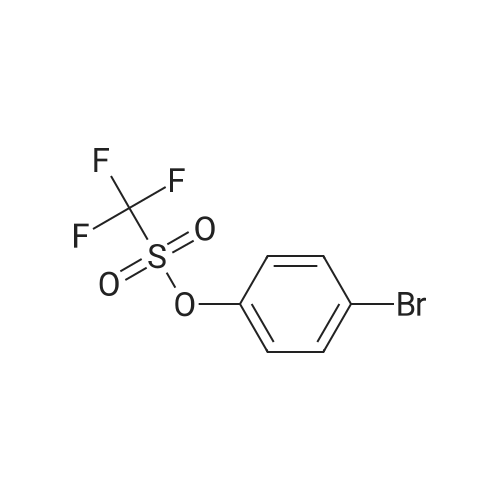
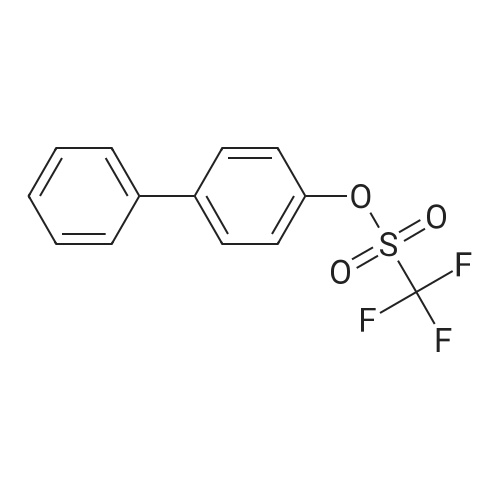
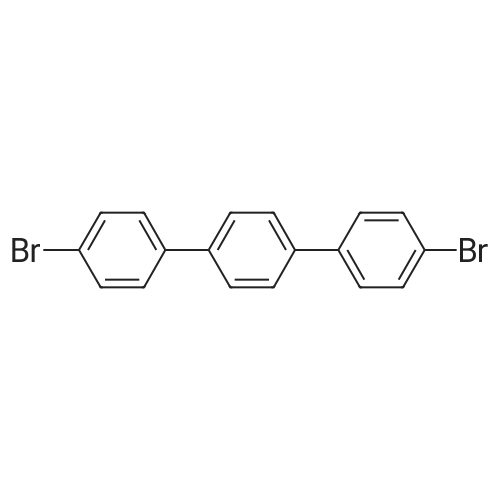
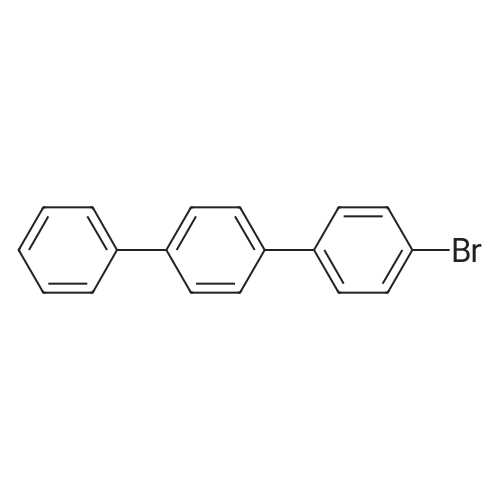
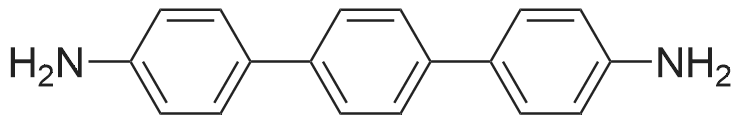
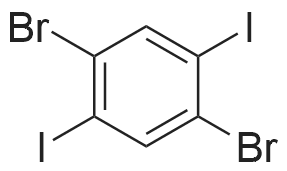
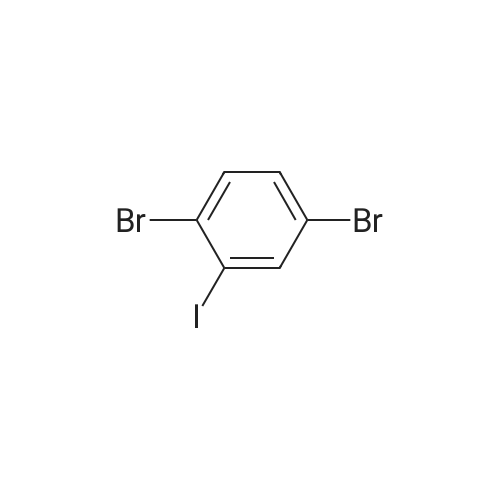
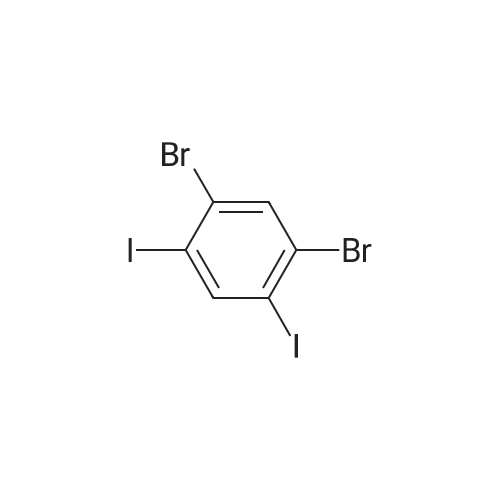
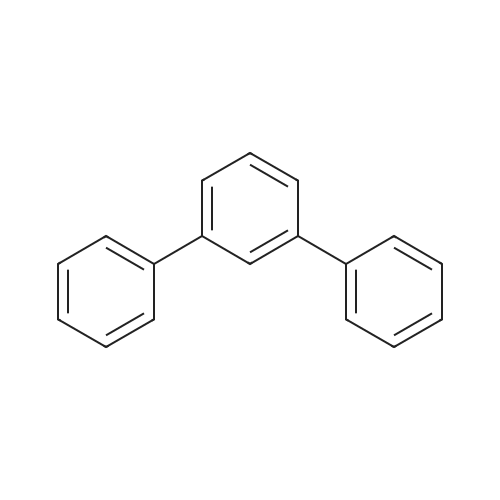
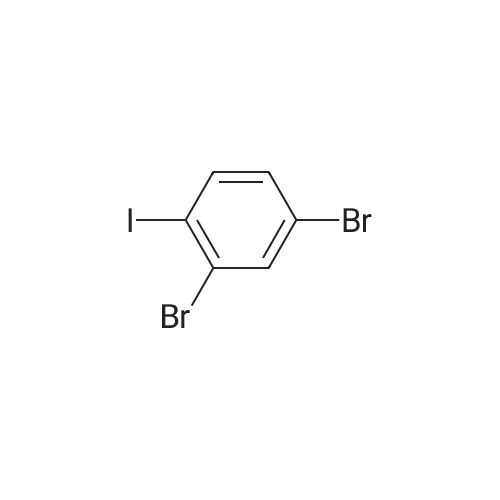
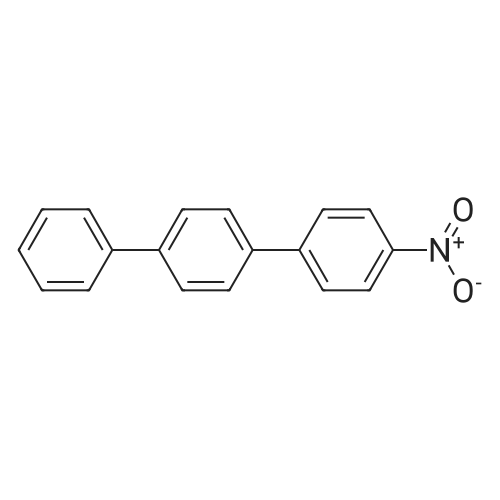
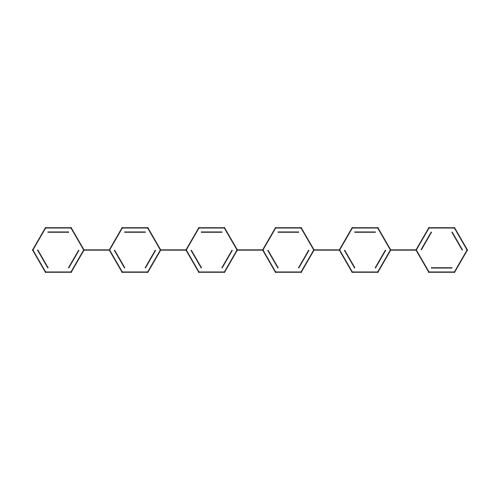
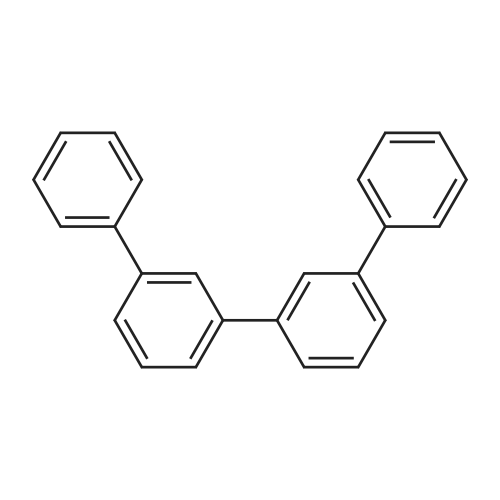
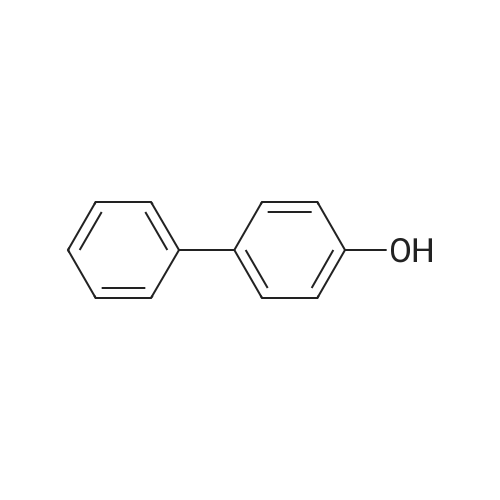
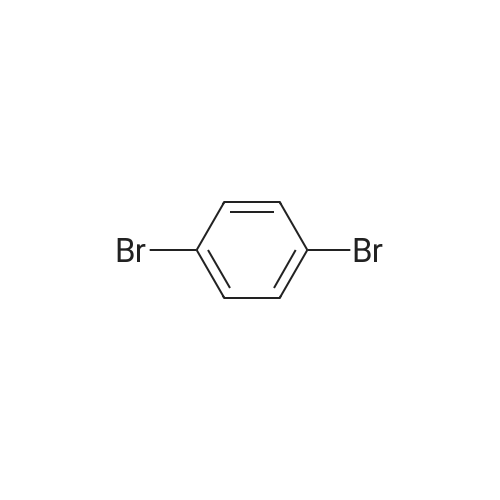
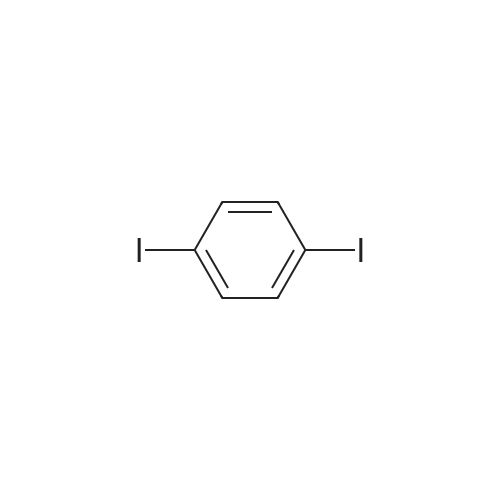
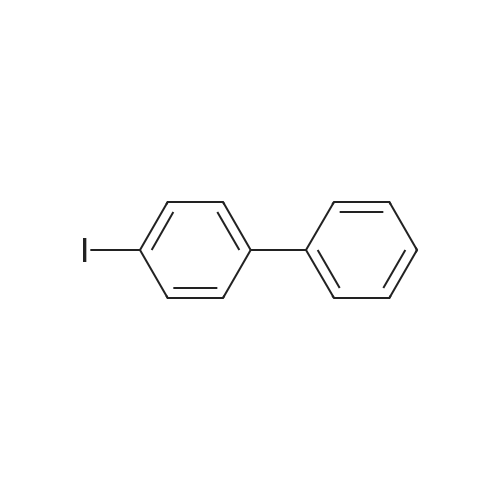
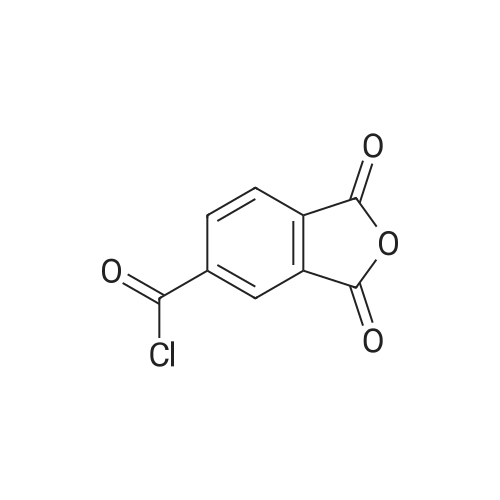
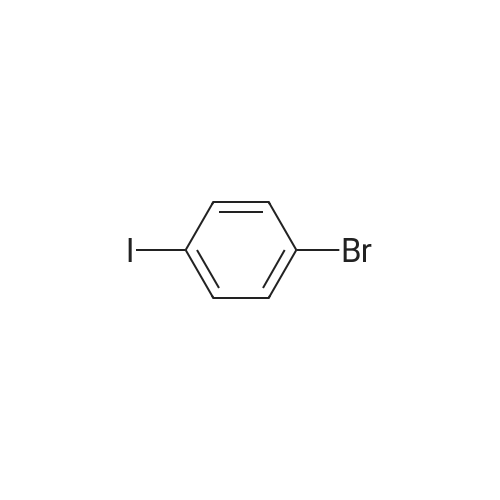
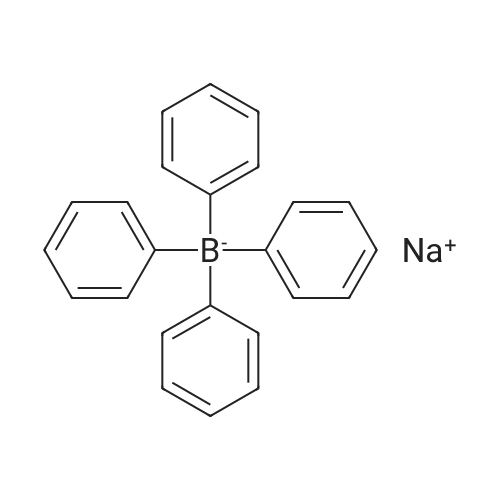
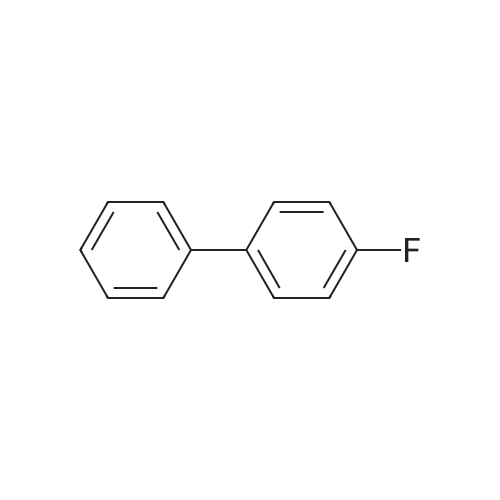
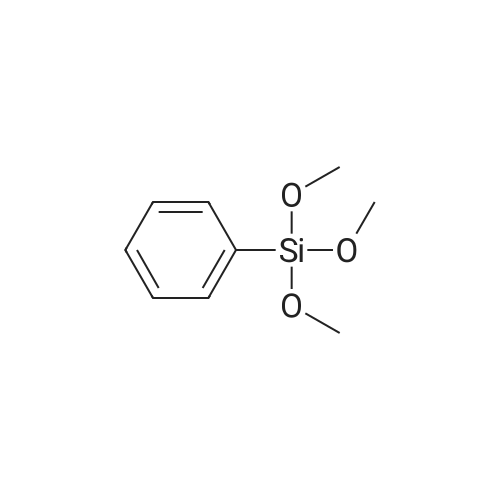
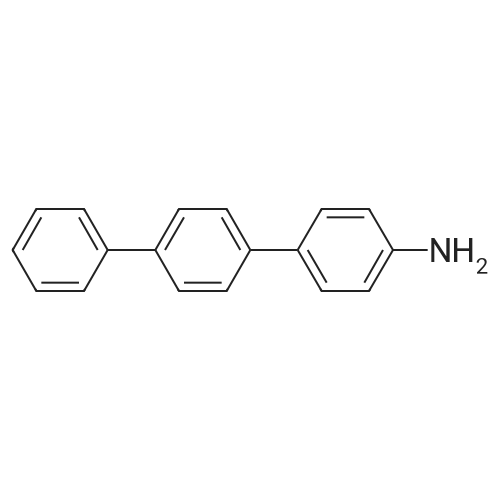
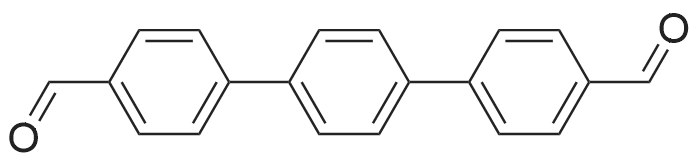
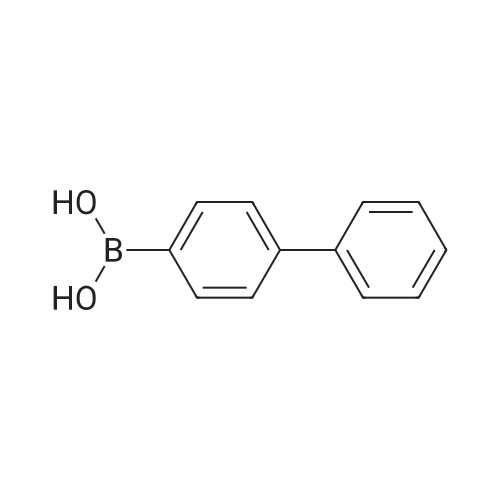
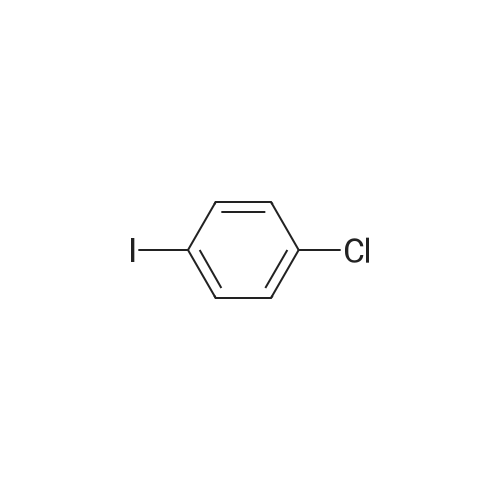
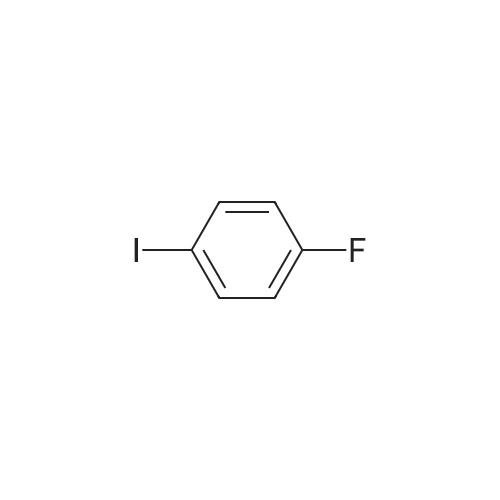
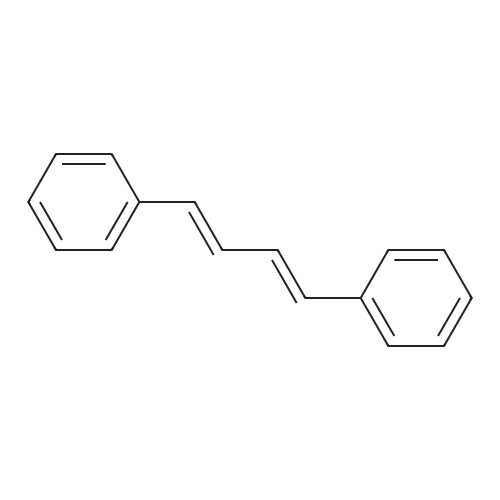
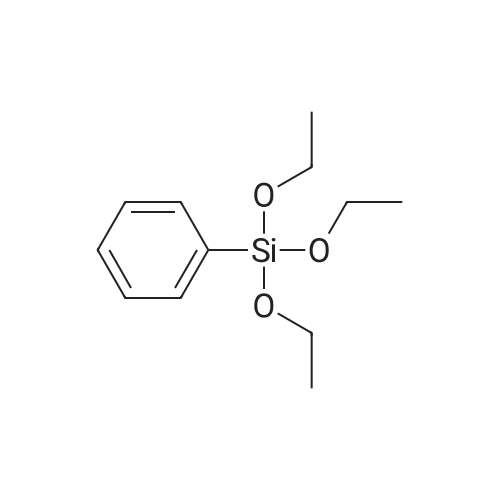
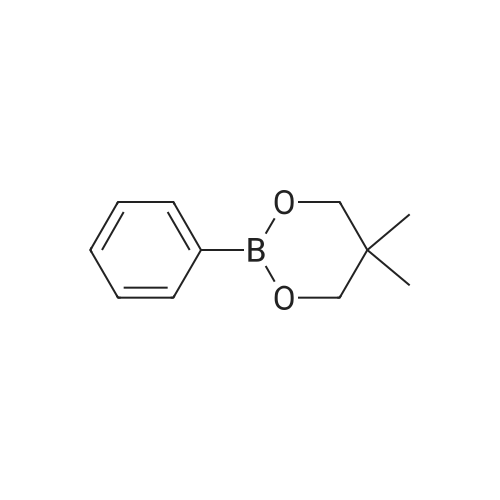
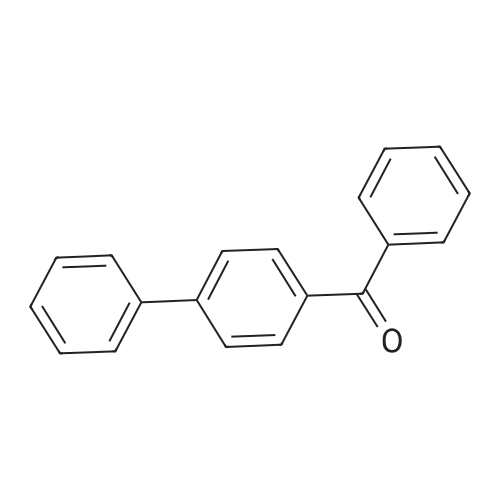
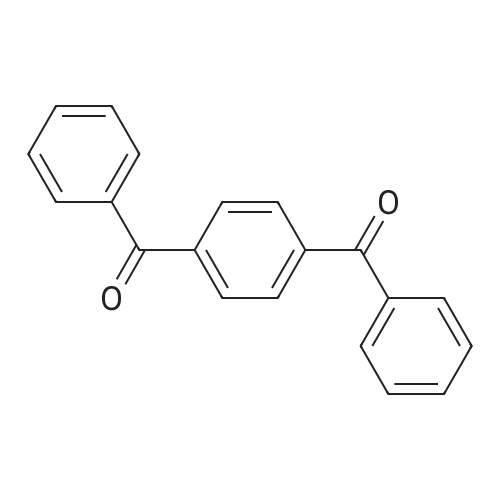
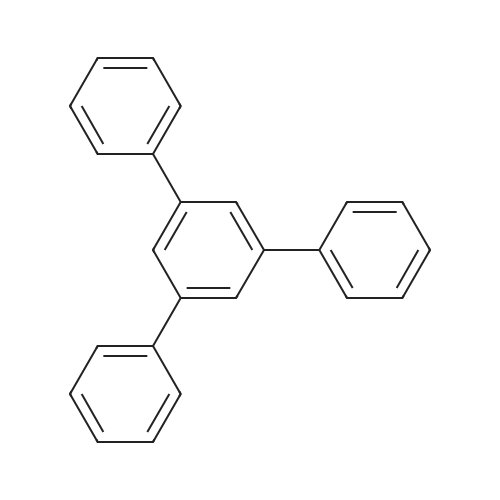
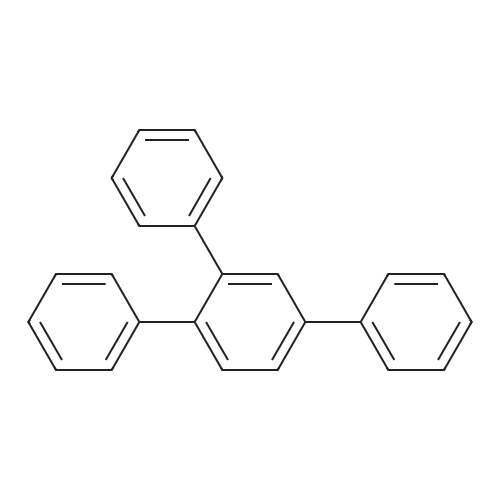
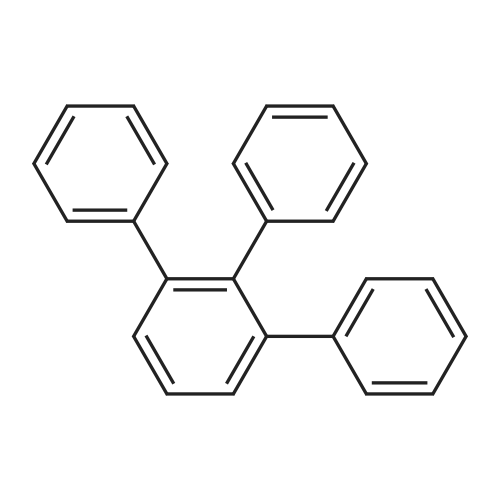
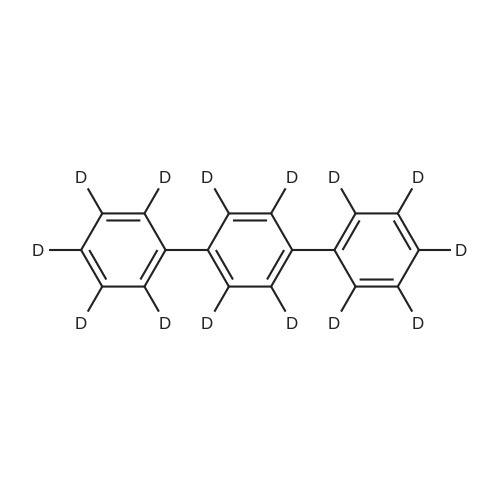
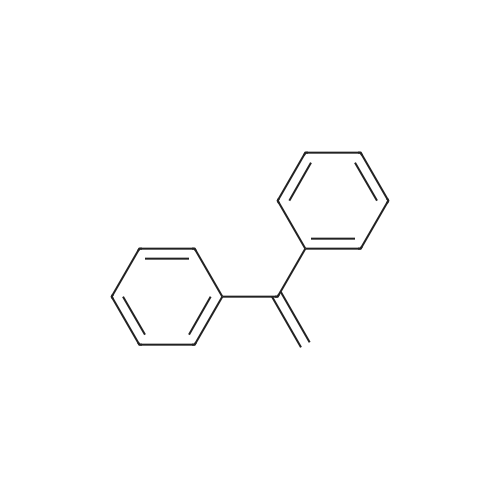
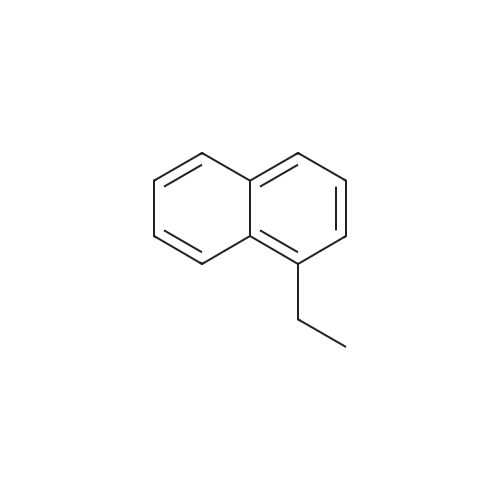
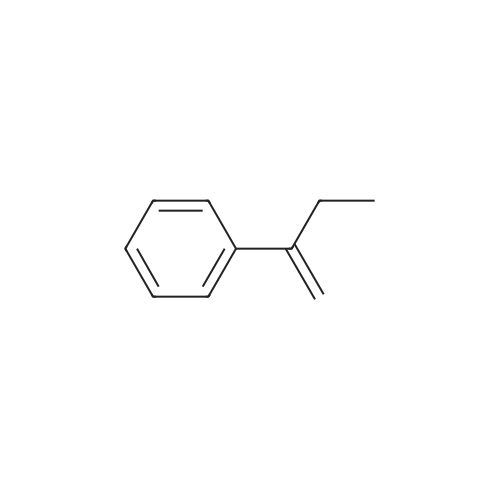
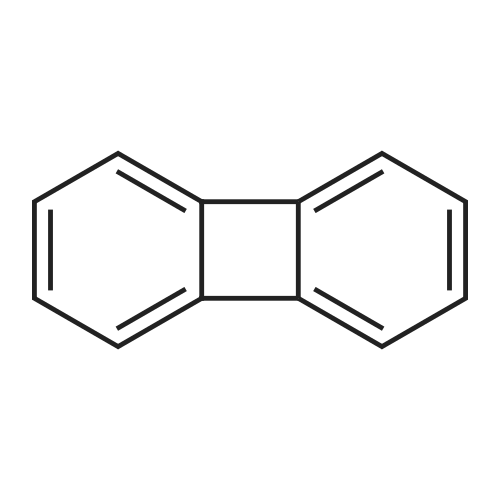
| 99% |
With tripotassium phosphate tribasic; C16H16ClN8Pd(1+)*F6P(1-) In ethanol at 60℃; for 5h; Inert atmosphere; |
|
| 98% |
With tripotassium phosphate tribasic In ethanol; lithium hydroxide monohydrate at 80℃; for 18h; Schlenk technique; Green chemistry; |
|
| 98% |
With C20H18I2N4Pd; potassium-t-butoxide In ethanol at 30℃; for 3h; |
General procedure for Suzuki-Miyaura coupling
General procedure: A mixture of aryl halide (100 mg scale, 1.0 eq), phenylboronic acid (1.2 eq),t-BuOK (1.5 eq), complex 5 (1 mol%with respect to substrate) in EtOH (5 mL) was stirred at room temperature (30 oC)until the starting aryl halide disappeared (checked by TLC). The reactionmixture was diluted with ice cold water (10 mL) and extracted with ethylacetate. Removal of solvent under vacuumgave the crude product which was purified either by chromatography on silicagel or by simply washing with hexane. The products and their spectral data arereported in literature and they were characterized by 1H and 13CNMR spectroscopic data in the present study. |
| 97% |
With C42H54Cl2N6Pd; triphenylphosphine; sodium hydroxide In 1,4-dioxane at 105 - 110℃; for 3h; Inert atmosphere; |
|
| 97% |
With potassium carbonate In ethanol; lithium hydroxide monohydrate for 0.583333h; Sonication; |
2.3. General procedure for the Suzuki-Miyaura coupling reaction
General procedure: A vial equipped with a magnetic stirrer bar and a condenser was charged with aryl halide (1 mmol), phenylboronic acid (1.2 mmol),K2CO3 (2 mmol), catalyst (8 mol% Ni) and Water/EtOH (1:1) (4 mL)and the reaction mixture was irradiated in ultrasonic apparatus. The reaction progress was followed using thin layer chromatography. After completion of the reaction, the mixture was cooled to room temperature,the catalyst was separated by an external magnet, and the product was extracted with ethyl acetate (15 mL) and dried over anhydrous MgSO4. The resulting solution was evaporated under vacuum to givethe crude product. The separation of product by column chromatography on silica gel using n-hexane or different mixtures of n-hexane,ethyl acetate as the eluents to afford the highly pure products (Table 4). The obtained pure products were characterized by physical methods and the IR, 1H NMR and 13C NMR spectra. |
| 96% |
With anhydrous sodium carbonate In lithium hydroxide monohydrate; N,N-dimethyl-formamide at 60℃; for 12h; |
|
| 96% |
With {2,6-bis[(di-1-piperidinylphosphino)amino]phenyl}palladium(II) chloride; potassium carbonate In 1,4-dioxane; lithium hydroxide monohydrate; butan-1-ol at 100℃; for 0.5h; |
|
| 96% |
With tripotassium phosphate tribasic; dichloro{bis[1-(dicyclohexylphosphanyl)(II) In toluene at 80℃; for 0.166667h; Air; |
|
| 95.4% |
With anhydrous sodium carbonate In lithium hydroxide monohydrate at 100℃; |
|
| 95% |
With C27H25Cl2N3PPd(1-)*H(1+); Cs2CO3 In lithium hydroxide monohydrate; N,N-dimethyl-formamide at 20℃; for 1h; Inert atmosphere; |
|
| 95% |
With sodium palladium(II) tetrachloride; C22H16O8S2(2-)*2Na(1+); sodium hydroxide In ethanol; lithium hydroxide monohydrate at 100℃; for 8h; Schlenk technique; |
|
| 95% |
With [Pd(N-(3-chloro-2-quinoxalinyl)-N'-(2,6-diisopropylphenyl)imidazolium)(PPh3)Cl2]; potassium carbonate In lithium hydroxide monohydrate at 70℃; for 3h; |
|
| 95% |
With potassium carbonate In ethanol at 80℃; for 0.5h; |
|
| 95% |
With potassium carbonate In lithium hydroxide monohydrate; N,N-dimethyl-formamide at 80℃; for 0.333333h; |
|
| 94% |
With C31H26N6O4Pd; potassium carbonate In lithium hydroxide monohydrate at 100℃; for 4h; |
|
| 94.5% |
With tetrabutylammonium bromide; potassium carbonate In 1,4-dioxane; lithium hydroxide monohydrate at 80℃; for 24h; |
2.1.1 Synthesis of Biphenyl (9)
General procedure: A round bottom flask was charged with iodobenzene (0.2 g,0.980 mmol), catalyst B-2 (0.061 g, 0.000098 mmol Pd, 0.01 mol %), phenyl boronic acid (0.143 g, 1.176 mmol),dry potassium carbonate (0.270 g, 1.960 mmol), TBAB(0.063 g, 0.196 mmol) and dioxane-water (1:1; 10 mL) assolvent. This mixture was heated to 80 C and continued for24 h. The reaction mixture was filtered to remove the catalystand was quenched with water and extracted with ethylacetate (3 9 25 mL). The combined organic phase waswashed with water and dried over anhydrous sodium sulfate.Solvent was removed in vacuum and the crude product waspurified by column chromatography on silica gel to affordbiphenyl (0.148 g, 98.7 %) as white solid (m.p. 66-67 C, lit[73] 68-70 C). |
| 94% |
With potassium carbonate In ethanol; lithium hydroxide monohydrate at 50℃; for 1h; |
2.4.1 Suzuki Cross-Coupling Reaction
General procedure: A flame-dried 50mL round-bottom flask equipped with amagnetic stir bar and a rubber septum was charged with arylhalide (1.0mmol), phenyl boronic acid (1.1mmol), K2CO3(2.0mmol) and CL-salen-Pd(II) (0.5% mmol). The mixturewas stirred in Ethanol: H2O= 1:1 (5.0mL) at 50 underair atmosphere for 1h. The mixture was cooled to roomtemperature, quenched with water (5mL), and diluted withethyl acetate (5mL). The layers were separated, and theaqueous layer was extracted with 2 × 5mL of ethyl acetate.The combined organic extracts were dried over anhydrousmagnesium sulfate, filtered, and concentrated in vacuo.Finally, the product was purified by column chromatography. |
| 93% |
With potassium carbonate In methanol at 20℃; for 10h; Green chemistry; |
|
| 93% |
With C34H32Cl2FeP2Pd In ethanol at 80℃; for 3h; Schlenk technique; |
General procedure for the Suzuki-Miyaura reaction
General procedure: An oven-dried Schlenk flask, equipped with a magneticstir bar, a septum and a condenser was charged with arylhalide (1.0 mmol), arylboronic acid (1.2 mmol), the gelentrappedbase (1 g, 2 mmol), Pd(dppf)Cl2 (0.0085 g,1 mol%) and 5 mL of 95% ethanol. The flask was immersedand stirred in an oil bath at 80 8C. Upon completeconsumption of starting materials as determined by TLCanalysis, the gel was separated by filtration and water(10 mL) was added. The filtrate was extracted with diethylether (3 5 mL). The combined organic layer was collected,dried over anhydrous Na2SO4 and concentratedunder vacuum to afford the product, which was purified bysilica gel column chromatography (n-hexane:ethyl acetate9:1) |
| 93% |
With tripotassium phosphate monohydrate; palladium (II) chloride; 3-(diphenylphosphanyl)propanoic acid In dimethyl sulfoxide at 100℃; for 12h; Schlenk technique; |
General procedure for Pd-catalyzed Suzuki-Miyaura reactions
The Schlenk tube (5 mL) equipped with a stir bar was charged with PdCl2 (1.0 mol%), Ph2P(CH2)2COOH (2.0 mol%), and K3PO4 ·H2O (4.0 equiv.), then 1,4-dibromobenzene (0.5 mmol), phenylboronic acid (1.20 mmol), and DMSO (2.0 ml) were added, and the mixture was stirred at 100 °C until the substrate was completely consumed. After cooling to room temperature, the solution was quenched with water and extracted with EtOAc (3 × 10 mL). The combined EtOAc extracts were dried over anhydrous Na2SO4 and filtrated and the solvent was removed under reduced pressure. The residue was purified by flash column chromatography on silica gel with PE as the eluent to obtain the desired products. |
| 92% |
With potassium carbonate In various solvent(s) at 110℃; for 24h; |
|
| 92% |
With sodium hydroxide In lithium hydroxide monohydrate for 5h; Reflux; |
2.4. General procedure for the synthesis of p-teraryls using PNP-SSS catalyst
General procedure: To a mixture of diarylhalide (1 mmol), aryl boronic acid (2.1 mmol), and NaOH (3 mmol) in 2 mL water, PNP-SSS catalyst (0.05 g) was added and heated in an oil bath at the refluxing temperature of water. The reaction was followed by TLC. After completion of the reaction, the mixture was cooled down to room temperature and filtered and the remaining solid was washed with dichloromethane (3 x 5 mL) in order to separate catalyst. After the extraction of dichloromethane from water, the organic extract was dried over Na2SO4. The products were purified by column chromatography (hexane or hexane/ethyl acetate) to obtain the desired purity. |
| 92% |
With potassium carbonate In ethanol at 80℃; for 2h; Schlenk technique; |
|
| 92% |
With potassium carbonate In lithium hydroxide monohydrate; N,N-dimethyl-formamide at 80℃; for 8h; Schlenk technique; |
19. General procedure for the Suzuki-Miyaura reaction:
General procedure: An oven-dried Schlenk flask, equipped with a magnetic stir bar, septum, and a condenser was charged with aryl halide (1.0 mmol), arylboronic acid (1.2 mmol), K2CO3 (2 mmol), 4 (0.143 g, 1 mol %), and 5 mL of solvent. The flask was immersed in an oil bath and stirred at 80 °C. Upon complete consumption of starting materials as determined by TLC analysis, the reaction mass was filtered and the solid washed with water (2Χ5 mL), and extracted with diethyl ether (3Χ5 mL). The combined organic layers were collected, dried over anhydrous Na2SO4, and concentrated in vacuum to afford product which was purified by silica gel column chromatography (n-hexane/EtOAc = 9:1) |
| 92% |
With C14H19NO3*C6H15N; potassium hydroxide; palladium (II) chloride In ethanol; lithium hydroxide monohydrate at 80℃; for 4h; Green chemistry; |
General procedure of aqueous Suzuki coupling reaction.
General procedure: In the first step,reference solution (CPd=0.5×10-4mmol/mL,CLigand=1.0×10-4mmol/mL) was prepared. A single-neckedground tube (rinner=1.1 cm, L=17.5 cm) was equipped with a magnetic stirbar. 0.5 mmol of aryl halides, 0.75 mmol of phenyl boronic acid and 1 mmol basewere added into the tube under atmospheric condition. Then, appropriate amountof reference solution and solventwere added into the tube. The reaction mixture was stirredat the pre-arranged temperature for appropriate reaction time in oil bath. Afterthe reaction was completed, 3 mL water was added, and the mixture was extractedwith EtOAc(10 mL×4). Then the organic phase was dried with Na2SO4and concentrated under reduced pressure. The residue was then purified by columnchromatography on silica gel. The pure product was obtained and was analyzed by1H NMR spectroscopy. |
| 91% |
With palladium diacetate; potassium carbonate; tris-(o-tolyl)phosphine In methanol; toluene at 75℃; |
|
| 91% |
With palladium diacetate; potassium carbonate; tris-(o-tolyl)phosphine In methanol; toluene at 75℃; Inert atmosphere; |
|
| 91% |
With potassium carbonate In ethyl acetate at 90℃; for 2h; |
|
| 90% |
With tripotassium phosphate tribasic In ethanol; lithium hydroxide monohydrate at 50℃; for 1h; |
|
| 90% |
With tripotassium phosphate tribasic In methanol at 60℃; for 6h; |
|
| 90% |
With C28H40Br4N4Pd2; potassium carbonate In lithium hydroxide monohydrate; propan-2-one at 20℃; for 5h; |
4.3 General procedure of Suzuki reaction
General procedure: A mixture of aryl halide (1 mmol), arylboronic acid (1.2 mmol), catalyst A (1 mol %, 0.0096 g), K2CO3 (2 mmol), and (1:1) acetone/water mixed solvent (3 mL) were taken in 25 mL round bottom flask and the mixture was stirred at room temperature (40 °C for heteroaryl halides) until the completion of reaction (required time given in Tables 3-5). The reaction mixture was then diluted with water (20 mL) and extracted three times with dichloromethane (3×10 mL). The combined organic layer was washed with brine (20 mL) and dried over anhydrous Na2SO4. After that it was concentrated under reduced pressure and the crude product was purified by column chromatography on silica gel (60-120 mesh) using petroleum ether (60-80 °C) and ethyl acetate were as the eluent. |
| 89% |
With anhydrous sodium carbonate In N,N-dimethyl-formamide at 20℃; for 24h; |
|
| 88% |
With C32H28Cl2N6Pd; triphenylphosphine; sodium hydroxide In 1,4-dioxane at 105℃; for 6h; |
Suzuki-Miyaura coupling of 1,4-dibromobenzene and phenylboronic acid
A mixture of 1,2-dibromobenzene (100 mg, 0.42 mmol), phenylboronic acid (124 mg, 1.02 mmol), triphenylphosphine (5 mg,0.018 mmol), NaOH (68 mg, 1.7 mmol) and Pd-complex 8 (5 mg,2 mol%) were taken in a 50 mL two necked round-bottom flask. Dioxane (4 mL) was added to the mixture and heated at 105 °C. After 6 h the reaction was complete (TLC). Removal of solvent unde rreduced pressure followed by aqueous work up gave the crude product which was purified by crystallization from CH2Cl2/hexane mixture to yield pure p-terphenyl as a colorless crystalline solid in 88% yield. The reaction was repeated with complex 10 and p-terphenyl was obtained in 75%. The product was confirmed by comparison with authentic sample. 1H NMR (CDCl3, 500 MHz) δ:7.35-7.38 (t, 2H, J = 7.5 Hz), 7.45-7.48 (t, 4H, J = 8 Hz), 7.65-7.66 (d,4H, J = 7.5 Hz), 7.69 (s, 4H); 13C NMR (CDCl3, 125 MHz) δ: 127.2,127.5, 127.6, 128.9, 140.2, 140.8. |
| 88% |
With C64H68Cl4N6O2Pd2; potassium hydroxide In 1,4-dioxane at 100℃; for 24h; Inert atmosphere; |
|
| 87% |
With anhydrous Sodium acetate In lithium hydroxide monohydrate; N,N-dimethyl-formamide at 80℃; for 1h; |
|
| 87% |
With palladium diacetate; 3,3'-((phenylmethylene)bis(4-methoxy-3,1-phenylene))dipyridine; potassium carbonate In N,N-dimethyl-formamide for 0.333333h; Sonication; |
General procedure for SMC reaction of aryl bromides with aryl boronic acid using Pd(OAc)2 / L1-4 catalyticsystem under US irradiation (Table 2)
General procedure: To a mixture of Pd(OAc)2 (0.5 mol%), L1-4 (0.5 mol%),aryl bromide 1a-k (1 mmol), and aryl boronic acids 4b-d(1.5 mmol) and K2CO3 (2 mmol) was added 4 mL of DMF.The resulting mixture was sonicated for appropriate time at 30% amplitude using the P-US irradiation mode (Pulse dmode US, the probe is on for 10 Sec / The probe is off for 10 Sec). The reaction mixture was purified as described in the 4.2.3 section to obtain the pure products 6a-m. The product 6 l was also synthesized using double SMC reaction ofsubstrate 1j with twofold amounts of phenyl boronic acid 4b under similar conditions. |
| 85% |
With potassium carbonate In N,N-dimethyl-formamide at 110℃; for 6h; Inert atmosphere; |
B) Typical experimental procedure for Suzuki-Miyaura cross-coupling reaction using SS-Pd catalyst
A mixture of 4-iodotoluene, 4 (100 mg, 0.45 mmol), phenylboronic acid (55.41 mg, 0.45 mmol), potassium carbonate (253 mg, 1.83 mmol), SS-Pd (207 mg, 2mol% Pd) and DMF (1 ml) was stirred at 110 oC for 6 h under nitrogen atmosphere. The progress of reaction was monitored by TLC. After completion of reaction, the reaction mixture was cooled, diluted with 3 ml of water and filtered through the cotton bed. The filtrate was extracted with ethyl acetate (3×2 ml) and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure and the crude residue was purified by silica gel (mesh 60-120) column chromatography (Hexane, 100%) afforded 14 as a white solid (69 mg, 90%); |
| 85% |
With anhydrous sodium carbonate In lithium hydroxide monohydrate at 80℃; for 1.08333h; |
|
| 84% |
With anhydrous sodium carbonate In N,N-dimethyl-formamide at 110℃; for 5h; Inert atmosphere; |
|
| 84% |
With palladium; potassium carbonate In methanol; acetonitrile at 20℃; for 2.5h; |
Typical procedure for synthesis of terphenyls
General procedure: To a freshly prepared solution of PdNPs (10 mL, 0.02 mmol), required amount of K2CO3 (2 mmol) was added followed by aryldihalides/ arylhalide (1 mmol) and arylboronic acid (3 mmol)/diboronic acid (0.75 mmol). Then, the reaction mixture was stirred at room temperature in open atmosphere. The reaction was monitored by TLC and was stopped after the complete consumption of starting material. The desired product got precipitated out which was separated by filtration and extracted with chloroform. The chloroform layer was evaporated to get the terphenyl in pure state. |
| 83% |
With 2-phenylbenzothiazole; Cs2CO3 In lithium hydroxide monohydrate; N,N-dimethyl-formamide at 60℃; for 0.166667h; Inert atmosphere; Schlenk technique; Glovebox; |
|
| 83% |
With C44H66Cl2N2Pd; potassium carbonate In lithium hydroxide monohydrate at 110℃; |
|
| 80% |
With Cs2CO3 In N,N-dimethyl-formamide at 20℃; for 3h; |
|
| 78% |
With anhydrous sodium carbonate In lithium hydroxide monohydrate; N,N-dimethyl-formamide at 90℃; for 2h; |
|
| 78% |
With tetrabutylammonium bromide; potassium carbonate In lithium hydroxide monohydrate at 100℃; for 0.333333h; Microwave irradiation; Green chemistry; |
2.6. General procedure for the Pd(0)-EDA/SC-2 catalyzed Suzukireaction
General procedure: To a mixture of aryl halide (1 mmol), aryl/heteroaryl boronicacid (1.2 mmol), TBAB (0.25 mmol), K2CO3(0.25 mmol) and Pd(0)-EDA/SC-2 (0.2 g, 2.5 mol% Pd), double distilled water (5 mL) wasadded and the reaction mixture was stirred in a microwave syn-thesizer at 100C for an appropriate time (monitored by TLC)(Scheme 2). After completion, the reaction mixture was cooledto room temperature and filtered. The catalyst was washed withEtOAc (3 × 5 mL) followed by double distilled water (3 × 10 mL). Itwas dried at 100C for 1 h and could be used in subsequent reac-tions. The organic layer was washed with water and dried overanhydrous Na2SO4. Finally, the product was obtained after removalof the solvent under reduced pressure followed by crystallizationfrom a suitable solvent or passing through column of silica gel(EtOAc-pet. ether). |
| 78% |
With tetrabutylammonium bromide; potassium carbonate In 1,4-dioxane for 10h; Inert atmosphere; |
|
| 78% |
With potassium carbonate In N,N-dimethyl-formamide at 60℃; for 22h; |
|
| 76% |
With 2C60H80NaO12*Cl6Pd2; potassium carbonate In methanol; lithium hydroxide monohydrate at 20℃; for 1h; |
3.3. General procedure for the Suzuki-Miyaura cross-coupling reaction of aryl bromides with aryl boronic acid
General procedure: A mixture of aryl bromides (0.5 mmol), aryl boronic acid (0.6 mmol), K2CO3 (0.6 mmol), catalyst 2 (0.2 mol%), and CH3OH/H2O (2/1, 2 mL) was stirred at room temperature under air. The reaction mixture was stirred for 10 min, and then diluted with water and CH2Cl2. The organic layer was separated and the aqueous layer was extracted with CH2Cl2 for three times. The combined organic phase was dried with MgSO4, filtrate, solvent was removed on a rotary evaporator, and the product was isolated by thin layer chromatography. The purified products were identified by 1H NMR, 13C NMR spectroscopy and melting points with the literature data. |
| 75% |
With tris-imidazolium-stabilized palladium; potassium carbonate In lithium hydroxide monohydrate; toluene at 90℃; for 15h; |
|
| 75% |
With bis(1,5-cyclooctadiene)nickel (0); tripotassium phosphate tribasic; tricyclohexylphosphine In tetrahydrofuran for 20h; Inert atmosphere; |
|
| 73% |
With Cs2CO3 In 1,4-dioxane at 80℃; for 15h; Inert atmosphere; |
|
| 72% |
Stage #1: phenylboronic acid With sodium hydroxide
Stage #2: 1.4-dibromobenzene In lithium hydroxide monohydrate; N,N-dimethyl-formamide for 12h; Reflux; |
|
| 72% |
With tetrakis-(triphenylphosphine)-palladium; anhydrous sodium carbonate In 1,4-dioxane at 70℃; for 72h; Inert atmosphere; |
|
| 70% |
With tetrabutylammonium bromide; triethylamine In lithium hydroxide monohydrate; N,N-dimethyl-formamide at 75℃; for 3h; |
2.5. General procedure for the Suzuki-Miyaura coupling reaction
General procedure: Aryl halide (1 mmol), arylboronic acid (1.2 mmol), Et3N (1.2 mmol), and the supported palladium catalyst (Fe3O4SiO2C22-Pd(II), 0.5 mol%) were mixed in DMF/H2O(1:1) (1 mL). The mixture was shaken at 75 °C in air atmosphere for an appropriate time. The progress of the reaction was monitored by thin-layer chromatography (TLC, silica gel), using n-hexane as eluent. After the completion and magnetic separation of the catalyst, the reaction mixture was treated with ethyl acetate (10 mL) and water (5 mL). The organic layer was separated and the aqueous layer was extracted with ethyl acetate (3 × 5 mL).The combined organic solution was washed with brine (3 × 5 mL).Drying (Na2SO4) and evaporation of the solvent provided a residue which was purified on preparative TLC, using hexane. All of these products have been previously reported and their identities have been confirmed by comparing their 1H and13C NMR spectral data with the values of the authentic samples. |
| 70% |
With potassium carbonate In lithium hydroxide monohydrate at 100℃; for 5h; |
2.3. General procedure for the CoGO/Fe3O4/L-dopa catalyzed Suzukicross-coupling
General procedure: A mixture of aryl halide (1 mmol), phenyl boronic acid (1.2 mmol),K2CO3 (1.2 eq.) and CoGO/Fe3O4/L-dopa (0.05 g, 1.84 mol% Co) in double distilled water (5 mL) was stirred in a round bottom flask (50 mL) at 100 °C till the completion of reaction (monitored by TLC) (Table 3). After that, the reaction mixture was cooled to room temperature.The catalyst was removed via external magnet and washed with EtOAc (3×5 mL) followed by deionized water (3×10 mL). It was dried under vacuum for 2 h. The organic fraction was washed with brine solution and dried over anhydrous Na2SO4. Finally, the product was obtained either by the exclusion of the solvent under reduced pressure or by passing through column of silica gel using EtOAc-pet.ether as eluting solvent. |
| 68.3% |
With C14H8F6O4; nickel(II) trifluoroacetate; potassium carbonate; triphenylphosphine; 1-n-butyl-3-methylimidazolium bistrifluoromethylsulfonylamide In lithium hydroxide monohydrate at 80℃; for 18h; Green chemistry; |
Typical procedure for synthesis of Suzuki-Miyaura coupling
General procedure: A 50mL round-bottomed flask was charged with aryl halides (0.5 mmol), arylboronic acid (0.6 mmol), K2CO3 (1.25 mmol), Ni(TFA)2 (0.025 mmol), β-diketone ligand (0.05 mmol), PPh3 (0.05 mmol), 1.5 g of the ionic liquid (IL) and 0.5 g of H2O. Then, the mixture was stirred at 80 open to the atmosphere. The reaction was monitored by TLC and then stopped after the starting material was completely consumed. Next, the mixture was diluted with water (10 mL) and extracted with ether (310 mL). The combined organic layers were washed with brine (310 mL), dried over MgSO4, and concentrated in vacuum. The cross coupling products were not the only product of the reaction. A small amount of homo-coupled products and removal boron product from boric acids were observed. The crude product was purified by column chromatography (silica gel, petroleum ether/ethyl acetate, 10:1). |
| 63% |
With potassium fluoride; palladium diacetate for 0.25h; microwave irradiation; |
|
| 57% |
With tri-tert-butyl(decyl)phosphonium tetrafluoroborate; palladium diacetate; Cs2CO3 In toluene at 30℃; for 12h; Inert atmosphere; Schlenk technique; |
|
| 40% |
With anhydrous sodium carbonate In ethanol; toluene for 12h; Heating; |
|
| 18% |
With disodium[(N,N’-bis(2-hydroxy-5-sulfonatobenzyl)-1,2-diphenyl-1,2-diaminoethano)palladate(II)]; Cs2CO3 In lithium hydroxide monohydrate at 80℃; for 1h; |
Catalysis experiments and gas chromatographic analysis of the reaction mixtures
General procedure: Stock solutions of the catalysts (Na2[Pd(HSS)], Na2[Pd(PrHSS)], Na2[Pd(BuHSS)],Na2[Pd(dPhHSS)], rac-Na2[Pd(CyHSS)], Na2[Pd(cis-CyHSS)]) Na2[Pd(trans-CyHSS)]) were prepared by dissolving 5.0×10-7 mol complex in 6 mL water. In general, 0.5 mmol aryl-halide, 0.75 mmol boronic acid derivative (1.5 mmol in the reactions of aryl-dihalides), and 0.5 mmolbase (Cs2CO3) were used in each reaction. Good quality distilled water was used as solvent, the organic phase was comprised of the substrates. 3 mL of water was used in each reaction. The reactions were carried out at 80 °C in 30-120 min reaction time. At the end of the reactions, the mixtures were allowed to cool to room temperature and then were extracted by chlorofom (2 mL). After separation of the phases (15-20 min) the organic phase was removed by a Pasteur pipette and filtered through a short MgSO4 plug. |
| 89 %Chromat. |
With potassium carbonate In ethanol; lithium hydroxide monohydrate at 70℃; for 1h; Green chemistry; |
|
| 100 %Chromat. |
With tripotassium phosphate tribasic In ethanol; lithium hydroxide monohydrate at 80℃; for 6h; Inert atmosphere; |
2.2. General procedure for Suzuki-Miyaura cross coupling reaction [22]
General procedure: Inside an argon atmosphere glove box, halide (0.5mmol), boronic acid (0.75mmol), K3PO4 (1.5mmol), PPPd (4mg, 1.4wt% Pd, 0.0005mmol), solvent (2ml, H2O/EtOH v/v=2:3) were mixed in a 5ml vessel and sealed. Then it was moved out of the glove box and heated in an oil bath to 80°C for 2-6h. After cooled down to r.t. for about half an hour, the mixture was filtered. Take a small amount of the liquid for GC analysis. The solid part was washed three times with ethanol. The combined liquid was dried and purified by column to give pure coupling product. For recycle experiments, the solid part after washing was degassed and used into the next cycle. Combing the recycled catalyst from X reaction times was marked as Re-PPPd-X. |
| 91 %Chromat. |
With C24H28N2O4Pd; potassium carbonate In chloroform; lithium hydroxide monohydrate at 75℃; for 4h; Sealed tube; |
|
| 99 %Spectr. |
With C78H66Al2Cl6N12O12Pd3*2C2H6OS; potassium carbonate In glycerol at 80℃; for 4h; |
2.4. General procedure for the Suzuki-Miyaura coupling reaction
General procedure: Aryl halide (1.0 mmol), aryl boronic acid (1.1 mmol), K2CO3 (1.5 mmol) and catalyst (0.02 mol%) were mixed together in 4.0 mL glycerol and allowed to react at 80 °C for 2 h. After that, the reaction mixture was cooled to room temperature and the organic residue was extracted from glycerol phase by CH2Cl2 (3×5 mL). Removal of solvent CH2Cl2, the crude residue was purified by silica gel chromatography to afford pure product, which was identified using 1H NMR and 13C NMR analyses (see ESI). |
| 84 %Chromat. |
With PdCl2{κ2−N,S−2−(4,5-dihydrothiazol-2-yl)aniline}; potassium carbonate In methanol at 70℃; for 0.5h; Reflux; |
2.9. Typical procedure for the coupling reactions between aryl bromides andboronic acid
General procedure: The round-bottom flask was placed with the appropriate amount ofcatalyst, and 5 mL of methanol was added to it. After stirring for 5 min,aryl bromide (0.5 mmol), phenylboronic acid (0.55 mmol), and K2CO3(1 mmol) were introduced into the reaction flask. The mixture washeated at 60 °C for the required time (the course of reaction wasmonitored by GC analysis), following which the solvent was removedunder reduced pressure. The residue was diluted with water (8 mL) andEtOAc (8 mL) followed by extraction twice (2-6 mL) with EtOAc. The combined organic fractions were dried (MgSO4), stripped of the solventunder vacuum, and the residue was dissolved in 5 mL of dichloromethane.An aliquot was taken with a syringe and subjected to GC analysis. Yields were calculated versus aryl bromides as an internalstandard. |
| 99 %Chromat. |
With C33H42Br3N2Pd(1-)*C33H43N2(1+); potassium carbonate In lithium hydroxide monohydrate; isopropanol at 20℃; for 0.166667h; |
3.4. Suzuki-Miyaura cross-coupling reaction
General procedure: In a typical run, in the air atmosphere, a reaction tube was charged with aryl halide (1.0 mmol), phenylboronic acid (1.0 mmol), ionic Pd-NHC complex (0.001 mmol), and K2 CO3 (1 mmol). Solvent (2-propanol/H2 O, 1:2 v/v) (3 mL) was added to tube and the mixture was vigorously stirred at room temperature for a specific time. After the desired reaction time, 5 mL of diethyl ether was added to the reaction mixture, and the organic phase was extracted with the appropriate volume of water and dried over MgSO4 . Next, the organic phase (1 L ) was injected to GC. The reactions were monitored with a Shimadzu GC-2010 Plus (FID) (Kyoto, Japan). The results were the average of the 2 runs. The yields were determined by GC with use of undecane as the internal standard. All of the coupling products were previously reported. The turn over frequency (TOF) was calculated using the following equations: TOF = TON/time of reaction and TON = moles of desired product formed/moles of the catalyst. |
| 87 %Chromat. |
With C30H44Br2N4Ni; potassium hydroxide In 1,4-dioxane; lithium hydroxide monohydrate at 120℃; for 3h; Inert atmosphere; |
Evaluation of the catalytic activity of nickel and palladium complexes in the Suzuki-Miyaura reaction (general procedure).
General procedure: The reaction was carried out under an argon atmosphere in a screw top glass vial (V = 7 mL) equipped with a magnetic stir bar. To a solution of the appropriate base (0.35 mmol), aryl halide 4a-p (0.25 mmol), and arylboronic acid 5a-c (0.35 mmol) in the appropriate solvent (0.5 mL), a solution of the appropriate catalyst (0.0625-7.5 mol, 0.025-3 mol.%) in the same solvent (1.5 mL) was added (see Tables 1, 3, and 4). The vial was herme-tically sealed and heated under stirring (see Tables 1, 3, and 4). The mixture was cooled to room temperature and an internal standard (a solution of naphthalene (16 mg, 0.125 mmol) in acetonitrile (2 mL)) was added. An aliquot (2 μL) of the result-ing mixture was taken, dissolved in acetonitrile (1 mL), and analyzed by GC/MS (see Tables 1, 3, and 4). |
| 92 %Chromat. |
With anhydrous Sodium acetate In lithium hydroxide monohydrate at 40℃; for 1h; Green chemistry; |
|
| 100 %Chromat. |
With potassium carbonate In ethanol; lithium hydroxide monohydrate for 0.833333h; Green chemistry; |
Suzuki-Miyaura cross-coupling reaction
General procedure: The mixture of various aryl halides (1 mmol), phenylboronic acid (1.3 mmol) and K2CO3 (2 mmol) was stirred at the round-bottom flask in the presence of SBAPr-3APPd catalyst (0.002 g) in H2O: EtOH (4 mL) under an air atmosphere at 100 °C. The reaction progress was checked by thin-layer chromatography (TLC). At the end of the reaction, the SBA-Pr-3APPd catalyst was filtered off, and the mixture was cooled down to ambient temperature. Next, the organic layer was extracted with chloroform and water. The resulting residue was purified by plate chromatography using n-hexane and ethyl acetate as eluents. The chemical structure of the desired products was confirmed by melting point and GC-MS analysis. |
| 89 %Chromat. |
With 2C27H33O6(3-)*3Pd(2+); potassium carbonate In toluene at 110℃; for 4h; |
|






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping