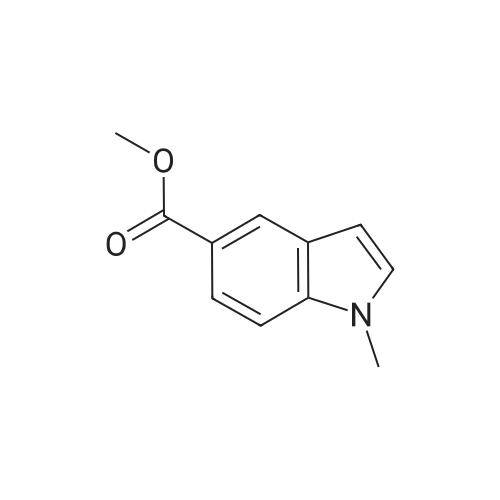
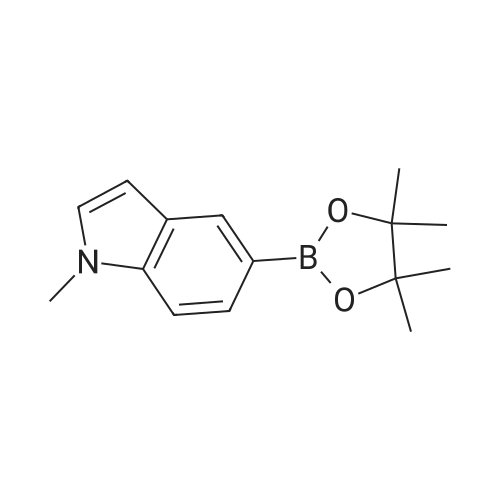
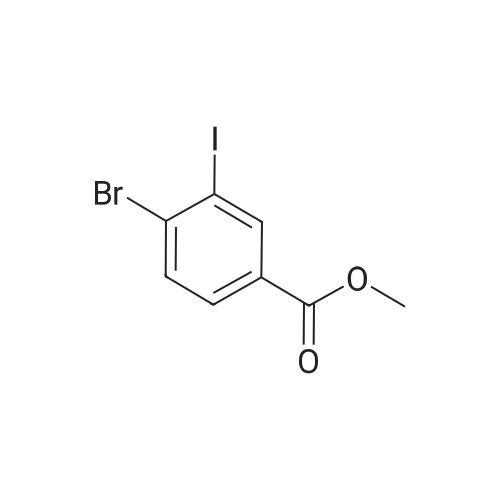
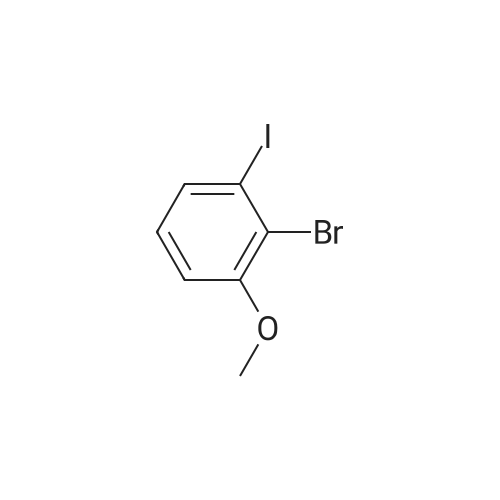
There will be a HazMat fee per item when shipping a dangerous goods. The HazMat fee will be charged to your UPS/DHL/FedEx collect account or added to the invoice unless the package is shipped via Ground service. Ship by air in Excepted Quantity (each bottle), which is up to 1g/1mL for class 6.1 packing group I or II, and up to 25g/25ml for all other HazMat items.
Type
HazMat fee for 500 gram (Estimated)
Excepted Quantity
USD 0.00
Limited Quantity
USD 15-60
Inaccessible (Haz class 6.1), Domestic
USD 80+
Inaccessible (Haz class 6.1), International
USD 150+
Accessible (Haz class 3, 4, 5 or 8), Domestic
USD 100+
Accessible (Haz class 3, 4, 5 or 8), International
USD 200+
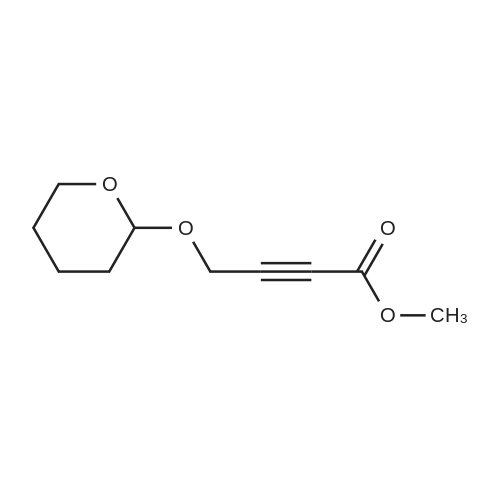
Structure of 6089-04-9 * Storage: {[proInfo.prStorage]}
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
With toluene-4-sulfonic acid In dichloromethane at 0 - 20℃; for 1 h;
O-THP Propargyl alcohol (18) Compound 18 was prepared according to a literature procedure.24 To the solution of propargyl alcohol 17 (2.6mL, 44.6mmol) and p-TsOH monohydrate (0.09g, 0.5mmol) in CH2Cl2 (45mL) DHP (4.3mL, 46.8mmol) was added dropwise at 0°C. After stirring for 5min at 0°C, the reaction mixture was allowed to warm up to RT and was stirred for 1h. Next, the reaction mixture was washed with saturated NaHCO3 (30mL) and the aqueous layer was extracted with CH2Cl2 (45mL). The combined organic layer dried over MgSO4, filtered, and concentrated under reduced pressure to give compound 18 (6.1g, 97.2percent) as a yellow oil. Rf=0.30 (n-hexane/Et2O 95/5). 1H NMR (400MHz, CDCl3): δ=1.50–1.67 (m, 4H), 1.70–1.89 (m, 2H), 2.41 (t, J=2.4Hz, 1H), 3.52–3.57 (m, 1H), 3.82–3.87 (m, 1H), 4.27 (ddd, J=2.4Hz, 11.3Hz, 15.8Hz, 2H), 4.83 (t, J=3.4Hz, 1H). 13C NMR (100MHz, CDCl3): δ=19.0, 25.3, 30.2, 53.9, 61.9, 74.0, 79.7, 96.8. MS-CI: m/z calcd for C8H12O2 [M+H]+, 141; found, 141. This compound has been also previously reported.25
93%
With toluene-4-sulfonic acid In dichloromethane at 0 - 20℃; Inert atmosphere
TsOH (840 mg, 4.40 mmol) was added to dihydropyran (18.6 g, 0.220 mol) in dichloromethane (20 mL) at 0 ºC and stirred until it dissolved. Propargyl alcohol 15 (12.3 g, 0.220 mol) was added dropwise at 0 ºC. The reaction solution was warmed to rt and stirred overnight. The reaction was quenched with saturated NaHCO3, extracted with dichloromethane. The organic layer was washed with brine, dried with MgSO4 and concentrated to give 30.0 g brown liquid. The colorless liquid 16 was obtained from distillation at 40-41 ºC, 5 mmHg (21.5 g, 70 percent) and at 25 - 40 ºC, 6 mmHg (7.2g, 23 percent). 1H NMR (400 MHz, CDCl3): δ 1.54 (m, 2 H), 1.63 (m, 2 H), 1.74 (m, 1 H), 1.84 (m, 1 H), 2.40 (t, J = 2.4 Hz, 1 H), 3.53 (m, 1 H), 3.84 (m, 1 H), 4.23 (dd, J = 15.6, 2.4 Hz, 1 H), 4.30 (dd, J = 15.6, 2.4 Hz, 1 H), 4.82 (br t, J = 3.2 Hz, 1 H).
87%
With toluene-4-sulfonic acid In dichloromethane at 29℃; for 2 h;
To a solution of propargyl alcohol (1 g, 16.93 mmol) in dichloromethane (15 mL), PTSA (3 mg, 0.16 mmol) and DHP (3 mL, 33.86 mmol) were added and the reaction mixture was stirred at room temperature for 2 h. After completion of starting materials, the reaction mixture was quenched with NaHC03 and extracted with dichloromethane (100 mL x 2), washed with water (100 mL x 2), and brine (100 n L x 1). The combined organic extracts were evaporated under reduced pressure to obtain the crude product which was purified by column chromatography (100-200 mesh silica gel, eluent 10 percent EtOAc in hexane) to furnish compound (ii) (2.078 g, 87percent) as a light brown liquid.
82%
at 25℃; for 3 h;
To a solution of 15 g (0.18 mol) of dihydropyrane and 2 mg of p-toluenesulfonic acid was added 10 g (0.18 mol) of 2-propyn-1-ol. After stirring at 25 °C for 3 h, the reaction solution was diluted with 100 mL of diethyl ether and washed with 10percent aqueous NaHCO3, water, and brine, dried (MgSO4), concentrated to give 20.5 g (82percent yield) of 2-(2-propynyloxy)-tetrahydropyrane. 1H NMR δ 4.83 (t, J = 3.6 Hz, 1H, CHO), 4.29 (dd, J = 16, 3 Hz, 1H, CH2O), 4.26 (dd, J = 16, 3 Hz, 1H, CH2O), 3.88-3.82 (m, 1H, CHO), 3.58-3.52 (m, 1H, CHO), 2.42 (d, J = 3 Hz, 1H, CH), 1.90-1.50 (m, 6H); 13C NMR δ 97.0, 79.9, 74.2, 62.1, 54.1, 30.4, 25.5, 19.1; MS (electrospray; negative mode) m/z 139.0 (M - H-, 100percent). MS (electrospray; positive mode) m/z 163.1 (M + Na+; 100percent).
78%
at 60℃; for 10 h; Sealed tube; Green chemistry
General procedure: A mixture of corresponding alcohol (1, 1 mmol) and 3,4-dihydro-2H-pyran (2, 100 mg, 1.2 mmol) was stirred in the presence of zwitterionic-salt A (10 mg, 10 molpercent) at 60-80 °C (oil bath) for 10-12 h in a seal tube. After completion of the reaction (TLC), the reaction mixture was cooled to room temperature and diluted with water (10 mL) and extracted with ethyl acetate (20 mL). Organic layer was dried over anhydrous Na2SO4. After evaporation of solvent the crude product was purified by column chromatography on silica gel using petroleum ether/ethyl acetate (4-5percent) as eluent.
Reference:
[1] Synlett, 2003, # 14, p. 2163 - 2166
[2] Tetrahedron, 2010, vol. 66, # 25, p. 4573 - 4576
[3] Synthesis, 2005, # 19, p. 3293 - 3296
[4] Journal of the Chinese Chemical Society, 2007, vol. 54, # 2, p. 525 - 532
[5] Journal of Organic Chemistry, 2009, vol. 74, # 4, p. 1531 - 1540
[6] Recueil des Travaux Chimiques des Pays-Bas, 1989, vol. 108, # 9, p. 304 - 313
[7] Gazzetta Chimica Italiana, 1980, vol. 110, # 9/10, p. 523 - 526
[8] Journal of the American Chemical Society, 1999, vol. 121, # 39, p. 9034 - 9042
[9] Tetrahedron Asymmetry, 2005, vol. 16, # 23, p. 3773 - 3784
[10] Synthesis, 2007, # 5, p. 779 - 790
[11] Journal of Molecular Catalysis A: Chemical, 2011, vol. 345, # 1-2, p. 96 - 100
[12] European Journal of Organic Chemistry, 2013, # 22, p. 4903 - 4908
[13] Tetrahedron, 1990, vol. 46, # 11, p. 4063 - 4082
[14] Organic Letters, 2011, vol. 13, # 21, p. 5924 - 5927
[15] Bioorganic and Medicinal Chemistry, 2017, vol. 25, # 18, p. 5008 - 5015
[16] Tetrahedron Letters, 2005, vol. 46, # 15, p. 2543 - 2545
[17] Organic Syntheses, 1999, vol. 76, p. 178 - 178
[18] Synthesis, 1993, # 11, p. 1069 - 1070
[19] Tetrahedron Letters, 1998, vol. 39, # 44, p. 8159 - 8162
[20] Tetrahedron, 2001, vol. 57, # 5, p. 889 - 898
[21] Synthetic Communications, 2002, vol. 32, # 10, p. 1549 - 1555
[22] Patent: US4186141, 1980, A,
[23] Journal of Labelled Compounds and Radiopharmaceuticals, 1994, vol. 34, # 8, p. 747 - 757
[24] Synthetic Communications, 2005, vol. 35, # 14, p. 1939 - 1945
[25] Journal of Organic Chemistry, 2001, vol. 66, # 22, p. 7511 - 7513
[26] European Journal of Organic Chemistry, 2013, # 29, p. 6710 - 6721
[27] Chemistry - A European Journal, 2015, vol. 21, # 4, p. 1482 - 1487
[28] Canadian Journal of Chemistry, 1987, vol. 65, p. 43 - 55
[29] Canadian Journal of Chemistry, 1987, vol. 65, p. 56 - 68
[30] Journal of Organic Chemistry, 1981, vol. 46, p. 5340 - 5343
[31] Journal of Organic Chemistry, 1985, vol. 50, # 24, p. 4762 - 4766
[32] Journal of Medicinal Chemistry, 1995, vol. 38, # 6, p. 875 - 882
[33] Journal of Chemical Research - Part S, 1996, # 11, p. 494 - 495
[34] Tetrahedron, 1999, vol. 55, # 17, p. 5449 - 5456
[35] Bioorganic and Medicinal Chemistry, 2013, vol. 21, # 7, p. 1811 - 1822
[36] Australian Journal of Chemistry, 1997, vol. 50, # 11, p. 1067 - 1079
[37] Synlett, 1999, # 8, p. 1261 - 1262
[38] Tetrahedron Letters, 2002, vol. 43, # 15, p. 2801 - 2805
[39] Tetrahedron Letters, 2006, vol. 47, # 14, p. 2341 - 2344
[40] Journal of the American Chemical Society, 2001, vol. 123, # 50, p. 12736 - 12737
[41] Journal of Organic Chemistry, 1983, vol. 48, # 13, p. 2151 - 2158
[42] Tetrahedron Letters, 1993, vol. 34, # 33, p. 5269 - 5272
[43] Organic Letters, 2017, vol. 19, # 19, p. 5340 - 5343
[44] Tetrahedron Letters, 1998, vol. 39, # 50, p. 9287 - 9288
[45] Australian Journal of Chemistry, 2002, vol. 55, # 12, p. 795 - 798
[46] Synthetic Communications, 2006, vol. 36, # 19, p. 2893 - 2900
[47] Organic Letters, 2008, vol. 10, # 6, p. 1075 - 1078
[48] Journal of Organic Chemistry, 2013, vol. 78, # 15, p. 7714 - 7726
[49] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1991, # 3, p. 581 - 587
[50] Organic Preparations and Procedures International, 1996, vol. 28, # 5, p. 613 - 617
[51] Synthetic Communications, 2000, vol. 30, # 23, p. 4323 - 4328
[52] Angewandte Chemie - International Edition, 2016, vol. 55, # 44, p. 13719 - 13723[53] Angew. Chem., 2016, vol. 128, p. 13923 - 13927,5
[54] European Journal of Organic Chemistry, 2014, vol. 2014, # 32, p. 7095 - 7098
[55] Patent: WO2013/71418, 2013, A1, . Location in patent: Page/Page column 19-20
[56] Bulletin of the Chemical Society of Japan, 1984, vol. 57, # 10, p. 2768 - 2776
[57] Journal of Organic Chemistry, 2004, vol. 69, # 21, p. 7157 - 7170
[58] Steroids, 1983, vol. 41, # 3, p. 339 - 348
[59] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1992, # 9, p. 1089 - 1100
[60] Chemistry - A European Journal, 2011, vol. 17, # 27, p. 7480 - 7491
[61] Synthetic Communications, 2013, vol. 43, # 9, p. 1237 - 1242
[62] Chemistry - A European Journal, 1998, vol. 4, # 11, p. 2342 - 2352
[63] Chemistry - A European Journal, 2003, vol. 9, # 11, p. 2419 - 2438
[64] New Journal of Chemistry, 2001, vol. 25, # 1, p. 40 - 54
[65] Canadian Journal of Chemistry, 2001, vol. 79, # 8, p. 1259 - 1271
[66] Angewandte Chemie - International Edition, 2017, vol. 56, # 3, p. 847 - 850[67] Angew. Chem., 2017, vol. 129, p. 865 - 868,4
[68] Advanced Synthesis and Catalysis, 2012, vol. 354, # 7, p. 1363 - 1372
[69] European Journal of Medicinal Chemistry, 2012, vol. 50, p. 311 - 318
[70] Tetrahedron, 1998, vol. 54, # 36, p. 10779 - 10788
[71] Journal of Organic Chemistry, 1998, vol. 63, # 18, p. 6128 - 6131
[72] European Journal of Organic Chemistry, 2001, # 13, p. 2577 - 2588
[73] Journal of the Chemical Society. Perkin Transactions 1, 2002, # 13, p. 1555 - 1563
[74] Synthetic Communications, 1996, vol. 26, # 24, p. 4539 - 4544
[75] Tetrahedron Letters, 2004, vol. 45, # 26, p. 5135 - 5138
[76] Synthetic Communications, 2017, vol. 47, # 20, p. 1905 - 1915
[77] European Journal of Organic Chemistry, 2014, vol. 2014, # 22, p. 4733 - 4740
[78] Natural Product Research, 2010, vol. 24, # 13, p. 1274 - 1281
[79] Bulletin of the Chemical Society of Japan, 1994, vol. 67, # 10, p. 2838 - 2849
[80] Bulletin of the Chemical Society of Japan, 1995, vol. 68, # 6, p. 1689 - 1705
[81] Journal of Photochemistry and Photobiology A: Chemistry, 2017, vol. 349, p. 162 - 170
[82] Synthetic Communications, 1989, vol. 19, # 5and6, p. 901 - 906
[83] Canadian Journal of Chemistry, 2006, vol. 84, # 9, p. 1174 - 1179
[84] Journal of the Chemical Society - Perkin Transactions 1, 1998, # 11, p. 1839 - 1858
[85] Journal of the American Chemical Society, 1953, vol. 75, p. 4048,4050
[86] Journal of the Chemical Society, 1950, p. 3646,3649
[87] Collection of Czechoslovak Chemical Communications, 1981, vol. 46, # 4, p. 917 - 925
[88] Journal of Organometallic Chemistry, 1987, vol. 319, p. 333 - 344
[89] Journal of Chemical Research, Miniprint, 1982, # 2, p. 321 - 351
[90] Journal of the American Chemical Society, 1994, vol. 116, # 26, p. 12093 - 12094
[91] Journal of the American Chemical Society, 1996, vol. 118, # 1, p. 275 - 276
[92] Tetrahedron: Asymmetry, 1997, vol. 8, # 20, p. 3393 - 3396
[93] Journal of Chemical Ecology, 1995, vol. 21, # 3, p. 299 - 311
[94] Synthetic Communications, 2003, vol. 33, # 22, p. 3929 - 3934
[95] Journal of Organic Chemistry, 2006, vol. 71, # 9, p. 3639 - 3642
[96] Journal of the American Chemical Society, 2006, vol. 128, # 9, p. 2772 - 2773
[97] Journal of Organic Chemistry, 2001, vol. 66, # 24, p. 8120 - 8126
[98] Journal of the Chemical Society. Perkin Transactions 1, 2002, # 22, p. 2485 - 2489
[99] Patent: US2003/3245, 2003, A1,
[100] Patent: US5238832, 1993, A,
[101] Patent: US5502073, 1996, A,
[102] Patent: US2003/220313, 2003, A1, . Location in patent: Page/Page column 5
[103] Patent: WO2004/46079, 2004, A1, . Location in patent: Page/Page column 14-16
2
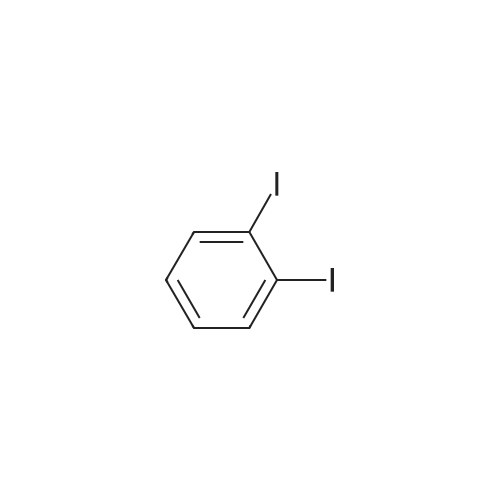
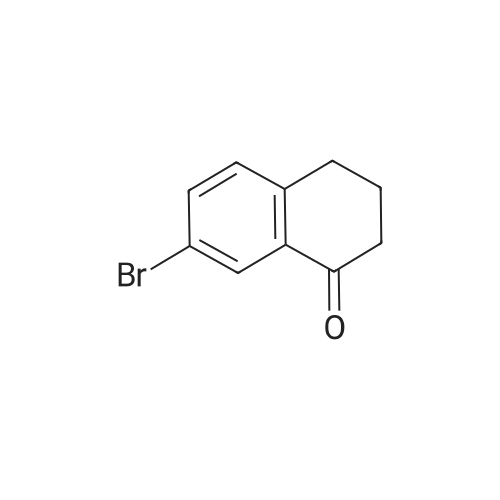
[ 110-87-2 ]
[ 104-15-4 ]
[ 6089-04-9 ]
Reference:
[1] Patent: US4254285, 1981, A,
[2] Patent: US4477388, 1984, A,
[3] Patent: US4194055, 1980, A,
[4] Patent: US4170597, 1979, A,
[5] Patent: US4218566, 1980, A,
[6] Patent: US4233231, 1980, A,
[7] Patent: US4085272, 1978, A,
With aluminium(III) triflate In dichloromethane at 20 - 25℃; for 2h; Inert atmosphere;
99%
With toluene-4-sulfonic acid for 0.75h;
99%
With pyridinium p-toluenesulfonate In dichloromethane
99%
With pyridinium p-toluenesulfonate In dichloromethane
98%
With toluene-4-sulfonic acid In dichloromethane for 2h; Ambient temperature;
98%
With Amberlyst H-15 In hexane at 0℃; for 0.666667h;
98%
With toluene-4-sulfonic acid In diethyl ether at 20℃; for 5h;
98%
With toluene-4-sulfonic acid for 2.5h;
98%
With N.N'-bis[3,5-bis(trifluoromethyl)phenyl]thiourea at 20℃; for 15h;
98%
With sulfonic acid functionalized polycyclic aromatic carbon catalyst from glycerol pitch In dichloromethane at 20℃; for 2h;
98%
Stage #1: 3,4-dihydro-2<i>H</i>-pyran With p-toluenesulfonic acid monohydrate at 20℃; for 0.166667h; Schlenk technique; Inert atmosphere;
Stage #2: propargyl alcohol for 0.666667h; Schlenk technique; Inert atmosphere;
Stage #3: With sodium hydrogencarbonate at 20℃; for 12h; Schlenk technique; Inert atmosphere;
98%
With toluene-4-sulfonic acid In dichloromethane at 20℃; for 1h; Inert atmosphere;
97%
With toluene-4-sulfonic acid for 0.5h;
97%
With toluene-4-sulfonic acid In dichloromethane at 0 - 23℃; Inert atmosphere;
97.2%
With toluene-4-sulfonic acid In dichloromethane at 0 - 20℃; for 1h;
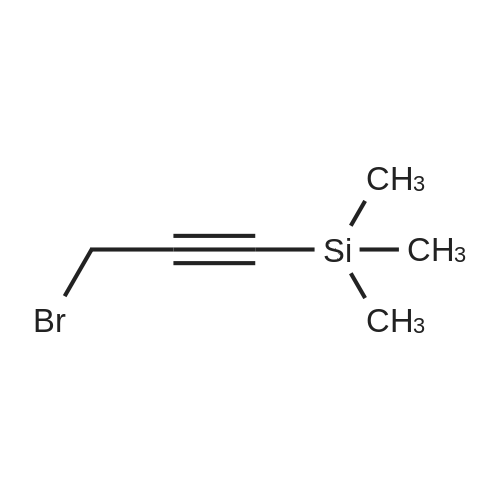
Preparation of silyl alkyne bromides
O-THP Propargyl alcohol (18) Compound 18 was prepared according to a literature procedure.24 To the solution of propargyl alcohol 17 (2.6mL, 44.6mmol) and p-TsOH monohydrate (0.09g, 0.5mmol) in CH2Cl2 (45mL) DHP (4.3mL, 46.8mmol) was added dropwise at 0°C. After stirring for 5min at 0°C, the reaction mixture was allowed to warm up to RT and was stirred for 1h. Next, the reaction mixture was washed with saturated NaHCO3 (30mL) and the aqueous layer was extracted with CH2Cl2 (45mL). The combined organic layer dried over MgSO4, filtered, and concentrated under reduced pressure to give compound 18 (6.1g, 97.2%) as a yellow oil. Rf=0.30 (n-hexane/Et2O 95/5). 1H NMR (400MHz, CDCl3): δ=1.50-1.67 (m, 4H), 1.70-1.89 (m, 2H), 2.41 (t, J=2.4Hz, 1H), 3.52-3.57 (m, 1H), 3.82-3.87 (m, 1H), 4.27 (ddd, J=2.4Hz, 11.3Hz, 15.8Hz, 2H), 4.83 (t, J=3.4Hz, 1H). 13C NMR (100MHz, CDCl3): δ=19.0, 25.3, 30.2, 53.9, 61.9, 74.0, 79.7, 96.8. MS-CI: m/z calcd for C8H12O2 [M+H]+, 141; found, 141. This compound has been also previously reported.25
96%
With iron(III) perchlorate In diethyl ether at 20℃; for 1h;
96%
With camphor-10-sulfonic acid In dichloromethane at 0 - 23℃; for 4h; Inert atmosphere;
95%
In hexane for 2h; Heating;
95%
With Zr(O3PCH3)1.2(O3PC6H4SO3H)0.8 In dichloromethane for 0.5h; Ambient temperature;
95%
With pyridinium p-toluenesulfonate In dichloromethane at 20℃; for 15h;
95%
With zirconium(IV) oxide for 0.116667h; microwave irradiation;
95%
With toluene-4-sulfonic acid
1 EXAMPLE 1
EXAMPLE 1 Propargyl alcohol (13.20 g) and dihydropyran (19.81 g) are placed into a 50-ml container, and 0.2 g of anhydrous p-toluenesulfonic acid is added. The mixture is cooled in an ice bath, then stirred at room temperature for 30 minutes and neutralized with anhydrous sodium carbonate. The resulting solids are separated off. Distillation of the remaining liquid in a vacuum gives 31.3 g of propargyl tetrahydropyranyl ether (yield: 95%), b.p. 75° to 80° C./37 mm Hg. IR (cm-1): 3277 (C C), 2940, 2865, 2055 (C C); NMR (CC14): δ 4.67 (s, 1H, O--CH--O); 4.11 (W, J=2, 5 Hz, 2H, C CCH2 --O); 3.13-3.94 (m, 2 H, CH2 --O); 2.32 (t, J=2, 5Hz, 1H, CH C).
95%
With camphor-10-sulfonic acid In dichloromethane at 0 - 20℃; for 3h; Inert atmosphere;
Preparation of THP ether 32
To the solution of propargylic alcohol (3.1 g, 50 mmol) in DCM (400 mL) cooled at 0oC was added DHP (4.6 mL, 50 mmol), CSA (580 mg, 2.5 mmol) and stirred for 3 h. The reaction mixure was washed with aq Na2CO3(10 mL), brine (5 mL), dried over Na2SO4and solvent was evaporated under reduced pressure to afford crude compound which was purified by column chromatography using 2% EtOAc/hexanes (v/v) as the eluent to yield ether32as a colourless oil (6.6 g, 47.5 mmol) in 95% yield. TLC, Rf0.52 (5% EtOAc/hexanes);1H NMR (200 MHz, CDCl3) δ 4.80 (t,J =3.2 Hz, 1H), 4.21 (t,J =2.7 Hz, 2H), 3.85-3.75 (m, 1H), 3.55-3.47 (m, 1H), 2.31 (t,J =2.2 Hz, 1H), 1.90-1.50 (m, 6H).
94%
With toluene-4-sulfonic acid In dichloromethane at 0℃; for 10h;
94%
With 1-butyl-3-methylimidazolium hydrogen sulfate at 20℃; for 0.0833333h;
94%
With ammonium cerium(IV) nitrate In acetonitrile at 20℃;
94%
With toluene-4-sulfonic acid In dichloromethane at 20℃; for 3h; Inert atmosphere; Cooling with ice;
94%
Stage #1: 3,4-dihydro-2<i>H</i>-pyran With toluene-4-sulfonic acid at 60℃;
Stage #2: propargyl alcohol at 60℃; for 2.58333h;
93%
With trifluoroacetic acid In dichloromethane for 18h;
93%
With trifluoroacetic acid In dichloromethane Ambient temperature;
93%
With toluene-4-sulfonic acid In tetrahydrofuran at 0℃; for 6h;
93%
With toluene-4-sulfonic acid
93%
With pyridinium p-toluenesulfonate In dichloromethane for 19h; Ambient temperature;
93%
With H-Rho zeolite In hexane for 4h; Heating;
93%
With toluene-4-sulfonic acid In dichloromethane at 0 - 20℃; for 3h;
93%
With toluene-4-sulfonic acid In dichloromethane at 0 - 20℃; Inert atmosphere;
Preparation of 2-(prop-2-ynyloxyl)-tetrahydro-2H-pyran (16).
TsOH (840 mg, 4.40 mmol) was added to dihydropyran (18.6 g, 0.220 mol) in dichloromethane (20 mL) at 0 ºC and stirred until it dissolved. Propargyl alcohol 15 (12.3 g, 0.220 mol) was added dropwise at 0 ºC. The reaction solution was warmed to rt and stirred overnight. The reaction was quenched with saturated NaHCO3, extracted with dichloromethane. The organic layer was washed with brine, dried with MgSO4 and concentrated to give 30.0 g brown liquid. The colorless liquid 16 was obtained from distillation at 40-41 ºC, 5 mmHg (21.5 g, 70 %) and at 25 - 40 ºC, 6 mmHg (7.2g, 23 %). 1H NMR (400 MHz, CDCl3): δ 1.54 (m, 2 H), 1.63 (m, 2 H), 1.74 (m, 1 H), 1.84 (m, 1 H), 2.40 (t, J = 2.4 Hz, 1 H), 3.53 (m, 1 H), 3.84 (m, 1 H), 4.23 (dd, J = 15.6, 2.4 Hz, 1 H), 4.30 (dd, J = 15.6, 2.4 Hz, 1 H), 4.82 (br t, J = 3.2 Hz, 1 H).
92%
With toluene-4-sulfonic acid for 2h; Ambient temperature;
92%
With lithium tetrafluoroborate In acetonitrile at 25 - 30℃; for 6h;
92%
With toluene-4-sulfonic acid In dichloromethane at 20℃;
92%
With lanthanum(III) nitrate at 20℃; for 2.5h;
92%
91%
With hydrogenchloride for 3h;
91%
In dichloromethane for 1h; Ambient temperature;
91%
Stage #1: propargyl alcohol With toluene-4-sulfonic acid In dichloromethane at 0℃; for 0.0833333h; Inert atmosphere;
Stage #2: 3,4-dihydro-2<i>H</i>-pyran In dichloromethane at 20℃; for 1h; Inert atmosphere;
90%
With lithium perchlorate In diethyl ether for 8h; Ambient temperature;
90%
With trichlorophosphate at 0 - 20℃;
90%
for 0.1h; sonication;
90%
With toluene-4-sulfonic acid In dichloromethane at 0 - 20℃; Inert atmosphere;
90%
With toluene-4-sulfonic acid In dichloromethane at 0 - 20℃; for 3.08333h; Inert atmosphere;
89%
With pyridinium p-toluenesulfonate In dichloromethane for 4.5h; Ambient temperature;
89%
With sulfated zirconia In dichloromethane for 1h; Ambient temperature;
89%
With lithium bromide In dichloromethane at 20℃; for 5.5h;
89%
With amberlyst15 In Petroleum ether at 20℃; for 6h;
88%
With hydrogenchloride In water at 20℃; for 1.5h;
87%
With toluene-4-sulfonic acid In dichloromethane at 29℃; for 2h;
1
To a solution of propargyl alcohol (1 g, 16.93 mmol) in dichloromethane (15 mL), PTSA (3 mg, 0.16 mmol) and DHP (3 mL, 33.86 mmol) were added and the reaction mixture was stirred at room temperature for 2 h. After completion of starting materials, the reaction mixture was quenched with NaHC03 and extracted with dichloromethane (100 mL x 2), washed with water (100 mL x 2), and brine (100 n L x 1). The combined organic extracts were evaporated under reduced pressure to obtain the crude product which was purified by column chromatography (100-200 mesh silica gel, eluent 10 % EtOAc in hexane) to furnish compound (ii) (2.078 g, 87%) as a light brown liquid.
86%
With toluene-4-sulfonic acid In diethyl ether 0 deg C, 0.5 h; 25 deg C, 1 h;
86%
With toluene-4-sulfonic acid In tetrahydrofuran at 0 - 20℃;
85%
With toluene-4-sulfonic acid for 5h; Ambient temperature;
85%
With toluene-4-sulfonic acid In dichloromethane at 0℃; for 2h;
85%
With hydrogenchloride In water at 20℃; for 3h; Inert atmosphere;
85%
With silica gel supported sodium hydrogen sulfate In neat (no solvent) at 25℃; for 0.0833333h;
84%
With pyridinium p-toluenesulfonate In dichloromethane for 16h;
84%
With toluene-4-sulfonic acid In tetrahydrofuran at 20℃; for 2h;
84%
In dichloromethane at 0 - 20℃; for 16h;
83%
With toluene-4-sulfonic acid In dichloromethane at 25℃; for 2h;
83%
Stage #1: propargyl alcohol With pyridinium p-toluenesulfonate In dichloromethane at 0℃; Inert atmosphere;
Stage #2: 3,4-dihydro-2<i>H</i>-pyran In dichloromethane at 0 - 20℃; Inert atmosphere;
82%
With toluene-4-sulfonic acid In dichloromethane at 0℃; for 2h;
82%
With toluene-4-sulfonic acid at 25℃; for 3h;
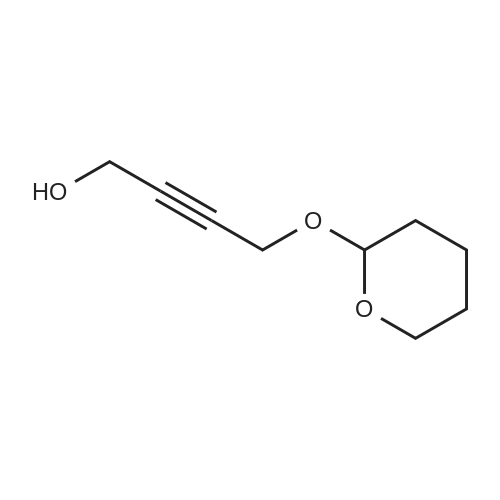
5.2.1. (E)-1-(2-Tetrahydropyranyl)oxy-nona-5-en-2-yne (3)
To a solution of 15 g (0.18 mol) of dihydropyrane and 2 mg of p-toluenesulfonic acid was added 10 g (0.18 mol) of 2-propyn-1-ol. After stirring at 25 °C for 3 h, the reaction solution was diluted with 100 mL of diethyl ether and washed with 10% aqueous NaHCO3, water, and brine, dried (MgSO4), concentrated to give 20.5 g (82% yield) of 2-(2-propynyloxy)-tetrahydropyrane. 1H NMR δ 4.83 (t, J = 3.6 Hz, 1H, CHO), 4.29 (dd, J = 16, 3 Hz, 1H, CH2O), 4.26 (dd, J = 16, 3 Hz, 1H, CH2O), 3.88-3.82 (m, 1H, CHO), 3.58-3.52 (m, 1H, CHO), 2.42 (d, J = 3 Hz, 1H, CH), 1.90-1.50 (m, 6H); 13C NMR δ 97.0, 79.9, 74.2, 62.1, 54.1, 30.4, 25.5, 19.1; MS (electrospray; negative mode) m/z 139.0 (M - H-, 100%). MS (electrospray; positive mode) m/z 163.1 (M + Na+; 100%).
80%
With Amberlyst A15 In dichloromethane for 2h; Ambient temperature;
80%
With pyridinium p-toluenesulfonate In dichloromethane for 3h;
80%
With camphor-10-sulfonic acid In dichloromethane at 20℃; for 12h;
80%
With trichlorophosphate at 20℃; for 3h;
78%
With natural kaolinitic clay In tetrachloromethane for 2h; Ambient temperature;
78%
In dichloromethane at 25℃; for 1h;
78%
With 4-(3-methylimidazolium)butanesulfonate at 60℃; for 10h; Sealed tube; Green chemistry; chemoselective reaction;
General procedure for the synthesis of 3
General procedure: A mixture of corresponding alcohol (1, 1 mmol) and 3,4-dihydro-2H-pyran (2, 100 mg, 1.2 mmol) was stirred in the presence of zwitterionic-salt A (10 mg, 10 mol%) at 60-80 °C (oil bath) for 10-12 h in a seal tube. After completion of the reaction (TLC), the reaction mixture was cooled to room temperature and diluted with water (10 mL) and extracted with ethyl acetate (20 mL). Organic layer was dried over anhydrous Na2SO4. After evaporation of solvent the crude product was purified by column chromatography on silica gel using petroleum ether/ethyl acetate (4-5%) as eluent.
76%
Stage #1: 3,4-dihydro-2<i>H</i>-pyran With toluene-4-sulfonic acid at 20℃; for 0.166667h;
Stage #2: propargyl alcohol In neat (no solvent) at 20℃; for 0.166667h;
Stage #3: With sodium hydrogencarbonate In neat (no solvent) at 20℃; for 12h;
(A) Synthesis of 2-(prop-2-yn-1-yloxy)tetrahydro-2H-pyran Int-1
p-Toluenesulfonic acid monohydrate (0.57g, 0.03 mol%) was added to 3,4-dihydro-2H-pyrane(9.08 g, 1.08 equiv.) and the mixture was stirred for 10 minutes at room temperature. After 10minutes, propargyl alcohol (5.61 g, 1 equiv.) was dropwisely added and stirred for further 10minutes followed by addition of NaHCO3 (as solid) (0.63 g, 7.5 mol%). The reaction mixture wasstirred for additional 12 hours at room temperature. After 12 hours, the reaction mixture wasfiltered with EtOAc, and then the filtrate was evaporated under reduced pressure. The resultingmixture was purified with column chromatography over silica gel (hexane/EtOAc = 40:1, v/v) toafford 2-(prop-2-yn-1-yloxy)tetrahydro-2H-pyran Int-1 (76%) as a colorless liquid.
75%
Stage #1: 3,4-dihydro-2<i>H</i>-pyran With toluene-4-sulfonic acid for 0.166667h;
Stage #2: propargyl alcohol for 0.666667h;
Stage #3: With sodium hydrogencarbonate In dichloromethane at 20℃;
71%
With pyridinium p-toluenesulfonate In dichloromethane
70%
With toluene-4-sulfonic acid In tetrahydrofuran for 2h; Ambient temperature;
70%
With pyridinium p-toluenesulfonate In dichloromethane at 25℃; for 12h;
70%
Stage #1: propargyl alcohol With toluene-4-sulfonic acid In dichloromethane at 0℃; for 0.0833333h; Inert atmosphere;
Stage #2: 3,4-dihydro-2<i>H</i>-pyran In dichloromethane at 0 - 20℃; for 1h; Inert atmosphere;
68%
With cobalt(II) chloride In acetonitrile Ambient temperature;
68%
With ruthenium(III) acetate at 20℃; for 18h;
11%
With toluene-4-sulfonic acid In dichloromethane Ambient temperature;
With hydrogenchloride
With trichlorophosphate
70 g
In dichloromethane at 15℃; for 4.5h;
With toluene-4-sulfonic acid
With trichlorophosphate at 0℃;
With pyridine; toluene-4-sulfonic acid In dichloromethane
With toluene-4-sulfonic acid
With pyridinium p-toluenesulfonate In dichloromethane at 0℃;
With toluene-4-sulfonic acid
100 % Chromat.
With aminosulfonic acid at 15℃; for 2h;
94 g
With toluene-4-sulfonic acid In dichloromethane for 1h; cooling;
With hydrogenchloride at 20℃; for 2h;
With hydrogenchloride
With trichlorophosphate In dichloromethane; 4-(dicyanomethylene)-2-methyl-6-(p-dimethylaminostyryl)-4H-pyran
1 1.
1. 2-Prop-2-ynyloxytetrahydropyran A solution of phosphoryl chloride (0.5 ml in 10 ml of dichloromethane [DCM])) was added with care to a solution of prop-2-yn-1-ol (7.00 g, 125 mmol) and dihydropyran (10.50 g, 125 mmol) in DCM (80 ml). The reaction mixture was stirred at room temperature for 16 h, washed with sodium hydrogen carbonate solution (3 times), dried (MgSO4) and the solvent removed in vacuo to yield a colorless oil. The crude product was purified by column chromatography [silica gel, eluted with hexane/ethyl acetate (4:1), Rf: 0.43] to yield a colorless oil. Yield: 12.78 g, 92.29 mmol, 74%
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran at -78 - -70℃; for 0.75h;
Stage #2: 1,4-dibromo-butane With N,N,N,N,N,N-hexamethylphosphoric triamide In tetrahydrofuran at -78 - 30℃;
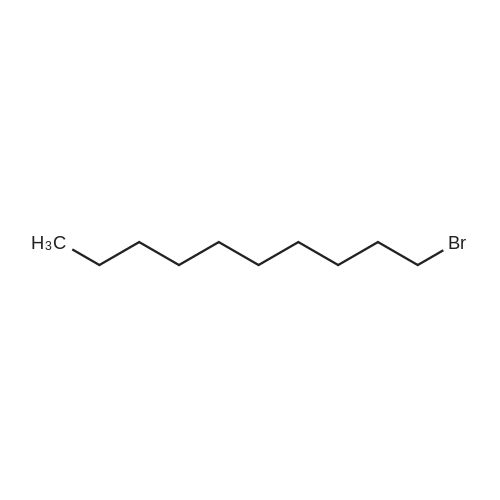
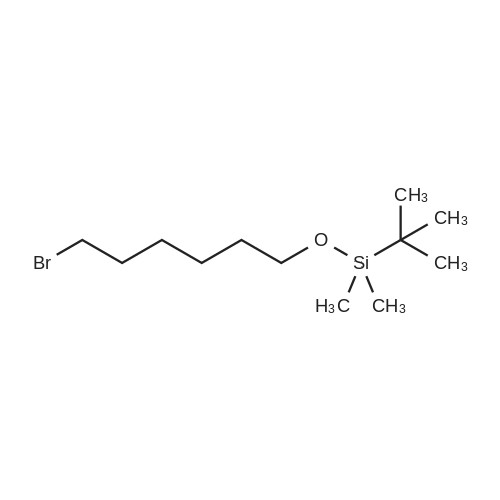
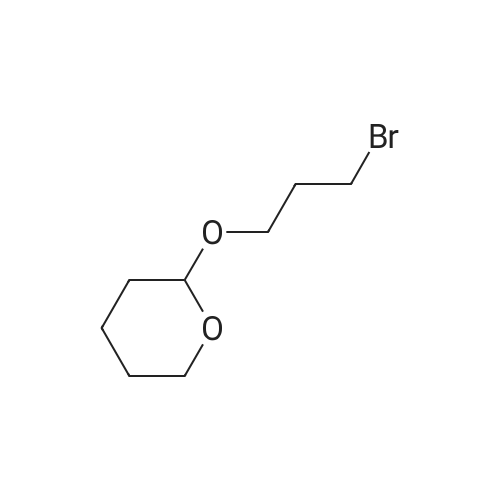
106 Example 106: Synthesis of 2-((7-bromohept-2-yn-1-yl)oxy)tetrahydro-2H-pyran
N-butyl lithium (12.6 mL) was cooled in a dry ice acetone bath at -78 °C.Add dropwise to piper (4g, 28.5mmol)In a solution of anhydrous tetrahydrofuran (40 mL),The internal temperature of the system does not exceed -70 ° C,Stir at this temperature for 45 minutes,Then 1,4-dibromobutane (18.5 g, 85.6 mmol)And hexamethylphosphoric triamide (8 mL)Anhydrous tetrahydrofuran (40 mL)The solution is added dropwise to the above reaction solution,Continue to stir at this temperature for 1 hour.The reaction system was then gradually warmed to room temperature and stirred overnight.The reaction was quenched with a saturated aqueous solution of ammonium chloride under ice bath.Dilute with water and extract with ethyl acetate (30 mL×3).Combine the organic phases and use water in turn,Washed with saturated saline,Separating the organic phase and concentratingThe residue was purified by silica gel column chromatography (EtOAc /EtOAc2-((7-Bromohept-2-yn-1-yl)oxy)tetrahydro-2H-pyran (7 g, yield: 87%)It is an oily liquid.
86%
With n-butyllithium In tetrahydrofuran; N,N,N,N,N,N-hexamethylphosphoric triamide; hexane 1.) -40 deg C; 2.) reflux, 30 h;
82%
With n-butyllithium; sodium iodide In tetrahydrofuran 1.) -80 deg C to room temperature, 2.) room temperature, 3 d;
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran at -40℃; for 2h; Inert atmosphere;
Stage #2: chloro-trimethyl-silane In tetrahydrofuran at -78 - 20℃; for 4h; Inert atmosphere;
94%
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran; hexane at 0℃; for 1h;
Stage #2: chloro-trimethyl-silane In tetrahydrofuran; hexane at -78 - 20℃;
87%
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran; hexane at -78℃; for 0.25h; Inert atmosphere;
Stage #2: chloro-trimethyl-silane In tetrahydrofuran; hexane at -78 - 20℃; for 4h; Inert atmosphere;
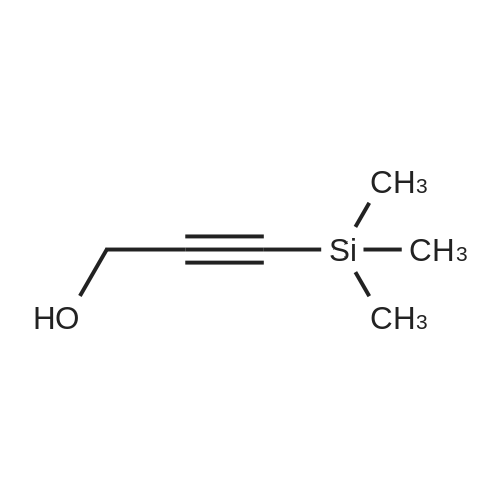
2-[(3-Trimethylsilylprop-2-yn-1-yl)oxy]tetrahydro-2H-pyran(5)28
To a solution of tetrahydro-2-(2-propynyloxy)-2Hpyran (4) (4.752 g, 33.94 mmol) in dry THF (34.0 mL) was added a 2.5 M solution of n-BuLi in hexanes (20.4 mL, 50.9 mmol) at -78 °C under N2. The mixture was stirred 15 min, and then TMSCl (5.529 g, 50.9 mmol) was added. The temperature was left to rise to rt and after 4 hr a 10 %aqueous solution of HCl (30 mL) was added at 0 °C. After separation of the phases, the aqueous phase was extracted with ethyl acetate (3 × 30 mL). The combined organiclayers were dried over Na2SO4. After concentration, the residue was purified by column chromatography (hexane/ethyl acetate 9:1) to afford compound 5 in 87 % yield (6.258 g, 29.52 mmol).
36%
With ethylmagnesium bromide In tetrahydrofuran 1.) 0 deg C, 10 min; r.t., 30 min, 2.) 50 deg C, 1 h;
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran; hexane at -78℃; for 2h; Inert atmosphere;
Stage #2: chloro-trimethyl-silane In tetrahydrofuran; hexane at -78 - 20℃; Inert atmosphere;
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran; hexane at -78℃; for 2.5h;
Stage #2: chloro-trimethyl-silane In tetrahydrofuran at -78 - 20℃; for 3.5h;
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In hexane at -78℃; for 2h; Inert atmosphere; Schlenk technique;
Stage #2: chloro-trimethyl-silane In hexane at -78 - 20℃; for 4h; Inert atmosphere; Schlenk technique;
(1)
General procedure: Schlenk tube was equipped with magnetic stirrer bar and purged with nitrogen. Freshly dryTHF (0.6 M) and 2-(prop-2-yn-1-yloxy)tetrahydro-2H-pyran Int-1 (1 equiv.) wassuccessively added to the Schlenk tube, and then cooled to -78. After cooling, n-BuLi (1.2equiv., 1.6 M in hexane) was dropwisely added followed by stirring for 2 hours. Thecorresponding silyl chloride (1.2 equiv.) was dropwisely added at -78, and then stirred forfurther 4 hours at room temperature. After 4 hours, the resulting mixture was quenched withwater and transferred to separate funnel. To the mixture was added sat. NH4Cl aqueous andbrine followed by extraction with EtOAc. The organic layer was dried over magnesiumsulfate (MgSO4) and evaporated under reduced pressure. The resulting mixture was purifiedwith column chromatography over silica gel (hexane/EtOAc = 40:1, v/v) to give thecorresponding THP-protected silylproparglic alcohol derivatives.
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran at -40℃; for 2h; Inert atmosphere;
Stage #2: triethylsilyl chloride In tetrahydrofuran at -78 - 20℃; for 4h; Inert atmosphere;
87.9%
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran; hexane at -78 - 20℃; for 1h; Inert atmosphere;
Stage #2: triethylsilyl chloride In tetrahydrofuran; hexane at 20℃; for 4h; Inert atmosphere;
Preparation of silyl alkyne bromides
O-THP(TES) Propargyl alcohol (19) Compound 19 was prepared according to a modified literature procedure.27 A solution of compound 18 (1.0g, 7.1mmol) in dry THF (20mL) was cooled to -78°C. Next, n-BuLi (3.0mL, 7.5mmol, 2.5M in hexane) was added dropwise and the reaction mixture was stirred under nitrogen atmosphere at -78°C for 1h before TES-Cl (1.3mL, 7.9mmol) was added. The reaction mixture was then allowed to warm up to RT and stirred under nitrogen atmosphere for an additional 4h before it was quenched by addition of saturated NH4Cl (15mL). The reaction mixture was extracted with Et2O (2×25mL), dried over MgSO4, filtered, and Et2O was carefully removed under reduced pressure with water bath kept at RT. The crude product was purified by column chromatography using (n-hexane to n-hexane/ Et2O 99/1) to afford 19 (1.6g, 87.9%) as a clear oil. Rf=0.42 (n-hexane/Et2O 95/5). 1H NMR (400MHz, CDCl3): δ=0.57-0.64 (m, 6H), 0.95-1.02 (m, 9H), 1.50-1.67 (m, 4H), 1.70-1.87 (m, 2H), 3.50-3.55 (m, 1H), 3.82-3.88 (m, 1H), 4.29 (s, 2H), 4.86 (t, J=3.4Hz, 1H). 13C NMR (100MHz, CDCl3): δ=4.3, 7.4, 19.1, 25.4, 30.3, 54.7, 62.0, 82.3, 96.5, 102.7. HRMS-ESI: m/z calcd for C14H26NaO2Si [M+Na]+, 277.1594; found, 277.1584. Although compound 8 has been also previously reported,28 no spectroscopic data of compound 8 are available
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran at -40℃; for 0.5h;
Stage #2: triethylsilyl chloride In tetrahydrofuran at 20℃;
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran at -78℃; for 1h;
Stage #2: triethylsilyl chloride In tetrahydrofuran at -78 - 20℃; for 1h; Further stages.;
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran; hexane at -78℃; for 1h; Inert atmosphere;
Stage #2: triethylsilyl chloride In tetrahydrofuran; hexane at -78 - 20℃; for 1h; Inert atmosphere;
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In hexane at -78℃; for 2h; Inert atmosphere; Schlenk technique;
Stage #2: triethylsilyl chloride In hexane at -78 - 20℃; for 4h; Inert atmosphere; Schlenk technique;
(1)
General procedure: Schlenk tube was equipped with magnetic stirrer bar and purged with nitrogen. Freshly dryTHF (0.6 M) and 2-(prop-2-yn-1-yloxy)tetrahydro-2H-pyran Int-1 (1 equiv.) wassuccessively added to the Schlenk tube, and then cooled to -78. After cooling, n-BuLi (1.2equiv., 1.6 M in hexane) was dropwisely added followed by stirring for 2 hours. Thecorresponding silyl chloride (1.2 equiv.) was dropwisely added at -78, and then stirred forfurther 4 hours at room temperature. After 4 hours, the resulting mixture was quenched withwater and transferred to separate funnel. To the mixture was added sat. NH4Cl aqueous andbrine followed by extraction with EtOAc. The organic layer was dried over magnesiumsulfate (MgSO4) and evaporated under reduced pressure. The resulting mixture was purifiedwith column chromatography over silica gel (hexane/EtOAc = 40:1, v/v) to give thecorresponding THP-protected silylproparglic alcohol derivatives.
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With N,N,N,N,N,N-hexamethylphosphoric triamide; n-butyllithium In tetrahydrofuran at -78℃; for 0.5h; Inert atmosphere;
Stage #2: pentadecyl bromide In tetrahydrofuran at 20℃; for 5h; Inert atmosphere;
1.1; 2.1 Example 2,
1) Add 1.2g of compound A-0 into a dry 100ml four-necked flask, add 10ml of anhydrous THF, under the protection of argon, reduce to -78, add 4.8ml of n-butyllithium, and 2.5ml of hexamethylphosphorus Triamide; After the addition is complete, stir at -78°C for 0.5h; then add 1-bromopentadecane in tetrahydrofuran; react at room temperature for 5h; after quenching with water, concentrate, wash and extract the crude compound Purified by column chromatography to obtain compound A-2a (2.22 g, yield 84%)
76%
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran; hexane at -78℃; for 0.5h;
Stage #2: pentadecyl bromide With N,N,N,N,N,N-hexamethylphosphoric triamide In tetrahydrofuran; hexane at -78℃; for 24h;
2.9. Synthesis of 2-(2-alkynyloxy)-tetrahydro-2H-pyrancompounds
General procedure: To a stirred solution of tetrahydro-2-(2-proponyloxy)-2H-pyran(7.13 mmol) in dry THF (20 mL), n-BuLi (2.5 M, 17.83 mmol) in dryhexane (7.10 mL) was added dropwise while keeping the temperatureat-78 C. After 30 min, HMPA (5.0 mL) and 1-bromotridecane(7.13 mmol) or 1-bromopentadecane (7.13 mmol) was added dropwiseto the reaction mixture while maintaining the temperature at78 C. After 24 h, the reaction mixture was worked up by pouring alarge volume of water and extracting with diethyl ether (2×20 mL).The organic layer was washed with brine (1×20 mL) before drying(MgSO4). Filtration, rotoevaporation of the solvent and columnpurification with hexane-diethyl ether (9:1, v/v) solution afforded1.38 g (60% yield) of 2-(2-hexadecynyloxy)-tetrahydro-2H-pyranand 1.89 g (76% yield) of 2-(2-octadecynyloxy)-tetrahydro-2Hpyran. 2-(2-Octadecynyloxy)-tetrahydro-2H-pyran was obtained in a76% yield as colorless oil from the reaction 1.00 mL of tetrahydro-2-(2-propynoloxy)-2H-pyran (1.00 g, 7.13 mmol) and 2.00 mL of1-bromopentadecane (7.13 mmol) according to the general proceduredescribed above. IR (neat) max: 2925, 2854, 2222, 1466,1345, 1201, 1132, 1118, 1079, 1053, 1025 cm-1; 1H NMR (300 MHz,CDCl3) 4.81 (1H, t, J = 3.27 Hz), 4.24 (2H, m), 3.84 (1H, m), 3.52(1H, m), 2.20 (2H, m); 1.89-1.25 (32H, m), 0.87 (3H, t, J = 6.69 Hz);13C NMR (75 MHz) 96.57, 86.77 (s, C-3), 75.64 (s, C-2), 61.95,54.62 (t, C-1), 31.90, 30.27, 29.67 (×3), 29.61, 29.52, 29.348, 29.12,28.87, 28.75, 28.59, 28.16, 25.36, 22.67, 19.10, 18.80, 14.10 (q, C-18).GC-MS (70 eV) m/z (relative intensity); 350 (M+, 0.55), 295 (0.68),279 (1), 209 (2), 149 (5), 135 (14), 121 (17), 111 (39), 95 (59), 85(100), 81 (55), 77 (9), 67 (47), 55 (81).
63%
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With N,N,N,N,N,N-hexamethylphosphoric triamide; n-butyllithium In tetrahydrofuran at -78℃; for 1h; Inert atmosphere;
Stage #2: pentadecyl bromide In tetrahydrofuran at -78 - 20℃; Inert atmosphere;
25.A A. Synthesis of 2-octadec-2-ynoxytetrahydropyran (NP-PD-023)
To a 250 mL oven-dried flask equipped with a stir bar was added 2-(2- propynyloxy)tetrahydro-2H-pyran (5.02 mL, 35.7 mmol, 1.00 eq), hexamethylphosphoramide (21.7 mL, 125 mmol, 3.50 eq) and THF (100 mL). The reaction mixture was cooled to -78 °C and stirred vigorously under Ar. n-Butyllithium (2.00 M in THF, 23.2 mL, 46.4 mmol, 1.30 eq) was added dropwise over a period of 15 minutes by way of an oven-dried pressure equalizing dropping funnel and the reaction was vigorously stirred at -78 °C for approximately 1 hour. Subsequently, 1-bromopentadecane (13.4 mL, 46.4 mmol, 1.30 eq) was added slowly dropwise over 10 minutes by way of a pressure equalizing dropping funnel after which the resulting reaction mixture was allowed to warm to room temperature overnight while stirring vigorously under Ar. The following day TLC indicated that all of the starting alkyne had been consumed. The reaction mixture was quenched with a saturated solution of ammonium chloride and then extracted three times into EtOAc. The organic phases were then combined, washed with brine, dried over anhydrous magnesium sulfate and concentrated in vacuo. Subsequent purification of the resulting crude material by column chromatography (2%-10% EtOAc/hexane) afforded a clear oil (7.86 g, 22.4 mmol, 63% yield).
63%
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran; hexane at -78 - -30℃; for 0.916667h; Inert atmosphere;
Stage #2: pentadecyl bromide In tetrahydrofuran; hexane at -30 - 20℃; Inert atmosphere;
With N,N,N,N,N,N-hexamethylphosphoric triamide; n-butyllithium In tetrahydrofuran; hexane 1.) -70 deg C, 1 h, 2.) -50 deg C, 5 h;
With N,N,N,N,N,N-hexamethylphosphoric triamide; n-butyllithium 1.) THF; Multistep reaction;
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran; hexane at -78℃; for 1h; Inert atmosphere;
Stage #2: chloroformic acid ethyl ester In tetrahydrofuran; hexane at -78 - -10℃; for 2h; Inert atmosphere;
86%
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran; hexane at -78℃; for 0.333333h;
Stage #2: chloroformic acid ethyl ester In tetrahydrofuran; hexane at -78 - -10℃; for 1.75h; Further stages.;
85%
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In diethyl ether; hexane at -78℃; for 0.5h;
Stage #2: chloroformic acid ethyl ester In diethyl ether; hexane at -78 - 20℃;
75%
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In diethyl ether at -78℃; for 1.6h;
Stage #2: chloroformic acid ethyl ester In diethyl ether at -78 - 20℃; for 20h;
31.1
Step 1 : 4-Tetrahydro-pyran-2-yloxy-but-2-ynoic acid ethyl ester To a solution of tetrahydro-2-(2-propynloxy)-2H-pyran (17.44 g, 124.4 mmol) in 400 ml of anhydrous ethyl ether cooled to -78°C was added a 2.5N n-butyl lithium solution (52.3 ml, 130.7 mmol) dropwise over 10 minutes. The mixture was stirred for 1.5 hours at this temperature. To this mixture was added a solution of ethyl chloro formate (20.4 g, 188 mmol) in 25 ml of ether dropwise over 15 minutes. The mixture was stirred at this temperature for 2 hours and was allowed to stir at room temperature for 18 hours. The mixture was added to aqueous ammonium chloride and was extracted with ethyl acetate (50 ml). The organic layer was washed with water, dried over magnesium sulfate and filtered. The concentrated filtrate was purified by flash chromatography using 0-10% ethyl acetate/heptane to afford the title compound (19.7 g, 75%).
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran; hexane at -78℃; Inert atmosphere;
Stage #2: chloroformic acid ethyl ester In tetrahydrofuran; hexane at -78 - 20℃; Inert atmosphere;
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran
Stage #2: chloroformic acid ethyl ester
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In hexane at -78℃; Inert atmosphere;
Stage #2: chloroformic acid ethyl ester In hexane at -78 - 20℃;
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In diethyl ether at -78℃; for 1h; Inert atmosphere;
Stage #2: chloroformic acid ethyl ester In diethyl ether at -78℃; for 1h; Inert atmosphere;
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran; hexane at -70℃; for 1h; Inert atmosphere;
Stage #2: methyl chloroformate In tetrahydrofuran; hexane at -70℃; for 2h; Inert atmosphere;
s11.1 Step 1: Synthesis of methyl 4-(oxan-2-yloxy)but-2-ynoate.
To a solution of 2-(prop-2-yn-1-yloxy)oxane (6.0 g, 43.37 mmol) in THF (80.0 mL) was added n-BuLi in hexanes (18.0 mL, 2.5 mol/L) at -70°C under N2. The mixture was stirred at -70°C for 1 h. Then methyl chloroformate (6.30 g, 66.67 mmol) was added dropwise to the mixture at -70°C. The mixture was stirred at -70°C for another 2 h. After the reaction was completed, the reaction mixture was quenched with NH4Cl and extracted with ethyl acetate. The combined organic layer was washed with brine, dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated under vacuum. The residue was purified by flash column chromatography with petroleum ether/ethyl acetate (82/18, v/v) to afford methyl 4-(oxan-2-yloxy)but-2-ynoate (7.4 g, 86%) as a colorless oil.
86%
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran; hexane at -70℃; for 1h; Inert atmosphere;
Stage #2: methyl chloroformate In tetrahydrofuran; hexane at -70℃; for 2h; Inert atmosphere;
s11.1 Step 1: Synthesis of methyl 4-(oxan-2-yloxy)but-2-ynoate.
To a solution of 2-(prop-2-yn-1-yloxy)oxane (6.0 g, 43.37 mmol) in THF (80.0 mL) was added n-BuLi in hexanes (18.0 mL, 2.5 mol/L) at -70°C under N2. The mixture was stirred at -70°C for 1 h. Then methyl chloroformate (6.30 g, 66.67 mmol) was added dropwise to the mixture at -70°C. The mixture was stirred at -70°C for another 2 h. After the reaction was completed, the reaction mixture was quenched with NH4Cl and extracted with ethyl acetate. The combined organic layer was washed with brine, dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated under vacuum. The residue was purified by flash column chromatography with petroleum ether/ethyl acetate (82/18, v/v) to afford methyl 4-(oxan-2-yloxy)but-2-ynoate (7.4 g, 86%) as a colorless oil.
84%
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran; hexane at -78℃; for 0.75h; Inert atmosphere;
Stage #2: methyl chloroformate In tetrahydrofuran; hexane at -78℃; for 2h; Inert atmosphere;
80%
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With methylmagnesium chloride In tetrahydrofuran
Stage #2: methyl chloroformate
68%
With n-butyllithium In tetrahydrofuran; hexane at -78℃; for 1h;
With n-butyllithium 1) THF, hexane <-70 deg C to -20 deg C, 1 h, 2) <-70 deg C to r.t., 3) r.t., overnight; Yield given. Multistep reaction;
With n-butyllithium 1.) hexane, THF, -78 deg C, 1 h, 2.) hexane, THF, a) -78 deg C, 2 h, b) RT, 2 h; Yield given. Multistep reaction;
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran; hexane at -78℃; for 1h;
Stage #2: methyl chloroformate In tetrahydrofuran; hexane at -78 - -10℃; for 2h;
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium
Stage #2: methyl chloroformate
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran; hexane at -78℃; for 0.833333h; Inert atmosphere;
Stage #2: methyl chloroformate In tetrahydrofuran; hexane at 20℃; for 2h; Inert atmosphere;
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With ethynylmagnesium bromide In tetrahydrofuran; diethyl ether for 0.2h; Inert atmosphere;
Stage #2: methyl chloroformate In tetrahydrofuran; diethyl ether at -20 - 0℃; for 3.5h; Inert atmosphere;
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In diethyl ether at -78℃; for 1h; Inert atmosphere;
Stage #2: methyl chloroformate In diethyl ether at -78℃; for 1h; Inert atmosphere;
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran; N,N,N,N,N,N-hexamethylphosphoric triamide at -78℃;
Stage #2: 1-bromo dodecane In tetrahydrofuran
86%
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium; N,N,N,N,-tetramethylethylenediamine In tetrahydrofuran at -78 - 20℃; for 1h; Inert atmosphere;
Stage #2: 1-bromo dodecane With potassium iodide In tetrahydrofuran at 80℃; for 8h; Inert atmosphere;
Preparation of 2-(tridec-2-ynyloxy)-tetrahydro-2H-pyran (17).
BuLi (3.94 mL, 8.73 mmol) was slowly added into a solution of 16 (1.11 g, 7.93 mmol) in THF (15 mL) at -78 ºC and followed by addition of TMEDA (1.80 g, 15.9 mmol). The reaction solution was warmed to rt and stirred for 1 h. 1-Bromodecane (2.46 mL, 11.9 mmol) was added dropwise at rt and followed by KI (132 mg, 0.793 mmol). The reaction mixture was heated at 80 ºC for 8 h then quenched with aqueous Na2CO3 (10 %). The resulted mixture was diluted with diethyl ether, and washed with aqueous Na2CO3 (10 %). The aqueous portion was back extracted with diethyl ether. The organic portions were combined, washed with brine, dried over Na2SO4, and concentrated to a brown liquid, which was purified by column chromatography (ethyl acetate / hexane, 1 : 9) to gave compound 17 (1.91 g, 86 %) as a colorless liquid. 1H NMR (400 MHz, CDCl3) δ 4.81 (t, J = 3.2 Hz, 1 H), 4.29 (dt, J = 15.2, 2.0 Hz, 1 H), 4.20 (dt, J = 15.2, 2.0 Hz, 1 H), 3.85 (m, 1 H), 3.53 (m, 1 H), 2.21 (tt, J =7.2, 2.4 Hz, 2 H), 1.84 (m, 1 H), 1.73 (m, 1 H), 1.65 (m, 1 H), 1.61 (m, 1 H), 1.50 (m, 4 H), 1.36 (m, 2 H), 1.26 (m, 12 H), 0.88 (t, J = 6.4 Hz, 3 H). GC: 26.78 min.
83%
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran; hexane at -78 - 20℃;
Stage #2: 1-bromo dodecane With sodium iodide In tetrahydrofuran for 40h; Heating;
82%
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran; pentane at -50℃; for 1h;
Stage #2: 1-bromo dodecane With N,N,N,N,N,N-hexamethylphosphoric triamide In tetrahydrofuran; pentane at -50 - 20℃; for 5.5h;
65%
With n-butyllithium In tetrahydrofuran; hexane; dimethyl sulfoxide at 0 - 20℃; for 3.66667h; Inert atmosphere;
With n-butyllithium
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran at -78℃; for 0.25h; Inert atmosphere;
Stage #2: 1-bromo dodecane In tetrahydrofuran; N,N,N,N,N,N-hexamethylphosphoric triamide at -78 - 20℃; Inert atmosphere;
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran at -78℃; for 0.5h; Inert atmosphere;
Stage #2: bromoundecane With 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone In tetrahydrofuran at -78 - 20℃; for 24h; Inert atmosphere;
3-6 Example 3
Under the protection of argon, add 0.19173 mol of compound 2 and 250 mL of dry tetrahydrofuran into a 1000 mLthree-necked flask, and cool to -78°C. Add 71mL of n-BuLi solution with a concentration of 2.7mol/L dropwise, stir for half an hour after the addition is complete, add 0.1743mol 1-bromoundecane dropwise successively at -78, 40g 1,3-dimethyl propylene urea, after the dripping is completed, slowly rise to room temperature and stir for 24h, the gas chromatograph monitors to the end of the reaction. After the reaction is complete, the reaction was quenched by adding saturated aqueous ammonium chloride solution, and the organic phase was extracted with ethyl acetate. After washing with water, brine, drying with anhydrous sodium sulfate, and purification by column chromatography, compound 3 was obtained with a yield of 80%.
63%
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With N,N,N,N,N,N-hexamethylphosphoric triamide; n-butyllithium In tetrahydrofuran at -78℃; for 1h; Inert atmosphere;
Stage #2: bromoundecane In tetrahydrofuran at -78 - 20℃; Inert atmosphere;
29.A A. Synthesis of 2-tetradec-2-ynoxytetrahydropyran (NP-PD-121)
To a 250 mL oven-dried flask equipped with a stir bar was added 2-(2- propynyloxy)tetrahydro-2H-pyran (5.02 mL, 35.7 mmol, 1.00 eq), 1,3-dimethyl-3,4,5,6- tetrahydro-2(1H)-pyrimidinone (12.9 mL, 107 mmol, 3.00 eq) and THF (100 mL). The reaction mixture was cooled to -78 °C and stirred vigorously under Ar. n-Butyllithium (2.30 M in THF, 20.2 mL, 46.4 mmol, 1.30 eq) was added dropwise over a period of 15 minutes by way of an oven-dried pressure equalizing dropping funnel and the reaction was vigorously stirred at -78 °C for approximately 1 hour. Subsequently, 1-bromoundecane (10.4 mL, 46.4 mmol, 1.30 eq) was added slowly dropwise over 10 minutes by way of a pressure equalizing dropping funnel after which the resulting reaction mixture was allowed to warm to room temperature overnight while stirring vigorously under Ar. The following day TLC indicated that all of the starting alkyne had been consumed. The reaction mixture was quenched with a saturated solution of ammonium chloride and then extracted three times into EtOAc. The organic phases were then combined, washed with brine, dried over anhydrous magnesium sulfate and concentrated in vacuo. Subsequent purification of the resulting crude material by column chromatography (2%-10% EtOAc/hexane) afforded a clear oil (6.64 g, 22.5 mmol, 63%).
63%
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran; hexane at -78 - -30℃; for 0.916667h; Inert atmosphere;
Stage #2: bromoundecane In tetrahydrofuran; hexane at -30 - 20℃; Inert atmosphere;
With N,N,N,N,N,N-hexamethylphosphoric triamide; n-butyllithium 1.) THF; Multistep reaction;
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran at -78℃; for 0.25h; Inert atmosphere;
Stage #2: bromoundecane In tetrahydrofuran; N,N,N,N,N,N-hexamethylphosphoric triamide at -78 - 20℃; Inert atmosphere;
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone; n-butyllithium In tetrahydrofuran at -78℃; for 1.5h;
Stage #2: 1-bromotridecane In tetrahydrofuran at 20℃; for 21h;
5.A A. Synthesis of2-(hexadec-2-yn-1-yloxy)tetrahydro-2H-pyran (MD-5-67)
In a 250 mL three neck round bottom flask, to a mixture of 2-(2-propynyloxy)tetrahydro- 2H-pyran (2.0 mL, 14.2 mmol, 1.0 eq) and 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (5.1 mL, 42.8 mmol, 3.0 eq) was added anhydrous THF (40 mL) and cooled to -78 oC. After 30 min, n-butyllithium (2.5 M in hexanes, 7.4 mL, 18.5 mmol, 1.3 eq) was added dropwise via a syringe pump at a rate of 20 mL/hr. The mixture was stirred for 1 h and then added 1- bromotridecane (4.7 mL, 18.5 mmol, 1.3 eq) dropwise over 10 min. The reaction was allowed to warm to room temperature and stirred overnight. After 21 h, TLC analysis indicated complete conversion to product (Rf = 0.43, 7.5% EtOAc/hexanes, PMA stain). The reaction mixture was quenched by the addition of saturated ammonium chloride and extracted three times with EtOAc. The combined organic layers were dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure to give a crude oil. Purification by silica gel chromatography eluting with 0-5% EtOAc/hexanes yielded product as an oil (3.5 g, 10.8 mmol, 76%).
76%
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran; hexane at -78 - -30℃; for 0.916667h; Inert atmosphere;
Stage #2: 1-bromotridecane In tetrahydrofuran; hexane at -30 - 20℃; Inert atmosphere;
60%
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran; hexane at -78℃; for 0.5h;
Stage #2: 1-bromotridecane With N,N,N,N,N,N-hexamethylphosphoric triamide In tetrahydrofuran; hexane at -78℃; for 24h;
2.9. Synthesis of 2-(2-alkynyloxy)-tetrahydro-2H-pyrancompounds
General procedure: To a stirred solution of tetrahydro-2-(2-proponyloxy)-2H-pyran(7.13 mmol) in dry THF (20 mL), n-BuLi (2.5 M, 17.83 mmol) in dryhexane (7.10 mL) was added dropwise while keeping the temperatureat-78 C. After 30 min, HMPA (5.0 mL) and 1-bromotridecane(7.13 mmol) or 1-bromopentadecane (7.13 mmol) was added dropwiseto the reaction mixture while maintaining the temperature at78 C. After 24 h, the reaction mixture was worked up by pouring alarge volume of water and extracting with diethyl ether (2×20 mL).The organic layer was washed with brine (1×20 mL) before drying(MgSO4). Filtration, rotoevaporation of the solvent and columnpurification with hexane-diethyl ether (9:1, v/v) solution afforded1.38 g (60% yield) of 2-(2-hexadecynyloxy)-tetrahydro-2H-pyranand 1.89 g (76% yield) of 2-(2-octadecynyloxy)-tetrahydro-2Hpyran. 2-(2-Hexadecynyloxy)-tetrahydro-2H-pyran was obtained ina 60% yield as colorless oil from the reaction of 1.00 mL of tetrahydro-2-(2-propynoloxy)-2H-pyran (1.00 g, 7.13 mmol) and1.82 mL of 1-bromotridecane (7.13 mmol) according to the generalprocedure described above. IR (neat) max: 2926, 2854, 2225, 1466,1345, 1202, 1118, 1133, 1079, 1054, 1025 cm-1; 1H NMR (300 MHz,CDCl3) 4.81 (1H, t, J = 3.3 Hz), 4.24 (2H, m), 3.84 (1H, m); 3.52 (1H,m), 2.20 (2H, m); 1.89-1.25 (33H, m), 0.87 (3H, t, J = 6.4 Hz); 13CNMR (75 MHz) 96.58, 86.78 (s, C-3), 75.64 (s, C-2), 61.96, 54.62(t, C-1), 31.90, 30.27, 29.64 (×3), 29.61, 29.52, 29.34, 29.12, 28.87,28.58, 25.36, 22.67, 19.11, 18.80, 14.10 (q, C-16). GC-MS (70 eV)m/z (relative intensity); 322 (M+, 0.11), 279 (0.13), 209 (0.55), 149(2), 135 (4), 121 (7), 111 (17), 95 (46), 85 (100), 81 (49), 77 (11), 67(57), 55 (90).
With N,N,N,N,N,N-hexamethylphosphoric triamide; n-butyllithium 1.) THF; Multistep reaction;
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran; hexane at 0℃;
Stage #2: 1-bromotridecane In tetrahydrofuran; 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone; hexane at 40℃; for 24h;
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran; hexane at -78℃; for 1h;
Stage #2: (R)-propylene oxide With boron trifluoride diethyl etherate In tetrahydrofuran; hexane at -78℃; for 1h; Further stages.;
75%
With n-butyllithium In tetrahydrofuran; N,N,N,N,N,N-hexamethylphosphoric triamide at -20℃; for 0.5h;
75%
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran; hexane for 0.5h; Inert atmosphere;
Stage #2: (R)-propylene oxide With boron trifluoride diethyl etherate In tetrahydrofuran; hexane at -78℃; Inert atmosphere; regioselective reaction;
Compound 4
To a cooled (-780C ) solution of 2-prop-2-ynyloxy-tetrahydro-pyran (2 g, 14.28 mmol) in dry THF (20 mL) under N2 was added n-BuLi (11.77 mL, 14.21mmol, 1.6 N hexane solution) drop wise and stirred for 30 min. The reaction mixture was sequentially reacted with BF3.OEt2 (1.76 mL, 14.23 mmol) and a solution of (R)-propylene oxide 3(0.91g, 15.71 mmol) in dry THF (2 mL) after10 min interval and stirred for an additional 3 h at -780C. After completion of the reaction as noticed by TLC, the reaction was quenched with saturated NaHCO3 solution (5 mL) followed by saturated NH4Cl solution (5 mL) and extracted with EtOAc (3 x 10 mL), washed with water (1 x 5 mL), dried over Na2SO4, solvent was removed under reduced pressure. The residue thus obtained was purified by column chromatography on silica gel (2/8 v/v ethyl acetate/hexane) to yield pure compound as a yellow syrup 4 (2.11g, 75%). [α]D25 = -2.0 (c 0.3, CHCl3). IR (Neat): υ 3447, 2930, 2857, 1465, 1381 cm-1. 1HNMR (300MHz, CDCl3): δ 4.79-4.73 (m, 1H ), 4.21(d, J= 6.0, 2H), 3.93-3.98 (m, 1H), 3.45-3.40 (m, 2H), 2.40-2.25(m, 2H), 1.90-1.45 (m, 6H) 1.22 (d, J=6.0, 3H). 13CNMR (75MHz, CDCl3): δ 96.8, 82.9, 78.3, 66.1, 61.9, 54.5, 30.2, 29.2, 25.2, 22.2, 18.9; EIMS: m/z 198 [M]+.
60%
With lithium amide In diethyl ether; ammonia at -70℃; for 72h;
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran at -40℃; for 2h; Inert atmosphere;
Stage #2: triisopropylsilyl chloride In tetrahydrofuran at -78 - 20℃; for 4h; Inert atmosphere;
85.9%
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran; hexane at -78℃; for 1h; Inert atmosphere;
Stage #2: triisopropylsilyl chloride In tetrahydrofuran; hexane at -78 - 20℃; for 4h; Inert atmosphere;
Preparation of silyl alkyne bromides
O-THP(TIPS) Propargyl alcohol (20) Compound 20 was prepared according to a modified literature procedure.26 A solution of compound 18 (1.0g, 7.1mmol) in dry THF (20mL) was cooled to -78°C. Next, n-BuLi (3.0mL, 7.5mmol, 2.5M in hexane) was added dropwise and the reaction mixture was stirred under nitrogen atmosphere at -78°C for 1h before TIPS-Cl (1.7mL, 7.9mmol) was added. The reaction mixture was then allowed to warm up to RT and stirred under nitrogen atmosphere for additional 4h before it was quenched by addition of saturated NH4Cl (15mL). The reaction mixture was extracted with Et2O (2×25mL), dried over MgSO4, filtered, and Et2O was carefully removed under reduced pressure with water bath kept at RT. The crude product was purified by column chromatography (n-hexane to n-hexane/ Et2O 99/1) to afford 20 (1.8g, 85.9%) as a clear oil. Rf=0.44 (n-hexane/Et2O 95/5). 1H NMR (400MHz, CDCl3): δ=1.07 (m, 21H), 1.51-1.65 (m, 4H), 1.71-1.87 (m, 2H), 3.50-3.55 (m, 1H), 3.83-3.89 (m, 1H), 4.31 (ddd, J=2.4Hz, 6.3Hz, 16.1Hz, 2H), 4.91 (t, J=3.4Hz, 1H). 13C NMR (100MHz, CDCl3): δ=11.2, 18.6, 19.2, 25.4, 30.4, 54.7, 62.2, 87.0, 96.3, 103.4. HRMS-ESI: m/z calcd for C17H32NaO2Si [M+Na]+, 319.2064; found, 319.2049. This compound has been also previously reported.27
84%
With n-butyllithium In tetrahydrofuran
79%
With n-butyllithium In tetrahydrofuran; hexane 1.) -78 deg C, 10 min; -78 to -20 deg C, 2.) -78 deg C, 20 min; 0 deg C, 5 h;
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran; hexane at -78℃; for 2h; Inert atmosphere;
Stage #2: triisopropylsilyl chloride In tetrahydrofuran; hexane at -78 - 20℃; Inert atmosphere;
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran at -78℃; for 1h;
Stage #2: triisopropylsilyl chloride In tetrahydrofuran at -78 - 20℃; for 2h;
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran; hexane at -78℃; for 2.5h;
Stage #2: triisopropylsilyl chloride In tetrahydrofuran at -78 - 20℃; for 3.5h;
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In hexane at -78℃; for 2h; Inert atmosphere; Schlenk technique;
Stage #2: triisopropylsilyl chloride In hexane at -78 - 20℃; for 4h; Inert atmosphere; Schlenk technique;
(1)
General procedure: Schlenk tube was equipped with magnetic stirrer bar and purged with nitrogen. Freshly dryTHF (0.6 M) and 2-(prop-2-yn-1-yloxy)tetrahydro-2H-pyran Int-1 (1 equiv.) wassuccessively added to the Schlenk tube, and then cooled to -78. After cooling, n-BuLi (1.2equiv., 1.6 M in hexane) was dropwisely added followed by stirring for 2 hours. Thecorresponding silyl chloride (1.2 equiv.) was dropwisely added at -78, and then stirred forfurther 4 hours at room temperature. After 4 hours, the resulting mixture was quenched withwater and transferred to separate funnel. To the mixture was added sat. NH4Cl aqueous andbrine followed by extraction with EtOAc. The organic layer was dried over magnesiumsulfate (MgSO4) and evaporated under reduced pressure. The resulting mixture was purifiedwith column chromatography over silica gel (hexane/EtOAc = 40:1, v/v) to give thecorresponding THP-protected silylproparglic alcohol derivatives.
With N,N,N,N,N,N-hexamethylphosphoric triamide; n-butyllithium In tetrahydrofuran at 0℃; for 1h;
87%
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran; hexane at -10℃; for 0.5h;
Stage #2: With N,N,N,N,N,N-hexamethylphosphoric triamide In tetrahydrofuran; hexane at -10℃; for 0.5h;
Stage #3: 1-Bromotetradecane In tetrahydrofuran; hexane at -10℃; for 0.5h;
4.25. 2-(Heptadec-2-ynyloxy)tetrahydro-2H-pyran (30)
General procedure: At -10 °C, a solution of the THP ether of the propargyl alcohol (1 g, 7.1 mmol) in THF (10 mL) was treated with n-BuLi (3.66 mL, 8.6 mmol) (2.34 M in hexane) and stirred for 30 min. HMPA (1.53 mL, 8.6 mmol) was added and the reaction mixture was stirred at -10 C for another 30 min. Myristyl bromide (2.37 g, 8.6 mmol) was dissolved in THF (20 mL) and stirred at -10 °C to which the solution of alkynyl lithium in THF was added and stirred for further 30 min. The reaction mixture was quenched with saturated NH4Cl. The organic layer was separated and the aqueous layer was washed with ethyl acetate, the combined organic layers were washed with ethyl acetate, brine, dried, and concentrated. Purification of the crude product by column chromatography (90:10 petroleum ether/EtOAc) afforded 30 (2.1 g, 87%) as colorless oil:
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran; hexane at -78℃; for 1h; Inert atmosphere;
Stage #2: 1-Iodooctane In tetrahydrofuran; hexane at -78 - 20℃; for 2h; Inert atmosphere;
Preparation of Alkyne 33
To the solution of THP ether32(6.3 g, 45 mmol) in anhydrous THF (135 mL) cooled at -78oC was addedn-BuLi (45 mL, 45 mmol, 1 M in Hexane) and stirred for 1 h at the same temperature. The solution of octyl iodide (10.8 g, 45 mmol) in HMPA (8.0 mL) was added and the mixture stirred for 2 h allowing the temperature to raise to rt. The mixture was cooled to 0oC, then quenched with aq saturated NH4Cl (30 mL). The aq layer was separated and extracted with ether (3x50 mL). The combined organic layers were washed with brine (25 mL), dried over Na2SO4, solvent was evaporated under reduced pressure to afford crude ether which was purified by column chromatography using 2% EtOAc/hexanes (v/v) as the eluent to yield pure33as an oil (9.6 g, 38 mmol) in 85% yield, TLC, Rf0.61 (5% EtOAc/Hexanes);1H NMR (200 MHz, CDCl3) δ 4.78 (t,J =2.8 Hz, 1H), 4.21 (td,J =4.4, 2.2 Hz, 2H), 3.87-3.74 (m, 1H), 3.56-3.44 (m, 1H), 2.25-2.15 (m, 6H), 1.75-1.24 (m, 14H), 0.90 (t,J =6.9 Hz, 3H).
With N,N,N,N,N,N-hexamethylphosphoric triamide; n-butyllithium In tetrahydrofuran at 0℃; for 1h;
(pentadec-2-yn-1-yloxy)tetrahydro-2H-pyran[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
80%
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With N,N,N,N,N,N-hexamethylphosphoric triamide; n-butyllithium In tetrahydrofuran; hexane at -78 - -30℃; for 0.75h; Inert atmosphere;
Stage #2: 1-Iodododecane In tetrahydrofuran at -30 - 20℃; Inert atmosphere;
1.A
2-(2-Propynyloxy)tetrahydro-2H-pyran (4.77 g, 34.0 mmol, 1.00 eq) was added to a 250 mL oven-dried flame-dried flask equipped with a stir bar, diluted with 40 mL THF and hexamethylphosphoramide (20.7 mL, 119 mmol, 3.50 eq), cooled to -78 °C, and stirred vigorously under Ar. After addition of n-butyllithium (2.5 M in hexane, 15.0 mL, 34.0 mmol, 1.00 eq) dropwise via syringe pump at a rate of 17 mL/hr, the resulting reaction mixture was stirred vigorously at -78 °C under Ar for 10 min, before warming to -30 °C and stirring for an additional 45 min. After addition of a solution of 1-iodododecane (10.1 g, 34.0 mmol, 1.00 eq) in 14 mL THF dropwise via syringe pump at a rate of 17 mL/hr, the resulting reaction mixture was allowed to slowly warm to room temperature and was stirred vigorously under Ar overnight. In the next morning, TLC indicated complete conversion of alkyl iodide and alkyne starting materials to one spot (stained with PMA). The reaction was quenched dropwise with saturated ammonium chloride, and the resulting aqueous layer was extracted 3 times with EtOAc. Combined organic layers were dried over anhydrous sodium sulfate, filtered, and evaporated under reduced pressure to yield 22.6 g of a yellow oil, which was purified via column chromatography eluting with 1:12 EtOAc to hexanes to yield a slightly yellow oil (8.40 g, 27.2 mmol, 80% yield).
80%
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran; hexane at -78 - -30℃; for 0.916667h; Inert atmosphere;
Stage #2: 1-Iodododecane In tetrahydrofuran; hexane at -30 - 20℃; Inert atmosphere;
With N,N,N,N,N,N-hexamethylphosphoric triamide; n-butyllithium In tetrahydrofuran at 0℃; for 1h;
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With N,N,N,N,N,N-hexamethylphosphoric triamide; n-butyllithium In tetrahydrofuran at -78℃; for 1h; Inert atmosphere;
Stage #2: hexadecanyl bromide In tetrahydrofuran at -78 - 20℃; Inert atmosphere;
27.A A. Synthesis of 2-nonadec-2-ynoxytetrahydropyran (NP-PD-092)
To a 250 mL oven-dried flask equipped with a stir bar was added 2-(2- propynyloxy)tetrahydro-2H-pyran (2.01 mL, 14.3 mmol, 1.00 eq), hexamethylphosphoramide (8.69 mL, 49.9 mmol, 3.50 eq) and THF (50.0 mL). The reaction mixture was cooled to -78 °C and stirred vigorously under Ar. n-Butyllithium (2.00 M in THF, 9.27 mL, 18.6 mmol, 1.30 eq) was added dropwise over a period of 15 minutes by way of an oven-dried pressure equalizing dropping funnel and the reaction was vigorously stirred at -78 °C for approximately 1 hour. Subsequently, hexadecylbromide (5.67 mL, 18.6 mmol, 1.30 eq) was added slowly dropwise over 10 minutes by way of a pressure equalizing dropping funnel after which the resulting reaction mixture was allowed to warm to room temperature overnight while stirring vigorously under Ar. The following day TLC indicated that all of the starting alkyne had been consumed. The reaction mixture was quenched with a saturated solution of ammonium chloride and then extracted three times into EtOAc. The organic phases were then combined, washed with brine, dried over anhydrous magnesium sulfate and concentrated in vacuo. Subsequent purification of the resulting crude material by column chromatography (2%-10% EtOAc/hexane) afforded a clear oil (3.89 g, 10.7 mmol, 75%).
75%
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran; hexane at -78 - -30℃; for 0.916667h; Inert atmosphere;
Stage #2: hexadecanyl bromide In tetrahydrofuran; hexane at -30 - 20℃; Inert atmosphere;
30 2-Nonadecyn-1-yl 2-tetrahydropyranyl ether
PREPARATION 30 2-Nonadecyn-1-yl 2-tetrahydropyranyl ether 50 ml of a 15% by weight solution of butyllithium in hexane was added dropwise at -50° C. to a solution of 11.48 g of 2-tetrahydropyranyl propargyl ether in 220 ml of tetrahydrofuran followed by 25 g of palmityl bromide, and the mixture was stirred for 30 minutes, during which time the temperature of the mixture was allowed to rise to room temperature. The reaction mixture was heated under reflux for 16 hours, poured into water and then extracted twice with diethyl ether. The combined extracts were washed with water, dried and concentrated by evaporation under reduced pressure. The residue was subjected to column chromatography through 500 g of silica gel. 14.59 g of the title compound were isolated as an oil from the fractions eluted with a 3:100 by volume mixture of diethyl ether and hexane. Nuclear Magnetic Resonance Spectrum (CDCl3) δ ppm: 0.7-2.0 (37H, multiplet); 2.20 (2H, multiplet); 3.3-4.1 (2H, multiplet); 4.21 (2H, triplet, J=2.5 Hz); 4.80 (1H, multiplet).
With copper(l) iodide; tetrakis(triphenylphosphine) palladium(0); triethylamine at 20℃; for 2.5h;
General procedure for Sonogashira coupling: Method 1 (for o-diiodobenzene):
1,2-Diiodobenzene (2.0 g, 6.06 mmol), Pd(PPh3)4 (210 mg, 0.18 mmol), CuI (230 mg, 1.21 mmol) and THP-protected propargyl alcohol (1.04 ml, 6.67 mmol) were added in succession to 30 mL of degassed triethylamine and left for 2.5 h at room temperature. The reaction mixture was then poured into ethyl acetate and the organic layer was washed with saturated NH4Cl solution and brine, dried over anhydrous sodium sulfate. Evaporation in vacuum gave an oily residue from which the product was isolated by column chromatography (Si-gel, petroleum ether-ethyl acetate mixture as eluent).
45%
With copper(l) iodide; tetrakis(triphenylphosphine) palladium(0); triethylamine at 20℃; for 0.5h; Inert atmosphere;
43%
With copper(l) iodide; N-butylamine In diethyl ether at 25℃; for 6h;
With copper(l) iodide; tetrakis(triphenylphosphine) palladium(0); triethylamine at 20℃;
With copper(l) iodide; tetrakis(triphenylphosphine) palladium(0); triethylamine In tetrahydrofuran Inert atmosphere;
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran; hexane at -78℃; for 0.5h;
Stage #2: pentadecyl bromide With 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone In tetrahydrofuran; hexane at -78 - 50℃; for 20h;
Stage #3: In methanol at 40℃; for 3h;
79%
Stage #1: pentadecyl bromide; 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran; N,N,N,N,N,N-hexamethylphosphoric triamide; hexane at 0 - 20℃;
Stage #2: With p-toluenesulfonic acid monohydrate In methanol at 20℃; Further stages.;
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran; hexane at -78℃; for 2h;
Stage #2: cyclopentanone In tetrahydrofuran; hexane at -78℃; for 1h; Further stages.;
95%
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran at -78℃; for 0.5h; Inert atmosphere;
Stage #2: cyclopentanone In tetrahydrofuran at 20℃; for 2h; Inert atmosphere;
59.1 First step: 1-(3-((tetrahydro-2H-pyran-2-yl)oxy)prop-1-yn-1-yl)cyclopentanol (59B)
2-(2-propargyloxy)tetrahydropyran (1a) (30 g, 214 mmol) was dissolved in tetrahydrofuran (150 mL) under reduced nitrogen, then cooled to -78 ° C, n-butyl lithium (103 mL, 257 mmol ) was added dropwise to the reaction, and the reaction was carried out for 0.5 hour after the addition.Then cyclopentanone (19 g, 225 mmol) was added dropwise to the reaction.The reaction was continued for 2 hours at room temperature.The reaction solution was quenched with saturated aqueous ammonium chloride (80 mL) and then evaporated.The aqueous phase was extracted with ethyl acetate (80 mL×2).Wash with saturated sodium chloride solution (80 mL), dry over anhydrous sodiumThe filtrate was concentrated to give 1-(3-((tetrahydro-2H-pyran-2-yl)oxy)prop-1-yn-1-yl)cyclopentanol (59B) (50 g, yield : 95%).
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium; N,N,N,N,-tetramethylethylenediamine In tetrahydrofuran at -78℃; for 0.5h;
Stage #2: cyclopentanone In tetrahydrofuran at -78 - 20℃; Further stages.;
(1R,2S,5R)-2-isopropyl-5-methyl-1-[3-(2H-tetrahydropyran-2-yloxy)-1-propynyl]cyclohexanol[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
1: 68%
2: 22%
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium; N,N,N,N,-tetramethylethylenediamine In tetrahydrofuran at -78℃; for 1h;
Stage #2: (2R,5S)-menthone In tetrahydrofuran at -78℃; for 1h;
With piperidine In tetrahydrofuran at 65℃; for 16h;
85%
With 4,4'-di-tert-butylbiphenyl; lithium; copper dichloride In tetrahydrofuran at 66℃; for 17h; Inert atmosphere;
82%
With copper(l) iodide; nickel(II) chloride hexahydrate; N,N,N,N,-tetramethylethylenediamine; oxygen In tetrahydrofuran at 20℃;
75%
With N,N'-di-tert-butyldiaziridinone; copper(I) bromide In chloroform-d1 at 20℃; for 5h;
71%
With Co(salen)complex; N,N'-di-tert-butyldiaziridinone In ethyl acetate at 80℃; for 6h; Schlenk technique; Inert atmosphere;
Cobalt-Catalyzed Homocoupling Reaction; General Procedure
General procedure: To a flame-dried Schlenk tube with a stir bar was added Co(salen)complex 4 (10 mol%). The contents were evacuated and backfilledwith N2 (3 ×). Di-tert-butyldiaziridinone (2; 0.3 mmol) and alkyne(0.2 mmol) were added via microinjection syringe dissolved in anhydEtOAc (0.2 M). The mixture was stirred at 80 °C (oil bath). When thereaction was complete (TLC monitoring), the mixture was directlyconcentrated by rotary evaporation. The coupling products wereisolated by column chromatography (silica gel, typically 0-30%EtOAc/PE)
64%
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran at -78℃;
Stage #2: With Triisopropyl borate In tetrahydrofuran at -78℃;
Stage #3: With copper(l) iodide; bis[2-(diphenylphosphino)phenyl] ether In tetrahydrofuran at 60℃; for 8h; Further stages.;
With zinc(II) oxide In neat (no solvent) at 0 - 20℃; for 3h; Green chemistry;
Typical experimental procedure for the synthesis of chloroesters by the cleavage of cyclic and acyclic ethers
General procedure: In the mixture of cyclic/acyclic ether (11 mmol) and acid chloride (10 mmol), nano-ZnO (5 mol%) was added at 0-5 °C and stirred at room temperature for an appropriate time. After the TLC monitoring reaction, the ZnO was removed by filtration and washed repeatedly with dichloromethane and water. It was then dried at 60 °C for 3 h and used for the next catalytic cycle. The solution was extracted three times with dichloromethane and water, and dried on anhydrous Na2SO4. The product was purified on a silica gel column chromatography using a mixture of petroleum ether/ethyl acetate (150:1, v/v). The product is obtained by vacuum distillation to remove the solvent. The compounds were characterized by 1H NMR and 13C NMR.
With n-butyllithium In tetrahydrofuran at -78 - 0℃;
78%
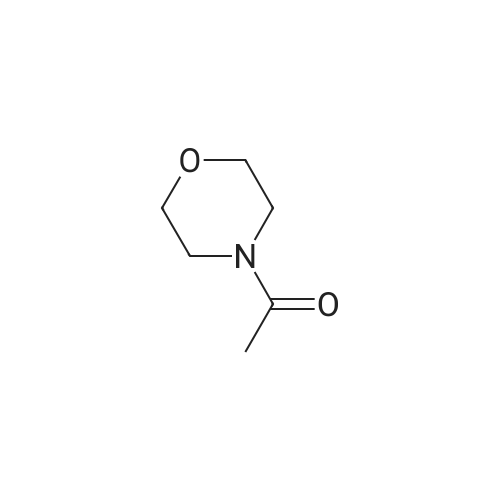
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran; hexane at 0℃; for 0.25h;
Stage #2: 4-acetylmorpholine In tetrahydrofuran; hexane at 0℃; for 1.4h;
78%
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran; hexanes at 0℃; for 0.583333h;
Stage #2: 4-acetylmorpholine In tetrahydrofuran; hexanes at 0℃; for 1.4h;
34
Example 34; To a stirred solution of tetrahydro-2-(prop-2-ynyloxy)-2H-pyran (21.01 g, 150 mmol) in THF (90 mL) was added nBuLi (1.6 M in hexanes, 92 mL) dropwise over 20 min at 0° C. under a nitrogen atmosphere. After an additional 15 min at the same temperature, N-acetylmorpholine (5.8 mL, 50 mmol) in THF (8 mL) was added dropwise to the reaction mixture, and then the container that initially contained N-acetylmorpholine was rinsed with THF (2×1 mL) and added to the reaction mixture at the same temperature. After an additional 1.4 h at 0° C., the reaction mixture was cannulated into a flask containing AcOH (120 mL) and H2O (60 mL) 0° C., the reaction container was rinsed with Et2O (50 mL), and then the resulting layers were separated. The organic layer was washed with saturated aqueous NaHCO3 (300 mL). The combined aqueous layers were extracted with Et2O (50 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was purified by flash chromatography (2.5×10% EtOAc in hexanes) on silica gel (600 mL) to afford 35 (7.141 g, 78%) as a colorless oil. Spectroscopic data for 35 were consistent with the literature (J. Org. Chem. 1983, 48, 2151-2158).
78%
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran; hexanes at 0℃; for 0.583333h;
Stage #2: 4-acetylmorpholine In tetrahydrofuran; hexanes at 0℃; for 1.4h;
34
To a stirred solution of tetrahydro-2-(prop-2-ynyloxy)-2H-pyran (21.01 g, 150 mmol) in THF (90 mL) was added "BuLi (1.6 M in hexanes, 92 mL) dropwise over 20 min at 0 0C under a nitrogen atmosphere. After an additional 15 min at the same temperature, JV-acetylmorpholine (5.8 mL, 50 mmol) in THF (8 mL) was added dropwise to the reaction mixture, and then the container that initially contained JV- acetylmorpholine was rinsed with THF (2 x 1 mL) and added to the reaction mixture at the same temperature. After an additional 1.4 h at 0 0C, the reaction mixture was cannulated into a flask containing AcOH (120 mL) and H2O (60 mL) 0 0C, the reaction container was rinsed with Et2O (50 mL), and then the resulting layers were separated. The organic layer was washed with saturated aqueous NaHCO3 (30O mL). The combined aqueous layers were extracted with Et2O (50 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was purified by flash chromatography (2.5 - > 10% EtOAc in hexanes) on silica gel (600 mL) to afford 35 (7.141 g, 78%) as a colorless oil.[0145] Spectroscopic data for 35 were consistent with the literature (J. Org. Chem. 1983, 45, 2151-2158).
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne; N-methoxy-N-methylpyridine-3-carboxamide With lithium hexamethyldisilazane In tetrahydrofuran at -10 - 10℃; for 1h;
Stage #2: With ammonium chloride In tetrahydrofuran Further stages.;
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne; N-methoxy-N-methylcyclohexanecarboxamide With lithium hexamethyldisilazane In tetrahydrofuran at -10 - 10℃; for 1h;
Stage #2: With ammonium chloride In tetrahydrofuran at 40℃; Further stages.;
With copper(l) iodide; triethylamine; triphenylphosphine at 120℃; for 0.416667h; microwave;
100%
With copper(l) iodide; triethylamine; triphenylphosphine In N,N-dimethyl-formamide at 120℃; for 0.416667h;
68.B
Example 68B 2-({3-[4-(benzyloxy)-3-nitrophenyl]prop-2-ynyl}oxy)tetrahydro-2H-pyran Example 68A (500 mg, 1.62 mmol), 2-prop-2-ynyloxy-tetrahydro-pyran (1.14 mL, 8.11 mmol), Pd(PPh3)2Cl2 (171 mg, 0.24 mmol), CuI (2 mg, 0.011 mmol), PPh3 (320 mg, 1.22 mmol), triethylamine (1 mL), and DMF (3 mL) were mixed and purged with N2. The mixture was heated at 120° C. for 25 minutes in a Smith Synthesizer. The solvents were removed via vacuum pump. The residue was dissolved in acetone. The solution was dried with silica gel powder (10 g). 10% Ethyl acetate in hexanes (2 L) was used to run flash chromatography to give the title compound (0.60 g, 100%). MS (DCI) m/z 385.10 (M+NH4)+; 1H NMR (300 MHz, CD3OD) δ ppm 1.45-1.92 (m, 6H) 3.48-3.60 (m, 2H) 3.88 (m, 1H) 4.45 (s, 1H) 4.46 (s, 1H) 5.29 (s, 2H) 7.29-7.42 (m, 4H) 7.43-7.48 (m, 2H) 7.61 (dd, J=8.82, 2.03 Hz, 1H) 7.87 (d, J=2.03 Hz, 1H).
2-amino-5-[3-(tetrahydro-pyran-2-yloxy)-prop-1-ynyl]-4-trifluoromethyl-benzoic acid methyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
72%
With copper(l) iodide; triethylamine;bis-triphenylphosphine-palladium(II) chloride; In 1,4-dioxane; for 16h;Heating / reflux;
Example 25:; N-[6-(3-Hydroxy-propyl)-2,4-dioxo-7-trifluoromethyl-l,4-dihydro-2H-quinazolin-3- yl]-methanesulfonamide; 2-Amino-5-[3-(tetrahydro-pyran-2-yloxy)-prop-l-ynyl]-4-trifluoromethyl-benzoic acid methyl ester; A solution of <strong>[872624-52-7]2-amino-5-iodo-4-trifluoromethyl-benzoic acid methyl ester</strong> (2.0 g, 5.80 mmol), tetrahydro- 2-(2-propynyloxy)-2H-pyran (2.44 mL, 17.4 mmol), copper iodide (55.0 mg, 0.29 mmol) and bis(triphenylphosphine)palladium (II) dichloride (407 mg, 0.58 mmol) in dioxane (15 mL) and Et3N (15 mL) was heated to reflux for 16 h under nitrogen. The mixture was then allowed to cool to r.t., filtered and the filtrate was diluted with AcOEt. This mixture was washed once with water. The organic phase was then dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The crude product was purified by flash chromatography (silica gel 60, hexanes/EtOAc 5: 1) to give an orange oil, which was recrystallized in hexanes to provide 2-amino-5-[3-(tetrahydro-pyran-2-yloxy)-prop-l-ynyl]-4- trifluoromethyl-benzoic acid methyl ester (1.50 g, 4.20 mmol, 72 %) as a pale brown powder, m.p. 88 0C, ESI-MS: m/z 358 [M+Eta]+.
With potassium hydroxide; iodine; In tetrahydrofuran; magnesium;
Step A To a Grignard mixture prepared in the conventional manner from 6.1 g. of magnesium (etched with iodine) and 27.2 g. of ethyl bromide in a total of 120 ml. of dry tetrahydrofuran, there is dropwise added a solution of 32.2 g. of 3-(2'-tetrahydropyranyloxy)-propyne in 32 ml. of dry tetrahydrofuran. After 1 hour stirring at room temperature, a solution of 36.6 g. of 6-methoxy-2-naphthaldehyde in 200 ml. of dry tetrahydrofuran is dropwise added. After 1 hour stirring at room temperature, the reaction is poured onto saturated ammonium chloride solution. The aqueous phase is extracted once with benzene. The combined organic solutions are washed with 500 ml. of 1N potassium hydroxide and then with 300 ml. of water, dried over sodium sulfate and evaporated to dryness to obtain 1-(6'-methoxy-2'-naphthyl)-4-(tetrahydropyran-2-yloxy)-2-butyn-1-ol.
With copper(l) iodide; triethylamine In acetonitrile at 20 - 30℃; for 1.5h;
67.1
To a solution of 4- (4-amino-3-chlorophenoxy) -6-iodo-N- methylpyrirαidine-5-amine (3.0 g, 7.97 mmol) in acetonitrile (90 mL) /triethylamine (90 mL) were added tetrahydro-2- (2- propynyloxy) -2H-pyran (1.34 g, 9.56 mmol), dichlorobis (triphenylphosphine) palladium (0.28 g, 0.40 mmol) and copper iodide (0.15 g, 0.80 mmol), and the mixture was stirred at room temperature for 30 min and then stirred at 600C for 1 hr. After cooling to room temperature, the solvent was evaporated under reduced pressure. Water was added to the residue, and the mixture was extracted with ethyl acetate. The mixture was washed with saturated brine, dried over anhydrous magnesium sulfate, and filtrated. The filtrate was concentrated under reduced pressure, and the residue was purified by column chromatography (silica gel, ethyl acetate/hexane) to give the title compound (2.11 g, 68%) as a brown liquid.1H-NMR (CDCl3, 300 MHz) δ 1-25 - 1.86 (6H, m) , 3.27 (3H, d, J = 5.4 Hz), 3.54 - 3.61 (IH, m) , 3.85 - 3.94 (IH, m) , 4.04 (2H, s), 4.42 - 4.49 (IH, m) , 4.56 (2H, s), 4.87 - 4.90 (IH, m) , EPO 6 . 83 ( IH, d, J = 8 . 7 Hz ) , 6 . 89 ( IH, dd, J = 8 . 7 , 2 . 7 Hz ) , 7 . 10 ( IH, d, J = 2 . 7 Hz ) , 8 . 05 ( IH, s ) .
68%
With bis-triphenylphosphine-palladium(II) chloride; copper(l) iodide; triethylamine In acetonitrile at 60℃; for 1h;
With triethylamine;tetrakis(triphenylphosphine) palladium(0); copper(I) iodide; In N,N-dimethyl-formamide;
Preparation 9 Preparation of 5-[3-(Tetrahydro-pyran-2-yloxy)-prop-1-ynyl]-3H-isobenzofuran-1-one To a mixture of <strong>[64169-34-2]5-bromophthalide</strong> (20.00 g, 9.39 mmol), tetrahydro-2-(2-propynyloxy)-2H-pyran (5.3 ml, 37.6 mmol), copper(I) iodide (0.645 g, 3.39 mmol), triethylamine (1.90 g, 18.8 mmol) in DMF (20 ml), was added tetrakis(triphenylphosphine)palladium (1.30 g, 1.13 mmol). The mixture was heated at 65 C. under argon for 16 hours, cooled to room temperature and diluted with diethyl ether (250 ml). The filtrate was washed with brine (4*100 ml), dried over anhydrous Na2SO4, and concentrated to give a dark-reddish residue. Purification of the residue mixture by silica gel column chromatography, eluted with a gradient of EtOAc, led to 5-[3-(tetrahydro-pyran-2-yloxy)-prop-1-ynyl]-3H-isobenzofuran-1-one as a white solid (2.23 g, 87%). 1H NMR (500 MHz, DMSO-d6) delta ppm 1.50 (m, 4H) 1.69 (m, 2H) 3.49 (m, 1H) 3.77 (m, 1H) 4.50 (m, 2H) 4.83 (m, 1H) 5.40 (s, 2H) 7.63 (d, J=6.83 Hz, 1H) 7.77 (s, 1H) 7.84 (d, J=7.81 Hz, 1H); LR MS (EI): 272 (M+).
With copper(l) iodide; palladium diacetate; triethylamine; triphenylphosphine; at 80℃; for 16h;Inert atmosphere;
General procedure: A mixture of bromide 13 (61.8 mmol), Pd(OAc)2 (140 mg, 0.624 mmol), PPh3 (0.330 g, 1.26 mmol), CuI (22 mg, 0.116 mmol), and 2-prop-2-ynyloxytetrahydropyran (14) (13.0 g, 92.8 mmol) in triethylamine (250 mL) was stirred at 80 C under an argon atmosphere for 16 h. After this period, the mixture was cooled to rt, the precipitate was filtered, and the filtrate was evaporated. The residue was dissolved in EtOAc (700 mL), the solution was washed with saturated aq NaHCO3 (2200 mL), brine (200 mL), dried over Na2SO4, and evaporated in vacuo. The crude product was purified by column chromatography (CHCl3 - MeOH (19:1) (15a-c) or hexanes - EtOAc (2:1) (15d,e) as eluent).
With copper(l) iodide; tetrakis(triphenylphosphine) palladium(0); triethylamine In N,N-dimethyl-formamide at 20℃; for 16h; Inert atmosphere;
4.4A 4A. 2-((3-(4-Bromophenyl)prop-2-yn-1-yl)oxy)tetrahydro-2H-pyran
To a solution of 1-bromo-4-iodobenzene (10.0 g, 35.3 mmol) in DMF (50 mL) was added TEA (25 mL, 177 mmol), CuI (0.40 g, 2.12 mmol), Pd(Ph3P)4 (0.82 g, 0.71 mmol) and 2-(prop-2-yn-1-yloxy)tetrahydro-2H-pyran (6.44 g, 46.0 mmol). The reaction mixture was stirred at RT under N2 for 16 h, then was concentrated in vacuo. The residue was chromatographed (120 g SiO2; isocratic hexanes/EtOAc=95:5) to afford the title compound (10.0 g, 33.9 mmol, 96% yield) as a colorless oil. LCMS, [M+Na]+=319.0. 1H NMR (500 MHz, CDCl3) δ 7.46-7.42 (m, 2H), 7.33-7.29 (m, 2H), 4.89 (t, J=3.4 Hz, 1H), 4.54-4.40 (m, 2H), 3.89 (ddd, J=11.5, 9.0, 2.9 Hz, 1H), 3.61-3.54 (m, 1H), 1.92-1.51 (m, 6H).
96%
With copper(l) iodide; tetrakis(triphenylphosphine) palladium(0); triethylamine In N,N-dimethyl-formamide at 20℃; for 16h; Inert atmosphere;
Intermediate 4 A. 2-((3-(4-Bromophenyl)prop-2-yn-l-yl)oxy)tetrahydro-2H-pyran
To a solution of l-bromo-4-iodobenzene (10.0 g, 35.3 mmol) in DMF (50 mL) was added TEA (25 mL, 177 mmol), Cul (0.40 g, 2.12 mmol), Pd(Ph3P)4 (0.82 g, 0.71 mmol) and 2-(prop-2-yn-l-yloxy)tetrahydro-2H-pyran (6.44 g, 46.0 mmol). The reaction mixture was stirred at RT under N2 for 16 h, then was concentrated in vacuo. The residue was chromatographed (120 g S1O2; isocratic hexanes/EtOAc = 95:5) to afford the title compound (10.0 g, 33.9 mmol, 96 % yield) as a colorless oil. LCMS, [M + Na]+ = 319.0. 1H NMR (500 MHz, CDCI3) d 7.46 - 7.42 (m, 2H), 7.33 - 7.29 (m, 2H), 4.89 (t, 7=3.4 Hz, 1H), 4.54 - 4.40 (m, 2H), 3.89 (ddd, 7=l l.5, 9.0, 2.9 Hz, 1H), 3.61 - 3.54 (m, 1H), 1.92 - 1.51 (m, 6H).
84%
With propylamine; copper(l) iodide at 20℃; for 24h;
With copper(l) iodide; dichlorobis(tri(2-furyl)phosphine)palladium(II); triethylamine In acetonitrile at 20℃; for 1h;
(Z)-5-(2-(tetrahydro-2H-pyran-2-yloxy)ethylidene)furan-2(5H)-one[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
72%
With copper(l) iodide; palladium 10% on activated carbon; triethylamine; triphenylphosphine; In 1,4-dioxane; at 25 - 80℃; for 3.5h;Inert atmosphere;
General procedure: (a) The reaction was performed in a bigger scale using 100 mg of 10% Pd/C (0.092 mmol), PPh3 (0.37 mmol), CuI (0.184 mmol), Et3N (10.68 mmol), compound 1a (3.56 mmol), and acetylenic compound 2a (5.32 mmol) in 1,4-dioxane (20.0 mL). After stirring at 80 C for 3 h under nitrogen the mixture was cooled to room temperature. The Pd/C was filtered off and washed with water (2 10 mL), acetone (2 10 mL), and EtOAc (2 10 mL). Then the catalyst was collected, dried at 100 C in an oven, and reused for the next run. The co-catalyst CuI along with PPh3 was added in every repeated run. (b) General method for the preparation of 3: A mixture of compound 1 (0.89 mmol), 10% Pd/C (0.023 mmol), PPh3 (0.092 mmol), CuI (0.046 mmol), and Et3N (2.67 mmol) in 1,4-dioxane (5.0 mL) was stirred at 25 C for 30 min under nitrogen. The acetylenic compound 2 (1.33 mmol) was added slowly with stirring. The mixture was then stirred at 80 C for 3 h, cooled to room temperature, diluted with EtOAc (30 mL), and filtered through celite. The filtrate was collected and concentrated. The residue was purified by column chromatography (2-15% EtOAc/hexane) to afford the desired product
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran; hexane at -78℃; Inert atmosphere;
Stage #2: 7-bromo-3,4-dihydro-2H-naphthalen-1-one In tetrahydrofuran; hexane at -78 - 20℃; Inert atmosphere;
With sodium hydrogencarbonate;bis-triphenylphosphine-palladium(II) chloride; copper(l) iodide; In tetrahydrofuran; water; at 20℃; for 48h;Inert atmosphere;
M. Synthesis of (6S)-6-({1-methyl-3-[4-(trifluoromethoxy)phenyl]-1H-pyrazol-5-yl}methoxy)-2-nitro-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazine (11) by the method of Scheme 8 A solution of 2-(2-propynyloxy)tetrahydro-2H-pyran (69) (0.758 g, 5.41 mmol), CuI (17 mg, 0.09 mmol) and PdCl2(PPh3)2 (0.158 g, 0.023 mmol) in THF (15 mL) was purged with N2. 1-Iodo-4-(trifluoromethoxy)benzene (70) (1.30 g, 4.51 mmol) in THF (10 mL) was added, followed by a solution of methylhydrazine sulfate (1.95 g, 13.5 mmol) and NaHCO3 (2.27 g, 27 mmol) in water (25 mL). The mixture was flushed with carbon monoxide and then stirred at room temperature for 2 days under one atmosphere of carbon monoxide. The resulting mixture was partitioned between CH2Cl2 and water, the CH2Cl2 fraction was dried, and the solvent was evaporated. Column chromatography of the residue on silica gel (eluting with CH2Cl2) gave 1-methyl-5-[tetrahydro-2H-pyran-2-yloxy)methyl]-3-[4-(trifluoromethoxy)phenyl]-1H-pyrazole (71) (1.034 g, 64%) as a brown solid: mp 40-42 C.; 1H NMR (CDCl3) δ 7.78 (d, J=8.8 Hz, 2H), 7.21 (d, J=8.0 Hz, 2H), 6.51 (s, 1H), 4.75 (d, J=12.8 Hz, 1H), 4.69 (t, J=3.3 Hz, 1H), 4.57 (d, J=12.8 Hz, 1H), 3.94 (s, 3H), 3.84-3.91 (m, 1H), 3.53-3.60 (m, 1H), 1.68-1.88 (m, 2H), 1.50-1.66 (m, 4H). APCI MS m/z 357 [M+H]+.
With triethylamine;bis-triphenylphosphine-palladium(II) chloride; copper(l) iodide; In tetrahydrofuran; at 100.0℃; for 0.5h;Inert atmosphere; Microwave irradiation;
Nitrogen was bubbled through a solution of Ethyl 5-bromo-2-phenylthiazole- 4-carboxylate (3.6 g, 11.5 mmol) and 2-(prop-2-ynyloxy)tetrahydro-2H-pyran (3.3 rnL, 23.1 mmol) in THF (30 mL). Dichlorobis(triphenylphosphine)palladium (II) (405 mg, 0.6 mmol), copper (I) iodide (55 mg, 0.3 mmol), and triethylamine (TEA) (8 mL, 57.7 mmol) were added and the reaction mixture was heated at 1000C for 30 min in a microwave reactor. The reaction mixture was poured into H2O and extracted with EtOAc. The combined organics were washed with brine, dried (MgSO4), and concentrated. The crude product was purified by MPLC eluting with pentane/EtOAc (0-50% gradient) to give ethyl 2-phenyl-5-(3-(tetrahydro-2H-pyran-2-yloxy)prop-l- ynyl)thiazole-4-carboxylate (3.8 g, 88 % yield)
With copper(l) iodide; sodium hydrogencarbonate;bis(triphenylphosphine)palladium(II) dichloride; In tetrahydrofuran; water; at 20℃; for 48h;
A solution of 2-(2-propynyloxy)tetrahydro-2/J-pyran (69) (0.758 g, 5.41 mmol), CuI (17 mg, 0.09 mmol) and PdCl2(PPh3)2 (0.158 g, 0.023 mmol) in THF (15 mL) was purged with N2. l-Iodo-4-(trifiuoromethoxy)benzene (70) (1.30 g, 4.51 mmol) in THF (10 mL) was added, followed by a solution of methylhydrazine sulfate (1.95 g, 13.5 mmol) and NaHCO3 (2.27 g, 27 mmol) in water (25 mL). The mixture was flushed with carbon monoxide and then stirred at room temperature for 2 days under one atmosphere of carbon monoxide. The resulting mixture was partitioned between CH2Cl2 and water, the CH2Cl2 fraction was dried, and the solvent was evaporated. Column chromatography of the residue on silica gel (eluting with CH2Cl2) gave 1 -methyl-5-[(tetrahydro-2//-pyran-2-yloxy)methyl]-3-[4-(trifluoromethoxy)phenyl]-lH-pyrazole (71) (1.034 g, 64%) as a brown solid: mp 40-42 0C; 1H NMR (CDCl3) δ 7.78 (d, J = 8.8 Hz, 2 H), 7.21 (d, J = 8.0 Hz, 2 H), 6.51 (s, 1 H), 4.75 (d, J = 12.8 Hz, 1 H), 4.69 (t, J= 3.3 Hz, 1 H), 4.57 (d, J- 12.8 Hz, 1 H), 3.94 (s, 3 H), 3.84-3.91 (m, 1 H), 3.53-3.60 (m, 1 H), 1.68-1.88 (m, 2 H), 1.50-1.66 (m, 4 H). APCI MS m/z 357 [M + H]+.
With copper(l) iodide; triethylamine In N,N-dimethyl-formamide at 20℃; for 3h; Inert atmosphere;
56.1
To a stirred solution of 5-bromo-2-iodopyrimidine (548 mg,1.92 mmol) and 2-prop-2-ynyloxytetrahydropyran (0.28 mL, 1.99 mmol) in DMF (2 mL) were added TEA (0.60 mL, 4.31 mmol) and Cul (20 mg, 0.11 mmol). Argon was bubbled through the solution for 5 min before PdCl2(PPh3)2 (65 mg, 0.093 mmol) was added. The mixture was stirred at rt for 3 h, whereupon analysis by LC-MS indicated the presence of desired product. The solution was partitioned between EtOAc (50 mL) and brine (50 mL), and the aqueous layer was extracted with EtOAc (3 x 40 mL). The combined organic phases were dried over MgS04, filtered, and concentrated under reduced pressure, and the residue was purified by silica gel flash chromatography eluting with 5-100 % EtOAc in hexanes to give 5-(4-bromophenyl)-2-(3-(tetrahydro- 2H-pyran-2-yloxy)prop-l-ynyl)pyrimidine (474 mg, 83%). LC-MS (ESI) m/z 298 (M + H)+.
With copper(l) iodide; trans-bis(triphenylphosphine)palladium dichloride; sodium acetate; triethylamine; at 70℃; for 3h;Inert atmosphere;
General procedure: To a stirred mixture of aniline 2b (0.100 g, 0.344 mmol), hex-1-yne 3g (0.084 g, 1.031 mmol) and Et3N (3 mL) under argon added were Pd(PPh3)2Cl2 (0.010 g, 0.014 mmol), CuI (0.006 g, 0.031 mmol) and, additionally, Et3N (3 mL). Stirring was continued at 70 C for 3 h, the mixture was cooled to r.t., diluted by CHCl3 (10 mL) and poured into a mixture of H2O (30 mL) and CHCl3 (20 mL). The aqueous layer was extracted with CHCl3 (2 × 30 mL), the collected organic solution was washed with H2O (3 × 30 mL) and dried (MgSO4). Evoparation of the solvent and chromatography (TLC) of the crude product (eluent: hexane-EtOAc, 10:1 ? 10:1 ? 10:1, Rf = 0.54) gave aniline 4bg (0.067 g, 80%) as an oil.
With copper(l) iodide; trans-bis(triphenylphosphine)palladium dichloride; triethylamine; In acetonitrile; at 60℃; for 2h;Inert atmosphere;
General procedure: To a stirred solution of aniline 1(2 mmol) and alkyne 2 (3 mmol) in MeCN (12 mL) were added Pd(PPh3)2Cl2 (56 mg, 0.08 mmol), CuI (34 mg,0.18 mmol) and Et3N (3 mL) at room temperature under an argon atmosphere. The reaction mixture was stirred at 60 C for 2 h. After the mixture was cooled to room temperature, diluted with CH2Cl2(10 mL), poured into H2O (40 mL) and extracted with CH2Cl2(3 × 50 mL). The combined organic layers were washed with H2O (30mL) and dried (MgSO4). After evaporation of the solvent in vacuo,the crude product 3 was obtained (the 1H and19FNMR spectra closely agree with the literature data [2]) and used further without purification.
3,3-Ethylenedioxy-17 -hydroxy-17-(3-tetrahydropyranyloxy-l-propynyl)-estra-5(10),9(l 1)- diene (28)To a solution of tetrahydro-2-(2-propynyloxy)-2H-pyran (13.4 g) in THF (50 mL) at 0 C, n- BuLi (2.5 M solution in hexane, 38 mL) was added dropwise and allowed to stir for 30 min at 0 C. A solution of 3,3-ethylenedioxy-estra-5(10),9(l l)-diene-17-one (13) (10 g) in THF (50 mL) was added dropwise to the reaction mixture and allowed to stir for 2 hrs at 0 C. Quenched with saturated ammonium chloride solution (100 mL) and extracted with ethyl acetate (3 X 25 mL). The collective organic layer was washed once with water, dried over sodium sulfate and concentrated under reduced pressure. Purification on silica gel column yielded 28 (13 g).XH NMR (delta, 300 MHz) 0.84 (s, 3H), 1.10-2.8 (m, 24H), 3.4-3.6 (m, 1H), 3.7-3.9 (m, 1H), 4.00 (s, 4H), 4.20-4.40 (m, 2H), 4.80 (s, 1H), 5.61 (s, 1H).
General procedure: BuLi (3.94 mL, 8.73 mmol) was slowly added into a solution of 16 (1.11 g, 7.93 mmol) in THF (15 mL) at -78 ºC and followed by addition of TMEDA (1.80 g, 15.9 mmol). The reaction solution was warmed to rt and stirred for 1 h. 1-Bromodecane (2.46 mL, 11.9 mmol) was added dropwise at rt and followed by KI (132 mg, 0.793 mmol). The reaction mixture was heated at 80 ºC for 8 h then quenched with aqueous Na2CO3 (10 percent). The resulted mixture was diluted with diethyl ether, and washed with aqueous Na2CO3 (10 percent). The aqueous portion was back extracted with diethyl ether. The organic portions were combined, washed with brine, dried over Na2SO4, and concentrated to a brown liquid, which was purified by column chromatography (ethyl acetate / hexane, 1 : 9) to gave compound 17 (1.91 g, 86 percent) as a colorless liquid.
CuNPs/MagSilica catalyzed multicomponent Huisgen 1,3-dipolar cycloaddition reaction.
General procedure: To a vigorously stirred suspension of the CuNPs/MagSilica catalyst (40 mg) and NaN3 (72 mg, 1.1 mmol) in water (2 mL), the corresponding alkyl halide (1.0 mmol) was added dropwise, followed by the addition of the terminal alkyne (1.0 mmol). The reaction mixture was stirred at 70 ◦C until total conversion of the starting alkyne (TLC, GC). The triazole product formation can be easily visualized as a solid floating on the water surface. Then, the catalyst was immobilized by means of a permanent magnet placed on the outer wall of the reaction flask, and was successively washed with water (2 mL) and CH2Cl2 (10 mL). Finally, the catalyst was dried under vacuum (5 Torr) for its recovery and reuse. The possibility of catalyst leaching in the reaction media was evaluated by measuring the copper concentration in the reaction mixture, by means of atomic absorption spectroscopy.
With hydrogen In toluene at 110℃; for 4h; Schlenk technique;
Semi-Hydrogenation of Alkynes 1a-l; General Procedure
General procedure: The starting alkyne 1 (0.5 mmol) and dodecane as internal standard(0.22 mmol, 50 μL) were added to a suspension of the Cu-Pd-NPs/MCM-48 catalyst (10 mg) in toluene (5 mL). All reactions werecarried out in a Schlenk-type flask, fitted with a reflux condenser andsealed with a rubber septum. The reaction flask was purged and filledwith H2 (1 atm) through a balloon connected to the flask by a needle,and then heated at 110 °C. Then, the reaction mixture was centrifugedand the supernatant removed. The solvent was evaporated invacuo, and the crude product was purified by flash column chromatography(silica gel, hexane/EtOAc). The recovered solid catalyst waswashed with toluene (3 × 2 mL), dried in an oven, calcined at 150 °C(4 h), and reduced in H2 atmosphere at 200 °C before its reuse. The following known compounds included in Table 2 were characterizedby comparison of their chromatographic and spectroscopic data (FTIR,1H NMR, 13C NMR, and MS) with those described in the literature.Styrene (2a)18Yield: 46 mg (0.44 mmol, 89%); colorless liquid.IR (film): 3082, 3060, 3027, 1630, 1496, 1449, 992, 909, 777, 698 cm-1.1H NMR (300 MHz, CDCl3): δ = 7.35 (m, 2 H), 7.25 (m, 3 H), 6.7 (dd, J =10.8, 17.1 Hz, 1 H), 5.71 (d, J = 17.1 Hz, 1 H), 5.20 (d, J = 10.8 Hz, 1 H).13C NMR (75 MHz, CDCl3): δ = 137.5, 128.5, 127.7, 126.1, 136.9, 113.7.MS: m/z (%) = 104 (100, [M+]), 103 (40), 78 (35), 77 (17), 51 (17).
88%
With dimethylamine borane In ethanol at 25℃; for 1h;
98 %Spectr.
With formic acid; Au9924Ag0.76; triethylamine In N,N-dimethyl-formamide at 70℃; for 5h; Inert atmosphere; Green chemistry; chemoselective reaction;
(5S)-9-((tetrahydro-2H-pyran-2-yl)oxy)non-1-en-7-yn-5-ol[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
87%
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran; cyclohexane at -78 - -65℃; for 1.16667h;
Stage #2: With boron trifluoride diethyl etherate In tetrahydrofuran; cyclohexane at -78℃; for 0.333333h;
Stage #3: (-)-(S)-2-(but-3-enyl)oxirane In tetrahydrofuran; cyclohexane at -78 - -65℃; for 1.5h;
6 (5S)-9-((tetrahydro-2H-pyran-2-yl)oxy)non-1-en-7-yn-5-ol
A solution of tetrahydro-2-(2-propynyloxy)-2/-/-pyran (27 g, 192.6 mmol) in THF (270 m L) was cooled to - 78 °C and treated with 2 M n-BuLi in cyclohexane (99 mL, 198.4 mmol) over 30 min, while maintaining the internal temperature below -65 °C. After stirring for 40 min at -78 °C, to the mixture was added BF3 Et2 (25.1 mL, 198.4 mmol) over 5 min, the resulting mixture was stirred at -78 for 15 min. A solution of (S)-2-(but-3-en-1 -yl)oxirane (20.79 g, 21 1 .9 mmol) in THF (54.0 m L) was added over 30 min, while maintaining the internal temperature below -65 °C, and stirring was continued at -78 °C for 1 h. The reaction was quenched with saturated aqueous NH4CI (270 m L) and warmed to rt. The organic layer was separated and the aqueous layer was extracted with MTBE (270 m L). The organic layers were combined, washed with saturated aqueous NaHC03 (81 mL) and brine (80 mL), and dried over MgS04 to give the title compound (40.1 g, 87%). 1 H NMR (400 MHz, CDCI3) δ 1 .45-1 .88 (m , 7H), 2.00-2.30 (m, 3H), 2.34-2.50 (m, 2H), 3.51 (m, 1 H), 3.70-3.88 (m , 2H), 4.25 (m, 2H), 4.80 (m, 1 H), 5.00 (m , 2H), 5.81 (m, 1 H).
82.8%
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In diethyl ether at -78℃; for 0.833333h; Inert atmosphere;
Stage #2: (-)-(S)-2-(but-3-enyl)oxirane With boron trifluoride diethyl etherate In diethyl ether for 1h; Inert atmosphere;
2 Preparation of compound III-2
Under argon protection, compound II-2 (257.04g, 1.84mol) was dissolved in 1000mL ether in a 2000mL three-necked flask, cooled to -78°C, and n-butyllithium (736.00ml, 1.84mol) was added dropwise, stirred for 50min, Compound I (60.00 g, 0.61 mol) was added, then boron trifluoride ether (86.90 g, 0.61 mol) was added, and the mixture was stirred for 60 min. Saturated aqueous sodium bicarbonate solution was added, the organic phase was separated, dried and concentrated to obtain the crude product. The crude product was subjected to silica gel column chromatography to obtain compound III-2 (120.50 g) with a yield of 82.8%.
Stage #1: 3-(tetrahydropyran-2'-yloxy)propyne With n-butyllithium In tetrahydrofuran; cyclohexane at -78℃; for 1.5h;
Stage #2: (-)-(S)-2-(but-3-enyl)oxirane With boron trifluoride diethyl etherate In tetrahydrofuran; cyclohexane at -78℃; for 2h;
(4-(4-(((tetrahydro-2H-pyran-2-yl)oxy)methyl)-1H-1,2,3 -triazol-1-yl)phenyl)methanamine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
66%
With copper(l) iodide; sodium azide; (1S,2S)-N,N'-dimethyl-1,2-diaminocyclohexane; sodium L-ascorbate; triethylamine; In methanol; water; at 55℃;
A mixture of <strong>[59528-27-7](4-iodophenyl)methanamine hydrochloride</strong> (5.0 g, 18.55 mmol, 1.0 eq), (1S,2S)-N1,N2- dimethylcyclohexane-l,2-diamine (0.59 mL 3.71 mmol, 0.2 eq), Sodium ascorbate (368 mg, 1.86 mmol, 0.1 eq), Copper Iodide (530 mg, 2.78 mmol, 0.15 eq), Sodium azide (2.41 g, 37.1 mmol, 2.0 eq) , Et3N (3.11 mL, 22.26 mmol, 1.2 eq) and 2-(prop-2-yn-l-yloxy)tetrahydro- 2H-pyran (2.6 g, 18.55 mmol, 1.0 eq) in Methanol (50 mL) and water (12 mL) were purged with Nitrogen for 5 minutes and heated to 55 C for overnight. The reaction mixture was cooled to room temperature and filtered through 413 filter paper. Celite was added and the solvent was removed under reduced pressure and the residue was purified over silica gel (120 g) using dichloromethane/(methanol containing 12 % (v/v) aqueous ammonium hydroxide) as mobile phase. The concentration of (methanol containing 12 % (v/v) aqueous ammonium hydroxide) was gradually increased from 0 % to 6.25 % to afford (4-(4-(((tetrahydro-2H-pyran-2- yl)oxy)methyl)-1H-1,2,3-triazol-1-yl)phenyl)methanamine (15, 3.54 g, 66%) as a white solid. LCMS m/z: [M + H]+ Calcd for Ci5H2oN402 289.2; Found 289.2.
66%
With copper(l) iodide; sodium azide; sodium ascorbate; (1S,2S)-N,N'-dimethyl-1,2-diaminocyclohexane; triethylamine; In methanol; water; at 55℃;Inert atmosphere;
A mixture of <strong>[59528-27-7](4-iodophenyl)methanamine hydrochloride</strong> (5.0 g, 18.55 mmol, 1.0 eq), (lS,2S)- Nl,N2- dimethylcyclohexane-l, 2-diamine (0.59 mL 3.71 mmol, 0.2 eq), Sodium ascorbate (368 mg, 1.86 mmol, 0.1 eq), Copper Iodide (530 mg, 2.78 mmol, 0.15 eq), Sodium azide (2.41 g, 37.1 mmol, 2.0 eq) , Et3N (3.11 mL, 22.26 mmol, 1.2 eq) and 2-(prop-2-yn-l-yloxy)tetrahydro- 2//-pyran (2.6 g, 18.55 mmol, 1.0 eq) in Methanol (50 mL) and water (12 mL) were purged with Nitrogen for 5 minutes and heated to 55 C for over night. The reaction mixture was cooled to room temperature and filtered through 413 filter paper. Celite was added and the solvent was removed under reduced pressure and the residue was purified over silica gel (120 g) using dichloromethane/(methanol containing 12 % (v/v) aqueous ammonium hydroxide) as mobile phase. The concentration of (methanol containing 12 % (v/v) aqueous ammonium hydroxide) was gradually increased from 0 % to 6.25 % to afford (4-(4-(((tctrahydro-2//-pyran-2- yl)oxy)mcthyl)- 1 H- 1 ,2,3-triazol- 1 -yl)phcnyl)mcthanaminc (15, 3.54 g, 66%) as a white solid. LCMS m/z: [M + H]+ Calcd for CI5H20N4O2 289.2; Found 289.2.
66%
With copper(l) iodide; sodium azide; (1S,2S)-N,N'-dimethyl-1,2-diaminocyclohexane; sodium L-ascorbate; triethylamine; In methanol; water; at 55℃; for 16h;Inert atmosphere;
A mixture of <strong>[59528-27-7](4-iodophenyl)methanamine hydrochloride</strong> (5.0 g, 18.55 mmol, 1.0 eq), (lS,2S)- Nl,N2- dimethylcyclohexane-l, 2-diamine (0.59 mL 3.71 mmol, 0.2 eq), Sodium ascorbate (368 mg, 1.86 mmol, 0.1 eq), Copper Iodide (530 mg, 2.78 mmol, 0.15 eq), Sodium azide (2.41 g, 37.1 mmol, 2.0 eq) , Et3N (3.11 mL, 22.26 mmol, 1.2 eq) and 2-(prop-2-yn-l-yloxy)tetrahydro- 2 /-pyran (2.6 g, 18.55 mmol, 1.0 eq) in Methanol (50 mL) and water (12 mL) were purged with Nitrogen for 5 minutes and heated to 55 C for over night. The reaction mixture was cooled to room temperature and filtered through 413 filter paper. Celite was added and the solvent was removed under reduced pressure and the residue was purified over silica gel (120 g) using dichloromethane/(methanol containing 12 % (v/v) aqueous ammonium hydroxide) as mobile phase. The concentration of (methanol containing 12 % (v/v) aqueous ammonium hydroxide) was gradually increased from 0 % to 6.25 % to afford (4-(4-(((tctrahydro-2/7-pyran-2- yljoxyjmcthyl)- 1 H- 1 ,2,3-triazol- 1 -yljphcnyljmcthanaminc (15, 3.54 g, 66%) as a white solid. LCMS m/z: [M + H]+ Calcd for C15H20N4O2 289.2; Found 289.2.
With trans-N,N'-dimethyl-1,2-cyclohexyldiamine; copper(l) iodide; sodium azide; sodium L-ascorbate; In methanol; at 55℃;
Z2-Y12 amine: 10g of 2-(2-Propynyloxy) tetrahydopyran (1eq. 71.36mmol) was added to a solution of 5.1g Sodium azide (1.1eq, 78.5mmol), 1.41g Sodium ascorbate (0.1 eq, 7.14mmol), 2.29ml Trans-N-N?-Dimethylcyclohexane- 1,2-diamine (0.25eq, 17.83mmol), 3.4g Copper(I)-iodide (.025eq, 17.83mmol) in 75ml methanol. To this mixture 19.97g of 4 Iodobenzylamide HCl was added. The reaction was stirred overnight at 55C. The solvent was removed under reduced pressure. The crude reaction was purified by liquid chromatography with dichloromethane:ultra (22% MeOH in DCM with 3% NH4OH) mixture 0% --> 40% on silica gel. The product was then reacted with alginate as described below.
With trans-N,N'-dimethyl-1,2-cyclohexyldiamine; copper(l) iodide; sodium azide; sodium L-ascorbate; In methanol; at 55℃;
10 g of 2-(2-propynyloxy) tetrahydropyran (1 equiv. 71.36 mmol) was added to a solution of 5.1g sodium azide (1.1 equiv, 78.5 mmol), 1.41 g sodium ascorbate (0.1 equiv, 7.14 mmol), 2.29 ml trans-N-N?-dimethylcyclohexane-1,2-diamine (0.25 equiv, 17.83 mmol), 3.4 g copper(I)-iodide (0.025 equiv, 17.83 mmol) in 75 ml methanol. To this mixture 19.97 g of 4 iodobenzylamide HCl was added. The reaction was stirred overnight at 55 C. The solvent was removed under reduced pressure. The crude reaction was purified by liquid chromatography with dichloromethane:ultra (22% MeOH in DCM with 3% NH4OH) mixture 0% to 40% on silica gel. The product was then reacted with alginate as described below.
N,N-dimethyl-3-(3-(3-(tetrahydro-2H-pyran-2-yloxy)prop-1-yn-1-yl)-10,11-dihydro-5H-dibenzo[b,f]azepine-5-yl)propane-1-amine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
63%
<strong>[303-49-1]Clomipramine</strong> 2 (0.503 g; 1.43 mmol; 1 equiv) was mixed with Cs2CO3 (1.13 g; 3.57 mmol; 2.5 equiv) and XPhos precatalyst PdG1 (0.0313 g; 0.0428 mmol; 3 mol %) in a glove box. To this anhydrous acetonitrile (4 mL) was added, and the reaction mixture was left with stirring, in the glove box for 25 min before THP protected propargyl alcohol (0.501 g; 3.57 mol; 2.5 equiv) was added. The reaction vessel was sealed, removed from the glove box, and then heated to 80 C for 20 h. After 20 h the reaction mixture was cooled to rt and quenched with saturated NaHCO3 (aq). The mixture was extracted with AcOEt, and the combined organic phases were dried over MgSO4, filtered and evaporated. The product was purified by column chromatography (first CH2Cl2/CH3OH 50:1 then CH2Cl2/CH3OH 20:1). Upon repeated column chromatography, the product was isolated as brown oil in 63% yield (0.375 g; 0.895 mmol). Rf (CH2Cl2/CH3OH 20:1) 0.45. 1H NMR (400 MHz, CDCl3) deltaH 7.16-7.02 (m, 4H), 6.99 (s, 2H), 6.94 (dt, 1H, J = 7.3, 1.1 Hz), 4.88 (t, 1H, J = 3.4 Hz) 4.46 (q, 2H, J = 15.7 Hz), 3.93-3.81 (m, 1H), 3.77 (t, 2H, J = 6.6 Hz), 3.61-3.50 (m, 1H), 3.12 (s, 4H), 2.60 (t, 2H, J = 5.6 Hz), 2.35 (s, 6H), 1.96-1.48 (m, 8H). 13C NMR (100 MHz, CDCl3) deltaC 147.9, 134.9, 134.4, 130.3, 129.7, 126.7, 125.9, 123.3, 123.2, 120.7, 120.4, 96.9, 86.0, 84.4, 62.1, 57.3, 54.9, 48.5, 44.9, 32.6, 31.8, 30.4, 25.5, 19.2 ppm. HRMS (ES+): Calcd for C27H34N2O2H: 419.2699; found 419.2699.
2-chloro-3-(3-((tetrahydro-2H-1-pyran-2-yl)oxy)prop-1-yn-1-yl)pyridine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
89%
To a sealed tubewas added in order 2-chioro-3-iodopyridine (1.25 g, 5.22. mmol), anhydrous lithium chloride(553mg, 13.05 inmol), DMF (13 mL), tetrahydro-2-(2-propynyloxy)-2H-pyran (0.8 mL, 5.7 mrnol), and PdCI2(PPh3)2 (183mg, 0.261 rnmoi). The solution was degassed by bubbling through nitrogen gas for 5 mm. Then TEA (2.2 rnL, 15.7 mrnol) was added, and the reaction mixture was degassed with nitrogen gas for 1 mm. The reaction mixture was sealed and stirred vigorously at 50 C for 15h. Upon completion, the reaction mixture was cooled to 20 C and poured into EtOAc (100 mL).The organic phase was washed five times with water (250 ml. total), once with brine, driedMgSO4, filtered, and concentrated under reduced pressure Purificatin (FCC, Si02, 0% to 20% EtOAc in hexanes) afforded the title compound as an orange oil (1.17 g, 89%). MS (ESI): mass calcd. for C13H14C1N02 251.1, mlz found 252.1 [M÷Hf. ?H NMR (400 MI-k, CDCI3) d 8.33 (dd, J = 48, 2.0 Hz. 1H, 778 (dd, J= 7.7, 1.9 Hz, 1H), 7.20 (dd, J 7.7, 4.8 Hz, IH), 4.94 (t, J 3.4 Hz,IH), 4.55 (ci, J= 0.6 Hz, 211), 390 (ddd, J= 11.5, 8.9, 3.1 Hz, 1ff), 3.65 - 3.51 (m, 1H, 1.93 1.73 (m, 211), 1.73 - 149 (m, 4H).
2-((7,7,7-trifluorohept-2-yn-1-yl)oxy)tetrahydro-2H-pyran[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
72%
DMPU (1.10 g, 8.56 mmol) followed by n-BuLi (548 mg, 8.56 mmol) was added dropwise at -78C to a stirred solution of 2-(prop-2-yn-1-yloxy)tetrahydro-2H-pyran (1.00 g, 7.13 mmol) in THF(20 mL). The solution was stirred for 30 minutes at -78 C, then 4-bromo-1 ,1 ,1-trifluorobutane(1 .43 g, 7.49 mmol) was added dropwise. The cold bath was removed and the reaction mixturewas allowed to attain rt. After 18 NaHCO3 was added and the mixture was diluted with EtOAc(30 mL). The organic phase was separated and washed with water and brine, dried (Na2SO4),filtered and concentrated. The afforded crude was filtered through a silica plug eluting with 4:1Heptane EtOAc, which gave the title compound (1 .29 g 72%).
3-bromo-2-methyl-6-(3-((tetrahydro-2H-pyran-2-yl)oxy)prop-1-yn-1-yl)pyridine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
96%
With bis-triphenylphosphine-palladium(II) chloride; copper(l) iodide; triethylamine; In acetonitrile; at 20℃; for 14h;Inert atmosphere;
Example 1 (1S,3S)-3-((6-(5-(((cyclopentyl(methyl)carbamoyl)oxy)methyl)-1-methyl-1H-1,2,3-triazol-4-yl)-2-methylpyridin-3-yl)oxy)cyclohexane-1-carboxylic acid 1A 3-bromo-2-methyl-6-(3-((tetrahydro-2H-pyran-2-yl)oxy)prop-1-yn-1-yl)pyridine To a solution of <strong>[39919-65-8]2,5-dibromo-6-methyl-pyridine</strong> (5 g, 21.11 mmol) and 2-(prop-2-yn-1-yloxy) tetrahydro-2H-pyran (4.44 g, 31.7 mmol) in MeCN (42.2 mL) was added Et3N (8.83 mL, 63.3 mmol). The solution was degassed under N2, then trans-dichlorobis (triphenylphosphine) palladium (II) chloride (0.74 g, 1.06 mmol) and CuI (0.20 g, 1.06 mmol) were added. The reaction was stirred at rt for 14 h, after which the reaction mixture was filtered through a Celite plug and the plug was washed with EtOAc (2×10 mL). The filtrate was concentrated in vacuo and the residue was chromatographed (SiO2; continuous gradient from 0% to 100% EtOAc in Hexanes for 20 min) to give the title compound as a white solid (6.0 g, 20.3 mmol, 96% yield). 1H NMR (400 MHz, CDCl3) delta 8.65 (d, J=2.0 Hz, 1H), 7.80 (dd, J=8.3, 2.3 Hz, 1H), 7.35 (dd, J=8.4, 0.4 Hz, 1H), 4.91 (t. J=3.3 Hz, 1H), 4.61-4.45 (m, 2H), 3.98-3.81 (m, 1H), 3.66-3.44 (m, 1H), 1.92-1.73 (m, 2H), 1.72-1.52 (m, 2H). LCMS, [M+H]+=298.0.
96%
With bis-triphenylphosphine-palladium(II) chloride; copper(l) iodide; triethylamine; In acetonitrile; at 20℃; for 14h;Inert atmosphere;
To a solution of <strong>[39919-65-8]2,5-dibromo-6-methyl-pyridine</strong> (5 g, 21.11 mmol) and 2-(prop-2-yn-1-yloxy) tetrahydro-2H-pyran (4.44 g, 31.7 mmol) in MeCN (42.2 mL) was added Et3N (8.83 mL, 63.3 mmol). The solution was degassed under N2, then (Ph3P)2PdCl2 (0.74 g, 1.06 mmol) and CuI (0.20 g, 1.06 mmol) were added. The reaction was stirred at RT for 14 h, after which the reaction mixture was filtered through a Celite plug and the plug was washed with EtOAc (2×10 mL). The combined filtrates were concentrated in vacuo and the residue was chromatographed (SiO2; continuous gradient from 0% to 100% EtOAc in hexanes for 20 min) to give the title compound as a white solid (6.0 g, 20.3 mmol, 96% yield). 1H NMR (400 MHz, CDCl3) delta 8.65 (d, J=2.0 Hz, 1H), 7.80 (dd, J=8.3, 2.3 Hz, 1H), 7.35 (dd, J=8.4, 0.4 Hz, 1H), 4.91 (t, J=3.3 Hz, 1H), 4.61-4.45 (m, 2H), 3.98-3.81 (m, 1H), 3.66-3.44 (m, 1H), 1.92-1.73 (m, 2H), 1.72-1.52 (m, 2H). LCMS, [M+H]+=298.0.
96%
With bis-triphenylphosphine-palladium(II) chloride; copper(l) iodide; triethylamine; In acetonitrile; at 20℃; for 14h;Inert atmosphere;
To a solution of <strong>[39919-65-8]2,5-dibromo-6-methyl-pyridine</strong> (5 g, 21.11 mmol) and 2-(prop-2- yn-l-yloxy) tetrahydro-2H-pyran (4.44 g, 31.7 mmol) in MeCN (42.2 mL) was added Et3N (8.83 mL, 63.3 mmol). The solution was degassed under N2, then (Ph3P)2PdCh (0.74 g, 1.06 mmol) and Cul (0.20 g, 1.06 mmol) were added. The reaction was stirred at RT for 14 h, after which the reaction mixture was filtered through a plug of Celite and the plug was washed with EtOAc (2 X 10 mL). The combined filtrates were concentrated in vacuo and the residue was chromatographed (S1O2; continuous gradient from 0% to 100% EtOAc in Hexanes for 20 min) to give the title compound as a white solid (6.0 g, 20.3 mmol, 96 % yield). NMR (400 MHz, CDCl3) d 8.65 (d, 7=2.0 Hz, 1H), 7.80 (dd, 7=8.3, 2.3 Hz, 1H), 7.35 (dd, 7=8.4, 0.4 Hz, 1H), 4.91 (t, 7=3.3 Hz, 1H), 4.61 - 4.45 (m,2H), 3.98 - 3.81 (m, 1H), 3.66 - 3.44 (m, 1H), 1.92 - 1.73 (m, 2H), 1.72 - 1.52 (m, 2H). LCMS, [M+H]+ = 298.0.
96%
With bis-triphenylphosphine-palladium(II) chloride; copper(l) iodide; triethylamine; In acetonitrile; at 20℃; for 14h;Inert atmosphere;
To a solution of <strong>[39919-65-8]2,5-dibromo-6-methyl-pyridine</strong> (5 g, 21.11 mmol) and 2-(prop-2-yn-1-yloxy) tetrahydro-2H-pyran (4.44 g, 31.7 mmol) in MeCN (42.2 mL) was added Et3N (8.83 mL, 63.3 mmol). The solution was degassed under N2, then (Ph3P)2PdCh (0.74 g, 1.06 mmol) and Cul (0.20 g, 1.06 mmol) were added. The reaction was stirred at RT for 14 h, after which the reaction mixture was filtered through a plug of Celite and the plug was washed with EtOAc (2 X 10 mL). The combined filtrates were concentrated in vacuo the residue was chromatographed (SiCh; continuous gradient from 0% to 100% EtOAc in hexanes over 20 min) to give the title compound as a white solid (6.0 g, 20.3 mmol, 96 % yield). NMR (400 MHz, CDCh) d 8.65 (d, J=2.0 Hz, 1H), 7.80 (dd, =8.3,2.3 Hz, 1H), 7.35 (dd, J=8.4, 0.4 Hz, 1H), 4.91 (t, J=3.3 Hz, 1H), 4.61 -4.45 (m, 2H), 3.98 -3.81 (m, 1H), 3.66 -3.44 (m, 1H), 1.92 -1.73 (m, 2H), 1.72 -1.52 (m,2H). LCMS, [M+H]+ = 298.0.
3-bromo-2-(3-((tetrahydro-2H-pyran-2-yl)oxy)prop-1-yn-1-yl)pyridine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
93%
Stage #1: 2,3-dibromopyridine With bis-triphenylphosphine-palladium(II) chloride; copper(l) iodide; triethylamine In tetrahydrofuran at 60℃; for 0.416667h; Inert atmosphere;
Stage #2: 3-(tetrahydropyran-2'-yloxy)propyne With triethylamine In tetrahydrofuran at 60℃; for 20h; Inert atmosphere;
3-Bromo-2-(3-((tetrahydro-2H-pyran-2-yl)oxy)prop-1-yn-1-yl)pyridine (S38)
To a solution of 2,3-dibromopyridine (2.0 g, 8.44 mmol) in freshly distilled THF (24 mL) andEt3N (12 mL) under argon were added PdCl2(PPh3)2 (178 mg, 3 mol%) and CuI (96 mg, 6mol%). The mixture was then stirred at 60 °C. After 25 min, a solution of THP-protectedpropargylic alcohol (1.18 g, 8.44 mmol) in freshly distilled THF (6 mL) and Et3N (3 mL) wasadded to the mixture. After 20 h, the crude reaction mixture was cooled down to roomtemperature, diluted with Et2O, filtered through a pad of Celite and then the solvent wasevaporated under reduced pressure. The crude product was purified by flash chromatographyon silica gel (0-40% EtOAc/pentane) to give S38 (2.08 g, 93% based on the substrate reacted).
(4-iodo-2-(trifluoromethyl)phenyl)methanamine[ No CAS ]
(4-(4-(((tetrahydro-2H-pyran-2-yl)oxy)methyl)-1H-1,2,3-triazol-1-yl)-2-(trifluoromethyl)phenyl)methanamine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
71%
With copper(l) iodide; sodium azide; sodium L-ascorbate; triethylamine In methanol; water at 55℃;
3 Experimental Procedure for (4-(4-(((tetrahydro-2H-pyran-2-yl)oxy)methyl)-1H-1,2,3-triazol-1-yl)-2-(trifluoromethyl)phenyl)methanamine (28)
A mixture of (4-iodo-2-(trifluoromethyl)phenyl)methanamine (3.0 g, 9.97 mmol, 1.0 eq), (1S,2S)-N1,N2- dimethylcyclohexane-1,2-diamine (0.31 mL 1.99 mmol, 0.2 eq), Sodium ascorbate (197 mg, 1.00 mmol, 0.1 eq), Copper Iodide (285 mg, 1.49 mmol, 0.15 eq), Sodium azide (1.30 g, 19.93 mmol, 2.0 eq) , Et3N (1.67 mL, 11.96 mmol, 1.2 eq) and 2-(prop-2-yn-1-yloxy)tetrahydro-2H-pyran (1.40 g, 9.97 mmol, 1.0 eq) in Methanol (24 mL) and water (6 mL) were purged with Nitrogen for 5 minutes and heated to 55 °C for overnight. The reaction mixture was cooled to room temperature and filtered through a plug of Celite and rinsed with Methanol (3 x 50 mL). Celite was added to the filtrate and the solvent was removed under reduced pressure. The residue was purified over silica gel (120 g) using dichloromethane / (methanol containing 12 % (v/v) aqueous ammonium hydroxide) as mobile phase. The concentration of (methanol containing 12 % (v/v) aqueous ammonium hydroxide) was gradually increased from 0 % to 25 % to afford (4-(4-(((tetrahydro-2H-pyran-2-yl)oxy)methyl)-1H- 1,2,3-triazol-1-yl)-2-(trifluoromethyl)phenyl)methanamine (28, 2.53 g, 71%) as a green oil. LCMS m/z: [M + H]+ Calcd for C16H19N402F3 357.2; Found 357.1.
71%
With copper(l) iodide; sodium azide; (1S,2S)-N,N'-dimethyl-1,2-diaminocyclohexane; sodium L-ascorbate In methanol; water at 55℃; Inert atmosphere;
Experimental Procedure for (4-(4-(((tetrahydro-2H-pyran-2-yl)oxy)methyl)-1H-1,2,3-triazol-1-yl)-2-(trifluoromethyl)phenyl)methanamine (28)
(0585) A mixture of (4-iodo-2-(trifluoromethyl)phenyl)methanamine (3.0 g, 9.97 mmol, 1.0 eq), (lS,2S)-Nl,N2- dimethylcyclohexane-l, 2-diamine (0.31 mL 1.99 mmol, 0.2 eq), Sodium ascorbate (197 mg, 1.00 mmol, 0.1 eq), Copper Iodide (285 mg, 1.49 mmol, 0.15 eq), Sodium azide (1.30 g, 19.93 mmol, 2.0 eq) , Et3N (1.67 mL, 11.96 mmol, 1.2 eq) and 2-(prop-2-yn-1-yloxy)tetrahydro-2H-pyran (1.40 g, 9.97 mmol, 1.0 eq) in Methanol (24 mL) and water (6 mL) were purged with Nitrogen for 5 minutes and heated to 55 °C for over night. The reaction mixture was cooled to room temperature and filtered through a plug of Celite and rinsed with Methanol (3 x 50 mL). Celite was added to the filtrate and the solvent was removed under reduced pressure. The residue was purified over silica gel (120 g) using dichloromethane / (methanol containing 12 % (v/v) aqueous ammonium hydroxide) as mobile phase. The concentration of (methanol containing 12 % (v/v) aqueous ammonium hydroxide) was gradually increased from 0 % to 25 % to afford (4-(4-(((tctrahydio-2H-pyran-2-yl)oxy)methyl)-1H-1,2,3-triazol-1-yl)-2- (trifluoromethyl)phenyl)methanamine (28, 2.53 g, 71%) as a green oil. LCMS m/z: [M + H]+ Calcd for Ci6H19N402F3 357.2; Found 357.1.
71%
With copper(l) iodide; sodium azide; sodium ascorbate; (1S,2S)-N,N'-dimethyl-1,2-diaminocyclohexane; triethylamine In methanol; water at 55℃; Inert atmosphere;
1 Experimental Procedure for (4-(4-(((tetrahydro-2H-pyran-2-yl)oxy)methyl)-lH-l,2,3-triazol-l- yl)-2-(trifluoromethyl)phenyl)methanamine (28)
A mixture of (4-iodo-2-(trifluoromethyl)phenyl)methanamine (3.0 g, 9.97 mmol, 1.0 eq), (lS,2S)-Nl,N2- dimethylcyclohexane-l, 2-diamine (0.31 mL 1.99 mmol, 0.2 eq), Sodium ascorbate (197 mg, 1.00 mmol, 0.1 eq), Copper Iodide (285 mg, 1.49 mmol, 0.15 eq), Sodium azide (1.30 g, 19.93 mmol, 2.0 eq) , Et3N (1.67 mL, 11.96 mmol, 1.2 eq) and 2-(prop-2-yn-l- yloxy)tctrahydro-2/7-pyran (1.40 g, 9.97 mmol, 1.0 eq) in Methanol (24 mL) and water (6 mL) were purged with Nitrogen for 5 minutes and heated to 55 °C for over night. The reaction mixture was cooled to room temperature and filtered through a plug of Celite and rinsed with Methanol (3 x 50 mL). Celite was added to the filtrate and the solvent was removed under reduced pressure. The residue was purified over silica gel (120 g) using dichloromethane / (methanol containing 12 % (v/v) aqueous ammonium hydroxide) as mobile phase. The concentration of (methanol containing 12 % (v/v) aqueous ammonium hydroxide) was gradually increased from 0 % to 25 % to afford (4-(4-(((tetrahydro-2 -pyran-2-yl)oxy)methyl)-l - 1,2,3- triazol-l-yl)-2-(trifluoromethyl)phenyl)methanamine (28, 2.53 g, 71%) as a green oil. LCMS m/z: [M + H]+ Calcd for C16H19N4O2F3 357.2; Found 357.1.
71%
With copper(l) iodide; sodium azide; (1S,2S)-N,N'-dimethyl-1,2-diaminocyclohexane; sodium L-ascorbate; triethylamine In methanol; water at 55℃; Inert atmosphere;
Experimental Procedure for (4-(4-(((tetrahydro-2H-pyran-2-yl)oxy)methyl)-lH-l,2,3-triazol-l- yl)-2-(trifluoromethyl)phenyl)methanamine (28)
A mixture of (4-iodo-2-(trifluoromethyl)phenyl)methanamine (3.0 g, 9.97 mmol, 1.0 eq), (lS,2S)-Nl,N2- dimethylcyclohexane-l, 2-diamine (0.31 mL 1.99 mmol, 0.2 eq), Sodium ascorbate (197 mg, 1.00 mmol, 0.1 eq), Copper Iodide (285 mg, 1.49 mmol, 0.15 eq), Sodium azide (1.30 g, 19.93 mmol, 2.0 eq) , Et3N (1.67 mL, 11.96 mmol, 1.2 eq) and 2-(prop-2-yn-l- yloxy)tetrahydro-27/-pyran (1.40 g, 9.97 mmol, 1.0 eq) in Methanol (24 mL) and water (6 mL) were purged with Nitrogen for 5 minutes and heated to 55 °C for over night. The reaction mixture was cooled to room temperature and filtered through a plug of Celite and rinsed with Methanol (3 x 50 mL). Celite was added to the filtrate and the solvent was removed under reduced pressure. The residue was purified over silica gel (120 g) using dichloromethane / (methanol containing 12 % (v/v) aqueous ammonium hydroxide) as mobile phase. The concentration of (methanol containing 12 % (v/v) aqueous ammonium hydroxide) was gradually increased from 0 % to 25 % to afford (4-(4-(((tetrahydro-2 -pyran-2-yl)oxy)methyl)-l - 1,2,3- triazol-l-yl)-2-(trifluoromethyl)phenyl)methanamine (28, 2.53 g, 71%) as a green oil. LCMS m/z: [M + H]+ Calcd for C16H19N4O2F3 357.2; Found 357.1.
71%
With copper(l) iodide; sodium azide; (1S,2S)-N,N'-dimethyl-1,2-diaminocyclohexane; sodium L-ascorbate; triethylamine In methanol; water at 55℃; Inert atmosphere;
Experimental Procedure for (4-(4-(((tetrahydro-2H-pyran-2-yl)oxy)methyl)-lH-l,2,3-triazol-l-
A mixture of (4-iodo-2-(trifluoromethyl)phenyl)methanamine (3.0 g, 9.97 mmol, 1.0 eq), (1 S,2S)- N1,N2- dimethylcyclohexane- 1,2-diamine (0.31 mL 1.99 mmol, 0.2 eq), Sodium ascorbate (197 mg, 1.00 mmol, 0.1 eq), Copper Iodide (285 mg, 1.49 mmol, 0.15 eq), Sodium azide (1.30 g, 19.93 mmol, 2.0 eq) , Et3N (1.67 mL, 11.96 mmol, 1.2 eq) and 2-(prop-2-yn-l-yloxy)tetrahydro-2//-pyran (1.40 g, 9.97 mmol, 1.0 eq) in Methanol (24 mL) and water (6 mL) were purged with Nitrogen for 5 minutes and heated to 55 °C for overnight. The reaction mixture was cooled to room temperature and filtered through a plug of Celite and rinsed with Methanol (3 x 50 mL). Celite was added to the filtrate and the solvent was removed under reduced pressure. The residue was purified over silica gel (120 g) using dichloromethane / (methanol containing 12 % (v/v) aqueous ammonium hydroxide) as mobile phase. The concentration of (methanol containing 12 % (v/v) aqueous ammonium hydroxide) was gradually increased from 0 % to 25 % to afford (4-(4- (((tetrahydro-2//-pyran-2-yl)oxy)methyl )- 1 H- 1 ,2,3 -triazol- 1 -yl)-2-(trifluoromethyl)phenyl)methanamine (28, 2.53 g, 71%) as a green oil. LCMS m z: [M + H]+ Calcd for C16H19N4O2F3 357.2; Found 357.1.
71%
With copper(l) iodide; sodium azide; (1S,2S)-N,N'-dimethyl-1,2-diaminocyclohexane; sodium L-ascorbate; triethylamine In methanol; water at 55℃; Inert atmosphere;
3 Experimental Procedure for (4-(4-(((tetrahydro-2H-pyran-2-yl)oxy)methyl)-lH-l,2,3-triazol-l- yl)-2-(trifluoromethyl)phenyl)methanamine (28)
A mixture of (4-iodo-2-(trifluoromethyl)phenyl)methanamine (3.0 g, 9.97 mmol, 1.0 eq), (1 S,2S)- N1,N2- dimethylcyclohexane- 1,2-diamine (0.31 mL 1.99 mmol, 0.2 eq), Sodium ascorbate (197 mg, 1.00 mmol, 0.1 eq), Copper Iodide (285 mg, 1.49 mmol, 0.15 eq), Sodium azide (1 .30 g, 19.93 mmol, 2.0 eq) , Et3N (1.67 mL, 11.96 mmol, 1.2 eq) and 2-(prop-2-yn-l-yloxy)tetrahydro- 2H -pyran (1.40 g, 9.97 mmol, 1.0 eq) in Methanol (24 mL) and water (6 mL) were purged with Nitrogen for 5 minutes and heated to 55 °C for overnight. The reaction mixture was cooled to room temperature and filtered through a plug of Celite and rinsed with Methanol (3 x 50 mL). Celite was added to the filtrate and the solvent was removed under reduced pressure. The residue was purified over silica gel (120 g) using dichloromethane / (methanol containing 12 % (v/v) aqueous ammonium hydroxide) as mobile phase. The concentration of (methanol containing 12 % (v/v) aqueous ammonium hydroxide) was gradually increased from 0 % to 25 % to afford (4-(4- (((tetrahydro-2H -pyran-2-yl)oxy)methyl)- 1 H- 1 ,2,3-triazol- 1 -yl)-2- (trifluoromethyl)phenyl)methanamine (28, 2.53 g, 71%) as a green oil. LCMS m/z: [M + H]+ Calcd for C16H19N4O2F3 357.2; Found 357.1.
3-(4-(((tetrahydro-2H-pyran-2-yl)oxy)methyl)-1H-1,2,3-triazol-1-yl)propan-1-amine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
66%
With copper(l) iodide; triethylamine; tris[(1-benzyl-1H-1,2,3-triazol-4yl)methyl]amine In methanol; water at 55℃;
3 Experimental Procedure for 3-(4-(((tetrahydro-2H-pyran-2-yl)oxy)methyl)-1H-1,2,3-triazol-1-yl)propan-1-amine (30)
A mixture of 3-azidopropan-1-amine hydrochloride (1.5 g, 14.98 mmol, 1.0 eq), Tris[(l-benzyl- lH-l,2,3-triazol-4-yl)methyl]-amine (1.99 g, 3.75 mmol, 0.25 eq), Copper Iodide (0.29 g, 1.50 mmol, 0.1 eq), and Triethylamine (0.52 mL, 3.75 mmol, 0.25 eq) in Methanol (50 mL) and water (6 mL) were purged with Nitrogen for 5 minutes and cooled to 0 C. 2-(prop-2-yn-l- yloxy)tetrahydro-2H-pyran (2.10 g, 14.98 mmol, 1.0 eq) was added and the reaction mixture was warmed to 55 °C and stirred overnight under Nitrogen atmosphere. The reaction mixture was cooled to room temperature, filtered over a plug of Celite and rinsed with Methanol (3 x 50 mL). Celite (20 g) was added to the filtrate the solvent was removed under reduced pressure. The residue was purified over silica gel (120 g) using dichloromethane/(methanol containing 12 % (v/v) aqueous ammonium hydroxide) as mobile phase. The concentration of (methanol containing 12 % (v/v) aqueous ammonium hydroxide) was gradually increased from 0 % to 20 % to afford 3-(4-(((tetrahydro-2H-pyran-2-yl)oxy)methyl)- 1H- 1 ,2,3-triazol- l-yl)propan- 1 -amine (30, 2.36 g, 66%). LCMS m/z: [M + H]+ Calcd for CiiH2oN402 241.2; Found 241.2.
66%
Stage #1: 3-azido-1-propanamine hydrochloride With copper(l) iodide; triethylamine; tris[(1-benzyl-1H-1,2,3-triazol-4yl)methyl]amine In methanol; water at 0℃; for 0.0833333h; Inert atmosphere;
Stage #2: 3-(tetrahydropyran-2'-yloxy)propyne In methanol; water at 55℃; Inert atmosphere;
1 Experimental Procedure for 3-(4-(((tetrahydro-2H-pyran-2-yl)oxy)methyl)-lH-l,2,3-triazol-l- yl)propan-l -amine (30)
A mixture of 3-azidopropan-l-amine hydrochloride (1.5 g, 14.98 mmol, 1.0 eq), Tris[(l-benzyl- lH-l,2,3-triazol-4-yl)methyl]-amine (1.99 g, 3.75 mmol, 0.25 eq), Copper Iodide (0.29 g, 1.50 mmol, 0.1 eq), and Triethylamine (0.52 mL, 3.75 mmol, 0.25 eq) in Methanol (50 mL) and water (6 mL) were purged with Nitrogen for 5 minutes and cooled to 0 C. 2-(prop-2-yn-l- yloxy)tctrahydro-2/7-pyran (2.10 g, 14.98 mmol, 1.0 eq) was added and the reaction mixture was warmed to 55 °C and stirred over night under Nitrogen atmosphere. The reaction mixture was cooled to room temperature, filtered over a plug of Celite and rinsed with Methanol (3 x 50 mL). Celite (20 g) was added to the filtrate the solvent was removed under reduced pressure. The residue was purified over silica gel (120 g) using dichloromethane/(methanol containing 12 % (v/v) aqueous ammonium hydroxide) as mobile phase. The concentration of (methanol containing 12 % (v/v) aqueous ammonium hydroxide) was gradually increased from 0 % to 20 % to afford 3-(4-(((tetrahydro-27/-pyran-2-yl)oxy)methyl)- IH- 1 ,2,3-triazol- l-yl)propan- 1 -amine (30, 2.36 g, 66%). LCMS m/z: [M + H]+ Calcd for C11H20N4O2 241.2; Found 241.2.
66%
With copper(l) iodide; triethylamine; tris[(1-benzyl-1H-1,2,3-triazol-4yl)methyl]amine In methanol; water at 0 - 55℃; Inert atmosphere;
Experimental Procedure for 3-(4-(((tetrahydro-2H-pyran-2-yl)oxy)methyl)-lH-l,2,3-triazol-l- yl)propan-l -amine (30)
30 A mixture of 3-azidopropan-l-amine hydrochloride (1.5 g, 14.98 mmol, 1.0 eq), Tris[(l-benzyl- lH-l,2,3-triazol-4-yl)methyl]-amine (1.99 g, 3.75 mmol, 0.25 eq), Copper Iodide (0.29 g, 1.50 mmol, 0.1 eq), and Triethylamine (0.52 mL, 3.75 mmol, 0.25 eq) in Methanol (50 mL) and water (6 mL) were purged with Nitrogen for 5 minutes and cooled to 0 C. 2-(prop-2-yn-l- yloxy)tctrahydro-2/7-pyran (2.10 g, 14.98 mmol, 1.0 eq) was added and the reaction mixture was warmed to 55 °C and stirred over night under Nitrogen atmosphere. The reaction mixture was cooled to room temperature, filtered over a plug of Celite and rinsed with Methanol (3 x 50 mL). Celite (20 g) was added to the filtrate the solvent was removed under reduced pressure. The residue was purified over silica gel (120 g) using dichloromethane/(methanol containing 12 % (v/v) aqueous ammonium hydroxide) as mobile phase. The concentration of (methanol containing 12 % (v/v) aqueous ammonium hydroxide) was gradually increased from 0 % to 20 % to afford 3-(4-(((tetrahydro-27/-pyran-2-yl)oxy)methyl)- IH- 1 ,2,3-triazol- l-yl)propan- 1 -amine (30, 2.36 g, 66%). LCMS m/z: [M + H]+ Calcd for C11H20N4O2 241.2; Found 241.2.
66%
Stage #1: 3-azido-1-propanamine hydrochloride With copper(l) iodide; triethylamine; tris[(1-benzyl-1H-1,2,3-triazol-4yl)methyl]amine In methanol; water at 0℃; for 0.0833333h; Inert atmosphere;
Stage #2: 3-(tetrahydropyran-2'-yloxy)propyne In methanol; water at 55℃; Inert atmosphere;
Experimental Procedure for 3-(4-(((tetrahydro-2H-pyran-2-yl)oxy)methyl)-lH-l,2,3-triazol-l- yl)propan-l -amine (30)
30 A mixture of 3-azidopropan-l-amine hydrochloride (1.5 g, 14.98 mmol, 1.0 eq), Tris[(l -benzyl- lH-l,2,3-triazol-4-yl)methyl]-amine (1.99 g, 3.75 mmol, 0.25 eq), Copper Iodide (0.29 g, 1.50 mmol, 0.1 eq), and Triethylamine (0.52 mL, 3.75 mmol, 0.25 eq) in Methanol (50 mL) and water (6 mL) were purged with Nitrogen for 5 minutes and cooled to 0 C. 2-(prop-2-yn-l- y 1 o x y ) t et rah y dro-2 //- py ran (2.10 g, 14.98 mmol, 1.0 eq) was added and the reaction mixture was warmed to 55 °C and stirred overnight under Nitrogen atmosphere. The reaction mixture was cooled to room temperature, filtered over a plug of Celite and rinsed with Methanol (3 x 50 mL). Celite (20 g) was added to the filtrate the solvent was removed under reduced pressure. The residue was purified over silica gel (120 g) using dichloromethane/(methanol containing 12 % (v/v) aqueous ammonium hydroxide) as mobile phase. The concentration of (methanol containing 12 % (v/v) aqueous ammonium hydroxide) was gradually increased from 0 % to 20 % to afford 3-(4- (((tetrahydro-2i/-pyran-2-yl)oxy)methyl)-17T-l,2,3-triazol-l-yl)propan-l-amine (30, 2.36 g,66%). LCMS m/z: [M + H]+ Calcd for C11H20N4O2 241.2; Found 241.2.
66%
With copper(l) iodide; triethylamine; tris[(1-benzyl-1H-1,2,3-triazol-4yl)methyl]amine In methanol; water at 0 - 55℃; Inert atmosphere;
3 Experimental Procedure for 3-(4-(((tetrahydro-2H-pyran-2-yl)oxy)methyl)-lH-l,2,3-triazol-l- yl)propan-l -amine (30)
A mixture of 3-azidopropan-l-amine hydrochloride (1.5 g, 14.98 mmol, 1.0 eq), Tris[(l-benzyl- lH-l,2,3-triazol-4-yl)methyl]-amine (1.99 g, 3.75 mmol, 0.25 eq), Copper Iodide (0.29 g, 1.50 mmol, 0.1 eq), and Triethylamine (0.52 mL, 3.75 mmol, 0.25 eq) in Methanol (50 mL) and water (6 mL) were purged with Nitrogen for 5 minutes and cooled to 0 C. 2-(prop-2-yn-l- yloxy)tetrahydro-2H -pyran (2.10 g, 14.98 mmol, 1.0 eq) was added and the reaction mixture was warmed to 55 °C and stirred overnight under Nitrogen atmosphere. The reaction mixture was cooled to room temperature, filtered over a plug of Celite and rinsed with Methanol (3 x 50 mL). Celite (20 g) was added to the filtrate the solvent was removed under reduced pressure. The residue was purified over silica gel (120 g) using dichloromethane/(methanol containing 12 % (v/v) aqueous ammonium hydroxide) as mobile phase. The concentration of (methanol containing 12 % (v/v) aqueous ammonium hydroxide) was gradually increased from 0 % to 20 % to afford 3 -(4- (((tetrahydro-2H -pyran-2-yl)oxy)methyl)-lH -l,2,3-triazol-l-yl)propan-l-amine (30, 2.36 g, 66%). LCMS m/z: [M + H]+ Calcd for C11H20N4O2 241.2; Found 241.2.
1‐[(tert‐butyldiphenylsilyl)oxy]‐5‐(oxan‐2‐yloxy)pent‐3‐yn‐2‐one[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
90%
To a solution of diisopropylamine (1.6 mL, 11.2 mmol) in THF(8 mL), 1.6M solution of n-BuLi in hexane (6.3 mL, 10.7 mmol) wasadded dropwise at -78 C and the mixture stirred at 0 C for 15 min. At-78 C, a solution of propyne 3 (1.57 g, 11.2 mmol) in THF (8 mL) wasadded and the mixture stirred for 30 min. Still at -78 C, a solution ofWeinreb amide 4 (2.00 g, 5.6 mmol) in THF (10 mL) was added. Themixture was stirred at -78 C for 20 min and then at 0 C until allstarting material had been consumed (TLC), approximately 2 h. Themixture was quenched with a saturated aqueous NH4Cl solution(20 mL) and extracted with CH2Cl2 (3×20 mL). The combined organiclayers were dried with anhydrous Na2SO4, filtered and concentratedunder vacuum. The residue was purified by flash column chromatography(90:10 Hexane/EtOAc) to afford the ketone 5 (2.20 g, 90%) as acolourless viscous oil
With potassium carbonate In N,N-dimethyl-formamide at 20℃;
General procedure for the synthesis of alkynes (6a-e)
General procedure: A solution of propargyl bromide (2 equiv.) in dry DMF was added dropwise to a stirred suspension of substituted phenol (1 equiv.) and anhydrous K2CO3 (6 equiv.) in dry DMF at room temperature for 8-10 h. The progress of the reaction was monitored by thin layer chromatography and after completion of the reaction; it was poured into ice-cold water (100 mL) and extracted with chloroform (3 × 60 mL). The combined organic layer was washed with water (5 × 100). The chloroform layer was driedover anhydrous Na2SO4 and excess of solvent was removed under reduced pressure. The crude product, thus obtained was purified over a silica gel column using EtOAc/hexane as eluent (Scheme-I).
(1R,5S,6R)-2,7,7-trimethyl-6-(3-((tetrahydro-2H-pyran-2-yl)oxy)prop-1-yn-1-yl)bicyclo[3.1.1]hept-2-en-6-ol[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
67%
Stage #1: (-)-Verbenone In cyclohexane for 3h; Schlenk technique; Inert atmosphere; UV-irradiation;
Stage #2: 3-(tetrahydropyran-2'-yloxy)propyne With ethylmagnesium bromide In tetrahydrofuran at 50℃; for 20h;






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +
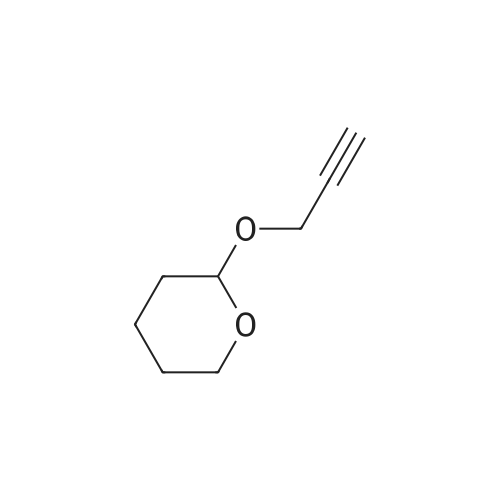

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping