved conditions for the utilization of the alveolar air.” [quotation from Euler and Liljestrand (20)]. Ever since, this phenomenon has been known as the Euler-Liljestrand-mechanism. In 1955, Blakemore and co-workers demonstrated the existence of this same mechanism in humans. In healthy human subjects, they ventilated one lobe of the lung with physiologically oxygenated air and the other lobe with only 5% oxygen. They observed a redistribution of pulmonary blood flow toward the better oxygenated lobe of the lung (21).
Clinical Relevance of Hypoxic Pulmonary Vasoconstriction
Physiologically, hypoxic pulmonary vasoconstriction (HPV) is a mechanism maintaining ventilation-perfusion matching and ensuring optimal oxygenation of blood. Table 1 summarizes clinical conditions in which HPV plays a pathophysiological role. Redirection of blood flow within the lung may become relevant to limit the detrimental influence of a pathogen in pneumonia, where HPV helps to divert blood flow away from regions of inflammatory infiltration toward healthy lung areas (34). However, the vasoconstrictor mechanism may become diminished in chronic pulmonary infection, and patients may experience hypoxemia in severe pneumonia (35). In bronchial asthma, bronchoconstriction may be spatially distributed in different parts of the lung; again, HPV helps to maintain ventilation-perfusion matching and minimize hypoxemia (31, 41).
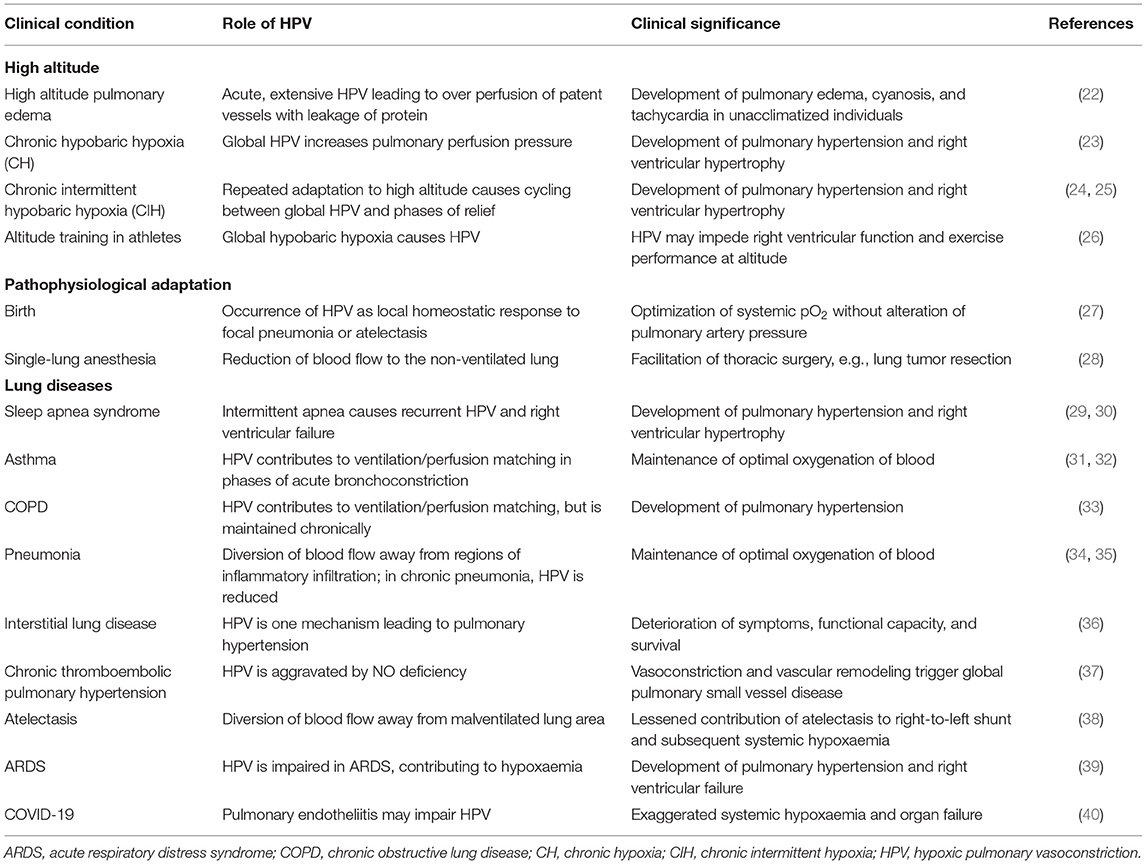
Table 1. Clinical conditions associated with pulmonary hypoxia.
HPV is also a mechanism keeping blood flow away from the still collapsed lungs in the fetus (32). However, after birth, focal atelectasis and pneumonia may occur. HPV helps to optimize systemic arterial oxygen pressure without altering pulmonary artery pressure (42).
Chronification of Hypoxic Pulmonary Vasoconstriction
When ventilation obstacles become chronic like in chronic obstructive lung disease, hypoxic pulmonary vasoconstriction often persists. Acting together with inflammatory and adaptative processes that stipulate remodeling of and fibrosis in the pulmonary vasculature (43), this may lead to persistently elevated pulmonary vascular resistance and structural changes in the pulmonary vascular walls during the progression of the disease and be a cause of pulmonary hypertension, right ventricular hypertrophy, and—finally—failure (33, 44). In chronic thromboembolic pulmonary hypertension (CTEPH, also classified as group IV of the WHO classification of pulmonary hypertension), thrombotic occlusion of a segmental pulmonary artery per se increases total pulmonary vascular resistance; However, secondary mechanisms may be triggered in the non-occluded pulmonary vessels that cause vascular remodeling and lead to a progressive further increase in total pulmonary vascular resistance (45, 46).
Global Pulmonary Hypoxia
Another cause of pathological consequences of HPV is exposure to global pulmonary hypoxia (Figure 2C). This may occur at high altitude, when hypoxia results from the low ambient pressure (hypobaric hypoxia). Acute exposure of non-acclimatized individuals to high altitude, as it can be seen in unexperienced climbers and tourists engaging in mountaineering activities, can lead to high-altitude pulmonary oedema (22). This oedema results from global but heterogeneous HPV with increased pulmonary perfusion pressure acting on the capillary bed, which becomes leaky to protein (47). High altitude pulmonary oedema can be resolved by returning to sea level (22). Residents of high altitude of different ethnic origins show different levels of adaptation to the consequences of chronic global pulmonary hypoxia. Indians native to the Andean highlands at 3,500–4,000 m have a high prevalence of hypoxic pulmonary hypertension (23), whilst inhabitants of the Tibetan plateau living at altitudes of ≥ 3,500 m rarely develop polycythaemia and pulmonary hypertension (48). One major factor contributing to altitude adaptation in Tibetans was reported to be accumulation of genetic polymorphisms in EGLN1, the gene encoding for HIF-2α (49–51). This is in accordance with the important role of HIF-2α in hypoxia-induced upregulation of erythropoietin expression (52).
The main desired effects of high altitude training also depend on hypoxia-inducible factor-2α (HIF2α)-mediated regulation of gene expression, e.g., transcriptional upregulation of erythropoiesis and subsequent improvement in oxygen transport capacity of the blood. However, the combined decreases in arterial oxygen saturation and cardiac output at altitude may limit aerobic exercise capacity, which can be resolved when lowering pulmonary arterial pressure, e.g., by treatment with an ET-1 antagonist (53), but not by acetazolamide treatment (54).
Diminished HPV may be a common mechanism of adaptation to life at high altitude: Cattle native to lowlands exhibit marked hypoxic pulmonary vasoconstriction when exposed to high altitude, resulting in an incidence of about 20% of pulmonary hypertension, pulmonary oedema, and right ventricular failure (55), a condition named brisket disease after the resulting oedema in the cows' necks (56). Interestingly, neonatal calves chronically exposed to high altitude progressively loose the vasodilator response of pulmonary arteries to acetylcholine, a well-characterized stimulus of endothelial NO release (57). This finding points to diminished NO-mediated pulmonary arterial vasodilation as a possible contributor to HPV. By contrast, yaks native to the high altitude of the Himalayan region exhibit diminished HPV and maintain low pulmonary arterial pressure (58). A recent study showed that yaks differ from cattle by lower circulating levels of ADMA and higher protein expression and activity of DDAH, the enzyme inactivating ADMA (59), supporting a role for modulation of the NO pathway in adaptation of the pulmonary circulation to high altitude.
A clinical condition that has been more recently defined is called chronic intermittent hypobaric hypoxia. Workers in mines of the Andean plateau at altitudes above 3,500 m, frontier officials, and other individuals may be exposed to working shifts alternating between several days at high altitude, followed by a few days of rest at sea level (60, 61). This leads to frequent cycling of affected individuals between the acute adaptation to hypoxia at high altitude and relief. In consequence, changes to the pulmonary circulation may occur that are very similar and may be as severe as in chronic hypobaric hypoxia (24, 62). The prevalence of elevated mean pulmonary arterial pressure (mPAP) with mPAP ≥ 25 mm Hg was reported to be as high as 26% and the prevalence of high altitude pulmonary hypertension [the threshold of which has been defined at mPAP ≥ 30 mm Hg (63)] was about 9% in chronic intermittent hypobaric hypoxia (24). Based on a meta-analysis of multiple large cohorts, systolic pulmonary arterial pressure (sPAP) at sea level was calculated to be (median [95% CI]) 18.4 [17.1–19.7] mm Hg, whilst sPAP at high altitude was 25.3 [24.0–26.7] mm Hg (64). As the threshold of mPAP for the definition of pulmonary arterial hypertension in lowlanders has recently been reduced to mPAP ≥ 20 mm Hg (65), an updated, evidence-based definition of pulmonary arterial hypertension at high altitude appears urgently needed (66).
Pulmonary hypertension is also one pathological consequence of chronic intermittent hypoxia in obstructive sleep apnoea syndrome (OSAS); increased pulmonary arterial pressure may occur during sleep, but also during waking hours (29). Whilst clinically relevant pulmonary hypertension is rare in pure OSAS, it may occur much more frequently in the so-called overlap syndrome, i.e., the combined occurrence of OSAS and chronic obstructive pulmonary disease (COPD) (30). Although there still remain gaps in our understanding of the pathophysiology of this relationship (67), one relevant observation helping us to understand the association of OSAS with vascular disease in both, the pulmonary and systemic circulation, is the presence of endothelial dysfunction, i.e., the inability of the vascular endothelium to generate physiological amounts of NO as required to maintain vasodilator tone (68).
Recent interest has focussed on the role of pulmonary vascular damage and endothelial dysfunction in COVID-19 pneumonia and ensuing hypoxaemia and organ failure (69, 70). We have reported that high ADMA and SDMA serum levels are superior biomarkers to predict COVID-19-associated in-hospital mortality (71), suggesting that NO deficiency may aggravate pulmonary and systemic vascular dysfunction in this disease. Accordingly, several small trials investigated the effects of inhaled NO (72, 73) or the phosphodiesterase V inhibitor sildenafil on COVID-19-associated hypoxaemia and outcome (74). However, the reported results of these studies have so far been inconclusive.
Mechanisms of Hypoxic Pulmonary Vasoconstriction
The best known transcriptional regulators of the physiological responses are the hypoxia-inducible factors (HIF). HIF-1α is activated acutely upon oxygen deficiency, whilst HIF-2α mediates the sustained responses to prolonged hypoxia (75). By this mechanism, hypoxia elicits a systemic hemodynamic response via activation of the carotid chemokine receptors and systemic humoral mechanisms. In addition, hypoxia also acts locally on the pulmonary vessels, thereby modulating the relation between pulmonary blood flow and alveolar ventilation. Although HIF-1 target genes have been shown to be involved in the pulmonary arterial response to hypoxia (76), the cellular crosstalk in the hypoxic lungs appears to be more complex, and the exact molecular and cellular nature of this local mechanism of HPV has remained elusive so far. A number of determinants can be defined, however, that are prerequisites of a locally functioning physiological mechanism:
A) There must be an oxygen sensor at the level or in the immediate adjacency of the pulmonary alveoli and pulmonary blood vessels.
B) There must be a locally functioning vasoconstrictor mechanism activated and / or vasodilator mechanism diminished by hypoxic signaling. This mechanism must be rapidly activated, reversible in nature, and evocable by mild hypoxia.
There are three major cell types in the lung, of which each may be responsible for initiating HPV: endothelial cells and vascular smooth muscle cells of the pulmonary arterioles, and alveolar epithelial cells lining the bronchioli and alveoli. The endothelial cells form the physiological barrier between the circulating blood and the adjacent vascular tissue, they are the major source of effectors influencing the vasoconstrictor and vasodilator properties of blood vessels. As such, they are predisposed to interlace between changes in tissue oxygen content and vascular tone by generating vasoactive mediators (see below). The vascular smooth muscle cell is less easily capable of sensing the blood oxygen content due to its more distant spatial localization. However, a hypothetical oxygen sensor located in the vascular smooth muscle cell itself could directly modulate the cell's contractile properties. The alveolar epithelial cells, on their turn, are the primary cells exposed to low oxygen content in the breathing air, and therefore predisposed to act as sensor cells. Thus, the complexity of this intercellular cross-talk may at least partly explain that the exact molecular mechanism of HPV has not yet been unraveled. Finally, different cell types or signaling mechanisms may be involved in mediating the early and late phases of HPV.
Oxygen Sensing
One of the most extensively studied sites of oxygen sensing is the carotid body, which regulates major neuroendocrine responses to hypoxemia. Carotid body glomus cells respond to hypoxemia by inhibition of K+ channels, leading to membrane depolarization, calcium influx via voltage-gated Ca2+ channels, and neuroendocrine secretion (77, 78). In the pulmonary circulation, the cellular and molecular identity of the oxygen sensor has remained much less clear. Experiments demonstrating that redox agents and certain inhibitors of complexes I and III of the mitochondrial electron transport chain cause vasoconstriction in the pulmonary vascular bed, but vasodilation in the fetal ductus arteriosus (79)—mimicking the differential responses to hypoxia in these two vascular beds—suggest that redox mechanisms may be involved. Thus, research to identify the pulmonary oxygen sensor has focused on NADPH oxidases and on the mitochondrial respiratory chain (78), and models aiming to explain HPV based on mitochondrial oxygen sensing have been proposed (80–82). In line with this, knockdown of NADH dehydrogenase ubiquinone iron-sulfur protein-2 (Ndufs-2) within the mitochondrial complex I significantly decreased hypoxic vasoconstriction in pulmonary artery smooth muscle cells (83). Another source of oxygen-derived radicals during hypoxia and ischemia episodes is accumulation of succinate, an intermediate metabolite in the mitochondrial citric acid cycle (84). Accumulation of succinate stimulates mitochondrial production of reactive oxygen species by reversing electron transport at mitochondrial complex I (85). Through this mechanism, succinate overload in hypoxia is known to activate HIF-1α (86). During normoxia, the HIF-1α protein is hydroxylated by prolyl hydroxylases that are absolutely dependent on the presence of oxygen. Hydroxylation enables binding of HIFs to the ubiquitin proteasome system and subsequent degradation; inhibition of this degradation pathway in hypoxia activates HIF-mediated gene transcription (75, 76).
Recent studies also suggest that pulmonary and systemic arteries share the same oxygen sensing mechanism within mitochondria, whilst differences in downstream signaling of reactive oxygen species released from hypoxic mitochondria cause site-specific vascular responses (87). As the three major cell types present in the lung have all been shown to be responsive to hypoxia (81, 88, 89), the cellular location of the oxygen sensor has remained controversial.
Signal Transduction and Effector Mechanisms: The Vascular Smooth Muscle Cell
HPV is brought about by a contractile response of the pulmonary vascular smooth muscle cells (VSMC). Smooth muscle cell contraction is highly dependent on elevated cytosolic calcium concentration; therefore, the effector mechanisms responsible for HPV likely involve modulation of VSMC calcium handling. Sarcoplasmic calcium channels, voltage-dependent potassium channels, transient receptor potential channels, and L-type calcium channels are the main regulators of cytosolic calcium (90). The coordinated response of these ion channels is influenced by protein kinases and reactive oxygen species (ROS). The Ca2+ influx directly triggers a conformational change of the myosin light chain, thereby facilitating interaction with actin filaments and contraction. Several studies have provided evidence for an involvement of ion channels in HPV: For example, inhibition of voltage-dependent potassium channels caused vasoconstriction in the isolated perfused rat lung (91). Furthermore, inhibition of L-type calcium channels diminished whereas activation of these channels enhanced the vasoconstrictor response to hypoxia (92, 93). However, the modulation of vascular tone by these channels does not differ between systemic and pulmonary arteries. Therefore, this mechanism cannot explain the heterogeneous response to hypoxia (vasoconstriction vs. vasodilation) in pulmonary and systemic arteries, respectively.
Signal Transduction and Effector Mechanisms: The Vascular Endothelial Cell
Endothelium-derived vasoactive mediators are major regulators of vascular tone in the systemic circulation. The endothelium-dependent vasoconstrictor substances include the peptide endothelin-1 (ET-1) (94), superoxide anions (95), and arachidonic acid-derived endoperoxides and/or thromboxane A2 (96). The endothelium-derived relaxing factors include NO, prostacyclin, and endothelium-derived hyperpolarizing factor (EDHF) (97). Both endothelial vasoconstrictor and vasodilator mediators are finely tuned to maintain the homeostasis of local blood flow and its adaptation to varying needs of oxygen and nutrient demand (Figure 3). Less information is available about the role of endothelium-derived mediators in the regulation of pulmonary vascular tone.
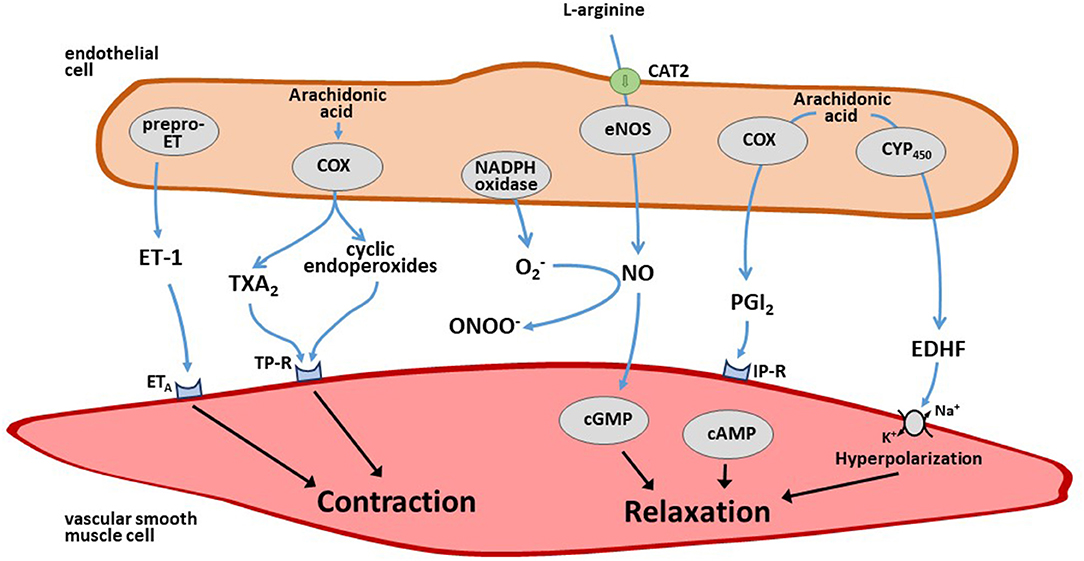
Figure 3. Schematic overview of endothelium-derived vasoconstrictor and vasodilator mediators. The endothelium produces several vasoconstrictor mediators like endothelin-1 (ET-1) and thromboxane (TX) A2 as well as vasodilator mediators like nitric oxide (NO), prostacyclin (PGI2), and endothelium-derived hyperpolarizing factor (EDHF) that diffuse to the adjacent smooth muscle cells that effect changes in vascular tone upon this stimulation. For further details see text.
ET-1 is the most potent vasoconstrictor peptide released by endothelial cells (94). Human ET-1 is synthesized as a 212-amino acid peptide (prepro-ET-1); it exerts a long-lasting vasoconstrictor effect by activating ETA receptors (98). By contrast, binding of ET-1 to ETB receptors, which are located on the endothelial cell membrane, causes vasodilation and anti-mitogenic effects through the release of NO and/or prostacyclin (PGI2) (99, 100). The lung is an important site of ET-1 production, with ET-1 mRNA being five times more abundant in the lung than in other organs (101). Lowering oxygen levels in cultured endothelial cells rapidly increases the mRNA expression of prepro-ET-1 (102). This effect persists for at least 48 h when hypoxia is maintained, and it is reversible after increasing oxygen tension to normal ambient pressure. These experimental findings are in line with in vivo observations from animal studies (103–105), and with the observation that circulating ET-1 is elevated in COPD patients with chronic hypoxia (106). However, the endothelin receptor antagonist bosentan had variable effects on HPV in animal models and clinical studies (107–109). This may be due to the fact that bosentan is a dual blocker of both ETA and ETB receptors. Hypoxia enhances the expression of ETA and ETB receptors in the lung, but there is evidence for a predominant upregulation of ETB receptors. Thus, under hypoxic conditions, the effect of bosentan in the pulmonary circulation may be dominated by blocking ETB-mediated vasodilation (110).
Arachidonic acid metabolites are released from endothelial cells upon stimulation with acetylcholine, serotonin, adenosine diphosphate (ADP), and other substances. Based on the expression of cyclooxygenase and the spectrum of prostaglandin synthases in a specific cell type, either the vasodilator metabolites prostacyclin and PGE2, or the vasoconstrictor endoperoxides and thromboxane A2 may be released. For example, stimulation of isolated aortic rings from Wistar rats with acetylcholine results in endothelium-dependent vasodilation, whereas aortic rings from spontaneously hypertensive rats (SHR) respond with vasoconstriction (111). Aortic vasoconstriction in SHR is enhanced when endothelial NO production is blocked, whilst vasodilation is unmasked when cyclooxygenase activity is blocked (112). During chronic hypoxia, mouse pulmonary arteries release less prostacyclin and more 8-iso-prostaglandin F2α [a lipid peroxide product derived from non-enzymatic oxidation of arachidonic acid by superoxide anion (113)]. Cyclooxygenase-2 is upregulated, and endothelium-dependent relaxation in normoxia is shifted to an endothelium-independent, thromboxane receptor-dependent contraction (114).
NO is the major endothelial vasodilator mediator in the systemic and in the pulmonary circulation. In most arterial beds, it is only under pathophysiological conditions when NO signaling is impaired or under experimental conditions when NO production is pharmacologically or genetically inhibited that a significant role can be determined for other endothelial mediators. During the recent years, our research has focused on the regulation of the NO pathway by endogenous, methylated analogs of L-arginine, the physiological precursor of NO (115, 116). Evidence has accumulated that dysregulation of the NO pathway by ADMA may be involved in HPV and pulmonary hypertension (117).
Signal Transduction and Effector Mechanisms: The Alveolar Epithelial Cell
Alveolar epithelial cells are the cell type most directly exposed to decreased oxygen content in the inspired air. Type II alveolar epithelial cells make up about two thirds of the alveolar epithelial surface in the normal human lung; they play an important role in surfactant production and recycling (118). Early experiments had shown that in the isolated perfused cat lung, ventilation with low oxygen gas increased, but perfusion with partially deoxygenated blood did not increase pulmonary vascular resistance, suggesting that oxygen content in the inspired air, but not hypoxemia in the pulmonary blood vessels stipulates HPV (119). More recent experiments showed differential effects of hypoxia on human alveolar epithelial cells and human pulmonary microvascular endothelial cells, respectively, with the alveolar epithelial cells displaying a more sensitive response to hypoxia (120). Others revealed that acute changes in inspired oxygen tension are sensed by large conductance calcium-activated potassium channels of human alveolar epithelial cells (121), causing membrane hyperpolarization. Beyond that, alveolar epithelial cells are capable of secreting paracrine mediators which may influence the function of adjacent endothelial and vascular smooth muscle cells; amongst such mediators, NO derived from inducible NOS in type II alveolar epithelial cells (122), interleukin-33, and the receptor for advanced glycation end products (RAGE) have been identified [for review, cf. (89)]. Thus, alveolar epithelial cells may be involved in sensing hypoxia and mediating this signal to vascular endothelial and smooth muscle cells, thereby contributing to pulmonary vascular contraction and remodeling in hypoxia (123).
Dysregulation of the Endothelial NO Pathway in the Hypoxic Pulmonary Circulation
Acute and chronic hypobaric hypoxia at high altitude result in endothelial dysfunction, a situation defined by impaired endothelium-dependent, NO-mediated vasodilation in response to brief phases of ischemia in the forearm or in response to local infusion of acetylcholine. Endothelium-dependent vasodilation is acutely impaired in lowlanders after arrival to high altitude hypoxia (124) as well as in Tibetan inhabitants of the Himalaya region, despite the good genetic adaptation of this population to chronic hypobaric hypoxia (125). Inhabitants of the Andean high altitude region also show distinct endothelial dysfunction, which is more pronounced in individuals with cardiovascular risk factors or overt cardiovascular disease than in controls (126).
The underlying mechanisms leading to dysfunction of the NO pathway have been extensively studied and are considered to be multifactorial. Changes in eNOS gene expression, reduced eNOS catalytic activity, altered L-arginine metabolism, and increased NO consumption by reaction with superoxide anion may all contribute to a lack of bioactive NO.
There is evidence of markedly decreased eNOS gene expression in the endothelium of patients with pulmonary hypertension (127). However, subsequent studies found pulmonary expression of eNOS unchanged in pulmonary hypertension (128), and some studies even reported increased expression of eNOS and/or the inducible isoform of NOS (129). Thus, NOS gene expression does not always correspond to NO production, as NOS activity may be influenced by several factors relevant to pulmonary hypoxia.
Endothelial NOS needs a variety of co-factors to function normally [reviewed in Förstermann and Sessa (130) and Moncada and Higgs (131)]. When the endothelial cell is depleted of co-factors, eNOS becomes “uncoupled,” i.e., its catalytic activity is driven toward the generation of superoxide anions (130). Specifically, oxidation of the essential eNOS co-factor tetrahydrobiopterin has been shown to cause uncoupling of eNOS activity and endothelial dysfunction.
Another cause of diminished eNOS activity may be the presence of endogenous NOS inhibitors. Table 2 summarizes experimental evidence from animal models for a link between dimethylarginine metabolism, hypoxia, and pulmonary arterial hypertension. ADMA is produced during the post-translational methylation of arginine residues within specific proteins (13, 144). When methylated proteins are cleaved, ADMA is released instead of L-arginine. ADMA competes with L-arginine for binding to the NOS catalytic site and thus competitively inhibits NOS activity. Another dimethylarginine, symmetric dimethylarginine (SDMA), is unable to directly interfere with NOS activity, but like ADMA, it may inhibit CAT-2, the cellular uptake transporter for L-arginine (145, 146). We have recently reviewed in detail the transcriptional and post-translational mechanisms of regulation of dimethylarginine metabolism (9). Dimethylation of proteins occurs as a process of posttranslational protein modification and leads to increased hydrophobicity of the respective protein moieties. This process is ubiquitously present in all tissues investigated so far, although the specific types of protein arginine N-methyltransferases (PRMT) may vary in a tissue-specific manner. Amongst highly dimethylated proteins are heterogeneous nuclear ribonucleoproteins. Histone proteins are activated by asymmetric dimethylation and repressed by symmetric dimethylation, this affects their regulatory roles in gene expression (147, 148). Myelin basic protein is a neuronal protein that is known to be highly symmetrically dimethylated (149), a fact that may explain why high SDMA concentrations can be found in cerebral ischemic stroke (150, 151). Physiological turnover of proteins releases either ADMA or SDMA, depending on the type of methylation of the degraded protein. Although several PRMT enzymes are expressed in the lungs, it is not known whether asymmetric or symmetric demethylation plays a functional role in the lungs or in the vascular system.
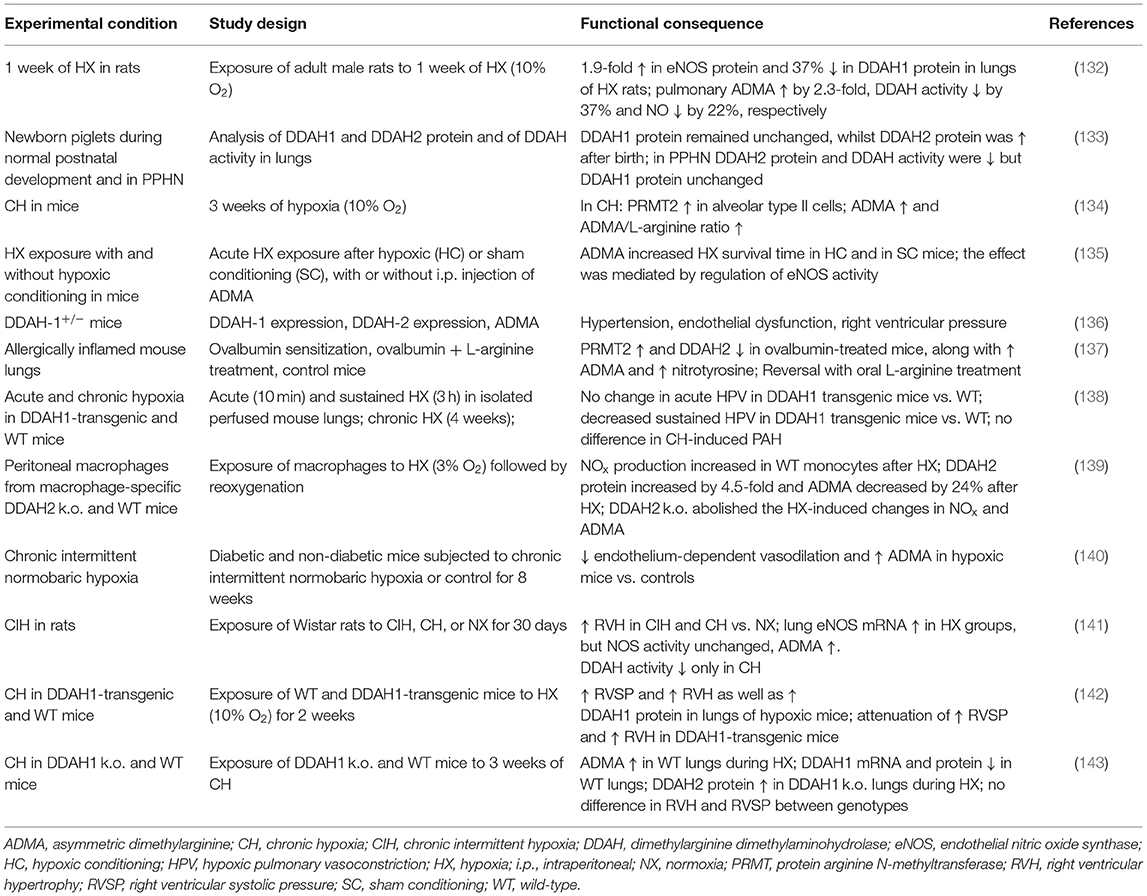
Table 2. Experimental models linking derangement of the ADMA/DDAH pathway with pulmonary hypoxia and pulmonary vascular dysfunction.
ADMA is mainly degraded by the enzyme dimethylarginine dimethylaminohydrolase (DDAH), which exists in two isoforms. DDAH-1 has been described as the major isoform in the kidneys and liver, whilst DDAH-2 is expressed mainly in vascular tissues (16, 152). Derangement of DDAH, either genetically induced in knockout mouse models, pharmacologically caused by DDAH-inhibitory compounds, or biochemically caused by high glucose or oxidative stress, leads to elevated ADMA that impairs NO generation by eNOS and results, amongst other effects, in elevated pulmonary arterial pressure (136).
Evidence for Dysregulation of the Dimethylarginine Pathway in Pulmonary Hypoxia and Pulmonary Arterial Hypertension
In patients with different pulmonary diseases, ADMA levels are higher than in healthy controls (Table 3). Specifically, elevated ADMA has been reported in patients with obstructive sleep apnoea syndrome (OSAS) and in those with chronic obstructive lung disease (COPD). Both conditions are associated with hypoxemia, the development of elevated pulmonary artery pressure, pulmonary arterial hypertension, and right heart failure, as well as a high risk of systemic cardiovascular disease (183, 184). Multiple small cross-sectional studies reported higher plasma or serum ADMA in COPD than healthy controls; in addition, some studies reported an inverse correlation between ADMA and FEV1 or COPD severity grade (167, 172), or significantly higher ADMA in acutely exacerbated than in stable COPD (170, 173). High ADMA was associated with intima-media thickness in the brachial artery of COPD patients (169) and inversely associated with serum NO metabolites in another study (170). Lastly, ADMA and SDMA had prognostic relevance in a prospective study with 150 patients with acutely exacerbated COPD; the highest quartiles of ADMA and SDMA were significantly associated with all-cause mortality after 6 years of follow-up (mortality rate, 54%) (170).

Table 3. Clinical conditions of pulmonary hypoxia in which derangement of the ADMA / DDAH pathway was described.
Data on plasma or serum ADMA concentrations are more controversial in OSAS. Some case-control studies reported higher ADMA concentration in OSAS (157, 159, 161), along with lower NO metabolite levels (162) or impaired endothelium-dependent vasodilation (185). However, other investigators were unable to reproduce these findings (156, 177). Interpretation of these studies is hampered by methodological flaws in some studies, by lack of healthy controls in others, and by differences and—in some studies—uncertainties about analytical methods utilized for ADMA quantification.
Additionally, elevated ADMA has been measured in several types of pulmonary arterial hypertension (179, 181, 182). A prospective study reported that elevated ADMA is associated with impaired long-term survival of patients with primary pulmonary arterial hypertension (178), a finding in line with the reported role of ADMA as a marker of long-term cardiovascular events and mortality in the general population (11, 186, 187).
We and others have studied the effects of chronic hypobaric hypoxia and chronic intermittent hypobaric hypoxia on the regulation of the NO/ADMA pathway in a number of experimental models and clinical cohorts. Rats that were exposed to chronic hypobaric hypoxia for 30 days developed right ventricular hypertrophy, diminished DDAH activity, and elevated circulating ADMA levels (141). Despite upregulated eNOS mRNA expression, the biological activity of NO was unchanged, suggesting that NOS activity was inhibited by elevated ADMA. In young, healthy humans who were exposed to high altitude (3,500 m) for the first time in an intermittent, weekly exposure regimen for 3 months developed a progressive elevation of circulating ADMA levels that significantly correlated with the elevation of haematocrit (153). In a cross-sectional study of Chilean mining workers who had been exposed to intermittent work at elevations of 4,400–4,800 m for more than 5 years, elevated ADMA levels were also significantly associated with elevated mean pulmonary artery pressure (24). Recent genetic analyses performed in our laboratory revealed significant associations of single nucleotide polymorphisms (SNPs) in the NOS III, DDAH1, AGXT2, and ARG2 genes with high altitude pulmonary hypertension (188). Specifically, individuals homozygous for the minor allele of DDAH1 SNP rs233112 had higher baseline ADMA plasma concentration but no change in the ADMA response to hypoxia (188). By contrast, homozygous carriers of the minor allele of the rs805304 SNP in the DDAH2 gene had a diminished ADMA increase during hypoxia but no difference in baseline ADMA concentration. In a parallel animal study, DDHA1 ko mice showed no difference in hypoxia-induced pulmonary arterial pressure or right ventricular morphology as compared to wild-type littermates (143). DDAH1 knockout mice, however, displayed pulmonary upregulation of DDAH2 protein during chronic hypoxia, predominantly in alveolar epithelial cells, suggesting that DDAH2 upregulation may compensate for deficient DDAH1 expression and/or activity and thereby limit the pathophysiological consequences of chronic hypoxia on pulmonary vascular NO function. To a similar point, we observed a gradual decline of SDMA in humans exposed to chronic intermittent hypoxia at altitude, which paralleled the gradual increase in ADMA as reported above (60). Homozygous carriers of AGXT2 rs37369 showed a greater reduction in plasma SDMA than carriers of the minor allele of this SNP, suggesting an upregulation of AGXT2 in hypoxia (188).
Conclusions and Future Perspectives
Dysfunctional endothelium-dependent, NO-mediated vasodilation contributes to sustained HPV. There is accumulating evidence that elevated concentrations of the endogenous NOS inhibitor, ADMA, are involved in downregulating pulmonary vascular NO production in chronic hypoxia. Whilst studies in animal models and clinical cohort studies at high altitude are useful to dissect the molecular mechanisms of this regulation, it may have important clinical impact in understanding the pathophysiology of chronic pulmonary diseases like COPD and OSAS. Current evidence suggests that downregulation of DDAH mediates hypoxic accumulation of ADMA, but data are controversial as to which isoform is involved. Further, there may be compensatory regulation of one DDAH isoform when the other one is dysfunctional as suggested by a recent study in DDAH1 ko mice, as well as upregulation of AGXT2, as suggested by recent human studies. More studies are required to clarify the mechanism of this regulation. Information on a possible dysregulation of the L-arginine – dimethylarginine – NO pathway in chronic lung diseases like COPD, OSAS, overlap syndrome, and PAH are mostly derived from small, cross-sectional studies. Small patient numbers, heterogeneous patient populations and study designs, as well as methodological shortcomings contribute to current incertitude in this field. Large, prospective biomarker studies as well as mechanistic clinical studies in acute and chronic hypoxia using state-of-the-art methods are needed to shed light on the role of this pathway in chronic hypoxic lung diseases. This may open up new avenues for better treatment of chronic hypoxia and its pulmonary and systemic hemodynamic consequences.
Author Contributions
JH and RB contributed equally to data acquisition and writing. Both authors agreed to the final version of the manuscript.
Funding
This work was funded by the German Federal Ministry of Education and Research under Grant no. 01DN17046 (DECIPHER). Work by the authors was also funded by the Georg and Jürgen Rickertsen Foundation, Hamburg, Germany, the Joachim Herz Foundation, Hamburg, Germany, and the Werner Otto Foundation, Hamburg, Germany (Grant no. 02/96).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Rowell LB, Blackmon JR. Human cardiovascular adjustments to acute hypoxaemia. Clin Physiol. (1987) 7:349–76. doi: 10.1111/j.1475-097X.1987.tb00179.x
2. Tune JD. Control of coronary blood flow during hypoxemia. Adv Exp Med Biol. (2007) 618:25–39. doi: 10.1007/978-0-387-75434-5_3
3. Umbrello M, Dyson A, Feelisch M, Singer M. The key role of nitric oxide in hypoxia: hypoxic vasodilation and energy supply-demand matching. Antioxid Redox Signal. (2013) 19:1690–710. doi: 10.1089/ars.2012.4979
4. Hausenloy DJ, Barrabes JA, Bøtker HE, Davidson SM, Di Lisa F, Downey J, et al. Ischaemic conditioning and targeting reperfusion injury: a 30 year voyage of discovery. Basic Res Cardiol. (2016) 111:70. doi: 10.1007/s00395-016-0588-8
5. Heusch G. Molecular basis of cardioprotection: signal transduction in ischemic pre-, post-, and remote conditioning. Circ Res. (2015) 116:674–99. doi: 10.1161/CIRCRESAHA.116.305348
6. Manukhina EB, Downey HF, Mallet RT. Role of nitric oxide in cardiovascular adaptation to intermittent hypoxia. Exp Biol Med (Maywood). (2006) 231:343–65. doi: 10.1177/153537020623100401
7. Beyne J. Influence de l'anoxémie sur la grande circulation et sur la circulation pulmonaire. C R Soc Biol. (1942) 136:399–400.
9. Hannemann J, Böger R. Transcriptional and post-translational regulation of the dimethylarginines ADMA and SDMA and their impact on the l-arginine – nitric oxide pathway. In: Jez J, editor. Encyclopedia of Biological Chemistry III (Third Edition). Oxford: Elsevier (2021) p. 674–87. doi: 10.1016/B978-0-12-819460-7.00128-6
10. Böger RH, Bode-Böger SM, Szuba A, Tsao PS, Chan JR, Tangphao O, et al. Asymmetric dimethylarginine (ADMA): a novel risk factor for endothelial dysfunction: its role in hypercholesterolemia. Circulation. (1998) 98:1842–7. doi: 10.1161/01.CIR.98.18.1842
11. Böger RH, Sullivan LM, Schwedhelm E, Wang TJ, Maas R, Benjamin EJ, et al. Plasma asymmetric dimethylarginine and incidence of cardiovascular disease and death in the community. Circulation. (2009) 119:1592–600. doi: 10.1161/CIRCULATIONAHA.108.838268
12. Zoccali C, Bode-Böger S, Mallamaci F, Benedetto F, Tripepi G, Malatino L, et al. Plasma concentration of asymmetrical dimethylarginine and mortality in patients with end-stage renal disease: a prospective study. Lancet. (2001) 358:2113–7. doi: 10.1016/S0140-6736(01)07217-8
13. Fulton MD, Brown T, Zheng YG. The biological axis of protein arginine methylation and asymmetric dimethylarginine. Int J Mol Sci. (2019) 20:3322. doi: 10.3390/ijms20133322
14. Nicholson TB, Chen T, Richard S. The physiological and pathophysiological role of PRMT1-mediated protein arginine methylation. Pharmacol Res. (2009) 60:466–74. doi: 10.1016/j.phrs.2009.07.006
15. Kimoto M, Whitley GS, Tsuji H, Ogawa T. Detection of NG,NG-dimethylarginine dimethylaminohydrolase in human tissues using a monoclonal antibody. J Biochem. (1995) 117:237–8. doi: 10.1093/jb/117.2.237
16. Leiper JM, Santa Maria J, Chubb A, MacAllister RJ, Charles IG, Whitley GS, et al. Identification of two human dimethylarginine dimethylaminohydrolases with distinct tissue distributions and homology with microbial arginine deiminases. Biochem J. (1999) 343:209–14. doi: 10.1042/bj3430209
17. Caplin B, Wang Z, Slaviero A, Tomlinson J, Dowsett L, Delahaye M, et al. Alanine-glyoxylate aminotransferase-2 metabolizes endogenous methylarginines, regulates NO, and controls blood pressure. Arterioscler Thromb Vasc Biol. (2012) 32:2892–900. doi: 10.1161/ATVBAHA.112.254078
18. Rodionov RN, Murry DJ, Vaulman SF, Stevens JW, Lentz SR. Human alanine-glyoxylate aminotransferase 2 lowers asymmetric dimethylarginine and protects from inhibition of nitric oxide production. J Biol Chem. (2010) 285:5385–91. doi: 10.1074/jbc.M109.091280
19. Böger R, Hannemann J. Dual role of the L-arginine-ADMA-NO pathway in systemic hypoxic vasodilation and pulmonary hypoxic vasoconstriction. Pulm Circ. (2020) 10:2045894020918850. doi: 10.1177/2045894020918850
20. von Euler U, Liljestrand G. Observations on the pulmonary arterial blood pressure in the cat. Acta Physiol Scand. (1946) 12:301–20. doi: 10.1111/j.1748-1716.1946.tb00389.x
21. Blakemore WS, Carlens E, Bjorkman S. The effect of unilateral rebreathing of low oxygen gas mixtures upon the pulmonary blood flow in man. Surg Forum. (1955) 5:691–6.
22. Hultgren HN. High-altitude pulmonary edema: current concepts. Annu Rev Med. (1996) 47:267–84. doi: 10.1146/annurev.med.47.1.267
23. Penaloza D, Arias-Stella J. The heart and pulmonary circulation at high altitudes: healthy highlanders and chronic mountain sickness. Circulation. (2007) 115:1132–46. doi: 10.1161/CIRCULATIONAHA.106.624544
24. Brito J, Siques P, López R, Romero R, León-Velarde F, Flores K, et al. Long-term intermittent work at high altitude: right heart functional and morphological status and associated cardiometabolic factors. Front Physiol. (2018) 9:248. doi: 10.3389/fphys.2018.00248
25. Sarybaev AS, Palasiewicz G, Usupbaeva DA, Plywaczewski R, Maripov AM, Sydykov AS, et al. Effects of intermittent exposure to high altitude on pulmonary hemodynamics: a prospective study. High Alt Med Biol. (2003) 4:455–63. doi: 10.1089/152702903322616209
26. Bourdillon N, Fan JL, Uva B, Müller H, Meyer P, Kayser B. Effect of oral nitrate supplementation on pulmonary hemodynamics during exercise and time trial performance in normoxia and hypoxia: a randomized controlled trial. Front Physiol. (2015) 6:288. doi: 10.3389/fphys.2015.00288
27. Dawes GS, Mott JC, Widdicombe JG, Wyatt DG. Changes in the lungs of the new-born lamb. J Physiol. (1953) 121:141–62. doi: 10.1113/jphysiol.1953.sp004936
28. Nagendran J, Stewart K, Hoskinson M, Archer SL. An anesthesiologist's guide to hypoxic pulmonary vasoconstriction: implications for managing single-lung anesthesia and atelectasis. Curr Opin Anaesthesiol. (2006) 19:34–43. doi: 10.1097/01.aco.0000192777.09527.9e
29. Adir Y, Humbert M, Chaouat A. Sleep-related breathing disorders and pulmonary hypertension. Eur Respir J. (2021) 57:2258. doi: 10.1183/13993003.02258-2020
30. Suri TM, Suri JC. A review of therapies for the overlap syndrome of obstructive sleep apnea and chronic obstructive pulmonary disease. FASEB Bioadv. (2021) 3:683–93. doi: 10.1096/fba.2021-00024
31. Kelly VJ, Hibbert KA, Kohli P, Kone M, Greenblatt EE, Venegas JG, et al. Hypoxic pulmonary vasoconstriction does not explain all regional perfusion redistribution in asthma. Am J Respir Crit Care Med. (2017) 196:834–44. doi: 10.1164/rccm.201612-2438OC
32. Lumb AB, Slinger P. Hypoxic pulmonary vasoconstriction: physiology and anesthetic implications. Anesthesiology. (2015) 122:932–46. doi: 10.1097/ALN.0000000000000569
33. Wagner PD, Dantzker DR, Dueck R, Clausen JL, West JB. Ventilation-perfusion inequality in chronic obstructive pulmonary disease. J Clin Invest. (1977) 59:203–16. doi: 10.1172/JCI108630
34. Light RB, Mink SN, Wood LD. Pathophysiology of gas exchange and pulmonary perfusion in pneumococcal lobar pneumonia in dogs. J Appl Physiol Respir Environ Exerc Physiol. (1981) 50:524–30. doi: 10.1152/jappl.1981.50.3.524
35. McCormack DG, Paterson NA. Loss of hypoxic pulmonary vasoconstriction in chronic pneumonia is not mediated by nitric oxide. Am J Physiol. (1993) 265:H1523–8. doi: 10.1152/ajpheart.1993.265.5.H1523
36. Ryu JH, Krowka MJ, Pellikka PA, Swanson KL, McGoon MD. Pulmonary hypertension in patients with interstitial lung diseases. Mayo Clin Proc. (2007) 82:342–50. doi: 10.1016/S0025-6196(11)61030-6
37. Simonneau G, Torbicki A, Dorfmüller P, Kim N. The pathophysiology of chronic thromboembolic pulmonary hypertension. Eur Respir Rev. (2017) 26:143. doi: 10.1183/16000617.0112-2016
38. Morrell NW, Nijran KS, Biggs T, Seed WA. Magnitude and time course of acute hypoxic pulmonary vasoconstriction in man. Respir Physiol. (1995) 100:271–81. doi: 10.1016/0034-5687(95)00002-U
39. Gierhardt M, Pak O, Walmrath D, Seeger W, Grimminger F, Ghofrani HA, et al. Impairment of hypoxic pulmonary vasoconstriction in acute respiratory distress syndrome. Eur Respir Rev. (2021) 30:161. doi: 10.1183/16000617.0059-2021
40. Brosnahan SB, Jonkman AH, Kugler MC, Munger JS, Kaufman DA. COVID-19 and respiratory system disorders: current knowledge, future clinical and translational research questions. Arterioscler Thromb Vasc Biol. (2020) 40:2586–97. doi: 10.1161/ATVBAHA.120.314515
41. Harris RS, Winkler T, Tgavalekos N, Musch G, Melo MF, Schroeder T, et al. Regional pulmonary perfusion, inflation, and ventilation defects in bronchoconstricted patients with asthma. Am J Respir Crit Care Med. (2006) 174:245–53. doi: 10.1164/rccm.200510-1634OC
42. Dunham-Snary KJ, Wu D, Sykes EA, Thakrar A, Parlow LRG, Mewburn JD, et al. Hypoxic pulmonary vasoconstriction: from molecular mechanisms to medicine. Chest. (2017) 151:181–92. doi: 10.1016/j.chest.2016.09.001
43. Leopold JA, Maron BA. Molecular mechanisms of pulmonary vascular remodeling in pulmonary arterial hypertension. Int J Mol Sci. (2016) 17:761. doi: 10.3390/ijms17050761
44. Mélot C, Naeije R, Rothschild T, Mertens P, Mols P, Hallemans R. Improvement in ventilation-perfusion matching by almitrine in COPD. Chest. (1983) 83:528–33. doi: 10.1378/chest.83.3.528
45. Bochenek ML, Rosinus NS, Lankeit M, Hobohm L, Bremmer F, Schütz E, et al. From thrombosis to fibrosis in chronic thromboembolic pulmonary hypertension. Thromb Haemost. (2017) 117:769–83. doi: 10.1160/TH16-10-0790
46. Ghofrani HA, Voswinckel R, Reichenberger F, Weissmann N, Schermuly RT, Seeger W, et al. Hypoxia- and non-hypoxia-related pulmonary hypertension - established and new therapies. Cardiovasc Res. (2006) 72:30–40. doi: 10.1016/j.cardiores.2006.07.025
47. Hopkins SR, Levin DL. Heterogeneous pulmonary blood flow in response to hypoxia: a risk factor for high altitude pulmonary edema? Respir Physiol Neurobiol. (2006) 151:217–28. doi: 10.1016/j.resp.2005.10.007
48. Groves BM, Droma T, Sutton JR, McCullough RG, McCullough RE, Zhuang J, et al. Minimal hypoxic pulmonary hypertension in normal Tibetans at 3,658 m. J Appl Physiol. (1985) 74:312–8. doi: 10.1152/jappl.1993.74.1.312
49. Beall CM, Cavalleri GL, Deng L, Elston RC, Gao Y, Knight J, et al. Natural selection on EPAS1 (HIF2alpha) associated with low hemoglobin concentration in Tibetan highlanders. Proc Natl Acad Sci USA. (2010) 107:11459–64. doi: 10.1073/pnas.1002443107
50. Simonson TS, Yang Y, Huff CD, Yun H, Qin G, Witherspoon DJ, et al. Genetic evidence for high-altitude adaptation in Tibet. Science. (2010) 329:72–5. doi: 10.1126/science.1189406
51. Yi X, Liang Y, Huerta-Sanchez E, Jin X, Cuo ZX, Pool JE, et al. Sequencing of 50 human exomes reveals adaptation to high altitude. Science. (2010) 329:75–8. doi: 10.1126/science.1190371
52. Gruber M, Hu CJ, Johnson RS, Brown EJ, Keith B, Simon MC. Acute postnatal ablation of Hif-2alpha results in anemia. Proc Natl Acad Sci USA. (2007) 104:2301–6. doi: 10.1073/pnas.0608382104
53. Naeije R, Huez S, Lamotte M, Retailleau K, Neupane S, Abramowicz D, et al. Pulmonary artery pressure limits exercise capacity at high altitude. Eur Respir J. (2010) 36:1049–55. doi: 10.1183/09031936.00024410
54. Faoro V, Huez S, Giltaire S, Pavelescu A, van Osta A, Moraine JJ, et al. Effects of acetazolamide on aerobic exercise capacity and pulmonary hemodynamics at high altitudes. J Appl Physiol. (1985) 103:1161–5. doi: 10.1152/japplphysiol.00180.2007
55. Newman JH, Holt TN, Hedges LK, Womack B, Memon SS, Willers ED, et al. High-altitude pulmonary hypertension in cattle (brisket disease): Candidate genes and gene expression profiling of peripheral blood mononuclear cells. Pulm Circ. (2011) 1:462–9. doi: 10.4103/2045-8932.93545
56. Rhodes J. Comparative physiology of hypoxic pulmonary hypertension: historical clues from brisket disease. J Appl Physiol. (2005) 98:1092–100. doi: 10.1152/japplphysiol.01017.2004
57. Durmowicz AG, Orton EC, Stenmark KR. Progressive loss of vasodilator responsive component of pulmonary hypertension in neonatal calves exposed to 4,570 m. Am J Physiol. (1993) 265:H2175–83. doi: 10.1152/ajpheart.1993.265.6.H2175
58. Durmowicz AG, Hofmeister S, Kadyraliev TK, Aldashev AA, Stenmark KR. Functional and structural adaptation of the yak pulmonary circulation to residence at high altitude. J Appl Physiol. (1985) 74:2276–85. doi: 10.1152/jappl.1993.74.5.2276
59. Mizuno S, Ishizaki T, Toga H, Sakai A, Isakova J, Taalaibekova E, et al. Endogenous asymmetric dimethylarginine pathway in high altitude adapted yaks. Biomed Res Int. (2015) 2015:196904. doi: 10.1155/2015/196904
60. Siques P, Brito J, Schwedhelm E, Pena E, León-Velarde F, De La Cruz JJ, et al. Asymmetric dimethylarginine at sea level is a predictive marker of hypoxic pulmonary arterial hypertension at high altitude. Front Physiol. (2019) 10:651. doi: 10.3389/fphys.2019.00651
61. West JB. Intermittent exposure to high altitude. High Alt Med Biol. (2002) 3:141–3. doi: 10.1089/15270290260131858
62. Richalet JP, Donoso MV, Jiménez D, Antezana AM, Hudson C, Cortès G, et al. Chilean miners commuting from sea level to 4500 m: a prospective study. High Alt Med Biol. (2002) 3:159–66. doi: 10.1089/15270290260131894
63. León-Velarde F, Maggiorini M, Reeves JT, Aldashev A, Asmus I, Bernardi L, et al. Consensus statement on chronic and subacute high altitude diseases. High Alt Med Biol. (2005) 6:147–57. doi: 10.1089/ham.2005.6.147
64. Soria R, Egger M, Scherrer U, Bender N, Rimoldi SF. Pulmonary artery pressure and arterial oxygen saturation in people living at high or low altitude: systematic review and meta-analysis. J Appl Physiol. (2016) 121:1151–9. doi: 10.1152/japplphysiol.00394.2016
65. Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. (2019) 53:1913. doi: 10.1183/13993003.01913-2018
66. Naeije R. Pulmonary hypertension at high altitude. Eur Respir J. (2019) 53:985. doi: 10.1183/13993003.00985-2019
67. Sharma S, Stansbury R, Hackett B, Fox H. Sleep apnea and pulmonary hypertension: A riddle waiting to be solved. Pharmacol Ther. (2021) 227:107935. doi: 10.1016/j.pharmthera.2021.107935
68. Bironneau V, Tamisier R, Trzepizur W, Andriantsitohaina R, Berger M, Goupil F, et al. Sleep apnoea and endothelial dysfunction: An individual patient data meta-analysis. Sleep Med Rev. (2020) 52:101309. doi: 10.1016/j.smrv.2020.101309
69. Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med. (2020) 383:120–8. doi: 10.1056/NEJMoa2015432
70. Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. (2020) 395:1417–8. doi: 10.1016/S0140-6736(20)30937-5
71. Hannemann J, Balfanz P, Schwedhelm E, Hartmann B, Ule J, Müller-Wieland D, et al. Elevated serum SDMA and ADMA at hospital admission predict in-hospital mortality of COVID-19 patients. Sci Rep. (2021) 11:9895. doi: 10.1038/s41598-021-89180-w
72. Lotz C, Muellenbach RM, Meybohm P, Mutlak H, Lepper PM, Rolfes CB, et al. Effects of inhaled nitric oxide in COVID-19-induced ARDS - Is it worthwhile? Acta Anaesthesiol Scand. (2021) 65:629–32. doi: 10.1111/aas.13757
73. Tavazzi G, Pozzi M, Mongodi S, Dammassa V, Romito G, Mojoli F. Inhaled nitric oxide in patients admitted to intensive care unit with COVID-19 pneumonia. Crit Care. (2020) 24:508. doi: 10.1186/s13054-020-03222-9
74. Santamarina MG, Beddings I, Lomakin FM, Boisier Riscal D, Gutiérrez Claveria M, Vidal Marambio J, et al. Sildenafil for treating patients with COVID-19 and perfusion mismatch: a pilot randomized trial. Crit Care. (2022) 26:1. doi: 10.1186/s13054-021-03885-y
75. Lucero García Rojas EY, Villanueva C, Bond RA. Hypoxia inducible factors as central players in the pathogenesis and pathophysiology of cardiovascular diseases. Front Cardiovasc Med. (2021) 8:709509. doi: 10.3389/fcvm.2021.709509
76. Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. (2012) 148:399–408. doi: 10.1016/j.cell.2012.01.021
77. Buckler KJ, Vaughan-Jones RD. Effects of mitochondrial uncouplers on intracellular calcium, pH and membrane potential in rat carotid body type I cells. J Physiol. (1998) 513:819–33. doi: 10.1111/j.1469-7793.1998.819ba.x
78. Weir EK, López-Barneo J, Buckler KJ, Archer SL. Acute oxygen-sensing mechanisms. N Engl J Med. (2005) 353:2042–55. doi: 10.1056/NEJMra050002
79. Olschewski A, Hong Z, Peterson DA, Nelson DP, Porter VA, Weir EK. Opposite effects of redox status on membrane potential, cytosolic calcium, and tone in pulmonary arteries and ductus arteriosus. Am J Physiol Lung Cell Mol Physiol. (2004) 286:L15–22. doi: 10.1152/ajplung.00372.2002
80. Freund-Michel V, Khoyrattee N, Savineau JP, Muller B, Guibert C. Mitochondria: roles in pulmonary hypertension. Int J Biochem Cell Biol. (2014) 55:93–7. doi: 10.1016/j.biocel.2014.08.012
81. McElroy GS, Chandel NS. Mitochondria control acute and chronic responses to hypoxia. Exp Cell Res. (2017) 356:217–22. doi: 10.1016/j.yexcr.2017.03.034
82. Waypa GB, Chandel NS, Schumacker PT. Model for hypoxic pulmonary vasoconstriction involving mitochondrial oxygen sensing. Circ Res. (2001) 88:1259–66. doi: 10.1161/hh1201.091960
83. Dunham-Snary KJ, Wu D, Potus F, Sykes EA, Mewburn JD, Charles RL, et al. Ndufs2, a core subunit of mitochondrial complex i, is essential for acute oxygen-sensing and hypoxic pulmonary vasoconstriction. Circ Res. (2019) 124:1727–46. doi: 10.1161/CIRCRESAHA.118.314284
84. Chouchani ET, Pell VR, Gaude E, Aksentijević D, Sundier SY, Robb EL, et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. (2014) 515:431–5. doi: 10.1038/nature13909
85. Murphy MP. Understanding and preventing mitochondrial oxidative damage. Biochem Soc Trans. (2016) 44:1219–26. doi: 10.1042/BST20160108
86. Corcoran SE, O'Neill LA. HIF1α and metabolic reprogramming in inflammation. J Clin Invest. (2016) 126:3699–707. doi: 10.1172/JCI84431
87. Waypa GB, Schumacker PT. Hypoxia-induced changes in pulmonary and systemic vascular resistance: where is the O2 sensor? Respir Physiol Neurobiol. (2010) 174:201–11. doi: 10.1016/j.resp.2010.08.007
88. Caja S, Enríquez JA. Mitochondria in endothelial cells: Sensors and integrators of environmental cues. Redox Biol. (2017) 12:821–7. doi: 10.1016/j.redox.2017.04.021
89. Oczypok EA, Perkins TN, Oury TD. Alveolar epithelial cell-derived mediators: potential direct regulators of large airway and vascular responses. Am J Respir Cell Mol Biol. (2017) 56:694–9. doi: 10.1165/rcmb.2016-0151PS
90. Sylvester JT, Shimoda LA, Aaronson PI, Ward JP. Hypoxic pulmonary vasoconstriction. Physiol Rev. (2012) 92:367–520. doi: 10.1152/physrev.00041.2010
91. Hasunuma K, Rodman DM, McMurtry IF. Effects of K+ channel blockers on vascular tone in the perfused rat lung. Am Rev Respir Dis. (1991) 144:884–7. doi: 10.1164/ajrccm/144.4.884
92. McMurtry IF. BAY K 8644 potentiates and A23187 inhibits hypoxic vasoconstriction in rat lungs. Am J Physiol. (1985) 249:H741–6. doi: 10.1152/ajpheart.1985.249.4.H741
93. McMurtry IF, Davidson AB, Reeves JT, Grover RF. Inhibition of hypoxic pulmonary vasoconstriction by calcium antagonists in isolated rat lungs. Circ Res. (1976) 38:99–104. doi: 10.1161/01.RES.38.2.99
94. Yanagisawa M, Kurihara H, Kimura S, Tomobe Y, Kobayashi M, Mitsui Y, et al. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. (1988) 332:411–5. doi: 10.1038/332411a0
95. Rubanyi GM, Vanhoutte PM. Superoxide anions and hyperoxia inactivate endothelium-derived relaxing factor. Am J Physiol. (1986) 250:H822–7. doi: 10.1152/ajpheart.1986.250.5.H822
96. Auch-Schwelk W, Vanhoutte PM. Endothelium-derived contracting factor released by serotonin in the aorta of the spontaneously hypertensive rat. Am J Hypertens. (1991) 4:769–72. doi: 10.1093/ajh/4.9.769
97. Feletou M, Vanhoutte PM. Endothelium-dependent hyperpolarization of canine coronary smooth muscle. Br J Pharmacol. (1988) 93:515–24. doi: 10.1111/j.1476-5381.1988.tb10306.x
98. Houde M, Desbiens L, D'Orléans-Juste P. Endothelin-1: biosynthesis, signaling and vasoreactivity. Adv Pharmacol. (2016) 77:143–75. doi: 10.1016/bs.apha.2016.05.002
99. de Nucci G, Thomas R, D'Orleans-Juste P, Antunes E, Walder C, Warner TD, et al. Pressor effects of circulating endothelin are limited by its removal in the pulmonary circulation and by the release of prostacyclin and endothelium-derived relaxing factor. Proc Natl Acad Sci USA. (1988) 85:9797–800. doi: 10.1073/pnas.85.24.9797
100. Takayanagi R, Kitazumi K, Takasaki C, Ohnaka K, Aimoto S, Tasaka K, et al. Presence of non-selective type of endothelin receptor on vascular endothelium and its linkage to vasodilation. FEBS Lett. (1991) 282:103–6. doi: 10.1016/0014-5793(91)80454-B
101. Firth JD, Ratcliffe PJ. Organ distribution of the three rat endothelin messenger RNAs and the effects of ischemia on renal gene expression. J Clin Invest. (1992) 90:1023–31. doi: 10.1172/JCI115915
102. Kourembanas S, Marsden PA, McQuillan LP, Faller DV. Hypoxia induces endothelin gene expression and secretion in cultured human endothelium. J Clin Invest. (1991) 88:1054–7. doi: 10.1172/JCI115367
103. Elton TS, Oparil S, Taylor GR, Hicks PH, Yang RH, Jin H, et al. Normobaric hypoxia stimulates endothelin-1 gene expression in the rat. Am J Physiol. (1992) 263:R1260–4. doi: 10.1152/ajpregu.1992.263.6.R1260
104. Li H, Chen SJ, Chen YF, Meng QC, Durand J, Oparil S, et al. Enhanced endothelin-1 and endothelin receptor gene expression in chronic hypoxia. J Appl Physiol. (1994) 77:1451–9. doi: 10.1152/jappl.1994.77.3.1451
105. Li H, Elton TS, Chen YF, Oparil S. Increased endothelin receptor gene expression in hypoxic rat lung. Am J Physiol. (1994) 266:L553–60. doi: 10.1152/ajplung.1994.266.5.L553
106. Ferri C, Bellini C, De Angelis C, De Siati L, Perrone A, Properzi G, et al. Circulating endothelin-1 concentrations in patients with chronic hypoxia. J Clin Pathol. (1995) 48:519–24. doi: 10.1136/jcp.48.6.519
107. Chen SJ, Chen YF, Meng QC, Durand J, Dicarlo VS, Oparil S. Endothelin-receptor antagonist bosentan prevents and reverses hypoxic pulmonary hypertension in rats. J Appl Physiol. (1995) 79:2122–31. doi: 10.1152/jappl.1995.79.6.2122
108. Holm P, Liska J, Clozel M, Franco-Cereceda A. The endothelin antagonist bosentan: hemodynamic effects during normoxia and hypoxic pulmonary hypertension in pigs. J Thorac Cardiovasc Surg. (1996) 112:890–7. doi: 10.1016/S0022-5223(96)70088-0
109. Pham I, Wuerzner G, Richalet JP, Peyrard S, Azizi M. Endothelin receptors blockade blunts hypoxia-induced increase in PAP in humans. Eur J Clin Invest. (2010) 40:195–202. doi: 10.1111/j.1365-2362.2010.02254.x
110. Kelland NF, Bagnall AJ, Morecroft I, Gulliver-Sloan FH, Dempsie Y, Nilsen M, et al. Endothelial ET(B) limits vascular remodelling and development of pulmonary hypertension during hypoxia. J Vasc Res. (2010) 47:16–22. doi: 10.1159/000231717
111. Lüscher TF, Vanhoutte PM. Endothelium-dependent contractions to acetylcholine in the aorta of the spontaneously hypertensive rat. Hypertension. (1986) 8:344–8. doi: 10.1161/01.HYP.8.4.344
112. Boulanger CM, Morrison KJ, Vanhoutte PM. Mediation by M3-muscarinic receptors of both endothelium-dependent contraction and relaxation to acetylcholine in the aorta of the spontaneously hypertensive rat. Br J Pharmacol. (1994) 112:519–24. doi: 10.1111/j.1476-5381.1994.tb13104.x
113. Schwedhelm E, Böger RH. Application of gas chromatography-mass spectrometry for analysis of isoprostanes: their role in cardiovascular disease. Clin Chem Lab Med. (2003) 41:1552–61. doi: 10.1515/CCLM.2003.238
114. Delannoy E, Courtois A, Freund-Michel V, Leblais V, Marthan R, Muller B. Hypoxia-induced hyperreactivity of pulmonary arteries: role of cyclooxygenase-2, isoprostanes, and thromboxane receptors. Cardiovasc Res. (2010) 85:582–92. doi: 10.1093/cvr/cvp292
115. Böger RH. The emerging role of asymmetric dimethylarginine as a novel cardiovascular risk factor. Cardiovasc Res. (2003) 59:824–33. doi: 10.1016/S0008-6363(03)00500-5
116. Böger RH. Asymmetric dimethylarginine (ADMA): a novel risk marker in cardiovascular medicine and beyond. Ann Med. (2006) 38:126–36. doi: 10.1080/07853890500472151
117. Hannemann J, Zummack J, Hillig J, Böger R. Metabolism of asymmetric dimethylarginine in hypoxia: from bench to bedside. Pulm Circ. (2020) 10:2045894020918846. doi: 10.1177/2045894020918846
118. Crapo JD, Barry BE, Gehr P, Bachofen M, Weibel ER. Cell number and cell characteristics of the normal human lung. Am Rev Respir Dis. (1982) 126:332–7.
119. Duke HN. The site of action of anoxia on the pulmonary blood vessels of the cat. J Physiol. (1954) 125:373–82. doi: 10.1113/jphysiol.1954.sp005165
120. Signorelli S, Jennings P, Leonard MO, Pfaller W. Differential effects of hypoxic stress in alveolar epithelial cells and microvascular endothelial cells. Cell Physiol Biochem. (2010) 25:135–44. doi: 10.1159/000272066
121. Jovanović S, Crawford RM, Ranki HJ, Jovanović A. Large conductance Ca2+-activated K+ channels sense acute changes in oxygen tension in alveolar epithelial cells. Am J Respir Cell Mol Biol. (2003) 28:363–72. doi: 10.1165/rcmb.2002-0101OC
122. Asano K, Chee CB, Gaston B, Lilly CM, Gerard C, Drazen JM, et al. Constitutive and inducible nitric oxide synthase gene expression, regulation, and activity in human lung epithelial cells. Proc Natl Acad Sci USA. (1994) 91:10089–93. doi: 10.1073/pnas.91.21.10089
123. Wang Y, Li X, Niu W, Chen J, Zhang B, Zhang X, et al. The alveolar epithelial cells are involved in pulmonary vascular remodeling and constriction of hypoxic pulmonary hypertension. Respir Res. (2021) 22:134. doi: 10.1186/s12931-021-01708-w
124. Lewis NCS, Bain AR, Wildfong KW, Green DJ, Ainslie PN. Acute hypoxaemia and vascular function in healthy humans. Exp Physiol. (2017) 102:1635–46. doi: 10.1113/EP086532
125. Bruno RM, Cogo A, Ghiadoni L, Duo E, Pomidori L, Sharma R, et al. Cardiovascular function in healthy Himalayan high-altitude dwellers. Atherosclerosis. (2014) 236:47–53. doi: 10.1016/j.atherosclerosis.2014.06.017
126. Calderón-Gerstein WS, López-Peña A, Macha-Ramírez R, Bruno-Huamán A, Espejo-Ramos R, Vílchez-Bravo S, et al. Endothelial dysfunction assessment by flow-mediated dilation in a high-altitude population. Vasc Health Risk Manag. (2017) 13:421–6. doi: 10.2147/VHRM.S151886
127. Giaid A, Saleh D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N Engl J Med. (1995) 333:214–21. doi: 10.1056/NEJM199507273330403
128. Zhao YY, Zhao YD, Mirza MK, Huang JH, Potula HH, Vogel SM, et al. Persistent eNOS activation secondary to caveolin-1 deficiency induces pulmonary hypertension in mice and humans through PKG nitration. J Clin Invest. (2009) 119:2009–18. doi: 10.1172/JCI33338
129. Berger RM, Geiger R, Hess J, Bogers AJ, Mooi WJ. Altered arterial expression patterns of inducible and endothelial nitric oxide synthase in pulmonary plexogenic arteriopathy caused by congenital heart disease. Am J Respir Crit Care Med. (2001) 163:1493–9. doi: 10.1164/ajrccm.163.6.9908137
130. Förstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J. (2012) 33:829-37:37a−37d. doi: 10.1093/eurheartj/ehr304
131. Moncada S, Higgs A. The L-arginine-nitric oxide pathway. N Engl J Med. (1993) 329:2002–12. doi: 10.1056/NEJM199312303292706
132. Millatt LJ, Whitley GS, Li D, Leiper JM, Siragy HM, Carey RM, et al. Evidence for dysregulation of dimethylarginine dimethylaminohydrolase I in chronic hypoxia-induced pulmonary hypertension. Circulation. (2003) 108:1493–8. doi: 10.1161/01.CIR.0000089087.25930.FF
133. Arrigoni FI, Vallance P, Haworth SG, Leiper JM. Metabolism of asymmetric dimethylarginines is regulated in the lung developmentally and with pulmonary hypertension induced by hypobaric hypoxia. Circulation. (2003) 107:1195–201. doi: 10.1161/01.CIR.0000051466.00227.13
134. Yildirim AO, Bulau P, Zakrzewicz D, Kitowska KE, Weissmann N, Grimminger F, et al. Increased protein arginine methylation in chronic hypoxia: role of protein arginine methyltransferases. Am J Respir Cell Mol Biol. (2006) 35:436–43. doi: 10.1165/rcmb.2006-0097OC
135. Song MY, Zwemer CF, Whitesall SE, D'Alecy LG. Acute and conditioned hypoxic tolerance augmented by endothelial nitric oxide synthase inhibition in mice. J Appl Physiol. (2007) 102:610–5. doi: 10.1152/japplphysiol.00894.2006
136. Leiper J, Nandi M, Torondel B, Murray-Rust J, Malaki M, O'Hara B, et al. Disruption of methylarginine metabolism impairs vascular homeostasis. Nat Med. (2007) 13:198–203. doi: 10.1038/nm1543
137. Ahmad T, Mabalirajan U, Ghosh B, Agrawal A. Altered asymmetric dimethyl arginine metabolism in allergically inflamed mouse lungs. Am J Respir Cell Mol Biol. (2010) 42:3–8. doi: 10.1165/rcmb.2009-0137RC
138. Bakr A, Pak O, Taye A, Hamada F, Hemeida R, Janssen W, et al. Effects of dimethylarginine dimethylaminohydrolase-1 overexpression on the response of the pulmonary vasculature to hypoxia. Am J Respir Cell Mol Biol. (2013) 49:491–500. doi: 10.1165/rcmb.2012-0330OC
139. Lambden S, Martin D, Vanezis K, Lee B, Tomlinson J, Piper S, et al. Hypoxia causes increased monocyte nitric oxide synthesis which is mediated by changes in dimethylarginine dimethylaminohydrolase 2 expression in animal and human models of normobaric hypoxia. Nitric Oxide. (2016) 58:59–66. doi: 10.1016/j.niox.2016.06.003
140. Badran M, Abuyassin B, Golbidi S, Ayas N, Laher I. Uncoupling of vascular nitric oxide synthase caused by intermittent hypoxia. Oxid Med Cell Longev. (2016) 2016:2354870. doi: 10.1155/2016/2354870
141. Lüneburg N, Siques P, Brito J, Arriaza K, Pena E, Klose H, et al. Long-term chronic intermittent hypobaric hypoxia in rats causes an imbalance in the asymmetric dimethylarginine/nitric oxide pathway and ROS activity: A possible synergistic mechanism for altitude pulmonary hypertension? Pulm Med. (2016) 2016:6578578. doi: 10.1155/2016/6578578
142. Iannone L, Zhao L, Dubois O, Duluc L, Rhodes CJ, Wharton J, et al. miR-21/DDAH1 pathway regulates pulmonary vascular responses to hypoxia. Biochem J. (2014) 462:103–12. doi: 10.1042/BJ20140486
143. Hannemann J, Glatzel A, Hillig J, Zummack J, Schumacher U, Lüneburg N, et al. Upregulation of DDAH2 limits pulmonary hypertension and right ventricular hypertrophy during chronic hypoxia in ddah1 knockout mice. Front Physiol. (2020) 11:597559. doi: 10.3389/fphys.2020.597559
144. Bedford MT, Clarke SG. Protein arginine methylation in mammals: who, what, and why. Mol Cell. (2009) 33:1–13. doi: 10.1016/j.molcel.2008.12.013
145. Chafai A, Fromm MF, König J, Maas R. The prognostic biomarker L-homoarginine is a substrate of the cationic amino acid transporters CAT1, CAT2A and CAT2B. Sci Rep. (2017) 7:4767. doi: 10.1038/s41598-017-04965-2
146. Closs EI, Basha FZ, Habermeier A, Förstermann U. Interference of L-arginine analogues with L-arginine transport mediated by the y+ carrier hCAT-2B. Nitric Oxide. (1997) 1:65–73. doi: 10.1006/niox.1996.0106
147. Guccione E, Richard S. The regulation, functions and clinical relevance of arginine methylation. Nat Rev Mol Cell Biol. (2019) 20:642–57. doi: 10.1038/s41580-019-0155-x
148. Scaglione A, Patzig J, Liang J, Frawley R, Bok J, Mela A, et al. PRMT5-mediated regulation of developmental myelination. Nat Commun. (2018) 9:2840. doi: 10.1038/s41467-018-04863-9
149. McBride AE, Silver PA. State of the arg: protein methylation at arginine comes of age. Cell. (2001) 106:5–8. doi: 10.1016/S0092-8674(01)00423-8
150. Lüneburg N, von Holten RA, Töpper RF, Schwedhelm E, Maas R, Böger RH. Symmetric dimethylarginine is a marker of detrimental outcome in the acute phase after ischaemic stroke: role of renal function. Clin Sci (Lond). (2012) 122:105–11. doi: 10.1042/CS20110013
151. Schulze F, Carter AM, Schwedhelm E, Ajjan R, Maas R, von Holten RA, et al. Symmetric dimethylarginine predicts all-cause mortality following ischemic stroke. Atherosclerosis. (2010) 208:518–23. doi: 10.1016/j.atherosclerosis.2009.06.039
152. Wang D, Gill PS, Chabrashvili T, Onozato ML, Raggio J, Mendonca M, et al. Isoform-specific regulation by N(G),N(G)-dimethylarginine dimethylaminohydrolase of rat serum asymmetric dimethylarginine and vascular endothelium-derived relaxing factor/NO. Circ Res. (2007) 101:627–35. doi: 10.1161/CIRCRESAHA.107.158915
153. Lüneburg N, Siques P, Brito J, De La Cruz JJ, León-Velarde F, Hannemann J, et al. Long-term intermittent exposure to high altitude elevates asymmetric dimethylarginine in first exposed young adults. High Alt Med Biol. (2017) 18:226–33. doi: 10.1089/ham.2016.0123
154. Ali Z, Mishra A, Kumar R, Alam P, Pandey P, Ram R, et al. Interactions among vascular-tone modulators contribute to high altitude pulmonary edema and augmented vasoreactivity in highlanders. PLoS ONE. (2012) 7:e44049. doi: 10.1371/journal.pone.0044049
155. Tannheimer M, Hornung K, Gasche M, Kuehlmuss B, Mueller M, Welsch H, et al. Decrease of asymmetric dimethylarginine predicts acute mountain sickness. J Travel Med. (2012) 19:338–43. doi: 10.1111/j.1708-8305.2012.00652.x
156. Silva WA, Almeida-Pititto B, Santos RB, Aielo AN, Giatti S, Parise BK, et al. Obstructive sleep apnea is associated with lower adiponectin and higher cholesterol levels independently of traditional factors and other sleep disorders in middle-aged adults: the ELSA-Brasil cohort. Sleep Breath. (2021) 25:1935–44. doi: 10.1007/s11325-021-02290-7
157. Arlouskaya Y, Sawicka A, Głowala M, Giebułtowicz J, Korytowska N, Tałałaj M, et al. Asymmetric dimethylarginine (ADMA) and symmetric dimethylarginine (SDMA) concentrations in patients with obesity and the risk of obstructive sleep apnea (OSA). J Clin Med. (2019) 8:897. doi: 10.3390/jcm8060897
158. Oyama J, Nagatomo D, Yoshioka G, Yamasaki A, Kodama K, Sato M, et al. The relationship between neutrophil to lymphocyte ratio, endothelial function, and severity in patients with obstructive sleep apnea. J Cardiol. (2016) 67:295–302. doi: 10.1016/j.jjcc.2015.06.005
159. In E, Özdemir C, Kaman D, Sökücü SN. Heat shock proteins, L-Arginine, and asymmetric dimethylarginine levels in patients with obstructive sleep apnea syndrome. Arch Bronconeumol. (2015) 51:544–50. doi: 10.1016/j.arbr.2015.09.011
160. Barceló A, Piérola J, de la Peña M, Esquinas C, Sanchez-de la Torre M, Ayllón O, et al. Day-night variations in endothelial dysfunction markers and haemostatic factors in sleep apnoea. Eur Respir J. (2012) 39:913–8. doi: 10.1183/09031936.00039911
161. Barceló A, de la Peña M, Ayllón O, Vega-Agapito MV, Piérola J, Pérez G, et al. Increased plasma levels of asymmetric dimethylarginine and soluble CD40 ligand in patients with sleep apnea. Respiration. (2009) 77:85–90. doi: 10.1159/000165630
162. Ozkan Y, Firat H, Simşek B, Torun M, Yardim-Akaydin S. Circulating nitric oxide (NO), asymmetric dimethylarginine (ADMA), homocysteine, and oxidative status in obstructive sleep apnea-hypopnea syndrome (OSAHS). Sleep Breath. (2008) 12:149–54. doi: 10.1007/s11325-007-0148-4
163. Gozal D, Kheirandish-Gozal L, Serpero LD, Sans Capdevila O, Dayyat E. Obstructive sleep apnea and endothelial function in school-aged nonobese children: effect of adenotonsillectomy. Circulation. (2007) 116:2307–14. doi: 10.1161/CIRCULATIONAHA.107.696823
164. Ohike Y, Kozaki K, Iijima K, Eto M, Kojima T, Ohga E, et al. Amelioration of vascular endothelial dysfunction in obstructive sleep apnea syndrome by nasal continuous positive airway pressure–possible involvement of nitric oxide and asymmetric NG, NG-dimethylarginine. Circ J. (2005) 69:221–6. doi: 10.1253/circj.69.221
165. Csoma B, Bikov A, Nagy L, Tóth B, Tábi T, Szucs G, et al. Dysregulation of the endothelial nitric oxide pathway is associated with airway inflammation in COPD. Respir Res. (2019) 20:156. doi: 10.1186/s12931-019-1133-8
166. Telo S, Kirkil G, Kuluöztürk M, Balin M, Deveci F. Can ADMA play a role in determining pulmonary hypertension related to chronic obstructive pulmonary disease? Clin Respir J. (2018) 12:1433–8. doi: 10.1111/crj.12675
167. Parmaksiz ET, Inal A, Salepci B, Comert S, Fidan A, Kiral N, et al. Relationship of asymmetric dimethylarginine levels with disease severity and pulmonary hypertension in chronic obstructive pulmonary disease. Lung India. (2018) 35:199–203. doi: 10.4103/lungindia.lungindia_11_17
168. Tajti G, Gesztelyi R, Pak K, Papp C, Keki S, Szilasi ME, et al. Positive correlation of airway resistance and serum asymmetric dimethylarginine level in COPD patients with systemic markers of low-grade inflammation. Int J Chron Obstruct Pulmon Dis. (2017) 12:873–84. doi: 10.2147/COPD.S127373
169. Urban MH, Eickhoff P, Funk GC, Burghuber OC, Wolzt M, Valipour A. Increased brachial intima-media thickness is associated with circulating levels of asymmetric dimethylarginine in patients with COPD. Int J Chron Obstruct Pulmon Dis. (2017) 12:169–76. doi: 10.2147/COPD.S118596
170. Vögeli A, Ottiger M, Meier MA, Steuer C, Bernasconi L, Huber A, et al. Asymmetric dimethylarginine predicts long-term outcome in patients with acute exacerbation of chronic obstructive pulmonary disease. Lung. (2017) 195:717–27. doi: 10.1007/s00408-017-0047-9
171. Costanzo L, Pedone C, Battistoni F, Chiurco D, Santangelo S, Antonelli-Incalzi R. Relationship between FEV(1) and arterial stiffness in elderly people with chronic obstructive pulmonary disease. Aging Clin Exp Res. (2017) 29:157–64. doi: 10.1007/s40520-016-0560-3
172. Aydin M, Altintas N, Cem Mutlu L, Bilir B, Oran M, Tülübaş F, et al. Asymmetric dimethylarginine contributes to airway nitric oxide deficiency in patients with COPD. Clin Respir J. (2017) 11:318–27. doi: 10.1111/crj.12337
173. Ruzsics I, Nagy L, Keki S, Sarosi V, Illes B, Illes Z, et al. L-Arginine pathway in COPD patients with acute exacerbation: a new potential biomarker. Copd. (2016) 13:139–45. doi: 10.3109/15412555.2015.1045973
174. Zinellu A, Fois AG, Sotgia S, Sotgiu E, Zinellu E, Bifulco F, et al. Arginines plasma concentration and oxidative stress in mild to moderate COPD. PLoS ONE. (2016) 11:e0160237. doi: 10.1371/journal.pone.0160237
175. Scott JA, Duongh M, Young AW, Subbarao P, Gauvreau GM, Grasemann H. Asymmetric dimethylarginine in chronic obstructive pulmonary disease (ADMA in COPD). Int J Mol Sci. (2014) 15:6062–71. doi: 10.3390/ijms15046062
176. Wang Y, Su M, Zhang X. [Effects of continuous positive airway pressure treatment of inflammatory factors in patients with overlap syndrome]. Zhonghua Yi Xue Za Zhi. (2014) 94:416–9.
177. Nural S, Günay E, Halici B, Celik S, Ünlü M. Inflammatory processes and effects of continuous positive airway pressure (CPAP) in overlap syndrome. Inflammation. (2013) 36:66–74. doi: 10.1007/s10753-012-9520-z
178. Kielstein JT, Bode-Böger SM, Hesse G, Martens-Lobenhoffer J, Takacs A, Fliser D, et al. Asymmetrical dimethylarginine in idiopathic pulmonary arterial hypertension. Arterioscler Thromb Vasc Biol. (2005) 25:1414–8. doi: 10.1161/01.ATV.0000168414.06853.f0
179. Dimitroulas T, Giannakoulas G, Sfetsios T, Karvounis H, Dimitroula H, Koliakos G, et al. Asymmetrical dimethylarginine in systemic sclerosis-related pulmonary arterial hypertension. Rheumatology (Oxford). (2008) 47:1682–5. doi: 10.1093/rheumatology/ken346
180. Liu J, Fu Q, Jiang L, Wang Y. Clinical value of asymmetrical dimethylarginine detection in patients with connective tissue disease-associated pulmonary arterial hypertension. Cardiol Res Pract. (2019) 2019:3741909. doi: 10.1155/2019/3741909
181. Parikh RV, Scherzer R, Nitta EM, Leone A, Hur S, Mistry V, et al. Increased levels of asymmetric dimethylarginine are associated with pulmonary arterial hypertension in HIV infection. Aids. (2014) 28:511–9. doi: 10.1097/QAD.0000000000000124
182. Skoro-Sajer N, Mittermayer F, Panzenboeck A, Bonderman D, Sadushi R, Hitsch R, et al. Asymmetric dimethylarginine is increased in chronic thromboembolic pulmonary hypertension. Am J Respir Crit Care Med. (2007) 176:1154–60. doi: 10.1164/rccm.200702-278OC
183. Kendzerska T, Leung RS, Aaron SD, Ayas N, Sandoz JS, Gershon AS. Cardiovascular outcomes and all-cause mortality in patients with obstructive sleep apnea and chronic obstructive pulmonary disease (overlap syndrome). Ann Am Thorac Soc. (2019) 16:71–81. doi: 10.1513/AnnalsATS.201802-136OC
184. Marin JM, Soriano JB, Carrizo SJ, Boldova A, Celli BR. Outcomes in patients with chronic obstructive pulmonary disease and obstructive sleep apnea: the overlap syndrome. Am J Respir Crit Care Med. (2010) 182:325–31. doi: 10.1164/rccm.200912-1869OC
185. Kato M, Roberts-Thomson P, Phillips BG, Haynes WG, Winnicki M, Accurso V, et al. Impairment of endothelium-dependent vasodilation of resistance vessels in patients with obstructive sleep apnea. Circulation. (2000) 102:2607–10. doi: 10.1161/01.CIR.102.21.2607
186. Böger RH, Maas R, Schulze F, Schwedhelm E. Asymmetric dimethylarginine (ADMA) as a prospective marker of cardiovascular disease and mortality–an update on patient populations with a wide range of cardiovascular risk. Pharmacol Res. (2009) 60:481–7. doi: 10.1016/j.phrs.2009.07.001
187. Leong T, Zylberstein D, Graham I, Lissner L, Ward D, Fogarty J, et al. Asymmetric dimethylarginine independently predicts fatal and nonfatal myocardial infarction and stroke in women: 24-year follow-up of the population study of women in Gothenburg. Arterioscler Thromb Vasc Biol. (2008) 28:961–7. doi: 10.1161/ATVBAHA.107.156596
Keywords: high altitude, endothelium/physiopathology, asymmetric dimethylarginine (ADMA), hypoxaemia, chronic obstructive lung disease (COPD), obstructive sleep apnea syndrome (OSAS)
Citation: Hannemann J and Böger R (2022) Dysregulation of the Nitric Oxide/Dimethylarginine Pathway in Hypoxic Pulmonary Vasoconstriction—Molecular Mechanisms and Clinical Significance. Front. Med. 9:835481. doi: 10.3389/fmed.2022.835481
Received: 14 December 2021; Accepted: 27 January 2022;
Published: 17 February 2022.
Edited by:
Olivier Sitbon, Université Paris-Saclay, FranceReviewed by:
Cheng Jun Hu, University of Colorado, United StatesDavid Wu, University of Chicago, United States
Copyright © 2022 Hannemann and Böger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rainer Böger, boeger@uke.de
†ORCID: Juliane Hannemann orcid.org/0000-0003-3499-4827
Rainer Böger orcid.org/0000-0003-3770-5357