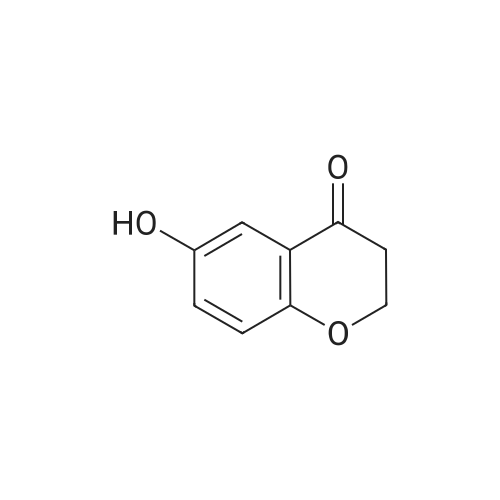
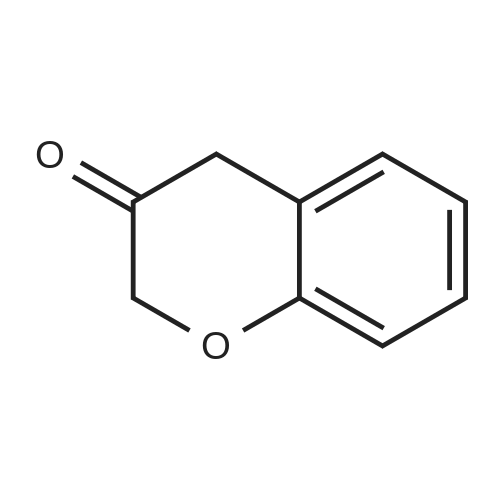
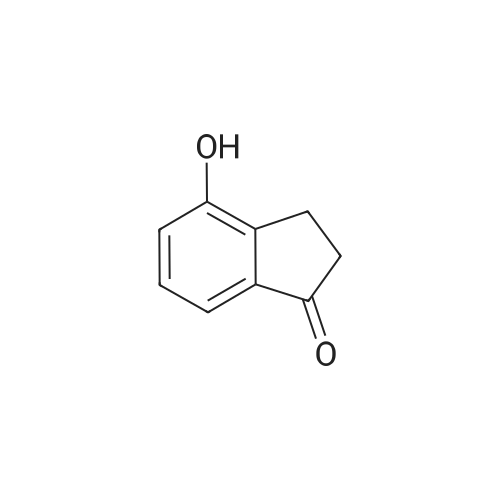
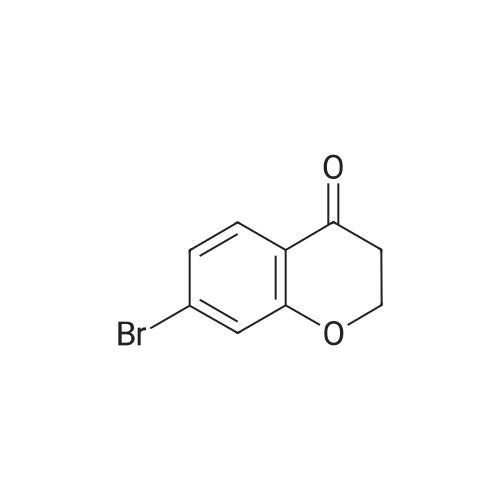
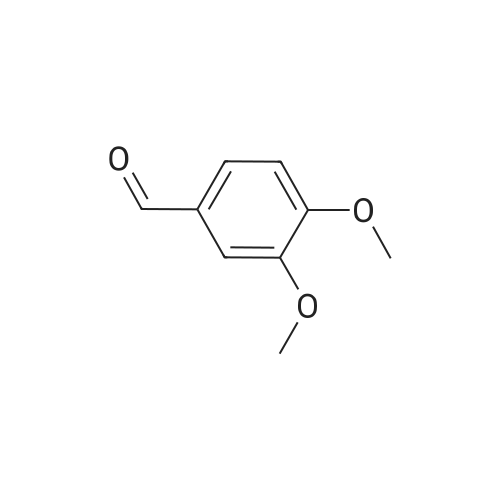
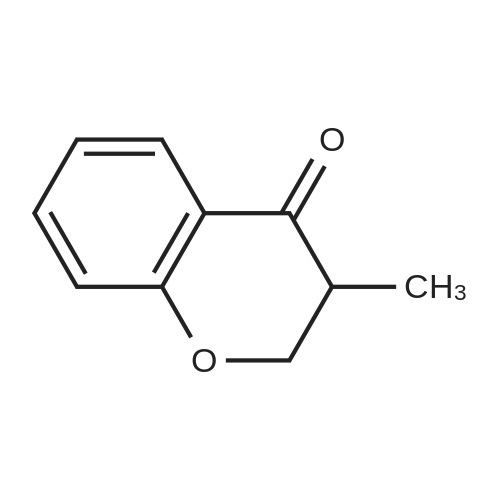
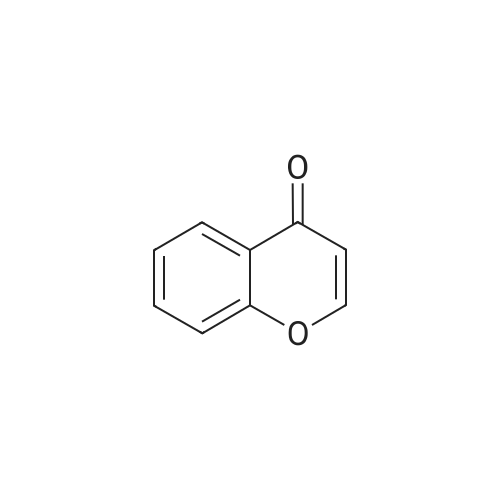
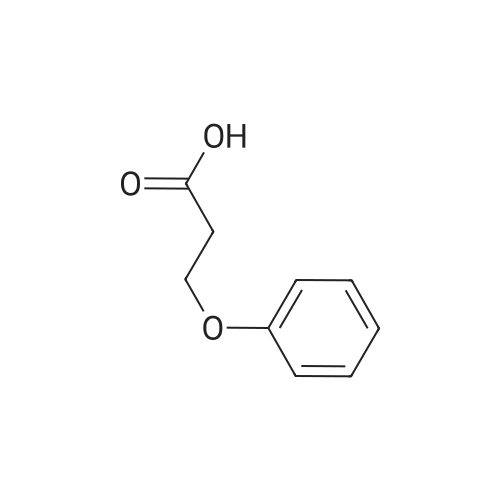
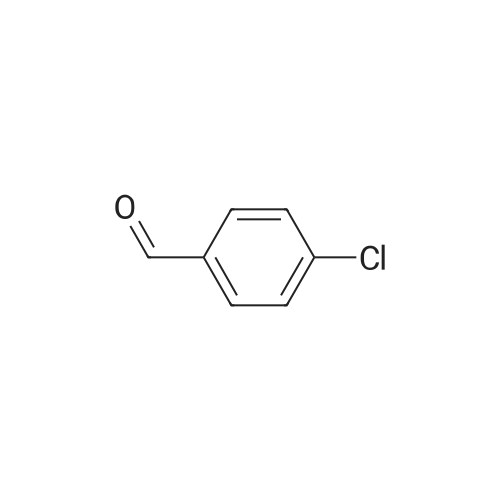
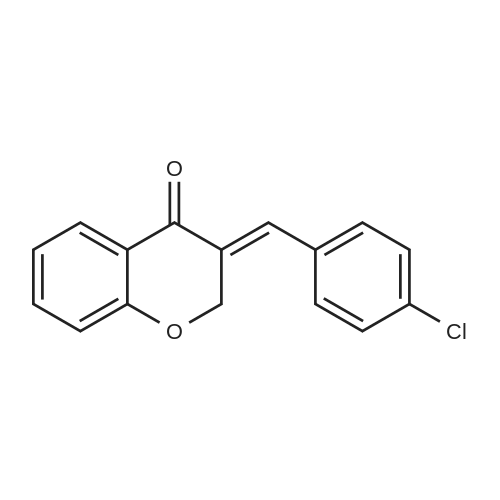
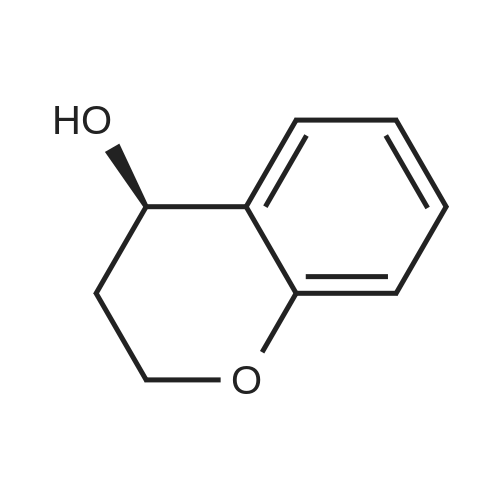
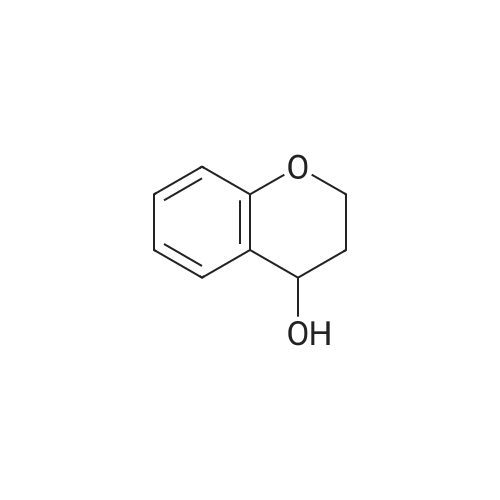
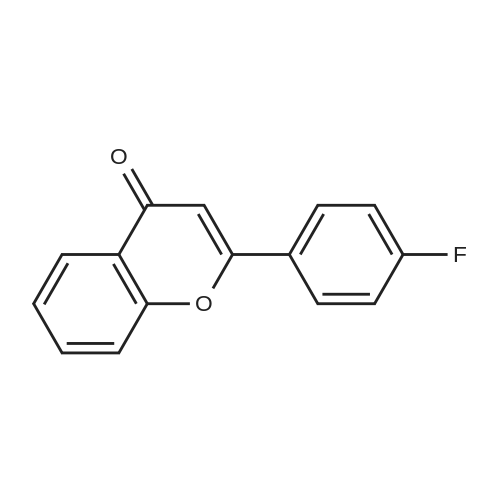
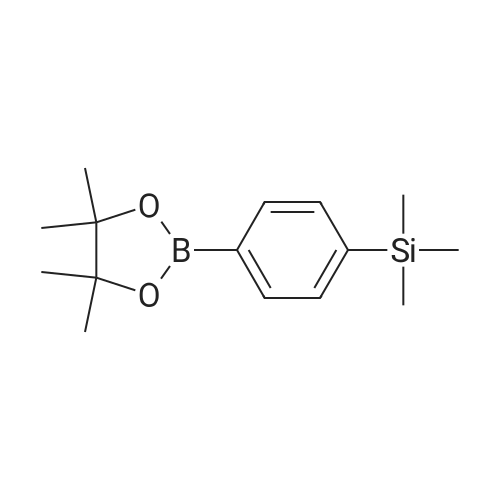
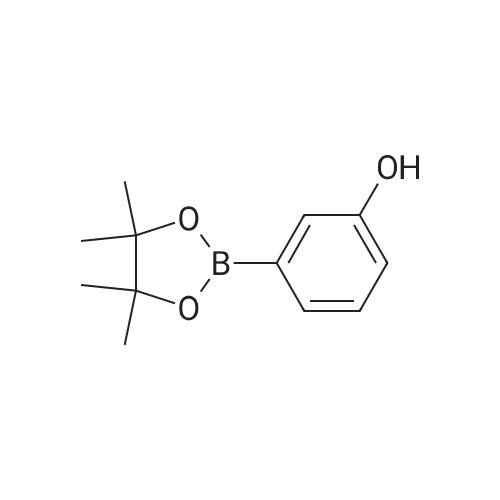
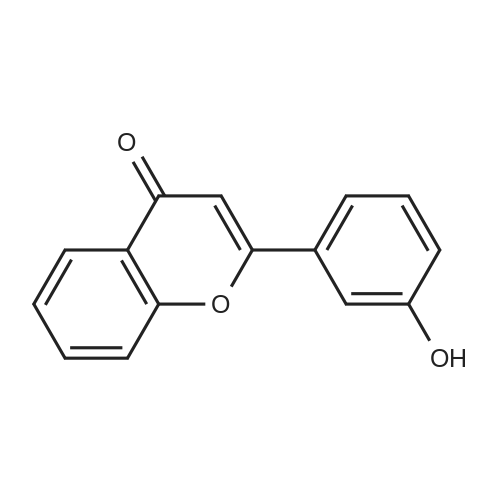
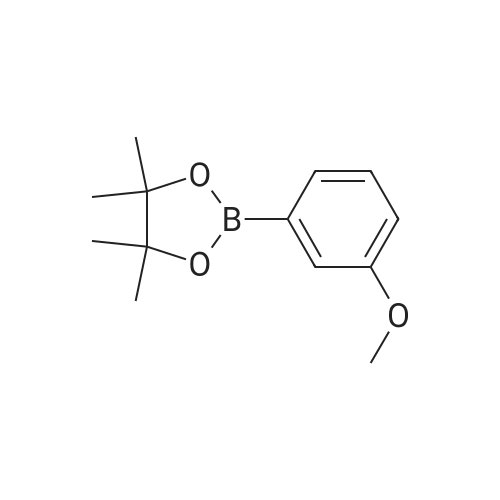
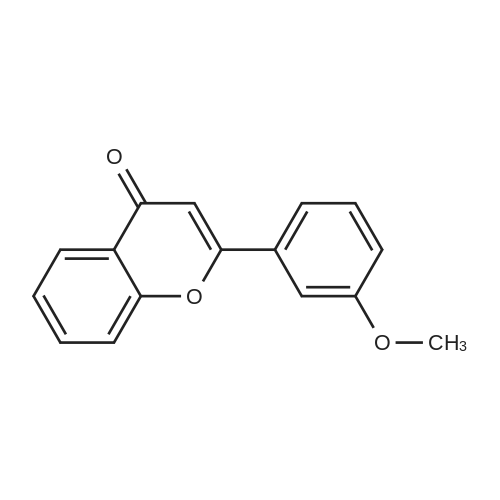
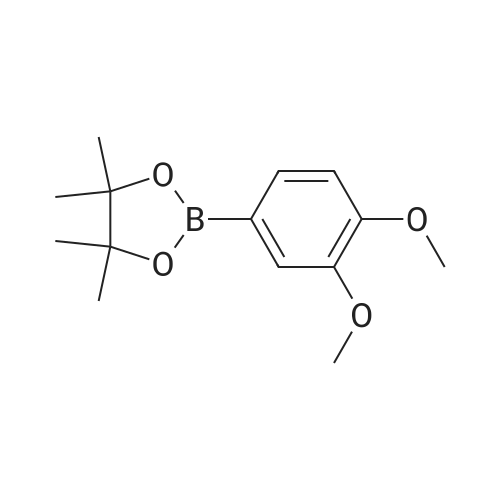
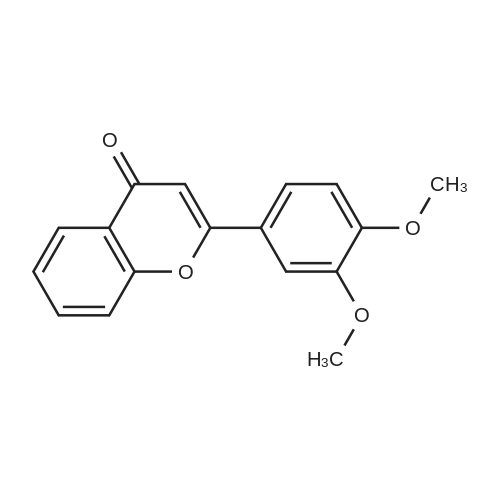
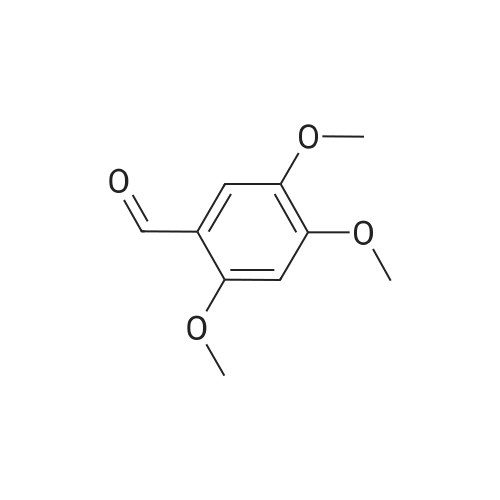
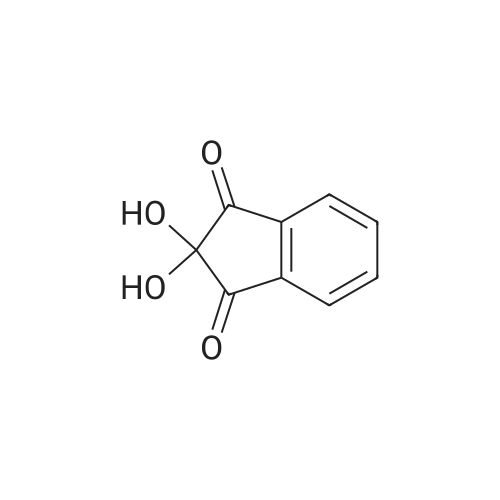
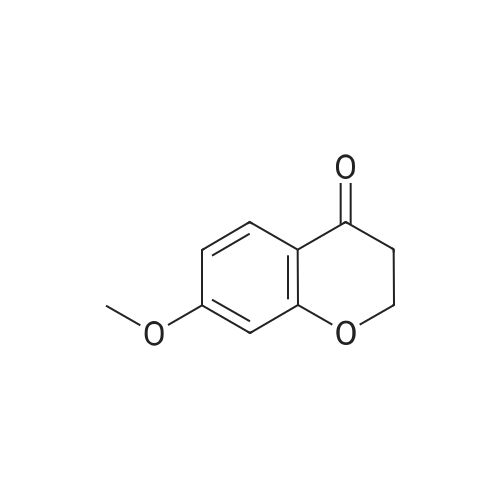
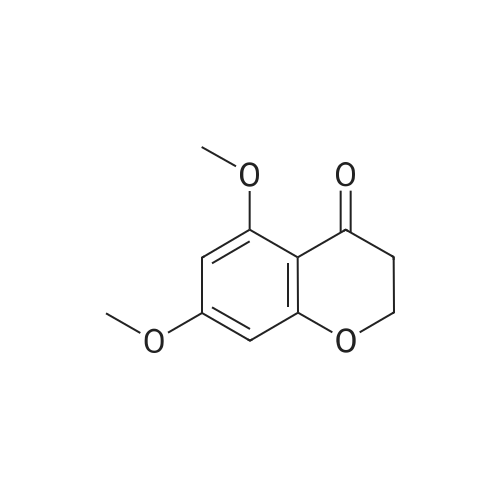
| 99% |
With formic acid; dichloro(hexamethylbenzene)ruthenium(II) dimer; (S)-2-piperidinemethanethiol hydrochloride; triethylamine at 30℃; for 24h; Inert atmosphere; optical yield given as %ee; enantioselective reaction; |
|
| 98% |
With hydrogen In methanol at 50℃; for 15h; |
17
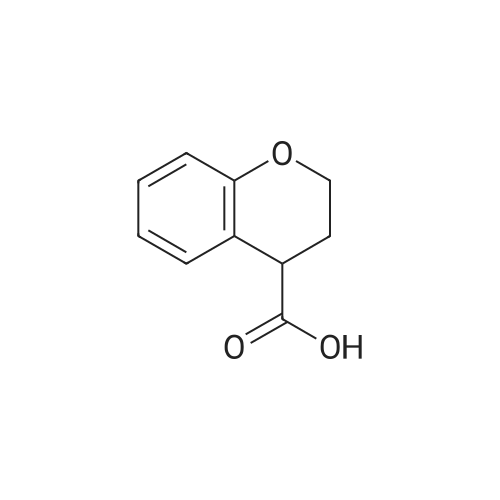
In a stainless steel autoclave, Ru(OTf)[(S,S)-Tsdpen](p-cymene) (2.4 mg, 3.3 μmol), Yb(OTf)3 (0.62 mg, 1 μmol), and 4-chromanone (1.48 g, 10 mmol) were charged, followed by purging with argon. Then, 0.7 ml of methanol was added, and the autoclave was pressurized with hydrogen, followed by ten times of purging. Then, hydrogen was charged to 15 atm to initiate reaction. After stirring at 50° C. for 15 hours, the reaction pressure was returned to normal pressure. 1HNMR and HPLC analysis of the product showed that (S)-4-chromanol with 97% ee was produced in a yield of 98%. |
| 98% |
With hydrogen In methanol at 50℃; for 15h; |
23 Synthesis of (S)-4-chromanol by Hydrogenation Reaction of 4-chromanone
Example 23 Synthesis of (S)-4-chromanol by Hydrogenation Reaction of 4-chromanone In a stainless steel autoclave, Ru(OSO2CH3) [(S,S)-Tsdpen](p-cymene) (1.4 mg, 2 μmol) and 4-chromanone (300 mg, 2 mmol) were charged, followed by purging with argon. Then, 0.1 ml of methanol was added, and the autoclave was pressurized with hydrogen, followed by ten times of purging. Then, hydrogen was charged to 15 atm to initiate reaction. After stirring at 50° C. for 15 hours, the reaction pressure was returned to normal pressure. 1H-NMR and HPLC analysis of the product showed that (S)-4-chromanol with 95% ee was produced in a yield of 98%. |
| 97% |
With formic acid; C29H38N3O2RuS; triethylamine at 60℃; for 30h; Inert atmosphere; enantioselective reaction; |
|
| 96% |
With potassium methylate In water monomer; toluene at 50℃; for 24h; |
F.1.2; F.1.3
The reaction was performed under the same conditions as those in Example F-1-1, except that 32 mg (100 μmol) of tetrabutylammonium bromide as the phase-transfer catalyst was added. HPLC analysis of the reactant confirmed that 4-chromanol with optical purity of 97% ee was produced in 99% yield, demonstrating that the activity and enantioselectivity of the main catalyst is improved by the addition of a phase-transfer catalyst.; Example F-1-3; Asymmetric Reduction of 4-chromanone Using Cp*IrCl[(S,S)-MsDPEN] Catalyst with Addition of Phase-Transfer Catalyst and Potassium Formate Solution as Hydrogen Source (Reaction at S/C=10,000); 2.02 g (24.0 mmol) of HCOOK as the hydrogen source, 1.305 mg (2.0 μmol) of Cp*IrCl[(S,S)-MsDPEN] as the catalyst, 64.5 mg (0.20 mmol) of tetrabutylammonium bromide as the phase-transfer catalyst, and 2.96 g (20.0 mmol) of 4-chromanone were introduced in a 20 mL Schlenk tube, and the mixture was subjected to argon substitution. 4 mL of water and 2 ml of toluene were added and the resulting mixture was maintained at 50° C. for 24 hr while stirring. The organic phase was washed three times with 5 mL of water, and the toluene was distilled off under reduced pressure to give an optically-active alcohol. HPLC analysis of the reactant confirmed that 4-chromanol with optical purity of 98% ee was produced in 96% yield, demonstrating that the use of a potassium formate solution as the hydrogen source and the catalyst system in which Cp*IrCl[(S,S)-MsDPEN] catalyst is combined with tetrabutylammonium bromide exhibits high efficiency. |
| 95% |
With hydrogen In methanol at 50℃; for 15h; |
15; 16; 18; 19; 20 Synthesis of (S)-4-chromanol by Hydrogenation Reaction of 4-chromanone
Example 15 Synthesis of (S)-4-chromanol by Hydrogenation Reaction of 4-chromanone In a stainless steel autoclave, Ru(OTf)[(S,S)-Tsdpen](p-cymene) (2.4 mg, 3.3 μmol) and 4-chromanone (1.48 g, 10 mmol) were charged, followed by purging with argon. Then, 0.5 ml of methanol was added, and the autoclave was pressurized with hydrogen, followed by ten times of purging. Then, hydrogen was charged to 15 atm to initiate reaction. After stirring at 50° C. for 15 hours, the reaction pressure was returned to normal pressure. 1H-NMR and HPLC analysis of the product showed that (S)-4-chromanol with 91% ee was produced in a yield of 100%. The spectral data of the resultant alcohol compound was as follows: 1H-NMR (400 MHz, CDCl3) δ 1.99 (m, 1H, CHCHOH), 2.08 (m, 1H, CHCHOH), 2.31 (br, 1H, OH), 4.23 (m, 2H, C2OC), 4.74 (m, 1H, COH), 6.81-6.92 (m, 2H, aromatic H), 7.17-7.30 (m, 2H, aromatic H); HPLC (CHIRALCEL OJ-H; solvent, hexane/2-propanol=99/1; flow rate, 1.5 ml/min; temperature, 35°; UV wavelength, 220 nm); tR of (S)-4-chromanol, 26.7 minutes; tR of (R)-4-chromanol, 30.8 minutes; tR of 4-chromanone, 11.8 minutes; specific rotatory power [α]25D-72° (c0.5, C2H5OH); document value, [α]25D+80.40 (c0.5, C2H5OH), 100% ee (R), J. Am. Chem. Soc., 1993, 115, 3318. Example 16Synthesis of (S)-4-chromanol by Hydrogenation Reaction of 4-chromanoneIn a stainless steel autoclave, Ru(OTf)[(S,S)-Tsdpen](p-cymene) (2.4 mg, 3.3 μmol) and 4-chromanone (1.48 g, 10 mmol) were charged, followed by purging with argon. Then, 0.7 ml of methanol was added, and the autoclave was pressurized with hydrogen, followed by ten times of purging. Then, hydrogen was charged to 15 atm to initiate reaction. After stirring at 50° C. for 15 hours, the reaction pressure was returned to normal pressure. 1HNMR and HPLC analysis of the product showed that (S)-4-chromanol with 93% ee was produced in a yield of 100%.; Examples 18 to 20(S)-4-chromanol was synthesized by reaction under the same conditions as in Example 15 except that the substrate/catalyst ratio and the hydrogen pressure were changed. The results are summarized in Table 3. |
| 95% |
With hydrogen In methanol at 50℃; for 15h; |
24 Synthesis of (S)-4-chromanol by Hydrogenation Reaction of 4-chromanone
In a stainless steel autoclave, Cp*Ir(OTf)[(S,S)-Tsdpen] (1.6 mg, 2 μmol) and 4-chromanone (300 mg, 2 mmol) were charged, followed by purging with argon. Then, 0.4 ml of methanol was added, and the autoclave was pressurized with hydrogen, followed by ten times of purging. Then, hydrogen was charged to 30 atm to initiate reaction. After stirring at 50° C. for 15 hours, the reaction pressure was returned to normal pressure. 1HNMR and HPLC analysis of the product showed that (S)-4-chromanol with 95% ee was produced in a yield of 95%. |
| 95% |
Stage #1: 2,3-dihydro-4H-1-benzopyran-4-one With 1-((1R,2R)-2-(benzylamino)cyclohexyl)-3-(3,5-bis(trifluoromethyl)phenyl)thiourea; benzo[1,3,2]dioxaborole In toluene at -46℃; for 24h; Molecular sieve; Inert atmosphere;
Stage #2: With methanol; sodium hydroxide In toluene at -46 - 20℃; optical yield given as %ee; enantioselective reaction; |
|
| 93% |
With hydrogen In methanol at 60℃; for 15h; |
|
| 93% |
With anhydrous sodium perchlorate; hydrogen In methanol at 30℃; for 23h; |
47
An example of synthesizing optically active 4-chromanol by hydrogenation of chromanone is described below. A 50 mL stainless steel autoclave was charged with RuCl[(S,S)-Tsdpen](p-cymene) (1.0 mg, 0.0016 mmol) and NaClO4 (10 mg, 0.08 mmol) under argon. Then 4-chromanone (1185 mg, 8.0 mmol) and methanol (16 mL) were added thereto. After pressurization with hydrogen, substitution was conducted five times. Hydrogen was charged to 50 atm to initiate reaction. After stirring for 23 hours at 30°C, the reaction pressure was reduced to normal. The product was analyzed by 1H-NMR and HPLC reporting synthesis of (S)-4-chromanol in 97% ee and 93% yield. |
| 89% |
With potassium methylate In water monomer; toluene at 50℃; for 24h; |
F.1.1
3.36 g (40.0 mmol) of HCOOK as the hydrogen source, 1.044 mg (1.6 μmol) of Cp*IrCl[(S,S)-MsDPEN] as the catalyst, and 1.185 g (8.0 mmol) of 4-chromanone were introduced in a 20 mL Schlenk tube, and the mixture was subjected to argon substitution. 2 mL of water and 2 ml of toluene were added and the resulting mixture was maintained at 50° C. for 24 hr while stirring. The organic phase was washed three times with 3 mL of water, and the toluene was distilled off under reduced pressure to give an optically-active alcohol. HPLC analysis of the reactant confirmed that 4-chromanol with optical purity of 95% ee was produced in 89% yield. |
| 85% |
With hydrogen In methanol at 30℃; for 23h; |
46
An example of synthesizing optically active 4-chromanol by hydrogenation of chromanone is described below. A 50 mL stainless steel autoclave was charged with RuCl[(S,S)-Tsdpen](p-cymene) (1.0mg, 0.0016 mmol) under argon. Then 4-chromanone (474 mg, 3.2 mmol) and methanol (6.4 mL) were added thereto. After pressurization with hydrogen, substitution was conducted five times. Hydrogen was charged to 50 atm to initiate reaction. After stirring for 23 hours at 30°C, the reaction pressure was reduced to normal. The product was analyzed by 1H-NMR and HPLC reporting synthesis of (S)-4-chromanol in 97% ee and 85% yield. |
| 83% |
With borane-N,N-diethylaniline complex; (R)-tetrahydro-1-methyl-3,3-diphenyl-1H,3H-pyrrolo[1,2-c]-[1,3,2]oxazaborole In toluene at 25℃; for 4h; Inert atmosphere; optical yield given as %ee; |
|
| 80% |
With 1,4-butenediol; Wills catalyst; potassium hydroxide In tetrahydrofuran at -10℃; for 4h; Inert atmosphere; enantioselective reaction; |
|
| 77% |
With hydrogen In methanol at 50℃; for 15h; |
22 Synthesis of (S)-4-chromanol by Hydrogenation Reaction of 4-chromanone
Example 22 Synthesis of (S)-4-chromanol by Hydrogenation Reaction of 4-chromanone In a stainless steel autoclave, Ru[OSO2(p-NO2Ph)][(S,S)-Tsdpen](p-cymene) (2.6 mg, 3.3 μmol) and 4-chromanone (1.48 g, 10 mmol) were charged, followed by purging with argon. Then, 0.5 ml of methanol was added, and the autoclave was pressurized with hydrogen, followed by ten times of purging. Then, hydrogen was charged to 15 atm to initiate reaction. After stirring at 50° C. for 15 hours, the reaction pressure was returned to normal pressure. 1H-NMR and HPLC analysis of the product showed that (S)-4-chromanol with 91% ee was produced in a yield of 77%. |
| 70% |
Stage #1: 2,3-dihydro-4H-1-benzopyran-4-one With dichloro(p-cymene)ruthenium(II) dimer; isopropanol; lithium chloride; Boc-L-alanine(2S)-hydroxypropylamide In tetrahydrofuran at 30℃; for 0.25h; Inert atmosphere;
Stage #2: With sodium isopropanolate In tetrahydrofuran at 30℃; for 0.5h; Inert atmosphere; optical yield given as %ee; enantioselective reaction; |
|
| 70% |
With di-μ-chlorobis-[(η6-p-cymene)chlororuthenium(II)]; N-(tert-butoxycarbonyl)-L-valine-(6-amido-1-O-benzyl-6-deoxy-2,3-O-isopropylidene-α-D-mannofuranose); potassium-t-butoxide; lithium chloride In tetrahydrofuran; isopropanol at 20℃; for 3h; enantioselective reaction; |
|
| 44% |
With Almag CRED A131; NADP; glucose dehydrogenase In water monomer; dimethyl sulfoxide at 30℃; Enzymatic reaction; |
4.4
General procedure: Almac CRED (200 mg),NADP or NAD (10 mg), GDH (20 mg) and glucose (1.5 equiv) weremeasured into a 100 mL round bottomed flask then dissolved in0.1 M potassium phosphate buffer (pH 7, ca. 50 mL) and stirredat 30 C. Next, a solution of ketone (900-1700 mg) in DMSO(2.5-5 mL, depending on solubility) was added to the reactionand this was allowed to stir overnight under pH-stat control (pH7.0, adjusted with 1 M NaOH solution). The following day, reactionprogress was checked by 1H NMR. If not complete, additional CRED(100-200 mg), NADP or NAD (10 mg) and GDH (10 mg) wereadded and stirring was continued. This was repeated until the reactionreached completion or had stalled and would not go any further. |
| 39% |
With potassium hydroxide; (+)-Norephedrine In isopropanol at 20℃; |
|
| 19% |
With potassium methylate In water monomer; toluene at 50℃; for 24h; |
F.9
The reaction was performed under the same conditions as those in Example F-1-1, except that 1.165 mg (1.6 μmol) of Cp*IrCl[(S,S)-TsDPEN] was used as the catalyst. HPLC analysis of the reactant confirmed that 4-chromanol with optical purity of 94% ee was produced in 19% yield. Comparison with Example F-1-1 demonstrated that it is superior to have a methyl group as the substituent on the sulfonyl group. |
| 10% |
With formic acid; triethylamine at 50℃; for 24h; |
F.2.1; F.2.2
A formic acid-triethylamine mixture (molar ratio of HCOOH:Et3N:substrate=3.1:2.6:1) as the hydrogen source, 1.044 mg (1.6 μmol) of Cp*IrCl[(S,S)-MsDPEN] as the catalyst, and 1.185 g (8.0 mmol) of 4-chromanone were introduced in a 20 mL Schlenk tube, and the mixture was subjected to argon substitution, then maintained at 50° C. for 24 hr while stirring. HPLC analysis of the reactant confirmed that 4-chromanol with optical purity of 88% ee was produced in 10% yield.; Example F-2-2; Asymmetric Reduction of 4-chromanone Using Cp*IrCl[(S,S)-MsDPEN] Catalyst and Formic Acid-Triethylamine Mixture as Hydrogen Source; The reaction was performed under the same conditions as those in Example F-2-1, except that the amount of the catalyst was 2.610 mg (4.0 μmol). HPLC analysis of the reactant confirmed that 4-chromanol with optical purity of 95% ee was produced in 89% yield. |
| 7% |
With hydrogen In methanol at 50℃; for 15h; |
1
In a stainless steel autoclave, Ru[(S,S)-Tsdpen](p-cymene) (1.2 mg, 2 μmol) and 4-chromanone (0.3 g, 2 mmol) were charged, followed by purging with argon. Then, 2 ml of methanol was added, and the autoclave was pressurized with hydrogen, followed by ten times of purging. Then, hydrogen was charged to 30 atm to initiate reaction. After stirring at 50° C. for 15 hours, the reaction pressure was returned to normal pressure. 1HNMR and HPLC analysis of the product showed that (S)-4-chromanol with 86% ee was produced in a yield of 7%. As a result, it was found that the sulfonate catalyst of the present invention exhibits excellent activity as compared with a conventional well-known ruthenium complex. |
|
With borane N,N-diethylaniline complex; Trimethyl borate; chiral 2-(diphenylhydroxymethyl)pyrrolidine In toluene at 23℃; |
|
|
With sodium isopropanolate; isopropanol; Boc-L-alanine(2S)-hydroxypropylamide at 20℃; for 1h; |
|
|
Multi-step reaction with 2 steps
1: [Rh(cod)Cl]2; asymmetric tetraphenyl-tetraoxa-phosphazulene derivative / toluene / 24 h / 0 °C
2: 1 percent p-TsOH / methanol |
|
|
With N,N-diethylaniline borane; (R)-(+)-α,α-diphenyl-2-pyrrolidinemethanol In tert-butyl methyl ether at 45℃; for 1.5h; |
|
| 97.9 % ee |
With (S,S)-RuCl(η6-CH3C6H4CH2CH2CH2CH2NHCH(C6H5)CH(C6H5)NSO2Ts); C32H35ClN2O2RuS; hydrogen In methanol at 60℃; for 19h; Inert atmosphere; Autoclave; enantiospecific reaction; |
39 Asymmetric hydrogenation of 4-chromanone using RuCl(Tol-C4-teth-(S,S)-Tsdpen)
Example 39 Asymmetric hydrogenation of 4-chromanone using RuCl(Tol-C4-teth-(S,S)-Tsdpen) In a 100-ml autoclave, 3.1 mg (0.00478 mmol, S/C=1000) of RuCl(Tol-C4-teth-(S,S)-Tsdpen) was placed, followed by purging with nitrogen. Subsequently, 0.72 g (5.0 mmol) of 4-chromanone and 4.4 ml of methanol were added thereto, and hydrogen was introduced to a pressure of 3.0 MPa, followed by stirring at 60° C. for 19 hours. The result of GC analysis of the reaction liquid showed that (S)-4-chromanol was obtained with a conversion of 100% and an optical purity of 97.9% ee.; Example 40 Asymmetric hydrogenation of 4-chromanone using RuCl(Tol-C4-teth-(S,S)-Msdpen) In a 100-ml autoclave, 2.8 mg (0.00489 mmol, S/C=1000) of RuCl(Tol-C4-teth-(S,S)-Msdpen) was placed, followed by purging with nitrogen. Subsequently, 0.72 g (5.0 mmol) of 4-chromanone and 4.4 ml of methanol were added thereto, and hydrogen was introduced to a pressure of 3.0 MPa, followed by stirring at 60° C. for 19 hours. The result of GC analysis of the reaction liquid showed that (S)-4-chromanol was obtained with a conversion of 100% and an optical purity of 97.6% ee. |
|
Multi-step reaction with 2 steps
1: sodium tetrahydridoborate
2: Burkholderia cepacia Amano-PS lipase; immobilized on diatomaceous earth / tert-butyl methyl ether / 14 h / 23 °C / Resolution of racemate; Enzymatic reaction |
|
| 96 % ee |
With di-μ-chlorobis-[(η6-p-cymene)chlororuthenium(II)]; anhydrous sodium formate; chitosan In water monomer; isopropanol at 20℃; for 46h; Green chemistry; enantioselective reaction; |
|
| 96 % ee |
With di-μ-chlorobis-[(η6-p-cymene)chlororuthenium(II)]; anhydrous sodium formate; chitosan In water monomer; isopropanol at 23℃; for 46h; Sealed tube; enantioselective reaction; |
|
|
Multi-step reaction with 3 steps
1: sodium tetrahydridoborate; methanol / 0.67 h / 0 - 20 °C / Inert atmosphere
2: (OC-6-23)-[2-[6-[(amino-κN)methyl]-2-pyridinyl-κN]-5-methylphenyl-κC][1,1'-(1,4-butanediyl)bis[1,1-diphenylphosphine-κP]]chlororuthenium(II); copper chloride (I); sodium tertiary butoxide; 1,2-bis((2R,5R)-2,5-diphenylphospholan-1-yl)ethane / toluene / 14 h / 20 °C / Glovebox; Inert atmosphere
3: N,N,N-tributylbutan-1-aminium fluoride / tetrahydrofuran / 0.5 h / 20 °C / Inert atmosphere |
|
| 96 % ee |
With di-μ-chlorobis-[(η6-p-cymene)chlororuthenium(II)]; anhydrous sodium formate; chitosan In water monomer; isopropanol at 20℃; for 24h; enantioselective reaction; |
|
| 99 % ee |
With di-μ-chlorobis-[(η6-p-cymene)chlororuthenium(II)]; anhydrous sodium formate In water monomer; isopropanol at 50℃; for 5h; |
|






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






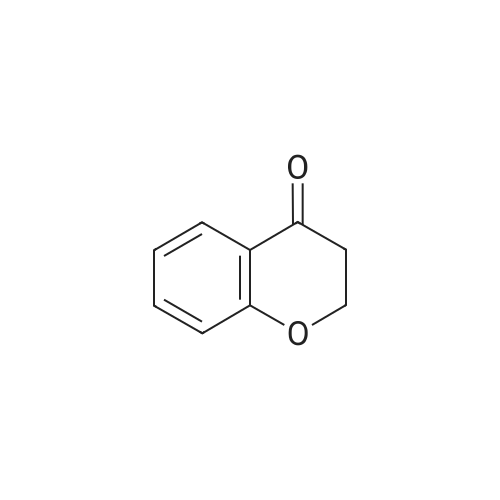

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping