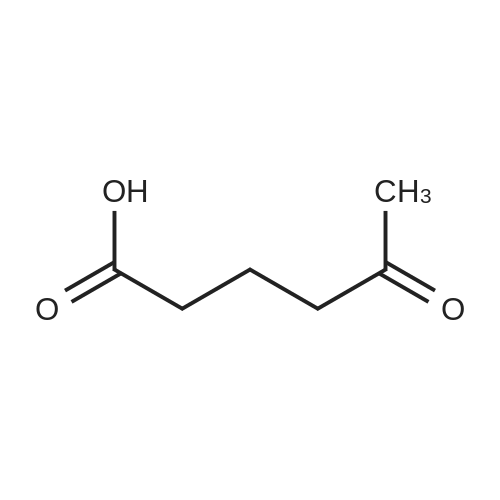
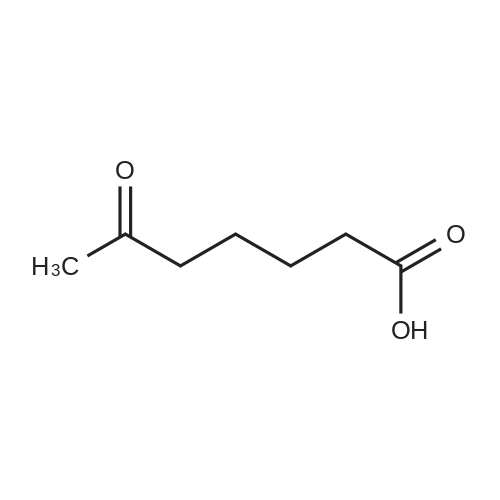
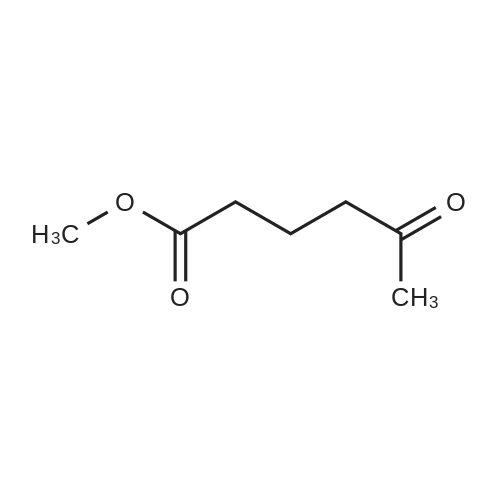
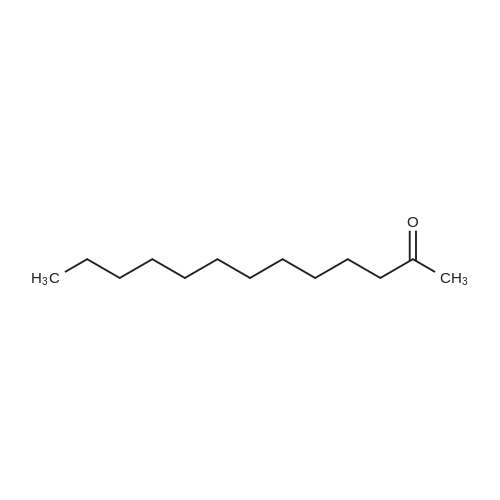
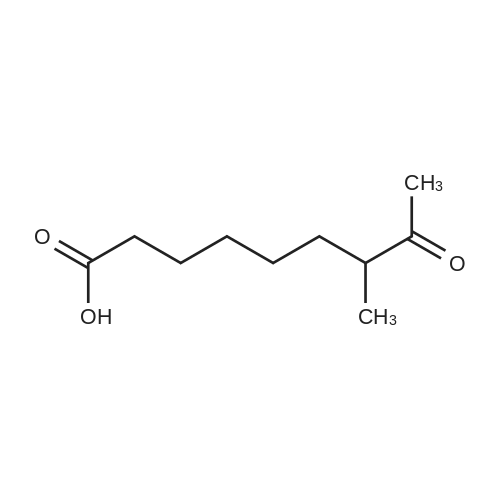
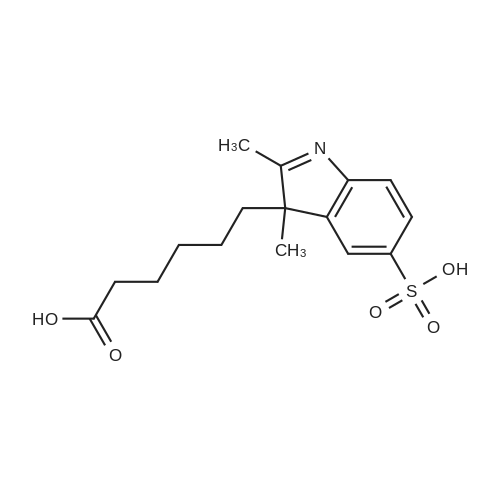
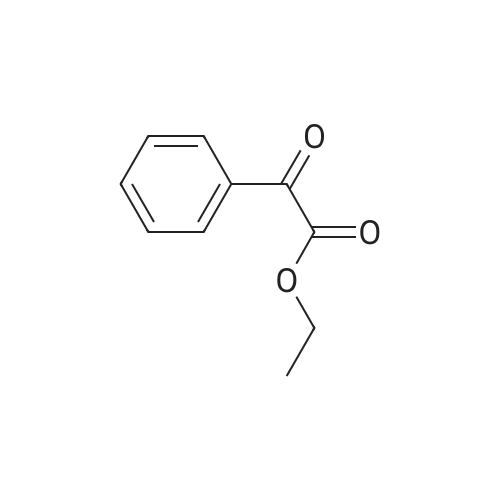
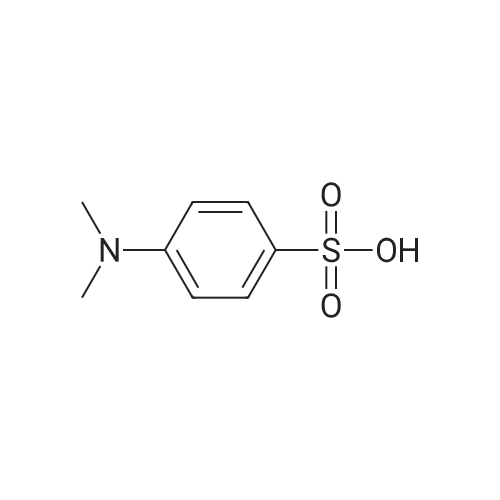
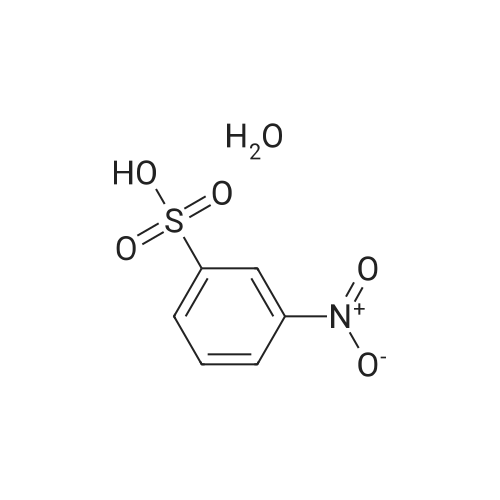
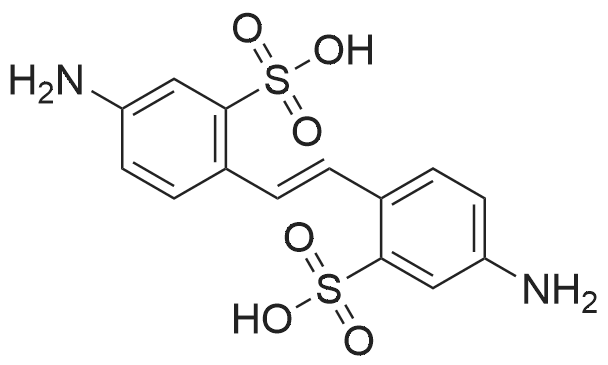
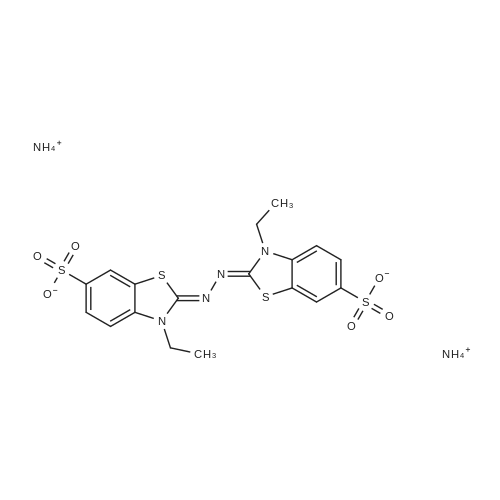
| 93.4% |
With acetic acid for 3h; Heating; |
|
| 91% |
With sodium acetate; acetic acid at 130℃; Inert atmosphere; Schlenk technique; |
|
| 90% |
In acetic acid for 14h; Reflux; |
|
| 89% |
With acetic acid for 4h; Reflux; |
1.1
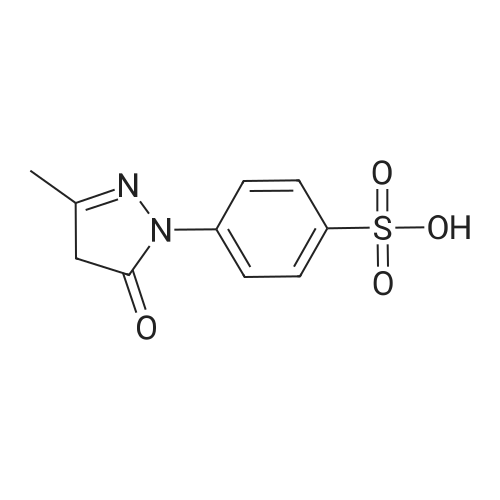
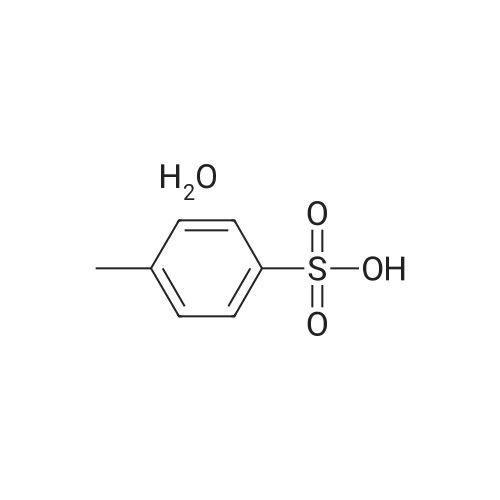
p-hydrazinobenzenesulfonic acid (10 g, 53 mmol, 1 eq, Aldrich) and 3-methyl-2-butanone (17.18 mL, 160 mmol, 3.02 eq, TCI) were added to acetic acid (30 mL), and the resulting mixture was heated under reflux for 4 hours. The reaction mixture was allowed to cool to ambient temperature and the resulting solid particles were filtered. The filtrate was washed with ethyl acetate two or three times and dried under reduced pressure (11.34 g, 89%). Rf=0.68 (RP-C18, acetonitrile/water 1:4 v/v). |
| 89% |
With acetic acid for 4h; Reflux; |
1.1
17.18 mL (160 mmol, 3.02 eq) of p-hydrazinobenzenesulfonic acid (10 g, 53 mmol, 1 eq) and 3-methyl-2-butanone Acetic acid (30 mL), and the mixture was heated and refluxed for 4 hours. The mixture was cooled to room temperature, and the resulting solid particles were filtered. After washing with ethyl acetate two or three times, it was dried under reduced pressure. (11.34 g, 89%). |
| 89% |
With acetic acid for 4h; Reflux; |
1.1
P-hydrazinobenzenesulfonic acid (10 g, 53 mmol, 1 eq, Aldrich) and 3-methyl-2-butanone (17.18 mL, 160 mmol, 3.02 eq, TCI) was added to 30 mL of acetic acid and the mixture was heated and refluxed for 4 hours. The mixture was cooled to room temperature, and the resulting solid particles were filtered. Washed with ethyl acetate two or three times, and then dried under reduced pressure. (11.34 g, 89%). |
| 89% |
With acetic acid for 4h; Reflux; |
1.1
p-hydrazinobenzenesulfonic acid (10 g, 53 mmol, 1 eq, Aldrich) and 3-methyl-2-butanone (17.18 mL, 160 mmol, 3.02 eq, TCI) was added to 30 mL of acetic acid, and the mixture was heated to reflux for 4 hours. The mixture was cooled to room temperature, and the resulting solid particles were filtered. After washing with ethyl acetate two or three times, it was dried under reduced pressure. (11.34 g, 89%). |
| 89% |
With acetic acid for 4h; Reflux; |
1.1 (1) Synthesis of Compound 4-1
p-hydrazinobenzenesulfonic acid (10 g, 53 mmol, 1 eq, Aldrich) and 3-methyl-2-butanone (17.18 mL, 160 mmol, 3.02 eq, TCI) was added to 30 mL of acetic acid, and the mixture was reacted by heating under reflux for 4 hours. The mixture was cooled to room temperature, and the resulting solid particles were filtered. After washing with ethyl acetate two or three times, it was dried under reduced pressure. (11.34 g, 89%). |
| 89% |
With acetic acid for 4h; Reflux; |
1.1 1. Synthesis of Compound 1-1
Hydrazinobenzenesulfonic acid (10 g, 53 mmol, 1 eq, Aldrich) and 3-methyl-2-butanone (17.18 mL, 160 mmol, 3.02 eq, TCI) was added to 30 mL of acetic acid, and the mixture was reacted by heating under reflux for 4 hours. After completion of the reaction, the reaction mixture was cooled to room temperature, and the resulting solid particles were filtered. The residue was washed with ethyl acetate two or three times, and dried under reduced pressure to obtain 11.34 g (89%) of a solid substance. |
| 86% |
With acetic acid at 120℃; for 3h; |
4.1; 12.1
Example 4. Synthesis of the compounds (32) and (42) of the present invention; [Show Image] (1) Synthesis of the indolenine compound (30); [Show Image] [Show Image] [Synthesis of the compound (27)]; As starting raw materials, 4-hydrazinobenzenesulfonic acid 0.5hydrate [compound(26)] (50.0 g, 0.253 mol), and 3-methyl-2-butanone (65.5 g, 0.759 mol) were used, which were subjected to stirring in acetic acid (200 mL) at 120°C for 3 hours. After completion of the reaction, the solvent was cooled, and was subjected to washing twice by the addition of diethyl ether (300 mL) to give the compound (27) (52.0 g, yield; 86%). Property data: Mass (nega=238); Example 12. Synthesis of the compound (43) of the present invention; [Show Image] (1) Synthesis of indolenine compound (30); [Show Image] [Show Image] [Synthesis of the compound (27)]; As starting raw materials, 4-hydrazinobenzene sulfonic acid 0.5hydrate (26) (50.0 g, 0.253mol) and 3-methyl-2-butanone (65.5 g, 0.759 mol) were used, which were subjected to stirring in acetic acid (200 mL) at 120°C for 3 hours. After completion of the reaction, the solvent was cooled, to be subjected to washing twice by the addition of diethyl ether (300 mL) to give the compound (27) (52.0 g, yield; 86%). Property data: Mass (nega=238) |
| 85% |
With acetic acid for 14h; Reflux; |
2 2.2 Synthesis of 2,5-bis[2,3,3-trimethyl-3H-indole-5-sulfonic acid]-croconaine
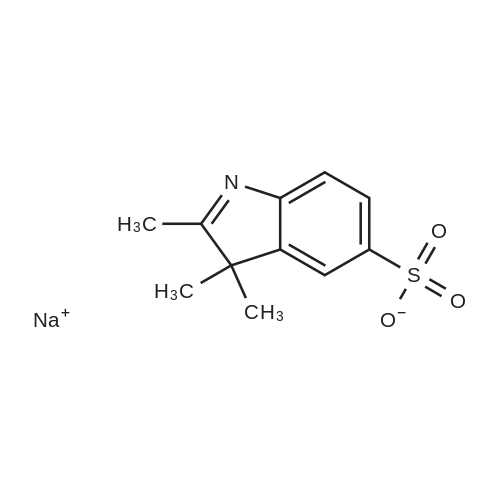
The synthetic route of 2,5-bis[2,3,3-trimethyl-3H-indole-5-sulfonic acid]-croconaine (TISC) was shown in Scheme 1. 4-Hydrazinobenzenesulfonic acid (2g, 10.627mmol) and 3-methyl-2-butanone (0.915g, 10.627mmol) were dissolved in acetic acid (30mL). The solution was refluxed for 14h, then allowed to cool to room temperature, and the precipitate was collected by filtration. The solid was washed with diethyl ether and was dried under reduced pressure to afford compound 1 as a pink solid [19] (2.16g, yield 85%). 1H NMR (300MHz, DMSO-d6): δ 7.64(d, 1H, J=1.5Hz), 7.56 (dd, 1H, J=7.9, 1.6Hz), 7.36 (d, 1H, J=8.0Hz), 2.23 (s, 3H), 1.25 (s, 6H). MS (ESI): m/z=240.00 [M+H]+, calcd m/z=239.06 for C11H13NO3S. |
| 83% |
With sodium acetate In acetic acid at 110℃; Inert atmosphere; Sealed tube; |
|
| 79% |
With acetic acid for 4h; Reflux; |
|
| 78% |
With acetic acid at 120℃; for 4h; |
|
| 75% |
With acetic acid for 3h; Heating / reflux; |
1.1 1.1 Synthesis of 5-sulfo-2, 3, 3-trimethylindolenine (I)
Conventional Fisher Indole synthesis was used as follows. In a 2-L three necked flask equipped with mechanical stirrer and reflux condenser was added acetic acid (300 mL), 3-methyl-2-butanone (168 mL, 1.59 mol) and p-hydrazinobenzenesulfonic acid (100 g, 0.53 mol). The mixture was heated to reflux for three hours and then cooled for several hours when pink solid separated. (93 g, 75%, mp. 290-95 °C). 1H NMR (D2O), δ, 1.4 (s, 6H C-(CH3)2); 6.5 (d, 1H, J = 7 Hz, 7-H); 7. 1 (dd, 1H, J = 7.0, 1.2 Hz, 6-H); 7.13 (s, 1H, 4-H). Singlet for 2-methyl is not seen in D2O, but it appears when nmr is recorded in DMSO d6. |
| 75% |
In acetonitrile for 7h; Reflux; |
The intermediate was synthesized following the report by Park et al. [29]. 5-Sulfo-2,3,3-trimethyl-3H-indole was synthesized by the reaction of 4-Hydrazino-benzenesulfonic acid hemihydrate 4 g (21.3 mmol) with 5 mL of 3-methyl-2-butanone in 20 mL acetonitrile under reflux for 7 h. Reaction monitoring was done by TLC and upon completion of reaction, solvent was evaporated and the crude was purified by column chromatography to give the product in 75 % yield. 2.5 g (10.4 mmol) of this compound was dissolved in 15 ml of acetonitrile and 2 equivalents of 1-Iodoethane (4 ml, 20.8 mmol) was added. The mixture was then refluxed for 12 h while monitoring the reaction progress by TLC. The solvent was evaporated under vacuum and ample diethyl ether was added for precipitation, which was filtered to get the titled compound as pinkish solid in 68 % yield (2.8 g). |
| 71.3% |
In acetic acid at 0℃; for 15h; Reflux; |
11
A mixture of phenylhydrazine-p-sulphonic acid (compound 7) (11.3 g; 0.06 mole), 3-methyl-2-butanone (6.2 g; 7.72 mL; 0.072 mole), and glacial acetic acid (34 mL) was refluxed for 3 h, then, allowed to hold for 12 h at 0°C. The precipitate formed was collected by filtration, washed with acetone, and dried in a vacuum desiccator over P2O5. 5-Sulfo-2,3,3-trimethylindolenyne was obtained with yield 5.1 g (71.3%). 1H- NMR (D2O), δ (ppm): 1.28 (6H, s, C(CH3)2); 7.47-7.79 (3H, m, ArH) |
|
With acetic acid |
|
|
In acetic acid at 20℃; for 5h; Heating / reflux; |
Preparation of 2,3,3-trimethylindole-5-sulfonic acid, potassium salt (1b)
Preparation of 2,3,3-trimethylindole-5-sulfonic acid, potassium salt (1b) 18.2 g (0.12 mol) of p-hydrazinobenzenesulfonic acid and 14.8 g (0.17 mol) of isopropylmethylketone were stirred in 100 ML of glacial acetic acid at room temperature for 1 h.The mixture was then refluxed for 4 h.The mixture was cooled to room temperature, and the resulting pink solid precipitate was filtered and washed with ether. |
| 64.5 g (87.5%) |
With acetic acid In methanol |
7 Synthesis of an Activated Ester of Cy5
EXAMPLE 7 Synthesis of an Activated Ester of Cy5 To a 500 ml round bottomed flask equipped with a stir bar and reflux condenser was added AcOH (150 ml), p-hydrazinobenzenesulfonic acid (50.0 g, 0.255 mol), and 3-methyl-2-butanone (84 ml, 0.785 mmol). The flask was then heated in an oil bath at 115° C. to reflux for 3 h until all the starting material consumed (monitored by TLC, 1:1 MeOH:CH2 Cl2). The oil bath was removed and the flask was cooled to room temperature. The pink solid was collected via filtration with the acid of EtOAc. The solid was then dissolved in MeOH (800 ml) and air dried. Further drying in an oven at 40° C. under high vacuum overnight provided 64.5 g (87.5%) of the desired product, 2,3,3-trimethylindoleninium-5-sulfonate, potassium salt. TLC: Rf =0.875 (1:1 CH2 Cl2:MeOH). |
|
With potassium hydroxide In methanol; acetic acid; isopropyl alcohol |
3 3.1 5-Sulphonato-2,3,3-trimethylindolenine
3.1 5-Sulphonato-2,3,3-trimethylindolenine To a stirred solution of 4-hydrazinobenzene sulphonic acid (68 g, 361 mmol) in acetic acid (205 ml) at ambient temperature was added 3-methyl-2-butanone (88.44 g, 1027 mmol). The reaction was heated under reflux. After 3 hours the solution was cooled and the resulting pink precipitate was filtered, washed with acetic acid (50 ml) and dried. The product was redissolved in methanol (800 ml) and a solution of potassium hydroxide (20.4 g, 364 mmol) in isopropanol (200 ml) was added. The yellow solid obtained was filtered and dried (48 g, 56%); m/z (FAB+): 240. |
|
With potassium hydroxide; acetic acid In methanol; dichloromethane; isopropyl alcohol |
1 Preparation of 2,3,3-Trimethylindoleninium-5-sulfonate, Potassium Salt (9)
Example 1 Preparation of 2,3,3-Trimethylindoleninium-5-sulfonate, Potassium Salt (9) Referring now to Scheme 2, a 500-mL round bottomed flask was equipped with a stir bar, reflux condenser, acetic acid (150 mL), p-hydrazinobenzenesulfonic acid (8, 50.0 g, 0.266 mol), and 3-methyl-2-butanone (84 mL, 0.785 mmol). The flask was then heated in an oil bath at 115° C. to reflux for 3 hours until all the starting material was consumed (determined by monitoring using TLC, 1:1 MeOH:CH2Cl2). The reaction flask was then cooled to room temperature. A pink solid was collected via filtration with the aid of ethylacetate. The pink solid was then dissolved in MeOH (800 mL) and passed through a pad of filter paper to remove some solid impurities. Potassium hydroxide (15 g) in isopropylalcohol (200 mL) was then added to the filtrate and the solution was stirred. A yellow solid precipitated which was collected, washed with methanol (2*50 mL), followed by washing with diethyl ether (2*50 mL), and then air dried. The yellow solid was further dried in an oven at 40° C. under high vacuum overnight, which provided 64.5 g (87.5%) of compound 9. TLC:Rf=0.875 (1:1 CH2Cl2:MeOH). |
|
With acetic acid for 3h; Heating / reflux; |
3.1 3.1 5-Sulphonato-2,3,3-trimethylindolenine
To a stirred solution of 4-hydrazinobenzene sulphonic acid (68 g, 361 mmol) in acetic acid (205 ml) at ambient temperature was added 3-methyl-2-butanone (88.44 g, 1027 mmol). The reaction was heated under reflux. After 3 hours the solution was cooled and the resulting pink precipitate was filtered, washed with acetic acid (50 ml) and dried. The product was redissolved in methanol (800 ml) and a solution of potassium hydroxide (20.4 g, 364 mmol) in isopropanol (200 ml) was added. The yellow solid obtained was filtered and dried (48 g, 56%); m/z (FAB+): 240. |
|
With acetic acid for 3h; Heating / reflux; |
1.a EXAMPLE 1; Preparation of Cy3 Labeling Reagent; (a) Preparation of Compound I (2,3,3- Trimethylindolinium 5-Sulfone)
P-Hydrazinobenzenesulfonic acid (250 g) was mixed with glacial acetic acid (750 ml) and 3-methyl-2- butanone (420 ml) and heated at reflux for 3 hr. The solution was poured into a 2 L beaker and allowed to cool overnight. The resultant suspension was filtered, washed with acetic acid and lyophylized to remove residual acetic acid. The resultant solid was dissolved in methanol (1.5 L) and a saturated solution of potassium hydroxide in 2-propanol (900 ml) was slowly added. The color of the solution turned progressively lighter as the potassium salt of 2,3,3-trimethylindolinium 5-sulfone precipitated. The precipitate was filtered by suction, washed with 2-propanol and lyophilized to dryness to give 238 g of Compound I. [0285] (b) Preparation of Compound II (1-Ethyl-2,3,3-Trimethylindolenineninium 5-Sulfone) [0286] A portion (78 g) of Compound I synthesized in step (a) was suspended in 1,2-dichlorobenzene (700 ml). Ethyl iodide (250 ml) was added and the mixture was heated at 90-100 C. for 12 hr while stirring. The mixture was poured into 3 L of a 1:1 mixture of ethylacetate/ether and stirred for 2 hours. The resulting precipitate was filtered, washed with a 1:1 mixture of ethylacetate/ether and air-dried to give 68 g of product, Compound II. [0287] (c) Preparation of Compound III (6-Bromohexanoyl Allyl Amide) [0288] 6-Bromohexanoic acid (20g) and N-hydroxysuccinimide (15 g) were dissolved in 200 ml of anhydrous dimethylformamide (DMF). Dicyclohexylcarbiimide (22 g) in anhydrous DMF (50 ml) was added and the mixture was left at room temperature overnight. The precipitated urea was removed by filtration and the DMF solution containing the product, N-hydroxysuccinimide-6-bromohexanoate, was cooled to -10 to -20° C. An equimolar amount of allylamine in H2O (11 ml) was first brought to pH 8-9 with glacial acetic acid and then added slowly with stirring to the active ester. Solid sodium bicarbonate (10 g) was added slowly to avoid excessive foaming and the mixture was left without covering until the temperature was raised to -10° C. in two hr. The mixture was poured into H2O (1 L) and the product was extracted twice with chloroform (300 ml). The extracts were washed once with 1 N HCl in H2O, once with 5% NaHCO3 (300 ml) and three times with 10% NaCl in water. The chloroform phase was dried by addition of solid MgSO4 and leaving it overnight under stirring. The chloroform was removed by evaporation under vacuum leaving a liquid that was used without any further purification for the next step. [0289] (d) Preparation of Compound IV (Addition of Linker Arm to Compound III) [0290] Compound I (11 g) from step (a) and Compound III (15 g) from step (c) were dissolved together in 1,2-dichlorobenzene (100 ml) and heated at 110° C. for 12 hours while stirring under argon. The mixture was slowly poured into ethylacetate a 1:1 mixture of ethylacetate/ether (700 ml) and after 30 minutes the solid precipitate was filtered, washed with a 1:1 mixture of ethylacetate/ether, air-dried and set aside. A glassy solid that was formed at the bottom of the flask was crushed in a mortar, triturated with a 1:1 mixture of ethylacetate/ether, filtered, washed with 2-propanol, dried in vacuum and combined with the precipitate from above to give Compound IV which was used without any further purification. [0291] (e) Synthesis of Cy3 Labeling Reagent (Compound V) [0292] A portion of Compound II (12 g) from step (b) and N,N'-diphenylformamidine (10 g) in acetic acid (60 ml) were heated at 100-110° C. for 90 min with stirring. During the reaction the absorption at 286 nm and 415 nm was measured. The ratio of 415/286 increased during the first 60 minutes then remained constant at 2.2 for the next 20 minutes. After 90 minutes, the hot mixture was poured slowly into 700 ml of a 1:1 mixture of ethylacetate/ether. The resultant solid precipitate was collected with a pressure filter funnel, washed with 1:1 mixture of ethylacetate/ether and dried by passing argon through the cake. The precipitate was collected from the pressure filter funnel and slowly added to a mixture of 6.5 g of Compound IV from step (d), 50 ml of pyridine and 50 ml of acetic anhydride. The progress of the reaction was monitored by the decrease of absorbance at 385 nm and an increase in absorbance at 550 nm. The reaction was carried out overnight under stirring at room temperature. The absorbance at 550 nm increased with time followed by a drop in absorbance as the product precipitated out of solution. At the end of the reaction, the brown precipitate was collected and put aside. The liquid portion was treated by the addition of a seven-fold volume of ethylacetate. The precipitate that formed was collected and combined with the first precipitate. Since pyridine would interfere with a later palladium catalyzed step, any remaining pyridine was removed by dissolving the combined precipitate in 100 ml of 0.5M Triethylammonium carbonate, pH 8.0 (TEAC). The TEAC was then removed by evaporation under vacuum leaving a solid pellet. This product (Compound V) was then dissolved in H2O and kept at -70° C. until ready to be used. |
|
With acetic acid for 3h; Reflux; |
1
Phenylhyrazine-4-sulfonic acid is commercially available from a number of fine chemical suppliers. 5 g of Phenylhyrazine-4-sulfonic acid was treated with 3-methyl-2-butanone (15 mL) in acetic acid (AcOH, 20 mL) and refluxed for 3 h. On cooling, 2,3,3-trimethylindolenine-5-sulfonic acid precipitated out and was filtered, washed with a little diethyl ether and dried. The free base of was then formed by the addition of potassium hydroxide in 2-propanol. This was then refluxed with dimethylsulfate for 3 h to afford after removal of the solvent, the N-methyl compound. This was then treated with malonaldehyde bisphenylimine (0.3 equivalents) and refluxed in an acetic anhydride/pyridine mixture (4:1, 10 mL) for 1 h until a deep blue solution formed. The solvent was removed on a rotory evaporator and the crude dye was purified by flash chromatography on silica gel eluting with a gradient of methylene chloride and methanol (A=methylene chloride, B=methanol, gradient was 0 to 100% B over 30 minutes) to afford 0.4 g of the bis sulfonic acid. This was then refluxed with POCl3 for 2 h to form a key bis sulfonyl chloride intermediate that can also be used in its own right as a bifunctional sulfonyl chloride dye for labeling analytes or biomolecules that contain primary or secondary amines or alcohols and also phenols (Ar-OH). 200 mg of this compound was treated with an excess of ethylenediamine (1 g) in dimethylformamide for 5 minutes at 5° C. and the solvent was evaporated under vacuum. The residue was then acidified with trifluoroacetic acid (TFA) and subjected to flash chromatography on silica gel with a gradient of methylene chloride and methanol (A=methylene chloride, B=methanol, gradient was 0 to 100% B over 30 minutes) to afford 1 g of the bifunctional amino dye. |
|
In acetic acid |
1 Synthesis of 2,3,3-trimethylindole-5-sulfonic acid 1a
Synthesis of 2,3,3-trimethylindole-5-sulfonic acid 1a 18.2 g (0.12 mol) of p-hydrazinobenzenesulfonic acid and 14.8 g (0.17 mol) of isopropylmethylketone were stirred in 100 mL of glacial acetic acid at room temperature for 1 h. The mixture was then refluxed for 4 h. The mixture was cooled to room temperature, and the resulting pink solid precipitate was filtered and washed with ether. The precipitate was dissolved in methanol, and a concentrated solution of potassium hydroxide in 2-propanol was added until a yellow solid completely precipitated. |
|
With acetic acid at 118℃; for 14h; |
1
4-hydrazinobenzenesulfonic acid and 3-methyl-2-butanone were added to a three-necked flask at a molar ratio of 1: 1,Dissolved in acetic acid, 118 ° C under reflux conditions 14h. After completion of the reaction, heating was stopped, and the mixture was cooled to room temperature, filtered with a Buchner funnel, washed with ether and dried under reduced pressure to obtain a pink solid.0.05 g of keto acid and 0.1685 g of 2,3,3-trimethyl-5-sulfonic acid-3H-1,Indole was added into a 100 mL three-neck flask, 30 mL of toluene and 30 mL of n-butanol were added, the condenser tube was connected,Stoke water separator, nitrogen, 105 heating reflux stirring 6 hours, the solution from red to deepBrown, the water generated in the reaction was separated by a water separator. After the end of the reaction, the heating was stopped and the solution was cooled toAt room temperature, the solvent was removed on a rotary evaporator under reduced pressure. Ethyl acetate to give a black powderend. The powder was placed in an evaporating dish, placed in an oven, and dried at 60 ° C. The resulting black powder was taken in two portions(Volume ratio V dichloromethane: V methanol = 4: 1) as the eluent, 300 mesh silica gelAs a stationary phase, column chromatography elution separation and purification, to get a black solid, 2,3,3-trimethyl-5-sulfonic acidYl-3H-indolidone cyanine dye. |
|
With acetic acid In methanol at 117℃; for 5h; |
1.1
A solution of phenylhydrazine in sulfonic acid (5 g) was added to a methyl isopropyl ketone in methanol (8.55 / 15 ml)The solution was heated to 117 ° C and stirred for 5 h. The solvent was evaporated.Further, 50 ml of diethyl ether was added to the oily product to give a pink powder, i.e., compound (3).Then, the reddish brown powder (6 g) was added to a solution of sodium hydroxide (1.5 g) in methanol (10 ml) and isopropyl alcohol (10 ml)The solution was stirred at 82 ° C for 15 minutes, then cooled to room temperature and a large amount of compound was isolated for the next step. |
|
With acetic acid for 10h; Reflux; Inert atmosphere; |
|
|
With acetic acid Reflux; |
1-2 Example 1
0.376 g of 4-hydrazinobenzenesulfonic acid(about 0.002 mol) and 0.258 g of isopentanone (about 0.003 mol) were added to a round bottom flask, 50 ml of glacial acetic acid was added, and glacial acetic acid was added. Stir under heating and reflux overnight, and the solution slowly changed from turbidity to clarification. And the color changes from light yellow to deep red. After the reaction is over, Heating and stirring were stopped, and the solvent was distilled off with a rotary evaporator. Using 200 ml of dichloromethane and 20 ml of methanol mixed solvent as eluent, Separation using a silica gel column, The separated product was suspended from the eluent by a rotary evaporator. Put the suspended product into an evaporating dish, After drying in an oven at 60 ° C, a yellow sulfonate oxime was obtained. |
|
With acetic acid Reflux; |
Compound 3 was synthetized from 4-Hydrazinobenzenesulfonic acid and 3-methyl-2-butanone, which cyclized to obtain 2,3,3-trimethyl indoline-5-sulfonic acid by Fisher method followed by potassium salinization. The detailed synthesis steps referred to reference |
|
With acetic acid for 8h; Reflux; Inert atmosphere; |
1.1 (1) Synthesis of potassium N-ethyl-2,3,3-trimethylsulfonium-5-sulfonate (compound (II))
Add 10 mL of acetic acid to a 50 mL single-mouth bottle,Benzopyrene-4-sulfonic acid (2.000g, 0.010mol)And methyl isopropyl ketone 4.00 mL (0.037 mol),It was refluxed for 8 hours under a nitrogen atmosphere, at which time the solution turned dark red.After cooling to room temperature,The reaction solution was slowly dropped into ethyl acetate, and a pink solid precipitated in the solution;And washed several times with diethyl ether, dried in vacuum;Dissolving the dried pink powder described above with an appropriate amount of methanol,0.561 g of potassium hydroxide was dissolved in isopropanol to prepare a saturated solution.Then, a saturated solution of potassium hydroxide in isopropanol was added dropwise to the methanol solution.Obtained a yellow solid, centrifuged,It was washed 3-4 times with diethyl ether and dried in vacuo.Take another 50mL single-mouth bottle and add the above yellow solid, 20mL acetonitrile and ethyl bromide 1.49mL (0.020mol) to the bottle.Reflow under nitrogen for 24 hours,A red solid precipitated. After cooling to room temperature, suction filtration,It was washed with diethyl ether (3 × 10 mL) and dried in vacuo to give Compound (II). |
|
With sodium acetate; acetic acid at 130℃; for 2h; |
|
|
With acetic acid for 10h; Reflux; |
|
|
With acetic acid at 120℃; for 6h; |
1.1
1. Take 4-hydrazinylbenzene-1-sulfonic acid10g,6g of methyl isopropyl ketone,70ml of acetic acid, heated and refluxed at 120 for 6h under constant stirring,It can be seen that the raw materials continue to dissolve and the solution turns brick red.After the reaction, the solution was spin-dried and washed with ether.Brick red solid 2,3,3-trimethyl-3H-indole-5-sulfonic acid can be obtained, |
|
With sodium acetate; acetic acid for 8h; Reflux; |
1 Synthesis of 2,3,3-trimethyl-5-sulfoindole
Dissolve p-sulfophenylhydrazine, 3-methyl-2-butanone, and anhydrous sodium acetate in a molar ratio of 1:1.1:1.5 in acetic acid, and react under reflux and stirring for 8 hours. The reaction solvent was removed by rotary evaporation, and then a mixed solution of water and methanol with a volume ratio of 9:1 was added to dissolve the remaining substances. The resultant was filtered, and then left open to crystallize at room temperature for 48 hours,The crystal 2,3,3-trimethyl-5-sulfoindole was obtained. |
|
With acetic acid at 120℃; for 18h; Inert atmosphere; |
1
4-hydrazinebenzenesulfonic acid (1.6 g, 31.9 mmol), 3-methyl-2-butanone (2.10 ml, 90 mmol) and glacial acetic acid (50ml) were mixed and heated to 120 C under nitrogen atmosphere for 18 h. After precipitation in ethyl acetate, filter and collect the crude product as a pink solid, the resulting product (6.5 g, 25.4 mmol) was dissolved in methanol (50 mL). Under mild conditions, the dissolved liquid was added dropwise to a solution of potassium hydroxide (1.7g, 30 mmol) and isopropanol (20 ml), and the crude mixture was filtered and washed to obtain a brown solid, product 1, with a yield of 97%. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping