| 100% |
|
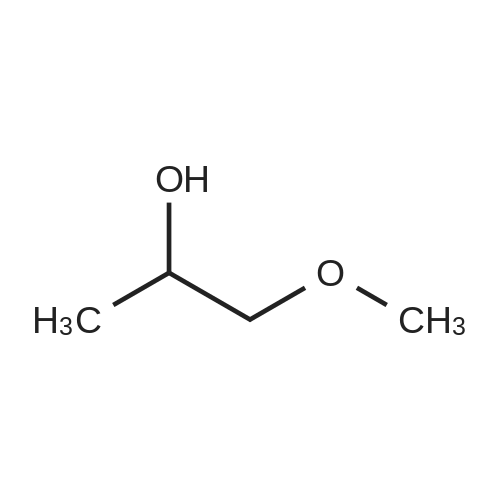
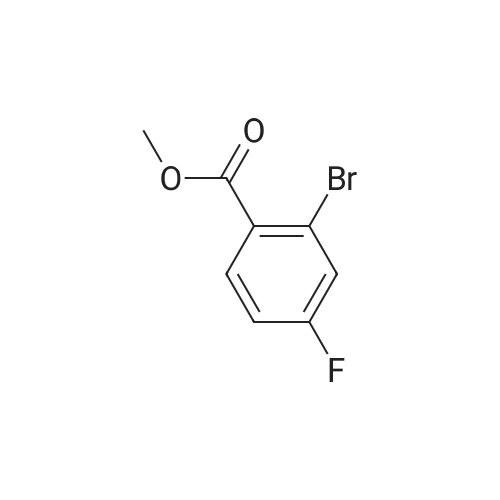
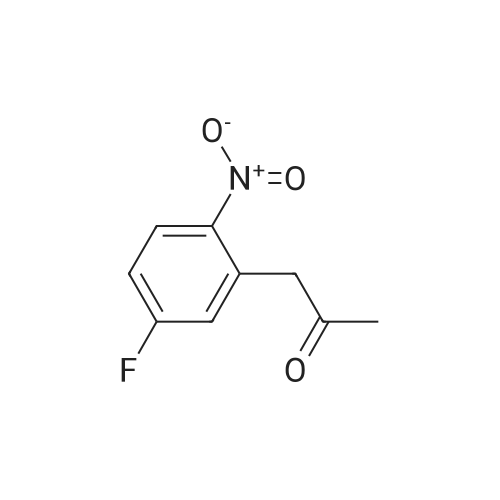
Example 35: 5-(2-Methoxy-l -methylethoxy)-N-l ,3-thiazol-2-yl-lH-indazol-3-amine[0372] To a stirred suspension of sodium hydride (60% oil dispersion, 1.36 g, 34.1 mraol) in DMF (50 mL) was added l-methoxy-2-propanol (3.4 mL, 34.1 mmol) at 00C, and the mixture was stirred for 30 min at 00C. To the mixture was added a solution of l-bromo-5- fluoro-2-nitrobenzene (5.0 g, 22.7 mmol) in DMF (10 mL) at 00C, and the mixture was stirred for 1 h at room temperature. To the mixture was carefully added IN HCl (50 mL) at 00C. The aqueous layer was extracted with EtOAc, and the organic layer was washed with IN HCl, H2O, and brine, dried (MgSO4), filtered, and concentrated in vacuo to give 6.60 g of 2-bromo-4-(2-methoxy-] -methylethoxy)-l -nitrobenzene (compound 35A) as a light yellow oil, which was used for the next step without further purification. 1H NMR (300 MHz, CDCl3) delta 1.34 (d, 3H, 7=6.40 Hz) 3.40 (s, 3H) 3.47 - 3.62 (m, 2H) 4.57 - 4.71 (m, IH) 6.94 (dd, IH, 7=9.23, 2.64 Hz) 7.25 - 7.28 (m, IH) 7.97 (d, IH, 7=9.23 Hz). [0373] To a stirred solution of 2-bromo-4-(2-methoxy-l-methylethoxy)-l -nitrobenzene (2.007 g, 6.92 mmol) in l-methyl-2-pyrrolidinone (20 mL) was added copper cyanide (0.75 g, 8.3 mmol) at room temperature. The mixture was stirred for 30 min at 1500C. After cooling, the mixture was diluted with EtOAc. The organic layer was washed successively with H2O, IN HCl, H2O, and brine, dried (MgSO4), filtered, and concentrated in vacuo. Purification by silica gel chromatography (hexane:EtOAc=30:l to 10:1 to 5:1 to 3:1) gave 1.46 g (89%) of 5- (2-methoxy-l-methylethoxy)-2-nitrobenzonitrile (compound 35B) as a brown oil. 1H NMR <n="124"/>(300 MHz, CDCl3) delta 1.37 (d, 3H, 7=6.40 Hz) 3.39 (s, 3H) 3.49 - 3.66 (m, 2H) 4.63 - 4.81 (m, IH) 7.24 (dd, IH, 7=9.42, 2.83 Hz) 7.37 (d, IH, 7=2.83 Hz) 8.28 (d, IH, 7=9.23 Hz). MS (ES) [m+H] calc'd for CnHi2N2O4, 237; found 237.[0374] 2-Amino-5-(2-methoxy-l-methylethoxy)benzonitxile (compound 35C) was prepared in 49% yield from 5-(2-methoxy-l-methylethoxy)-2-nitrobenzonitrile according to a procedure analogous to that outlined in Example 34. 1H NMR (300 MHz, CDCl3) delta 1.26 (d, 3H, 7=6.22 Hz) 3.40 (s, 3H) 3.42 - 3.57 (m, 2H) 4.13 (s, 2H) 4.27 - 4.40 (m, IH) 6.68 (d, IH, 7=8.85 Hz) 6.95 (d, IH, 7=2.83 Hz) 6.98 - 7.05 (m, IH). MS (ES) [m+H] calc'd for CnH14N2O2, 207; found 207.[0375] 5-(2-Methoxy-l-methylethoxy)-lH-indazol-3-amine (compound 35C) was prepared in 87% yield from 2-amino-5-(2-methoxy-l -methylethoxy)benzonitrile according to a procedure analogous to that outlined in Example 28. 1H NMR (300 MHz, DMSO-^6) delta 1.22 (d, 3H, 7=6.22 Hz) 3.30 (s, 3H) 3.40 - 3.55 (m, 2H) 4.35 - 4.54 (m, IH) 5.16 (s, 2H) 6.88 (dd, IH, 7=8.85, 2.45 Hz) 7.12 (d, IH, 7=8.85 Hz) 7.22 (d, IH, 7=2.26 Hz) 11.16 (s, IH). MS (ES) [m+H] calc'd for C11H13N3O2, 222; found 222.[0376] iV-[5-(2-Methoxy-l -methylethoxy)-l H-indazol-3-yl]thiourea (compound 35E) was prepared in 68% yield from 5-(2-methoxy-l-methylethoxy)-lH-indazol-3-amine according to a procedure analogous to that outlined in Example 9. 1H NMR (300 MHz, DMSO-^6) delta 1.27 (d, 3H, 7=6.22 Hz) 3.31 (s, 3H) 3.42 - 3.60 (m, 2H) 4.40 - 4.61 (m, IH) 7.01 (dd, IH, 7=8.95, 2.35 Hz) 7.33 (d, IH, 7=8.85 Hz) 7.77 (d, IH, 7=2.07 Hz) 8.68 (brs, IH) 9.27 (brs, IH) 10.70 (s, IH) 12.48 (s, IH). MS (ES) [m+H] calc'd for C12Hi6N4O2S, 281 ; found 281. [0377] The title compound was prepared in 49% yield from lambda/'-[5-(2-methoxy-l- methylethoxy)-lH-indazol-3-yl]thiourea according to a procedure analogous to that outlined in Example 28. 1H NMR (300 MHz, DMSO-J6) delta 1.27 (d, 3H, 7=6.44 Hz) 3.31 (s, 3H) 3.39 - 3.65 (m, 2H) 4.36 - 4.59 (m, IH) 6.96 (d, IH, 7=3.41 Hz) 7.00 (dd, IH, 7=9.09, 2.27 Hz) 7.30 (d, IH, 7=9.09 Hz) 7.34 (d, IH77=3.41 Hz) 7.66 (d, IH, 7=1.89 Hz) 11.10 (s, IH) 12.13 (s, IH). MS (ES) [m+H] calc'd for C14H16N4O2S, 305; found 305. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping