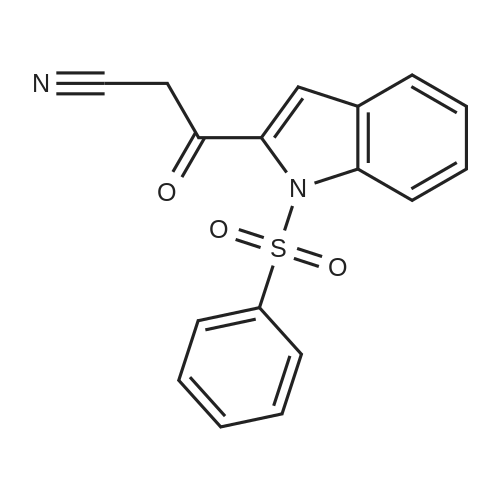
There will be a HazMat fee per item when shipping a dangerous goods. The HazMat fee will be charged to your UPS/DHL/FedEx collect account or added to the invoice unless the package is shipped via Ground service. Ship by air in Excepted Quantity (each bottle), which is up to 1g/1mL for class 6.1 packing group I or II, and up to 25g/25ml for all other HazMat items.
Type
HazMat fee for 500 gram (Estimated)
Excepted Quantity
USD 0.00
Limited Quantity
USD 15-60
Inaccessible (Haz class 6.1), Domestic
USD 80+
Inaccessible (Haz class 6.1), International
USD 150+
Accessible (Haz class 3, 4, 5 or 8), Domestic
USD 100+
Accessible (Haz class 3, 4, 5 or 8), International
USD 200+
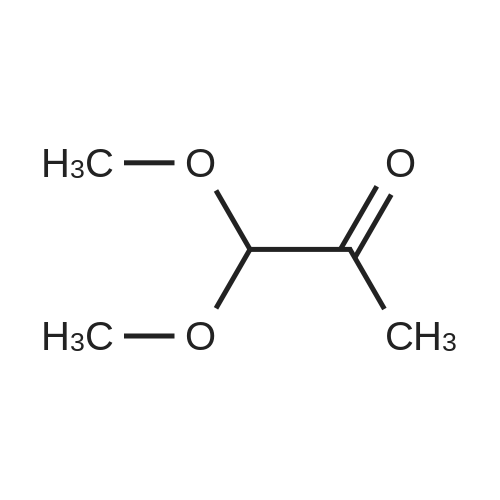
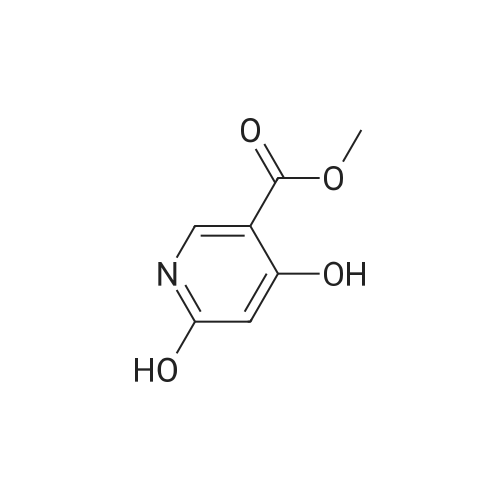
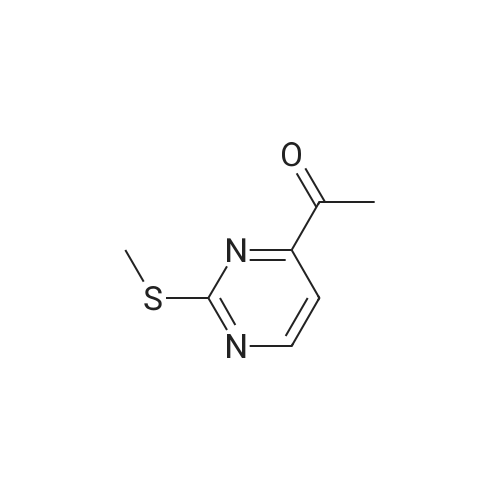
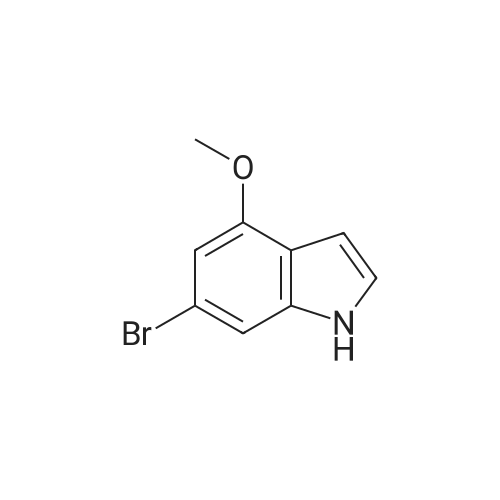
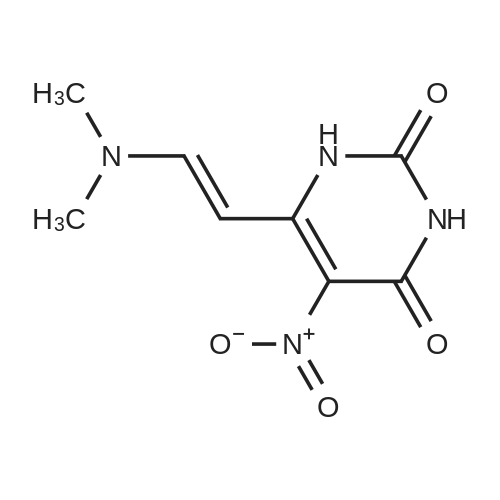
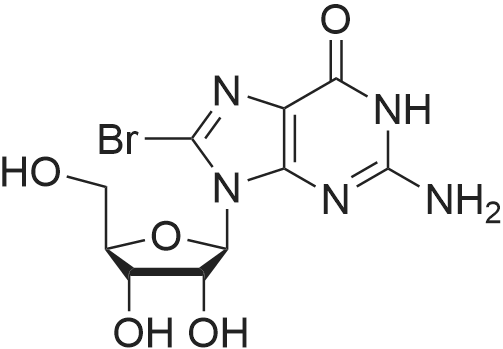
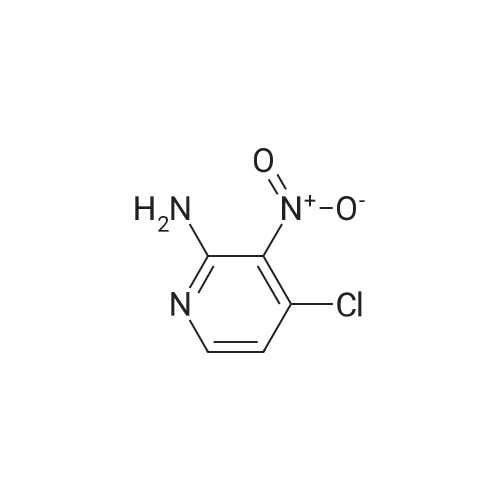
Structure of 4637-24-5 * Storage: {[proInfo.prStorage]}
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
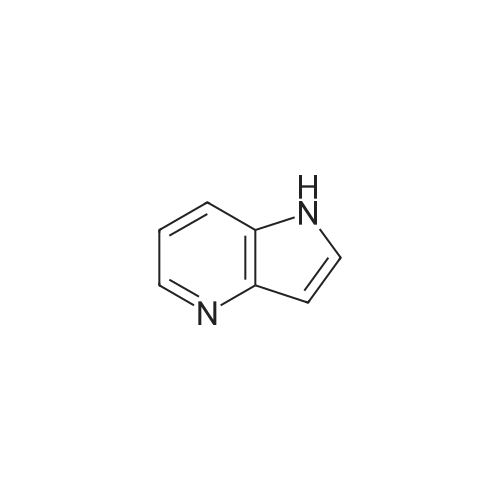
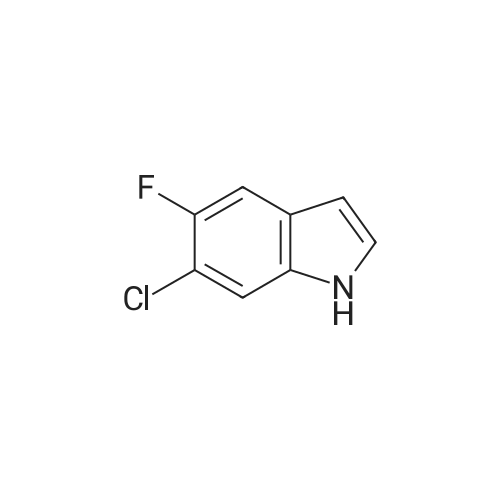
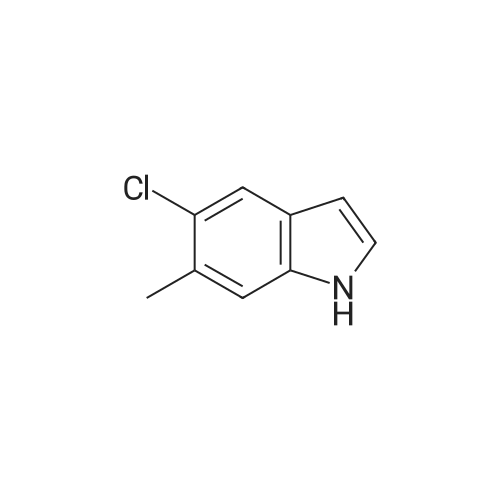
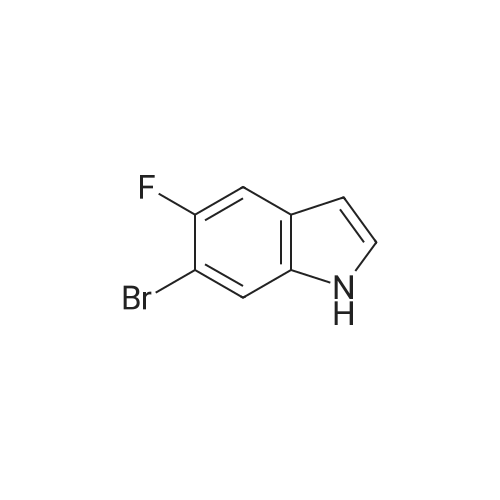
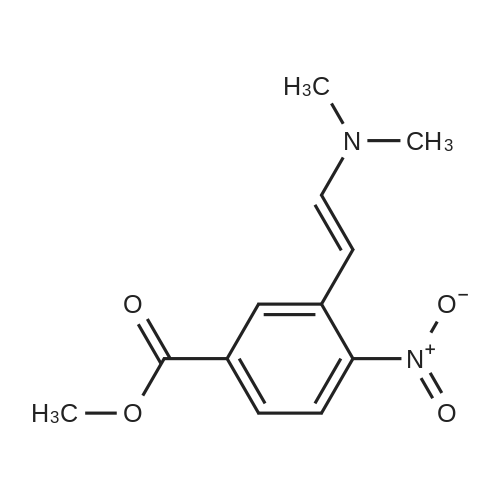
Stage #1: With pyrrolidine In DMF (N,N-dimethyl-formamide) at 20 - 115℃; Stage #2: With hydrogen In methanol at 50℃;
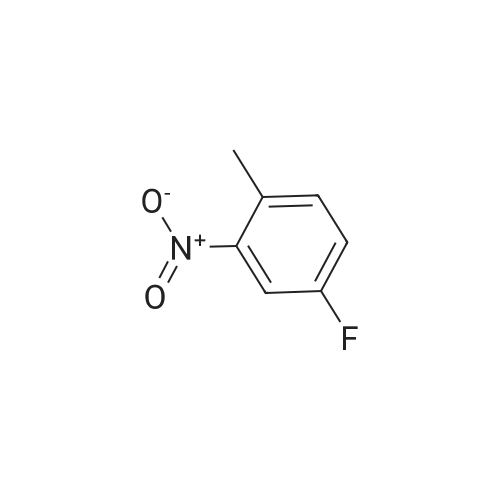
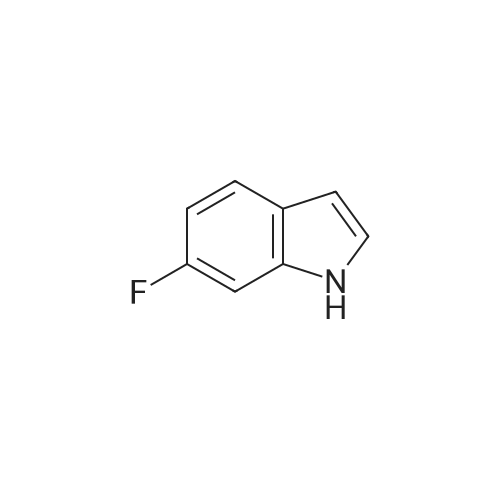
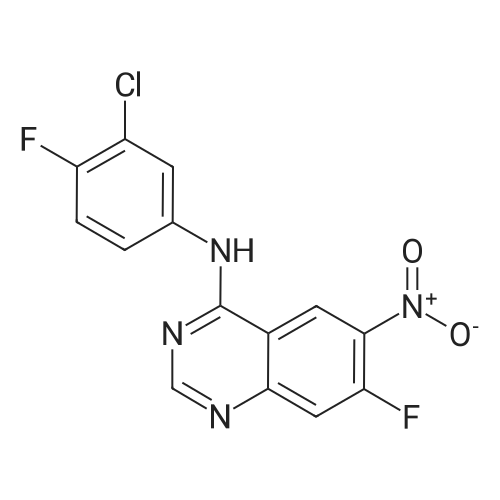
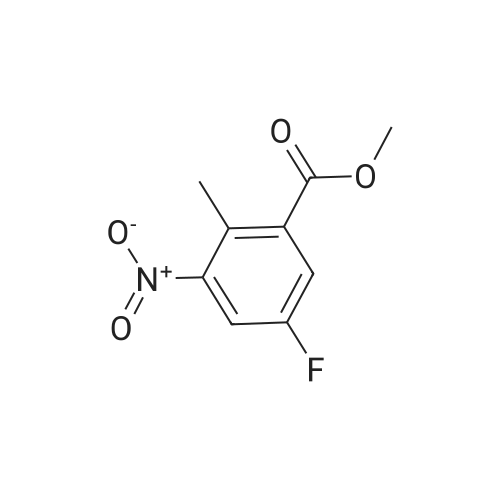
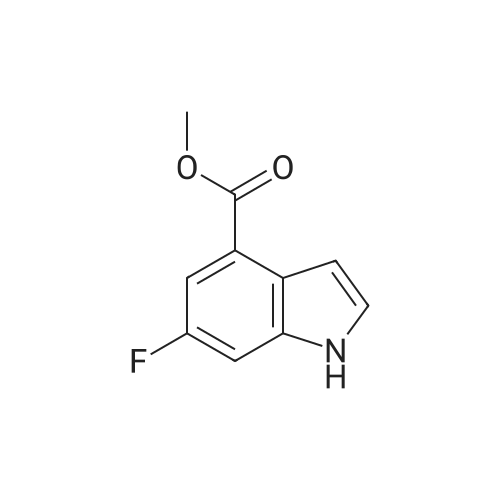
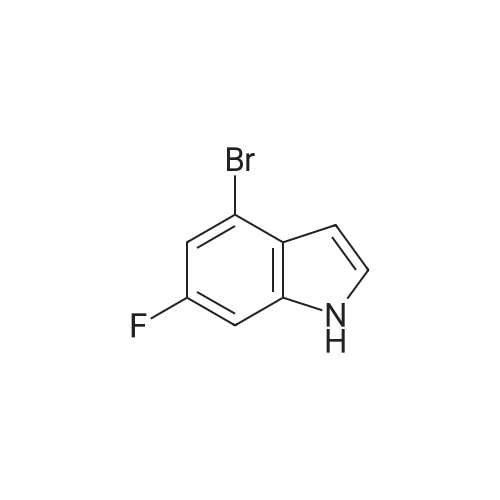
Example S40; Preparation of 6-fluoro-1H-indole-7-carboxylic acid; A solution of 70.3 g of 4-fluoro-1-methyl-2-nitro-benzene (453 mmol) in 330 ml DMF was treated at rt with 87.5 ml of N,N-dimethylformamide dimethylacetal (DMFDMA, 589 mmol) and 50 ml of pyrrolidine (544 mmol) and then heated in an oil bath at 115° C. under argon. The dark red solution was then cooled down to rt, and poured into 1500 ml brine, extracted twice with diethylether. The combined organic phases were washed with brine, dried over magnesium sulfate, filtered and concentrated in vacuo, leading to 106 g residue. About half of this residue (51.5 g) were dissolved in 800 ml methanol, treated with 12.5 g 10percent Pd/C and hydrogenated at 50° C. After removal of the catalyst by filtration, the residue was purified by silicagel chromatography (800 g silicagel, heptane/EtOAc 9:1) leading to 3.5 g of 6-fluoro-1H-indole as a light green solid (5.7percent).
Reference:
[1] European Journal of Organic Chemistry, 2006, # 13, p. 2956 - 2969
[2] Patent: US2006/30613, 2006, A1, . Location in patent: Page/Page column 28
4
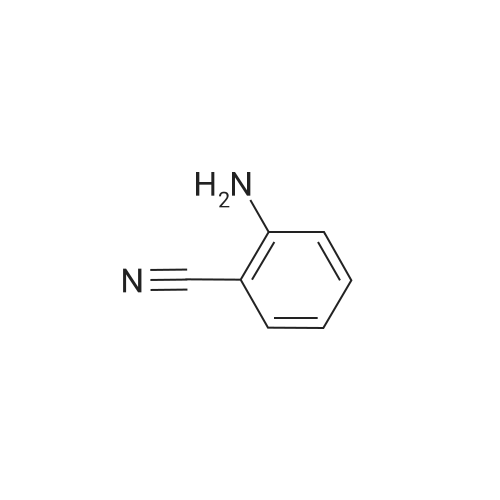
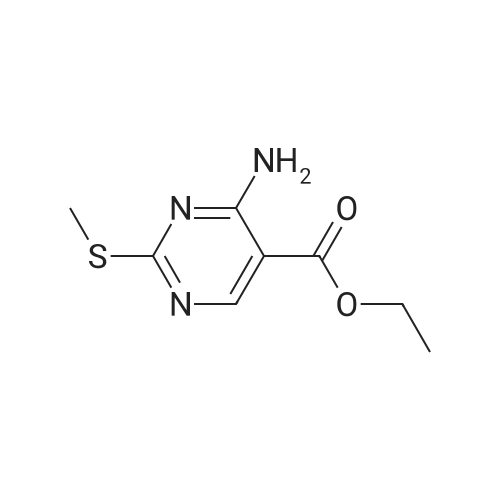
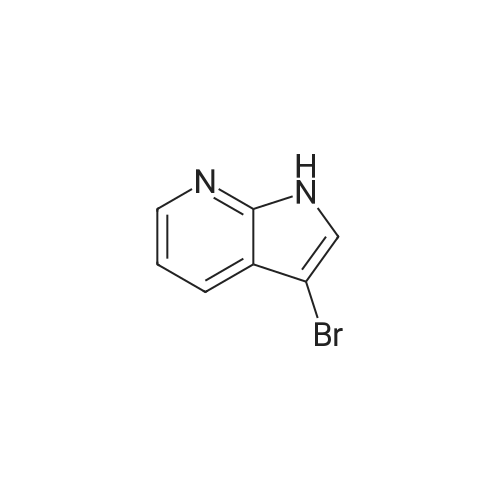
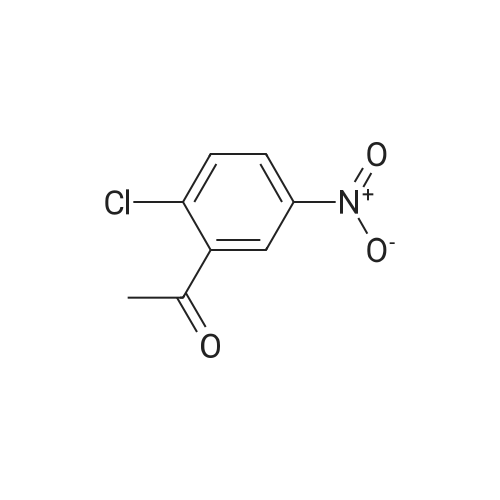
[ 123-75-1 ]
[ 89-59-8 ]
[ 4637-24-5 ]
[ 17422-33-2 ]
Reference:
[1] Patent: US5502071, 1996, A,
5
[ 89-59-8 ]
[ 4637-24-5 ]
[ 17422-33-2 ]
Reference:
[1] RSC Advances, 2014, vol. 4, # 9, p. 4672 - 4675
[2] Chemistry of Heterocyclic Compounds, 2011, vol. 47, # 4, p. 425 - 434
6
[ 60956-26-5 ]
[ 4637-24-5 ]
[ 52415-29-9 ]
Yield
Reaction Conditions
Operation in experiment
36%
Stage #1: With pyrrolidine In N,N-dimethyl-formamide at 110℃; for 1.5 h; Stage #2: With acetic acid; zinc In water at 75 - 85℃; for 5.5 h;
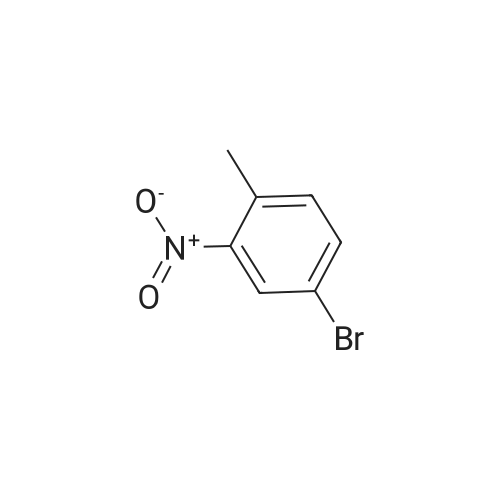
Step A: To a solution of 4-bromo-2-nitrotoluene (7.9 g, 36.6 mmol) in dimethylformamide (73 mL) were added N,N-dimethylformamide dimethylacetal (14.5 mL, 110 mmol) and pyrrolidine (4.7 mL), and the mixture was heated at 110° C. for 90 minutes. The cooled reaction mixture was diluted with diethyl ether, and washed with water. The aqueous layer was re-extracted with diethyl ether twice, and the combined organic extract was dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue obtained was dissolved in aqueous acetic acid (245 mL, 80percent), and heated to 75° C. Zinc powder (20.8 g, 318 mmol) was added to the hot solution in small portions over 2 hours. The reaction mixture was then heated at 85° C. for 3 hours and 30 minutes, cooled to room temperature and then to 0° C. The precipitate formed was removed by filtration, and the filtrate was diluted with ethyl acetate and washed with water twice. The organic extract was dried over sodium sulfate, filtered and concentrated under reduced pressure to give a brown oil. The crude product was purified by flash column chromatography (95:5 hexanes/ethyl acetate, then 90:10 hexanes/ethyl acetate) to give 6-bromoindole as a grey solid (2.61 g, 36percent): 1H NMR (500 MHz, CDCl3) δ 8.14 (br s, 1H), 7.53 (s, 1H), 7.49 (d, J=8.4 Hz, 1H), 7.21 (dd, J=8.4, 1.7 Hz, 1H), 7.17-7.15 (m, 1H), 6.53-6.51 (m, 1H).
Reference:
[1] Journal of Organic Chemistry, 2004, vol. 69, # 20, p. 6812 - 6820
[2] RSC Advances, 2014, vol. 4, # 9, p. 4672 - 4675
[3] Patent: US2006/52378, 2006, A1, . Location in patent: Page/Page column 154
[4] Tetrahedron, 1999, vol. 55, # 4, p. 935 - 942
[5] Patent: US2011/46370, 2011, A1, . Location in patent: Page/Page column 10
[6] Chemistry of Heterocyclic Compounds, 2011, vol. 47, # 4, p. 425 - 434
7
[ 123-75-1 ]
[ 60956-26-5 ]
[ 4637-24-5 ]
[ 52415-29-9 ]
Reference:
[1] Tetrahedron, 1992, vol. 48, # 14, p. 2919 - 2924
[2] Patent: US5252732, 1993, A,
[3] Patent: US5310901, 1994, A,
[4] Patent: US5349061, 1994, A,
8
[ 127693-25-8 ]
[ 4637-24-5 ]
[ 1074-86-8 ]
[ 127693-05-4 ]
Reference:
[1] Archiv der Pharmazie, 1990, vol. 323, # 3, p. 145 - 155
Reference:
[1] Journal of Heterocyclic Chemistry, 1996, vol. 33, # 2, p. 465 - 474
11
[ 4472-44-0 ]
[ 4637-24-5 ]
[ 14001-61-7 ]
[ 13012-25-4 ]
[ 179009-28-0 ]
Reference:
[1] Journal of Heterocyclic Chemistry, 1996, vol. 33, # 2, p. 465 - 474
12
[ 102871-92-1 ]
[ 4637-24-5 ]
[ 37865-86-4 ]
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2013, vol. 23, # 13, p. 3942 - 3946
13
[ 4637-24-5 ]
[ 1885-29-6 ]
[ 15018-66-3 ]
Reference:
[1] Patent: KR2016/21163, 2016, A, . Location in patent: Paragraph 0680; 0682
14
[ 58539-65-4 ]
[ 4637-24-5 ]
[ 23616-31-1 ]
Yield
Reaction Conditions
Operation in experiment
32%
Stage #1: at 50℃; for 2 h; Stage #2: With sodium hydride In N,N-dimethyl-formamide; mineral oil at 80℃; for 2.5 h;
[0621] A mixture of XI-2 (13 g, 95.6 mmol, 1 eq) and 18.2 mL of N,N-dimethylformamide dimethyl acetal was heated at 50° C. for 2 hrs. During the second hour, all the volatiles was removed. The residue was cooled to rt., diluted with 100 mL of anhydrous N,N-dimethylformamide, and then treated carefully with batch wise portions of sodium hydride (5 g, 124.3 mmol, 1.3 eq, 60percent oil dispersion; caution: vigorous evolution of hydrogen). The mixture was heated at 80° C. for 2.5 hrs, and then ice-cooled, treated cautiously with 25 mL of 2-propanol, and then maintained at 0-5° C. overnight. The solid were collected, and then dissolved in 10 mL of hot water. The solution was filtered, the filtrate was ice-cooled and then treated dropwise with concentrated hydrochloric acid to pH=7.0. After storage at 0-5° C. for 3 hrs, the precipitated solids were collected, washed with ice-cold water, and dried in vacuum to give XI-3 (3 g, 32percent yield). 1H NMR (DMSO-d6, 300 MHz): δ 8.90 (s, 1H), 8.49 (d, J=7.6 Hz, 1H), 7.51-7.43 (m, 2H), 6.61 (d, J=7.6 Hz, 1H).
32%
Stage #1: at 50℃; for 2 h; Stage #2: With sodium hydride In N,N-dimethyl-formamide; mineral oil at 80℃; for 2.5 h;
A mixture of X-2 (13 g, 95.6 mmol, 1 eq) and 18.2 mL of N,Ndimethylformamide dimethyl acetal was heated at 50°C for 2 hrs. During the second hour, all the volatiles was removed. The residue was cooled to rt., diluted with 100 mL of anhydrous N,Ndimethylformamide, and then treated carefully with batch wise portions of sodium hydride (5 g, 124.3 mmol, 1.3 eq, 60percent oil dispersion; caution: vigorous evolution of hydrogen). The mixture was heated at 80°C for 2.5 hrs, and then ice-cooled, treated cautiously with 25 mL of 2-propanol, and then maintained at 0-5°C overnight. The solid were collected, and then dissolved in 10 mL of hot water. The solution was filtered, the filtrate was ice-cooled and then treated dropwise with concentrated hydrochloric acid to pH=-7.0. After storage at 0-5°C for 3 hrs, the precipitated solids were collected, washed with ice-cold water, and dried in vacuum to give X-3 (3 g, 32percent yield). ‘H NMR (DMSO-d6, 300 MHz): ö 8.90 (s, 1H), 8.49 (d, J=7.6 Hz, 1H), 7.51-7.43 (m, 2H), 6.61 (d, J =7.6 Hz, 1H).
Reference:
[1] Journal of Heterocyclic Chemistry, 2006, vol. 43, # 5, p. 1311 - 1317
[2] Patent: US2014/94456, 2014, A1, . Location in patent: Paragraph 0619; 0621
[3] Patent: WO2015/153683, 2015, A1, . Location in patent: Paragraph 0367
15
[ 4637-24-5 ]
[ 2001-93-6 ]
[ 5909-26-2 ]
Reference:
[1] Australian Journal of Chemistry, 1981, vol. 34, # 8, p. 1729 - 1738
16
[ 504-29-0 ]
[ 4637-24-5 ]
[ 598-31-2 ]
[ 29096-64-8 ]
Yield
Reaction Conditions
Operation in experiment
64%
Stage #1: for 23 h; Reflux Stage #2: at 20℃; for 24 h;
2-Aminopyridine (1.88 g, 20 mmol) in 1,1-dimethoxyN,N-dimethylmethanamine (3.82 g, 32 mmol) was refluxed for 23 h and then evaporated. The resulting residue was dissolved in EtOH (35 mL), 1-bromoacetone (3.12 g, 22.75 mmol) was added and the reaction mixture was stirred at room temperature for 24 h. The reaction mixture was concentrated and the residue was purified by column chromatography (eluent: dichloromethane/methanol = 40:1) to afford 12s as a pale yellow solid (2.06 g, 64percent). Mp: 95-96 °C (Lit.Mp [20] 98-99 °C). 1H NMR (400 MHz, CDCl3): δ = 2.61 (s, 3H), 7.08 (t, J = 6.80 Hz, 1H), 7.50 (t, J = 8.00 Hz, 1H), 7.76 (d, J = 9.00 Hz, 1H), 8.34 (s, 1H), 9.65 (d, J = 6.84 Hz, 1H).
12%
Stage #1: at 90℃; for 3 h; Stage #2: at 20℃; for 4 h;
A solution of intermediate 1 (20 g, 0.21 mol) and DMF-DMA (38 g, 0.32 mol) was stirred at 90° C. for 3 hrs. The mixture was concentrated in vacuum, the residue was dissolved in EtOH (200 ml), 1-bromopropan-2-one (32 g, 0.23 mol) was added dropwise, and the mixture was stirred at room temperature for 4 hrs. The solvent was concentrated in vacuum and purified by flash column chromatography to give intermediate 23 (4.1 g, 12percent) as a yellow solid. (0384) 1H NMR (CDCl3, 400 MHz): δ (ppm) 9.649.66 (m, 1H), 8.35 (s, 1H), 7.76 (d, 1H, J=8.8 Hz), 7.487.52 (m, 1H), 7.077.11 (m, 1H), 2.62 (s, 3H)
Reference:
[1] European Journal of Medicinal Chemistry, 2015, vol. 93, p. 381 - 391
[2] Patent: US2018/214458, 2018, A1, . Location in patent: Paragraph 0382-0395
17
[ 552-41-0 ]
[ 4637-24-5 ]
[ 5751-52-0 ]
Yield
Reaction Conditions
Operation in experiment
70%
Stage #1: at 100℃; for 0.166667 h; Stage #2: With hydrogenchloride In dichloromethane; water at 40℃; for 0.5 h;
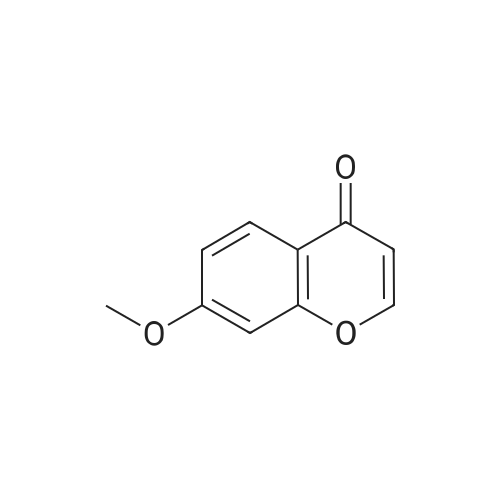
Example 10A 7-methoxy-4H-1-benzopyran-4-one 1-(2-Hydroxy-4-methoxyphenyl)ethanone (47 g, 283 mmol) was dissolved in N,N-dimethylformamide dimethyl acetal (47 mL, 351 mmol), and the solution was heated to >100° C. in a sand bath for 10 minutes, at which point a solid mass had formed. The flask was cooled to ambient temperature, and 200 mL of heptanes were added. The solids were broken up with a spatula and collected by filtration with a frilled funnel. The solid material was crushed with a pestle and then washed with heptanes. The solid was then dried on the filter to give about 60 g of the crude intermediate. This intermediate was dissolved in dichloromethane (1 L) and stirred with 150 mL of concentrated HCl at 40° C. for 30 minutes. The flask was cooled to ambient temperature, and about 100 mL of water was added. The layers were separated, and the aqueous layer was extracted with dichloromethane (2*100 mL). The combined organic extracts were washed with saturated sodium bicarbonate (100 mL) and brine (100 mL) and dried over sodium sulfate. The mixture was filtered, and the filtrate was concentrated in vacuo to give a solid. The solid was then precipitated from 500 mL of 1:1 cyclopentyl methyl ether:heptanes to give the title compound (35 g, 70percent yield). 1H NMR (400 MHz, CDCl3) δ ppm 8.11 (d, J=8.9 Hz, 1H), 7.77 (d, J=6.0 Hz, 1H), 6.97 (dd, J=8.9, 2.4 Hz, 1H), 6.84 (d, J=2.4 Hz, 1H), 6.28 (d, J=6.0 Hz, 1H), 3.90 (s, 3H); MS(ESI+) m/z 176.9 (M+H)+.
70%
Stage #1: at 100℃; for 0.166667 h; Stage #2: With hydrogenchloride In dichloromethane at 40℃; for 0.5 h;
1-(2-Hydroxy-4-methoxyphenyl)ethanone (47 g, 283 mmol) was dissolved in N,N-dimethylformamide dimethyl acetal (47 mL, 351 mmol), and the solution was heated at >100° C. in a sand bath for 10 minutes, at which point a solid mass formed. The flask was cooled to ambient temperature, and 200 mL of heptanes were added. The solids were broken up with a spatula and collected by filtration with a fritted funnel. The solid material was crushed with a pestle and washed with heptanes. The solid was dried on the filter to give about 60 g of the crude intermediate. The intermediate was dissolved in dichloromethane (1 L) and stirred with 150 mL of concentrated HCl at 40° C. for 30 minutes. The flask was cooled to ambient temperature, and about 100 mL of water was added. The layers were separated, and the aqueous layer was extracted with dichloromethane (2×100 mL). The combined organic extracts were washed with saturated sodium bicarbonate (100 mL) and brine (100 mL) and dried over sodium sulfate. The mixture was filtered, and the filtrate was concentrated in vacuo to give a solid. The solid was taken into 500 mL of 1:1 cyclopentyl methyl ether:heptanes and the precipitate was collected to provide the title compound (35 g, 70percent yield). 1H NMR (400 MHz, CDCl3) δ ppm 8.11 (d, J=8.9 Hz, 1H), 7.77 (d, J=6.0 Hz, 1H), 6.97 (dd, J=8.9, 2.4 Hz, 1H), 6.84 (d, J=2.4 Hz, 1H), 6.28 (d, J=6.0 Hz, 1H), 3.90 (s, 3H); MS(ESI+) m/z 176.9 (M+H)+.
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2018, vol. 28, # 11, p. 2098 - 2102
[2] Patent: US2017/15675, 2017, A1, . Location in patent: Paragraph 0970
[3] Patent: US2017/305891, 2017, A1, . Location in patent: Paragraph 0735
Reference:
[1] European Journal of Organic Chemistry, 2002, # 11, p. 1763 - 1769
20
[ 15128-52-6 ]
[ 4637-24-5 ]
[ 27387-31-1 ]
Yield
Reaction Conditions
Operation in experiment
71%
for 1 h; Reflux
A mixture of 2,3-Dihydro-1H-carbazol-4(9H)-one (1.0 mmol)and DMF-DMA (10 mmol) in DMF (5.0 mL) was refluxed for 1.0 h.The reaction mixture was cooled and extracted with ethyl acetate(2 25 mL). The organic layer was separated and washed with brine solution. The organic layer was dried over anhydrous Na2SO4.The solvent was removed under reduced pressure and the solidobtained was recrystallized from ethanol.White needles, yield 71percent, m.p. 188–90 C. IR (cm1) 1695(CO), 1H NMR (400 MHz, CDCl3) d 2.24–2.27 (t, 2H, CH2,J = 6.28 Hz), 2.56–2.59 (t, 2H, CH2, J = 6.0 Hz), 2.91–2.94 (t, 2H,CH2, J = 6.21 Hz), 3.70 (s, 3H, NCH3), 7.27–7.30 (m, 3H, Ph), 8.24–8.26 (m, 1H, Ph). Anal. Calc. for C13H13NO: C, 78.36; H, 6.58, N,7.03percent. Found: C, 78.29; H, 6.49; N, 6.92percent.
Reference:
[1] Indian Journal of Chemistry - Section B Organic and Medicinal Chemistry, 2014, vol. 53B, # 12, p. 1606 - 1610
[2] Journal of Molecular Structure, 2014, vol. 1080, p. 137 - 144
[3] Russian Chemical Bulletin, 2005, vol. 54, # 8, p. 1887 - 1891
Reference:
[1] Tetrahedron, 1985, vol. 41, # 14, p. 2919 - 2922
[2] Chemistry of Heterocyclic Compounds, 2011, vol. 47, # 4, p. 425 - 434
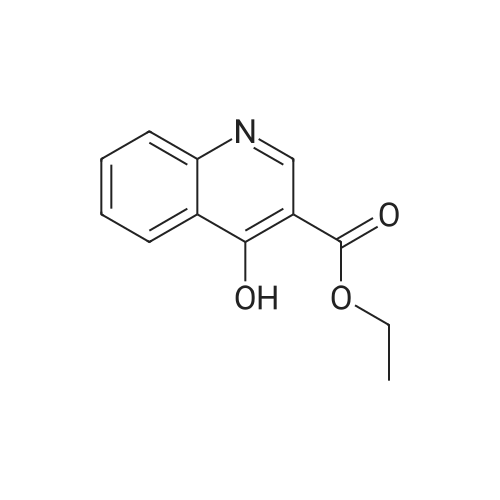
23
[ 4637-24-5 ]
[ 26892-90-0 ]
Yield
Reaction Conditions
Operation in experiment
71%
at 120℃; for 4 h;
Ethyl 3- (2-aminophenyl) -3-oxopropionate (20.7 g, 0.1 mol) was dissolved in 100 mL of N, N-dimethylformamide,N, N-dimethylformamide dimethyl acetal (24.0 g, 0.2 mol) was added thereto, and the reaction was carried out at 120 ° C for 4 hours.The reaction solution was cooled to 20 ° C and the solvent was concentrated under reduced pressure. The residue was added to 200 g of ice water and stirred for 2 h.Ethanol and water volume (30mLx2 times), 50 drying, with 160mL ethyl acetate and petroleum ether equal volume mixedWas purified by recrystallization to obtain 15.4 g of ethyl 4-hydroxyquinoline-3-carboxylate in 71percent yield as an off-white solid.
Reference:
[1] Patent: CN106187887, 2016, A, . Location in patent: Paragraph 0015; 0063; 0064; 0065; 0046; 0047
A solution of 4.0 g (33.0 mmol, 1.0 eq.) of 1-(pyridin-2-yl)ethan-1-one in 13.1 mL (99.1 mmol, 3.0 eq,) of DMF dimethylacetal was stirred at 90 °C for 12 h. The mixture wasallowed to cool to room temperature, and the resulting precipitate was collected by filtration. The solids were washed with 2 x 5 mL of ethyl acetate and dried under high vacuum to provide, 3.0 g (17.0 mmol, 51percent) of (E)-3 -(dimethylamino)- 1 -(pyridin-2-yl)prop-2-en- 1-one.
13 g
for 24 h; Reflux
12.1 g of 2-acetylpyridine was dissolved in 150 ml of toluene, 30 ml of DMF-DMA was added while stirring, and the mixture was heated to reflux for 24 hours, cooled to room temperature, concentrated to dryness under reduced pressure, and 150 ml of petroleum ether was added to the residue for stirring. Disperse, filter, filter cake washed with petroleum ether to give intermediate 13g, yellow crystals.
Reference:
[1] Molecules, 2007, vol. 12, # 8, p. 2061 - 2079
[2] European Journal of Organic Chemistry, 2012, # 32, p. 6407 - 6413,7
[3] Synthetic Communications, 1993, vol. 23, # 13, p. 1897 - 1903
[4] Organic Preparations and Procedures International, 1997, vol. 29, # 3, p. 285 - 292
[5] Journal of Chemical Research, Miniprint, 1997, # 3, p. 601 - 615
[6] Chemical Communications, 2017, vol. 53, # 91, p. 12286 - 12289
[7] Organic Letters, 2018, vol. 20, # 4, p. 1256 - 1260
[8] Journal of Heterocyclic Chemistry, 2007, vol. 44, # 2, p. 355 - 361
[9] European Journal of Organic Chemistry, 2016, vol. 2016, # 18, p. 3060 - 3064
[10] Tetrahedron Letters, 1999, vol. 40, # 26, p. 4779 - 4782
[11] Dalton Transactions, 2012, vol. 41, # 13, p. 3684 - 3694
[12] Patent: WO2018/172852, 2018, A1, . Location in patent: Page/Page column 204; 206
[13] European Journal of Organic Chemistry, 2005, # 17, p. 3775 - 3780
[14] Bioorganic and Medicinal Chemistry Letters, 1997, vol. 7, # 2, p. 187 - 192
[15] Synthesis, 2001, # 1, p. 55 - 62
[16] Bioorganic and Medicinal Chemistry Letters, 2007, vol. 17, # 6, p. 1641 - 1645
[17] Patent: US5859215, 1999, A,
[18] Patent: US4788195, 1988, A,
[19] Patent: US4281000, 1981, A,
[20] European Journal of Inorganic Chemistry, 2013, # 24, p. 4305 - 4317
[21] Patent: CN103936792, 2017, B, . Location in patent: Paragraph 0093; 0094; 0095
26
[ 1122-62-9 ]
[ 4637-24-5 ]
[ 98-86-2 ]
[ 1131-80-2 ]
[ 66521-54-8 ]
Reference:
[1] Synthesis, 2011, # 12, p. 1930 - 1935
27
[ 99-91-2 ]
[ 4637-24-5 ]
[ 107-91-5 ]
[ 23148-51-8 ]
Reference:
[1] Journal of Medicinal Chemistry, 1994, vol. 37, # 20, p. 3303 - 3312
28
[ 124-41-4 ]
[ 68-12-2 ]
[ 4637-24-5 ]
Yield
Reaction Conditions
Operation in experiment
85.4%
Stage #1: at 20 - 70℃; for 4 h; Stage #2: at 25℃; for 2 h;
A process for the synthesis of N, N-dimethylformamide dimethylacetal comprising the steps of:(1) N, N-dimethylformamide was heated to 70 ° C, incubated with dimethyl sulfate, and reacted at the end of the dropwise addition for 4 hours to obtain an imine complex,Cooled to room temperature, and the molar ratio of N, N-dimethylformamide and dimethyl sulfate is 1: 1;(2) the solid sodium methoxide added to 200 solvent oil, ultrasonic dispersion, adjust the temperature to 25 ,Insulation, dropping imine complex, after the end of the reaction for 2h, filtration, the filtrate under normal pressure fractionation,A fraction of 105-108 ° C was collected to give N, N-dimethylformamide dimethyl acetal in a yield of 75.3percentPurity of 97percent; where the molar ratio of sodium methoxide to imine complex is 1: The synthesis method of N, N-dimethylformamide dimethyl acetal of Example 6 was the same as in Example 2 except that the organic solvent of this example was a mixed solvent oil: D40 solvent oil, D60 solvent oil, 260 (Volume ratio: 2: 1: 1) to give N, N-dimethylformamide dimethylacetal as a yield of 85.4percent and a purity of 97percent.
Reference:
[1] Patent: CN106083611, 2016, A, . Location in patent: Paragraph 0034-0037; 0048-0049
29
[ 1196967-42-6 ]
[ 1196967-61-9 ]
[ 4637-24-5 ]
Reference:
[1] Angewandte Chemie, International Edition, 2009, vol. 48, # 41, p. 7600 - 7603
30
[ 21511-55-7 ]
[ 4637-24-5 ]
Yield
Reaction Conditions
Operation in experiment
59%
at 0 - 20℃; for 20 h; Inert atmosphere
Separately, 440 g of anhydrous methanol and 111 g (2.05 mol) of sodium methoxide are added to a 1-liter three-necked flask under a nitrogen atmosphere and cooled to 0 to 5 ° C. 400.4 g (2.01 mol) of the immune complex was dropped for 3 hours while maintaining the temperature at 5 DEG C or lower, and the mixture was stirred at room temperature for 20 hours. The methanol was removed by distillation under atmospheric pressure and distilled under vacuum to obtain 141.3 g (yield 59percent) of colorless liquid dimethylformamide dimethylacetal.
Reference:
[1] Patent: KR2016/120009, 2016, A, . Location in patent: Paragraph 0053
31
[ 68-12-2 ]
[ 77-78-1 ]
[ 4637-24-5 ]
Reference:
[1] Journal of Organometallic Chemistry, 1989, vol. 373, p. 1 - 10
[2] Journal of applied chemistry of the USSR, 1985, vol. 58, # 4 pt 1, p. 701 - 706
[3] Patent: WO2004/24727, 2004, A2, . Location in patent: Page 14
32
[ 124-41-4 ]
[ 21511-55-7 ]
[ 4637-24-5 ]
Reference:
[1] Patent: CN108516962, 2018, A, . Location in patent: Paragraph 0036; 0037; 0038
33
[ 1192-62-7 ]
[ 4637-24-5 ]
[ 109482-86-2 ]
Reference:
[1] Patent: US4374988, 1983, A,
34
[ 350-03-8 ]
[ 55314-16-4 ]
[ 4637-24-5 ]
Reference:
[1] Patent: US4374988, 1983, A,
35
[ 68-12-2 ]
[ 154147-42-9 ]
[ 4637-24-5 ]
Reference:
[1] Journal of Organic Chemistry, 1999, vol. 64, # 22, p. 8084 - 8089
36
[ 124-41-4 ]
[ 1071-38-1 ]
[ 1186-70-5 ]
[ 5762-56-1 ]
[ 4637-24-5 ]
Reference:
[1] Advanced Synthesis and Catalysis, 2004, vol. 346, # 9-10, p. 1081 - 1086
To 2-nitro-(1H)-imidazole (2.0 g, 17.7 mmol)N,N-dimethylformamide dimethyl acetal (8.4 g, 70.7 mmol) was added dropwise to a solution of DMF (10 ml).The reaction was carried out at 80 ° C for 2 h. Add water (30ml) and ethyl acetate (25ml),The mixture was extracted with ethyl acetate (25×2 mL).Dry over anhydrous sodium sulfate, filter, and concentrate the filtrate under reduced pressure.The crude product was separated by column chromatography (eluent: dichloromethane: anhydrous methanol = 30:1).A pale yellow solid (2.1 g, 93.5percent) was obtained.The product purity is 99.7percent (mass fraction)
Reference:
[1] Patent: CN108503583, 2018, A, . Location in patent: Paragraph 0051-0053; 0059; 0060
41
[ 4637-24-5 ]
[ 105-56-6 ]
[ 1260243-04-6 ]
Yield
Reaction Conditions
Operation in experiment
85%
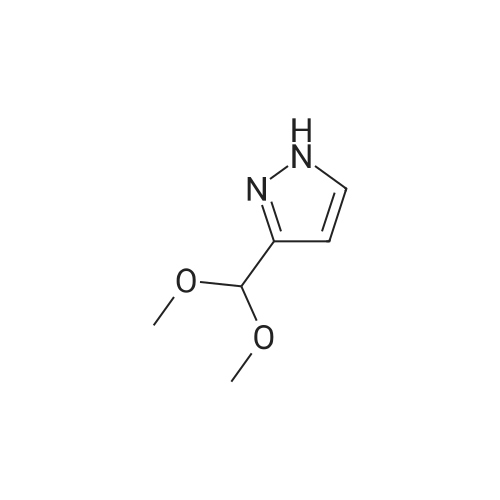
Stage #1: With acetic acid In N,N-dimethyl-formamide at 25℃; for 1 h; Stage #2: With hydrazine hydrate In N,N-dimethyl-formamide at 50℃; for 2 h;
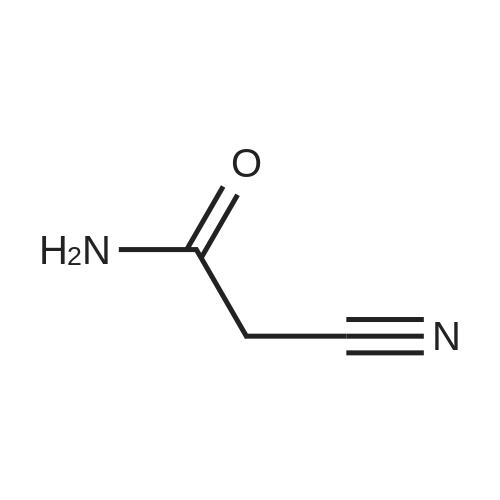
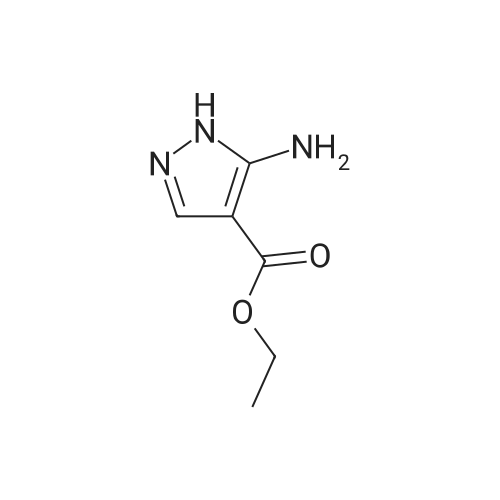
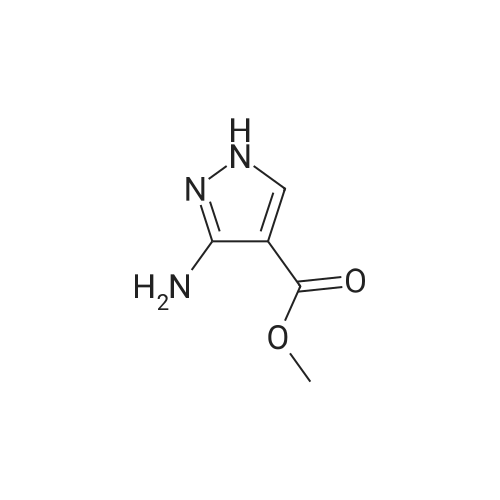
To a solution of 25 g of ethyl cyanoacetate (1, 0.221 mol)in 25 cm3 dimethylformamide was added 37.5 cm3 glacialacetic acid (0.077 mol) followed by 47.40 g of N,N-dimethylformamidedimethyl acetal (0.397 mol) dropwise at room temperature and stirred for 1 h. To the resultant pale yellow color solution was added 25 cm3 hydrazine hydrate (0.42 mol) drop wise at 0°C and the reaction medium was stirred for 2 h 50 °C. After completion of the reaction, the reaction medium was diluted with 2.5 dm3 of water and extracted with 2 9 700 cm3 of ethyl acetate. The combined organic layer was dried over sodium sulfate,filtered, and concentrated under reduced pressure to obtain the crude product as pale brown liquid. The crude product was further purified by column chromatography over silicagel, eluted with chloroform/methanol (95:5, v/v) to afford the compound 2 as a white solid (29 g, 85 percent). M.p.: 106.7–108.9 °C; TLC: Rf = 0.22 (CHCl3–MeOH 8:2); IR (ATR):v = 3193 (–NH), 2972 (=C–H), 1662 (ester –C=O), 1551(–C–C), 1496 (ester –C–O), 1337 (C–N) cm-1; 1H NMR(400 MHz, CDCl3): d = 1.23 (3H, t, J = 8 Hz, CH3), 4.15(2H, q, J = 8 Hz, OCH2CH3), 5.98 (2H, bs, ArNH2), 7.45(1H, bs, ArH), 12.04 (1H, bs, pyrazole NH) ppm; 13C NMR(100 MHz, DMSO-d6): d = 14.9, 59.1, 94.0, 139.9, 151.8,164.2 ppm; LC–MS: m/z = 156.7 [M+H]+.
70%
at 20℃; for 2.5 h; Inert atmosphere
To a solution of ethylcyanoacetate (3) (25 g, 221.0 mmol)in dimethylformamide (25 mL) and acetic acid(37.5 mL), dimethyl formamide dimethyl acetal(47.40 g, 397.8 mmol) was added drop wise at roomtemperature under nitrogen atmosphere over a period of30 min. The reaction medium was stirred at room temperaturefor 2 h. The reaction mixture was cooled to 0 °Cand hydrazine hydrate (80percent, 25 mL) was added drop wiseto it over a period of 45 min and then heated to 50 °C for2 h. Completion of the reaction was monitored by TLC.After completion of the reaction, the reaction mixture wasdiluted with ice cold water (1L) and then extracted to ethylacetate (2 × 250 mL). The combined ethyl acetate layerwas dried over anhydrous sodium sulfate, filtered andevaporated under reduced pressure. The crude materialobtained was purified by column chromatography oversilica gel, eluted with 2–5percent methanol in chloroform to getthe product (4) as off-white solid (24 g, 70percent). MP:106.7–108.9 °C. IR (ATR, cm−1) υ: 1125.41 (C–O),1337.61 (C–N), 1496.40 (C–C), 1615.86 (N–H bend),1662.37 (ester C=O), 2972.92 (C–H), 3193.79(N–H stretch). 1H NMR (DMSO-d6, 400 MHz) δ (ppm):1.23 (3H, t, J = 7.2 Hz, ester–CH3), 4.16 (2H, q, J = 6.9Hz, ester–CH2), 5.99 (2H, bs, pyrazole–NH2), 7.47 (1H,bs, pyrazole–CH), 11.84 (1H, bs, pyrazole–NH). 13CNMR (DMSO-d6, 100 MHz) δ (ppm): 14.92 (ester–CH3),59.10 (ester–CH2), 94.00 (pyrazole–C-3), 139.93(pyrazole–C-4), 151.86 (pyrazole–C-5), 164.27 (carbonylfrom ester). LC-MS (ESI, m/z): 153.7 (M-H) and 156.7(M+H).
Reference:
[1] Monatshefte fur Chemie, 2016, vol. 147, # 12, p. 2221 - 2234
[2] Medicinal Chemistry Research, 2017, vol. 26, # 4, p. 714 - 744
[3] Journal of Heterocyclic Chemistry, 2017, vol. 54, # 3, p. 1904 - 1924
[4] Journal of Heterocyclic Chemistry, 2018, vol. 55, # 1, p. 214 - 225
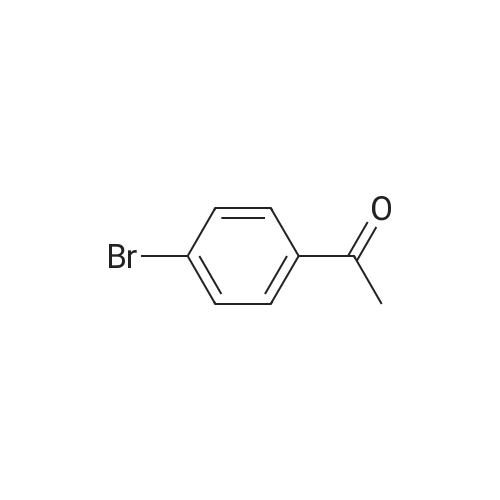
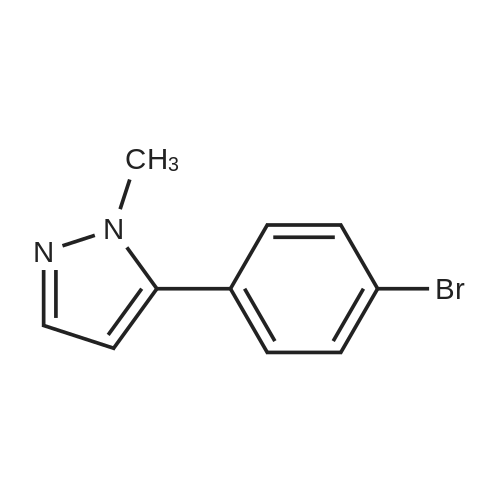
4-bromoacetophenone (20.0 g; 0.10 mole) and N,N-dimethylformamide dimethylacetal (28.5 mL; 0.20 mole) were mixed together in DMF (12 mL) and heated to 1100C for 4 hours. The methanol and water that were generated during the reaction were distilled (6.2 mL). The mixture was cooled to 25°C. Methyl t-butyl ether (100 mL) and methylhydrazine (21.2 mL; 0.40 moles) <n="70"/>were added and the mixture was stirred over night. The reaction mixture was washed with 1 M aqueous ammonium chloride (3 x 40 ml_) and water (40 ml_). The organic phase was dried by azeotropic distillation using a Dean-Stark apparatus. As an alternative to distillation, the solution was dried through an anhydrous magnesium sulfate cartridge. The solution was filtered through a silica gel cartridge (60 g). The product was flushed from the cartridge with methyl t-butyl ether. The fraction(s) containing product were combined and concentrated to about 70 ml_ by distillation. Heptane (120 ml_) was added and distillation was continued until the pot temperature reached 98.4 0C. About 100 ml_ of distillate was collected. The mixture was cooled to 40 0C. The mixture was seeded and the temperature was maintained at 40 0C for 30 minutes while crystallization was initiated. The mixture was slowly chilled to 0 0C over 90 minutes. The mixture was held at 0 0C for 30 minutes. The mixture was filtered and the solid was washed (3 x) with chilled (00C) heptane. The solid was dried on the filter. A cream-colored, crystalline solid (16.3 g; 68percent yield) was obtained. The NMR data of the title compound are as per alternative 1
Stage #1: at 80℃; Stage #2: With hydrazine hydrate In ethanol at 80℃; for 2 h;
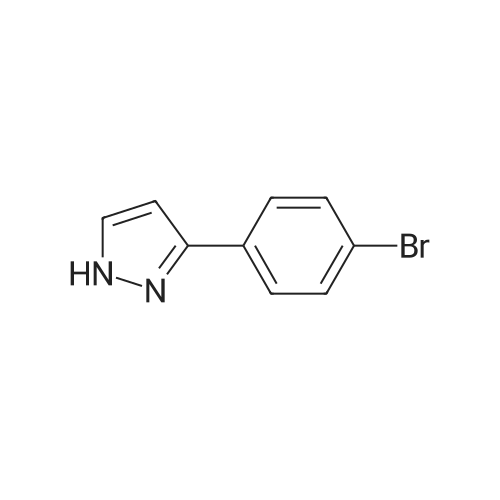
(1) 3-(4-bromophenyl)-1H-pyrazole 5 g (25.0 mmol) of 4-bromoacetophenone and 3.59 g (30.1 mmol) of 1,1-dimethoxy-N,N-dimethylmethanamine were added to 40 ml of DMF. The mixture was heated to 80° C. for overnight. After cooling, the mixture was poured to water (150 mL) and extracted with EA (100 mL*3). The combined organic phase was washed with brine, concentrated to give red liquid (6 g). The thus obtained product was redissolved in 50 ml of ethanol and treated with hydrazine monohydrate (3.5 ml, 75 mmol). After the reaction was stirred at 80° C. for 2 h, it was cooled to 23° C. and poured to ice-water. Solid was precipitated out of the solution and filtered, washed with water, and dried to give 3.6 g of compound 3-(4-bromophenyl)-1H-pyrazole as yellow solid (yield: 80percent). 1H NMR (400 MHz, CDCl3): δ=7.58-7.62 (m, 3H), 7.49-7.51 (m, 2H), 6.58 (d, J=2.4, 1H).
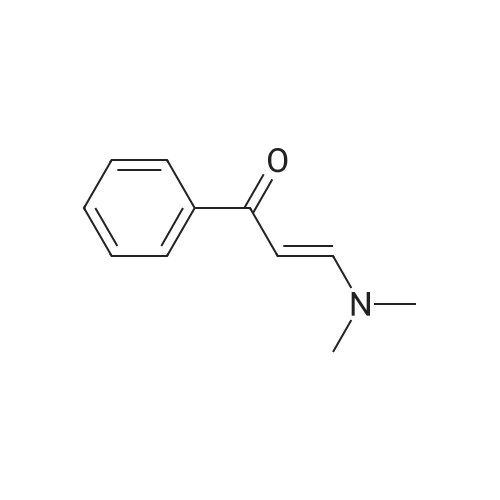
In a 100 mL dry round bottom flask, acetophenone 1.08 g (9 mmol) of N,N-Dimethylformamide dimethyl acetal 3.9 mL (30 mmol) and 30 mL of xylene were added as a solvent. The mixture was refluxed at 140 ° C for 12 hours. After the TLC detection reaction was completed, the heating was stopped, the solvent was removed under reduced pressure, and the crude product was separated by flash column chromatography to give a pale yellow solid N,N-(dimethylamino)-1-phenyl enaminoketones 1.48 g (97percent yield).
92%
Reflux
2-1 1: Preparation of 3-dimethylamino-1- Yield: 92percent. Appearance: yellow solid. phenylprop-2-en-1-one (Protocol SA and 1H NMR: 2.93-3.14 (m, 6H); 5.73 (d, 1H, PC) J = 12.4 Hz); 7.38-7.49 (m, 3H); 7.82 (d, 1H, J = 12.4 Hz); 7.89 (d, 2H, J = 7.9 Hz).; Protocol SA:The appropriate ketone (acetophenone for Ex. 2-1; 4-trifluoroacetophenone for Ex. 2-2; 2-phenylacetophenone for Ex. 2-3; 2-acetylfuran for Ex. 2-5; para-acetylbiphenyl for Example 2-6; 3-trifluoromethylacetophenone 2.7) is dissolved in dimethyl acetal of N,N-dimethylformamide (1.2 to 1.5 eq.). The reaction mixture is stirred under reflux. The estimated reaction time varies between 24 and 48 hours. Satisfactory results were obtained in 24 hours.
83%
at 115℃; for 20 h;
To a 100 mL round bottom flask, was added acetophenone (1.0 g, 0.0083 mol) followed by NN-dimethylformamide dimethyl acetal (1.48 g, 0.012 mol). The reaction mixture was stirred at 115 °C for 20 h. The reaction mixture was cooled to room temperature and ether was added to get the solid. The solid was collected by filtration to get the title compound [1.2 g, 83 percent]. JH NMR (300 MHz, CDC13): δ 7.89-7.86 (m, 2H), 7.80 (d, = 12.3 Hz, 1H), 7.44-7.39 (m, 3H), 5.72 (d, = 12.6 Hz, 1H), 3.11 (s, 3H), 2.91 (s, 3H); LC-MS: 176.0 [M+H]+.
82%
at 150℃; for 20 h; Inert atmosphere
A mixture of acetophenone 1 (5.0 g, 41.66 mmol) and DMF-DMA (17.71 mL, 49.9 mmol) in DMF (10 mL) was heated to 150 0C under N2 atmosphere. After stirring for 2Oh at 150 0C, the reaction mixture was concentrated in vacuo. The resulting residue was triturated with diethyl ether, filtered and dried under high vacuum to obtain compound 2 (6 g, 82percent) as a yellow solid. TLC Rf = 0.5 (CHCl3 - MeOH, 7:3); 1H NMR (CDCl3) δ 7.88 (dd, J= 7.8, 1.5 Hz, 2H), 7.8 (d, J= 12.3 Hz, IH), 7.42 (m, 3H), 5.71 (d, J= 12.3 Hz, IH), 3.1 (s, 6H); MS (ES) mlz 176 (M + H)+.
82%
at 150℃; for 20 h; Inert atmosphere
General procedure: A mixture of acetophenone 7a-7d (41.66 mmol) and DMF-DMA (49.9 mmol) in DMF (10 mL) was heated to 150 oC under N2 atmosphere. After being stirred for 20h at this temperature, the reaction mixture was concentrated in vacuo and the residue obtained was triturated with diethyl ether, filtered and dried under high vacuum to obtain compound 2a-2d (70-85percent) as a yellow solid.
81%
for 14 h; Reflux
General procedure: To a mixture of acetone 14b (5.8 g, 100 mmol), inxylene (200 mL), was added dimethylformamide dimethylacetal (DMF-DMA, 11.9 g, 100 mmol)and the reaction refluxed for 15 h (monitored by TLC). The xylene was distilled off and the resultingsolid residue was purified by column chromatography using PE and EtOAc (6:1) as eluent to afford4-(dimethylamino)but-3-en-2-one 15b (4.3 g, 38percent) as a yellow oil.
62%
for 21 h; Reflux
5 g of acetophenone, 15 g of N, N-dimethylformamide dimethylacetal, and 60 ml of xylene were refluxed overnight (21 hours), a little raw material was not reacted and the reaction was stopped. The reaction solution was cooled and concentrated to remove xylene. 150 ml of petroleum ether was added to the residue, and a solid precipitated under stirring, followed by filtration to obtain 4.5 g of 3-dimethylamino-1-phenyl-prop-2-en-1-one as a yellow solid, 62percent.
Reference:
[1] RSC Advances, 2017, vol. 7, # 45, p. 28483 - 28488
[2] Patent: CN108383763, 2018, A, . Location in patent: Paragraph 0044; 0046
[3] Patent: US2011/195993, 2011, A1, . Location in patent: Page/Page column 10; 14
[4] Journal of Organic Chemistry, 1980, vol. 45, # 24, p. 4857 - 4860
[5] Bioorganic and Medicinal Chemistry Letters, 2011, vol. 21, # 4, p. 1214 - 1217
[6] Organic Process Research and Development, 2008, vol. 12, # 3, p. 490 - 495
[7] Patent: WO2014/125426, 2014, A1, . Location in patent: Page/Page column 54; 55
[8] Bioorganic and Medicinal Chemistry Letters, 2008, vol. 18, # 20, p. 5609 - 5613
[9] Patent: WO2010/129049, 2010, A1, . Location in patent: Page/Page column 114; 118
[10] Bioorganic and Medicinal Chemistry Letters, 2011, vol. 21, # 15, p. 4528 - 4532
[11] Chemical Communications, 2016, vol. 52, # 83, p. 12306 - 12309
[12] Molecules, 2017, vol. 22, # 12,
[13] Patent: WO2006/123648, 2006, A1, . Location in patent: Page/Page column 20
[14] Journal of Heterocyclic Chemistry, 1996, vol. 33, # 6, p. 1707 - 1710
[15] Bioorganic and Medicinal Chemistry, 2011, vol. 19, # 8, p. 2742 - 2750
[16] Patent: EP3287456, 2018, A1, . Location in patent: Paragraph 0175
[17] Journal of Heterocyclic Chemistry, 1987, vol. 24, p. 837 - 843
[18] Journal of the Chinese Chemical Society, 2003, vol. 50, # 2, p. 283 - 296
[19] Bioorganic and Medicinal Chemistry Letters, 2003, vol. 13, # 18, p. 3055 - 3057
[20] Journal of the American Chemical Society, 2010, vol. 132, # 44, p. 15531 - 15533
[21] Journal of the Indian Chemical Society, 2010, vol. 87, # 6, p. 739 - 742
[22] ACS Combinatorial Science, 2011, vol. 13, # 4, p. 427 - 435
[23] Synthetic Communications, 2012, vol. 42, # 10, p. 1521 - 1531
[24] Tetrahedron, 2012, vol. 68, # 25, p. 5046 - 5052
[25] Organic Letters, 2012, vol. 14, # 15, p. 3920 - 3923
[26] Tetrahedron Letters, 2013, vol. 54, # 21, p. 2612 - 2614
[27] Bioorganic and Medicinal Chemistry Letters, 2014, vol. 24, # 6, p. 1581 - 1588
[28] Medicinal Chemistry Research, 2015, vol. 24, # 6, p. 2742 - 2755
[29] Synthetic Communications, 2015, vol. 45, # 16, p. 1902 - 1911
[30] Journal of Heterocyclic Chemistry, 2015, vol. 52, # 5, p. 1296 - 1301
[31] Synthetic Communications, 2015, vol. 45, # 23, p. 2683 - 2690
[32] Bioorganic and Medicinal Chemistry, 2016, vol. 24, # 8, p. 1749 - 1756
[33] New Journal of Chemistry, 2016, vol. 40, # 5, p. 4705 - 4709
[34] Chemistry - A European Journal, 2016, vol. 22, # 29, p. 9966 - 9970
[35] Molecules, 2015, vol. 20, # 5, p. 8800 - 8815
[36] Bioorganic and Medicinal Chemistry, 2016, vol. 24, # 23, p. 6166 - 6173
[37] European Journal of Medicinal Chemistry, 2017, vol. 128, p. 1 - 12
[38] Letters in Organic Chemistry, 2017, vol. 14, # 5, p. 353 - 360
[39] European Journal of Medicinal Chemistry, 2018, vol. 155, p. 545 - 551
[40] European Journal of Medicinal Chemistry, 2018, vol. 152, p. 307 - 317
[41] Patent: WO2018/142365, 2018, A1, . Location in patent: Page/Page column 66
[42] Bioorganic Chemistry, 2019, vol. 83, p. 549 - 558
[43] Chemical Communications, 2018, vol. 54, # 99, p. 13953 - 13956
48
[ 4637-24-5 ]
[ 118664-99-6 ]
[ 63762-72-1 ]
[ 122509-72-2 ]
Reference:
[1] Journal of Medicinal Chemistry, 2009, vol. 52, # 7, p. 1853 - 1863
Stage #1: at 140℃; for 16 h; Stage #2: With sodium periodate In tetrahydrofuran; water at 0 - 20℃; for 5.16667 h;
Example 110; This example concerns the synthesis of Aldehyde 5: To a stirred solution of 2 (5.248 g, 30.59 mmol) in dry DMF (172 mL) was added N,N-dimethylformamide dimethyl acetal (DMFDMA) (13.7 g, 12.0 mL, 88.5 mmol). After heating at 1400C for 16 h, the dark red solution was cooled to 0°C and added slowly, over 20 min via cannula, to a rapidly stirred solution OfNaIO4 (18.7 g, 87.4 mmol) in H2O (69 mL) and DMF (23 mL) at 0°C. The reaction flask was washed with DMF (20 mL) at 0°C and added to NaIO4 mixture. The reaction was stirred at 0°C for 30 min then allowed to warm to rt. After an additional 4 h, the orange solution was filtered and rinsed with PhMe (200 mL). The filtrate was then washed with H2O (2 x 200 mL) and sat. aq. NaCl (2 x 100 mL). The dried (MgSO4) extract was filtered, concentrated in vacuo to a dark red oil, and purified by flash chromatography over silica gel, eluting with 20-50percent EtOAc / Hexanes to give known aldehyde 5 (4.737 g, 25.53 mmol, 84percent). 1H NMR (400 MHz, CDCl3) δ 10.41 (s, IH), 8.13 (d, J- 2.0 Hz, IH), 7.97 (dd, J= 8.3 Hz, IH), 7.78 (dd, J= 2.0, 8.3 Hz, IH); 13C NMR (100 MHz, CDCl3) δ 186.9, 150.1, 140.2, 134.2, 130.9, 129.3, 124.8.
Stage #1: at 135℃; for 12 h; Stage #2: With sodium periodate In water; N,N-dimethyl-formamide at 20℃; for 3 h;
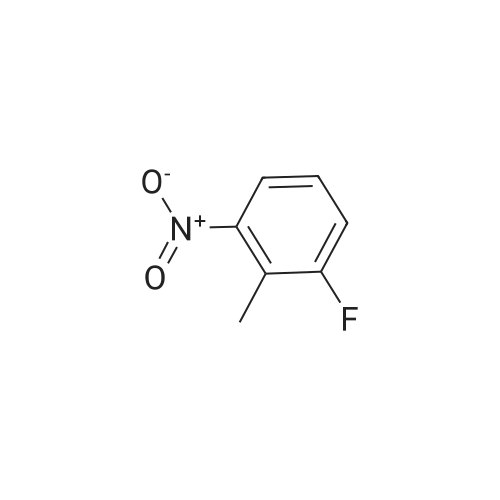
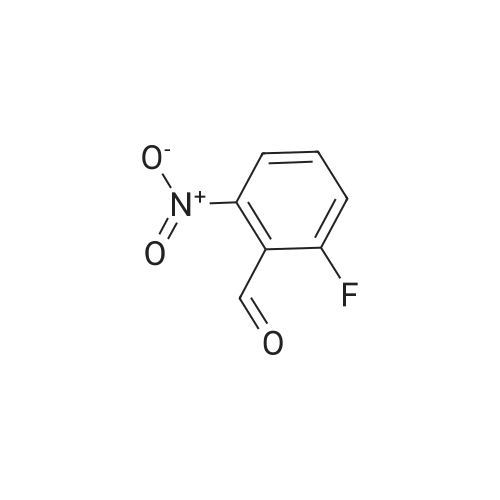
A mixture of l-fluoro-2-methyl-3-nitrobenzene and Λ/,Λ/-dimethylformamide dimethyl acetal (2.75 eq.) was heated to 135°C for 12 h. The reaction mixture was cooled to RT and added dropwise to a solution Of NaIO4 (2.75 eq.) in water/DMF (1 :1, 1.1 M). After the addition is finished, the reaction mixture was stirred at RT for 3 h. Then, it was filtered, and the solid was washed with toluene. The filtrate was transferred to a separatory funnel, and the layers were separated. The organic layer was washed with water and dried (Na2SO4). Evaporation of the solvent under reduced pressure gave a crude that was purified by flash chromatography on silica using a gradient of EtO Ac/Petroleum ether (1 :30) to afford (23percent) the title compound. MS (ES+) C7H4FNO3 required: 169, found: 170 (M+H)+.
Stage #1: at 140℃; for 16 h; Stage #2: With sodium periodate In water; N,N-dimethyl-formamide at 0 - 20℃; for 9 h;
Example 109; This example concerns the synthesis of Aldehyde 4: To a stirred solution of1 (18.53 g, 108.0 mmol) in dry DMF (240 mL) was added N,JV-dimethylformamide dimethyl acetal (DMF'DMA) (39.5 g, 44.0 mL, 331 mmol). After heating at 140°C for 16 h, the dark red solution was cooled to 0°C and added slowly, over 1 h via cannula, to a rapidly stirred solution OfNaIO4 (83.0 g, 388.0 mmol) in H2O (291 mL) and DMF (77 mL) at 0°C. The reaction flask was washed with DMF (20 mL) at 0°C and added to NaIO4 mixture. The reaction was stirred at 0°C for 2 h then allowed to warm to rt. After an additional 6 h, the orange solution was filtered and rinsed with PhMe/EtOAc (1 : 1 , 200 mL). The filtrate was then washed with H2O (3 x 150 mL) and sat. aq. NaCl (3 x 150 mL). The dried (MgSO4) extract was concentrated in vacuo to a dark red oil, and hexanes (40 mL) were added. Solids were isolated and recrystalized in PhMe to give the known aldehyde 4 (17.23 g, 92.88 mmol, 86percent). 1H NMR (400 MHz, CDCl3) δ 10.42 (s, IH), 8.01 (dd, J= 1.0, 8.2 Hz, IH), 7.79 (dd, J= 1.0, 8.1 Hz5 IH), 7.65 (t, J= 8.1 Hz, IH); 13C NMR (100 MHz, CDCl3) δ 188.6, 148.4, 138.6, 132.9, 132.4, 123.4, 121.9.
Reference:
[1] Chemical and Pharmaceutical Bulletin, 1981, vol. 29, # 1, p. 249 - 253
[2] Heterocycles, 1982, vol. 17, p. 417 - 423
57
[ 21423-81-4 ]
[ 4637-24-5 ]
[ 58588-64-0 ]
Reference:
[1] Patent: US2010/136142, 2010, A1,
58
[ 6342-56-9 ]
[ 4637-24-5 ]
[ 67751-23-9 ]
Yield
Reaction Conditions
Operation in experiment
91%
at 110℃; for 3 h;
1,1-dimethoxy-N,N-dimethylmethanamine (100 g, 839 mmol, 1.02 equiv.) and 1,1-dimethoxypropan-2-one (97 g, 821 mmol) were added and stirred at 110° C. for 3 hours. The produced methanol was removed by a Dean-Stark apparatus. After the solution was cooled to room temperature, the remaining volatile materials were removed in vacuo to provide 130 g of the crude product, (E)-4-(dimethylamino)-1,1-dimethoxybut-3-en-2-one (1) (130 g, 143 g theoretical, 91percent). LC-MS m/z 283 (M+1). Reference: WO 2006/0097341A1 (incorporated by reference), pg 67.
68%
for 20 h; Reflux
(3E)-4-(Dimethylamino)-1,1-dimethoxybut-3-en-2-one (C10) A solution of N,N-dimethylformamide dimethyl acetal (147 g, 1.23 mol) and 1,1-dimethoxyacetone (146 g, 1.24 mol) in 2-butanol (1 L) was heated at reflux for 20 hours. After removal of solvent in vacuo, the residue was distilled under vacuum to provide the product as an oil. Yield: 145 g, 0.837 mol, 68percent. Boiling point: 132-140° C./0.15 torr. NMR and MS data were obtained using the product of a reaction run under similar conditions. LCMS m/z 174.0 (M+1). 1H NMR (400 MHz, CDCl3) δ 2.77 (br s, 3H), 3.02 (br s, 3H), 3.30 (s, 6H), 4.47 (s, 1H), 5.23 (br d, J=12.6 Hz, 1H), 7.63 (d, J=12.6 Hz, 1H).
68%
at 110℃; for 3 h; Inert atmosphere
A stuffed solution of 1,1-dimethoxy-N,N-dimethylmethanamine (5 g, 42 mmol) was mixed with 1,1-dimethoxypropan-2-one (4.95 g, 42 mmol) under nitrogen atmosphere. The reaction mixture was heated to 110 °C for 3 h. The methanol produced was removed by Dean-Stark apparatus. After completion the solution was cooled to rt and volatiles were removed under reduced pressure to obtain oily residue. The crude material was purified by column chromatography (60-120 silica gel, 0-5percent Methanol/DCM) to afford 2.3g of the title compound (68percent). LCMS: mlz=174 [M+1]. ‘H-NMR (400MHz, CDC13) ö: 7.6 1-7.58 (d, 1H), 5.19-5.16 (d, 1H), 4.43 (s, 1H), 3.26 (s, 6H), 3.10-3.08 (d, 3H), 2.79 (s, 3H), 1.15 (s, 6H).
Reference:
[1] Journal of the American Chemical Society, 2002, vol. 124, # 51, p. 15302 - 15307
61
[ 55289-35-5 ]
[ 4637-24-5 ]
[ 20357-21-5 ]
Yield
Reaction Conditions
Operation in experiment
67%
Stage #1: at 135℃; for 16 h; Stage #2: With sodium periodate In water; N,N-dimethyl-formamide at 0 - 20℃; for 22 h;
Example 35; This example describes the synthesis of Aldehyde 10: To a stirred solution of 9 (4.94 g, 22.3 mmol) and DMF (22.3 mL) was added DMF'DMA (8.18 g, 9.59 mL, 68.6 mmol). After heating at 135°C for 16 h, the dark red solution was cooled to 0°C and added to a rapidly stirred solution OfNaIO4 (14.7 g, 68.6 mmol) in H2O (46 mL) and DMF (23 mL) at 0°C. The reaction flask was washed with DMF (23 mL) at 0°C and added to NaIO4 mixture. The reaction was stirred at O0C for 4 h then allowed to warm to rt. After an additional 18 h, the orange solution was filtered over a pad of celite-.(R). and rinsed with EtOAc (200 mL) to remove precipitate. The filtrate was then washed with H2O (3 x 150 mL) and sat. aq. NaCl (3 x 150 mL). The dried extract (MgSO4) was concentrated in vacuo and purified by chromatography over silica gel, eluting with 40percent EtOAc / Hexanes to give the known aldehyde 10 (3.44 g, 14.8 mmol, 67percent). 1H NMR (400 MHz, CDCl3) δ 10.3 (s, IH), 8.04 (dd, J= 1.0, 8.2 Hz, IH), 7.95 (dd, J= 1.0, 8.0 Hz, IH), 7.55 (t, J= 8.0 Hz, IH); 13C NMR (100 MHz, CDCl3) δ 188.6, 148.4, 138.6, 132.9, 132.4, 123.4, 121.9.
Reference:
[1] Zeitschrift fuer Naturforschung, B: Chemical Sciences, 1991, vol. 46, # 8, p. 1110 - 1112
[2] Zeitschrift fuer Naturforschung, B: Chemical Sciences, 1991, vol. 46, # 8, p. 1110 - 1112
[3] Journal of Heterocyclic Chemistry, 1991, vol. 28, # 7, p. 1715 - 1720
66
[ 1122-62-9 ]
[ 4637-24-5 ]
[ 75415-03-1 ]
Yield
Reaction Conditions
Operation in experiment
30%
Stage #1: at 100℃; for 16 h; Reflux Stage #2: With hydrazine hydrate In ethanol at 60℃; for 0.5 h;
A mixture of N-dimethoxymethyl-N,N-dimethylamine(10 mL) and 2-acetylpyridine (40 mL) was refluxed at 100 °C for 16 h, then concentrated by rotating evaporationto give dark brown solid. This was washed with hexane(100 mL) and diethyl ether (100 mL) to give the pure and bright yellow crystalline product. Hydrazine monohydrate (40 mL) and ethanol (15 mL) were mixed with the previously obtained solid, and the mixture was stirred at 60 °C for 30 min. After cooling, distilled water (75 mL) was added to the solution, which was kept at 0 °C for 24 h to produce a light yellow precipitate. This was recrystallized from a mixture of dichloromethane and hexane.
Reference:
[1] Transition Metal Chemistry, 2018, vol. 43, # 3, p. 211 - 220
67
[ 6342-56-9 ]
[ 4637-24-5 ]
[ 111573-59-2 ]
Reference:
[1] Chinese Journal of Catalysis, 2016, vol. 37, # 9, p. 1446 - 1450
Reference:
[1] Chemistry of Heterocyclic Compounds, 2011, vol. 47, # 4, p. 425 - 434
71
[ 41252-97-5 ]
[ 4637-24-5 ]
[ 115666-47-2 ]
Yield
Reaction Conditions
Operation in experiment
61%
Stage #1: With pyrrolidine In N,N-dimethyl-formamide at 110℃; for 4 h; Stage #2: With hydrogenchloride; titanium(III) chloride In water; N,N-dimethyl-formamide at 0℃; for 3 h;
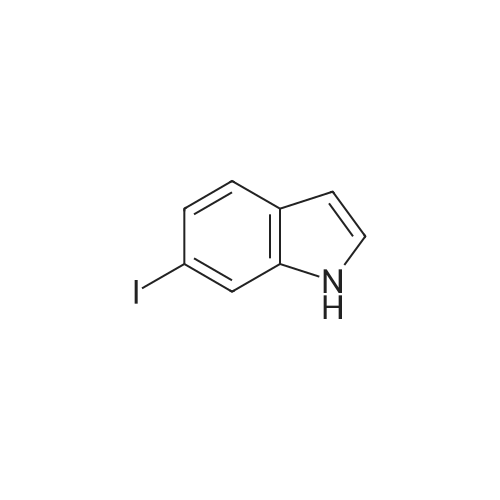
6-Iodoindole (4): Starting material 3 (5.00 g, 19.0 mmol, 1.0 equiv) was dissolved in DMF (50 mL). DMFDMA (3.05 mL, 22.8 mmol, 1.2 equiv) was added, followed by pyrrolidine (1.90 mL, 23.1 mmol, 1.2 equiv). The mixture was heated to 110 °C for 4 h until complete consumption of starting material as monitored by TLC, and allowed to cool to rt. DMF (100 mL) and 4 M aqueous NH4OAc buffer (83 mL, 17.5 equiv) were added and the solution was cooled with an ice bath. An aqueous solution of 20percent TiCl3 in 3percent HCl (73.3 mL, 114.06 mmol, 6.0 equiv) was added via a dropping funnel. The reaction mixture was stirred for 3 h followed by extraction with TBME (3 × 100 mL). The organic phase was washed with 2 M NaOH (100 mL), H2O (100 mL) and dried with MgSO4. After evaporation of the solvent the raw material was prepurified by short column chromatography [PE/EA (15:1)] to be sublimated in HV at 110–115 °C to give the product as a slightly yellowish solid (2.83 g, 11.6 mmol, 61percent). TLC [PE/EA (10:1)]: Rf = 0.25. Mp.: 103-105 °C. 1H NMR (400 MHz, CDCl3): = 8.10 (sbr, 1H, NH), 7.75 (dd, 1H, J = 2.0 Hz, J = 1.0 Hz, 4-H), 7.40-7.39 (m, 2H, 5-H and 7-H), 7.13 (dd, 1H, J = 3.2 Hz, J = 2.5 Hz, 2-H), 6.52 (ddd, 1H, J = 3.1 Hz, J = 2.0 Hz, J = 0.9 Hz, 3-H). 13C NMR (100 MHz, CDCl3): = 137.1 (1C, C-7a), 128.7 (1C, C-7), 127.2 (1C, C-3a), 124.5 (1C, C-2), 122.4 (1C, C-5), 120.0 (1C, C-4), 102.9 (1C, C-3), 85.8 (1C, C-6). IR (ATR): = 3888 (w), 3410 (s), 3094 (w), 3073 (w), 2925 (w), 1884 (w), 1726 (w), 1624 (w), 1597 (w), 1563 (w), 1493 (w), 1449 (m), 1392 (m), 1331 (m), 1309 (m), 1270 (w), 1232 (w), 1199 (w), 1090 (m), 1059 (w), 1043 (w), 998 (m), 944 (w), 878 (m), 858 (m), 802 (s), 759 (m), 727 (s), 651 (w), 607 (m), 586 (m), 565 (w). UV-Vis (MeOH): max (lg ) = 202 (4.32), 227 (4.62), 279 (3.86), 285 (3.86). MS (EI, 70 eV): m/z (percent) = 243 [M]+ (100), 116 [M–I]+ (56), 89 (26), 63 (12)
Reference:
[1] Organic Letters, 2017, vol. 19, # 23, p. 6296 - 6299
[2] Beilstein Journal of Organic Chemistry, 2015, vol. 11, p. 1700 - 1706
72
[ 1192-62-7 ]
[ 4637-24-5 ]
[ 17168-45-5 ]
Yield
Reaction Conditions
Operation in experiment
97%
at 100℃; for 9 h;
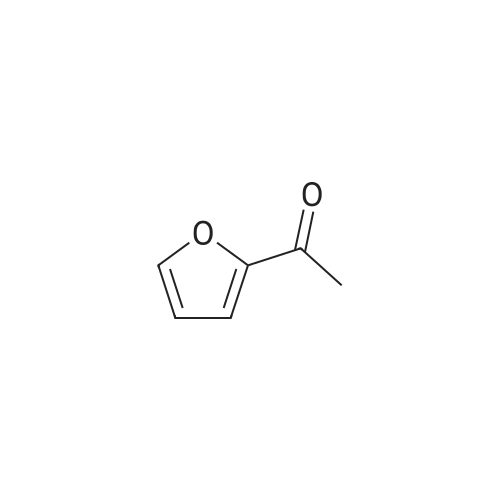
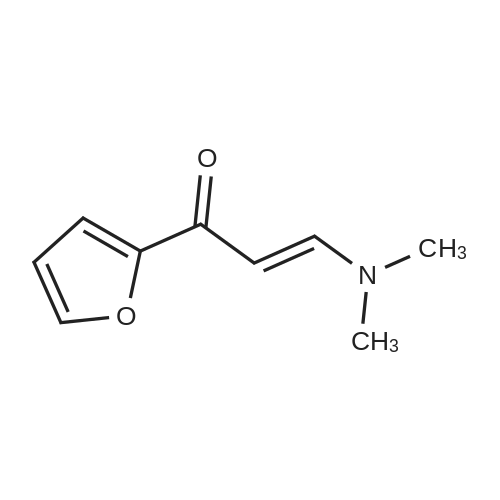
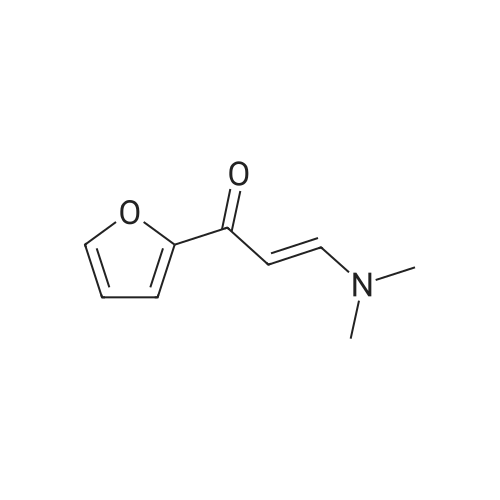
A mixture of 2-acetylfuran (25.0 g, 0.227 mmol) and N,N-dimethylformamide dimethylacetal (40 ml) was stirred at 100°C for 9 hours. After cooling as it was, the reaction mixture was concentrated. To the residue were added diethyl ether and hexane. The resulting solid was collected by filtration and washed with hexane, to give the title compound (36.5 g, 97percent) as a brown solid.1H NMR (400 MHz, DMSO-d6) δ ppm; 2.88 (3H, br s), 3.14 (3H, br s), 5.65 (1H, d, J= 12.6 Hz), 6.60 (1H, dd, J=2.0, 3.4 Hz), 7.10 (1H, dd, J = 0.8, 3.4 Hz), 7.68 (1H, d, J = 12.6 Hz), 7.79 (1H, dd, J = 0.8, 2.0 Hz).
97%
at 100℃; for 9 h;
Reference Example 5 3-(Dimethylamino)-1-(2-furyl)-2-propen-1-one A mixture of 2-acetylfuran (25.0 g, 0.227 mmol) and N,N-dimethylformamide dimethylacetal (40 ml) was stirred at 100° C. for 9 hours. After cooling as it was, the reaction solution was concentrated. Diethyl ether and hexane were added to the residue, and the resulting solid was collected by filtration and washed with hexane, to give the title compound (36.5 g, 97percent) as a brown solid. 1H NMR (400 MHz, DMSO-d6) δ ppm; 2.88 (3H, br s), 3.14 (3H, br s), 5.65 (1H, d, J=12.6 Hz), 6.60 (1H, dd, J=2.0, 3.4 Hz), 7.10 (1H, dd, J=0.8, 3.4 Hz), 7.68 (1H, d, J=12.6 Hz), 7.79 (1H, dd, J=0.8, 2.0 Hz).
Reference:
[1] Patent: EP1439175, 2004, A1, . Location in patent: Page 45-46
[2] Patent: US2004/6082, 2004, A1, . Location in patent: Page/Page column 22
[3] Journal of Heterocyclic Chemistry, 1996, vol. 33, # 6, p. 1707 - 1710
[4] Organic Process Research and Development, 2008, vol. 12, # 3, p. 490 - 495
[5] Journal of the Korean Chemical Society, 2011, vol. 55, # 2, p. 243 - 250
[6] Asian Journal of Chemistry, 2012, vol. 24, # 4, p. 1837 - 1843
[7] Journal of the Chinese Chemical Society, 2003, vol. 50, # 2, p. 283 - 296
[8] Bioorganic and Medicinal Chemistry Letters, 2010, vol. 20, # 21, p. 6282 - 6285
[9] Synthetic Communications, 2012, vol. 42, # 10, p. 1521 - 1531
[10] Bioorganic and Medicinal Chemistry Letters, 2014, vol. 24, # 6, p. 1581 - 1588
[11] Journal of Heterocyclic Chemistry, 2014, vol. 51, # SUPPL. 1, p. E384-E388
[12] Research on Chemical Intermediates, 2015, vol. 41, # 6, p. 3759 - 3765
[13] Chemical Biology and Drug Design, 2017, vol. 90, # 5, p. 936 - 942
[14] European Journal of Medicinal Chemistry, 2018, vol. 155, p. 545 - 551
[15] Bioorganic Chemistry, 2019, vol. 83, p. 549 - 558
73
[ 4637-24-5 ]
[ 118664-99-6 ]
[ 63762-72-1 ]
[ 122509-72-2 ]
Reference:
[1] Journal of Medicinal Chemistry, 2009, vol. 52, # 7, p. 1853 - 1863
Reference:
[1] Patent: EP1382603, 2004, A1, . Location in patent: Page 42; 43
[2] Journal of Organometallic Chemistry, 2010, vol. 695, # 14, p. 1753 - 1760
76
[ 34633-69-7 ]
[ 4637-24-5 ]
[ 162100-56-3 ]
Reference:
[1] Journal of Medicinal Chemistry, 1998, vol. 41, # 10, p. 1598 - 1612
77
[ 7494-45-3 ]
[ 4637-24-5 ]
[ 121859-57-2 ]
Reference:
[1] RSC Advances, 2014, vol. 4, # 9, p. 4672 - 4675
[2] Journal of Medicinal Chemistry, 1998, vol. 41, # 10, p. 1598 - 1612
78
[ 60956-25-4 ]
[ 4637-24-5 ]
[ 610794-15-5 ]
Yield
Reaction Conditions
Operation in experiment
5.5%
Stage #1: With pyrrolidine In 1,4-dioxane at 100℃; for 18 h; Inert atmosphere Stage #2: at 110℃; for 4 h;
ep 2: A mixture of 298 (3.0 g, 13.04 mmol), pyrrolidine (926 mg, 13.04 mmol), and DMF-DMA (7.76 g, 65.22 mmol) in 1,4-dioxane (20 mL) under nitrogen atmosphere was heated at 100 °C for 18 h. The reaction was concentrated under to dryness in vacuo and to the residue was added iron (3.65 g, 65.22 mmol) and HO Ac (40 mL). The resulting mixture was heated at 110 °C for 4 h, cooled to RT and filtered. The filtrate was concentrated in vacuo. The crude was purified by S1O2 chromatography eluting with petroleum ether/EtOAc (10:1) to afford 150 mg (5.5percent) of 4-bromo-5-methyl-lH-indole (300) as a yellow solid: MS (ESI) m/z = 210.1 (M+l).
Stage #1: at 110℃; for 15 h; Stage #2: With sodium hydroxide In water; N,N-dimethyl-formamide at 110℃; for 36 h;
Pyruvic aldehyde dimethyl acetal (10 g, 84 mmol) and N, N’-dimethyl formamide dimethyl diacetal (10 g, 84 mmol) in DMF (30 ml) were heated in a round bottom flask at 110 °C for 15 h. Guanidine hydrochloride (12 g, 127 mmol) and NaOH (6.7 g, 169 mmol) in water were then added to the reaction mixture. The mixture was refluxed for further 36 h. The reaction mass was then cooled and filtered. The product was recrystallized from hot ethyl acetate to yield white crystalline solid 2 (10 g, 70percent).
36%
Stage #1: at 100℃; for 16 h; Stage #2: With sodium hydroxide In water at 20℃; for 48 h;
Procedure H: Intermediate 8 (1-8) - 2-Aminopyrimidine-4-carboxaldehyde dimethylacetal.; [0093] A solution of 5.5 mL (41 mmol, 1.0 eq.) of dimethylformamide dimethyl acetal and 5.0 mL (41 mmol, 1.0 eq.) pyruvric aldehyde dimethyl acetal was heated at 100 0C for 16 h. Methanol was removed in vacuo to afford a brown oil. A solution of 1.8 g (45 mmol, 1.1 eq.) of sodium hydroxide in 5 mL of water was added to a solution of 4.3 g (45 mmol, 1.1 eq.) of guanidine HCl in 10 mL of water. The resulting solution was added to the above described oil. The resulting mixture was stirred at room temperature for 48 h. The mixture was filtered to provide 2.5 g (15 <n="38"/>mmol, 36percent) of 2-aminopyrimidine-4-carboxaldehyde dimethyl acetal (1-8).
50%
With sodium hydroxide In water
a) 2-Aminopyrimidine-4-carboxaldehyde dimethyl acetal Dimethylformamide dimethyl acetal (55 mL, 0.41 mol), and pyruvic aldehyde dimethyl acetal (50 mL, 0.41 mol) were combined and heated to 100° for 18 h. Methanol was removed in vacuo to afford an oil. A solution of NaOH (18 g, 0.45 mol) in H2 O (50 mL) was added to guanidine HCl (43 g, 0.45 mol) in H2 O (100 mL), and the resulting solution was added to the above described oil. The resulting mixture was stirred at 23° for 48 h. Filtration afforded 25 g (50percent) of the title compound.
Reference:
[1] Tetrahedron Letters, 2013, vol. 54, # 28, p. 3715 - 3717
[2] Patent: EP1227092, 2002, A2, . Location in patent: Page 25
[3] Patent: EP1229035, 2002, A1, . Location in patent: Page 25
[4] Patent: EP1227091, 2002, A2, . Location in patent: Page 25
[5] Patent: WO2007/104053, 2007, A2, . Location in patent: Page/Page column 36-37
[6] Patent: US5593992, 1997, A,
[7] Patent: US5670527, 1997, A,
80
[ 50-01-1 ]
[ 4637-24-5 ]
[ 165807-05-6 ]
Yield
Reaction Conditions
Operation in experiment
50%
With sodium hydroxide In water
a 2-Aminopyrimidine-4-carboxaldehyde dimethyl acetal Dimethylformamide dimethyl acetal (55 mL, 0.41 mol), and pyrrhic aldehyde dimethyl acetal (50 mL, 0.41 mol) were combined and heated to 100° for 18 h. Methanol was removed in vacuo to afford an oil. A solution of NaOH (18 g, 0.45 mol) in H2 O (50 mL) was added to guanidine HCl(43 g, 0.45 mol) in H2 O (100 mL), and the resulting solution was added to the above described oil. The resulting mixture was stirred at 23° for 48 h. Filtration afforded 25 g (50percent) of the title compound.
Reference:
[1] Patent: US5593991, 1997, A,
81
[ 6342-56-9 ]
[ 7732-18-5 ]
[ 4637-24-5 ]
[ 180869-36-7 ]
Yield
Reaction Conditions
Operation in experiment
93%
With sodium methylate; thiourea; methyl iodide In methanol
a 2-Methylthiopyrimidine-4-carboxaldehyde dimethyl acetal Pyruvaldehyde dimethyl acetal (19.2 mL, 159.1 mmol) and N,N-dimethylformamide dimethyl acetal (21.12 mL, 159.1 mmol) were combined in a 500 mL flask and heated at 100° C. After 4.5 h the flask was removed from the heat, thiourea (11.0 g, 144.5 mmol), NaOMe (25 wt. percent solution in MeOH, 39.7 mL, 173 mmol) and 30 mL of MeOH were added and heating was continued at 65° C. After 18 h the solution was cooled to 25° C. and MeI (10.8 mL, 173 mmol) was added over 5 min (exothermic). After 3 h, the solution was diluted with 250 mL of H2 O and extracted with EtOAc (3*100 mL). The organics were combined, dried over Na2 SO4 and concentrated to give the title compound (26.8 g, 93percent) as a brown oil.
93%
With sodium methylate; thiourea; methyl iodide In methanol
a) 2-Methylthiopyrimidine-4-carboxaldehyde dimethyl acetal Pyruvaldehyde dimethyl acetal (19.2 mL, 159.1 mmol) and N,N-dimethylformamide dimethyl acetal (21.12 mL, 159.1 mmol) were combined in a 500 mL flask and heated at 100° C. After 4.5 h the flask was removed from the heat, thiourea (11.0 g, 144.5 mmol), NaOMe (25 wt. percent solution in MeOH, 39.7 mL, 173 mmol) and 30 mL of MeOH were added and heating was continued at 65° C. After 18 h the solution was cooled to 25° C. and MeI (10.8 mL, 173 mmol) was added over 5 min (exothermic). After 3 h, the solution was diluted with 250 mL of H2 O and extracted with EtOAc (3 X 100 mL). The organics were combined, dried over Na2 SO4 and concentrated to give the title compound (26.8 g, 93percent) as a brown oil.
93%
With sodium methylate; thiourea; methyl iodide In methanol
a) 2-Methylthiopyrimidine-4-carboxaldehyde dimethyl acetal Pyruvaldehyde dimethyl acetal (19.2 mL, 159.1 mmol) and N,N-dimethylformamide dimethyl acetal (21.12 mL, 159.1 mmol) were combined in a 500 mL flask and heated at 100° C. After 4.5 h the flask was removed from the heat, thiourea (11.0 g, 144.5 mmol), NaOMe (25 wt. percent solution in MeOH, 39.7 mL, 173 mmol) and 30 mL of MeOH were added and heating was continued at 65° C. After 18 h the solution was cooled to 25° C. and MeI (10.8 mL, 173 mmol) was added over 5 min (exothermic). After 3 h, the solution was diluted with 250 mL of H2 O and extracted with EtOAc (3*100 mL). The organics were combined, dried over Na2 SO4 and concentrated to give the title compound (26.8 g, 93percent) as a brown oil.
93%
With sodium methylate; thiourea; methyl iodide In methanol
a 2-Methylthiopyrimidine-4-carboxaldehyde dimethyl acetal Pyruvaldehyde dimethyl acetal (19.2 mL, 159.1 mmol) and N,N-dimethylformamide dimethyl acetal (21.12 mL, 159.1 mmol) were combined in a 500 mL flask. After heating at 100° C. 4.5 h, thiourea (11.0 g, 144.5 mmol), NaOMe (25 wt. percent solution in MeOH, 39.7 mL, 173 mmol) and 30 mL of MeOH were added and heating was continued at 65° C. After 18 h, the solution was cooled to 25° C. and MeI (10.8 mL, 173 mmol) was added over 5 min (exothermic). After 3 h, the solution was diluted with 250 mL of H2 O and extracted with EtOAc (3*100 mL). The organics were combined, dried (Na2 SO4) and concentrated to give the title compound (26.8 g, 93percent) as a brown oil.
Reference:
[1] Patent: US5593991, 1997, A,
[2] Patent: US5593992, 1997, A,
[3] Patent: US5670527, 1997, A,
[4] Patent: US5739143, 1998, A,
[5] Patent: US5658903, 1997, A,
82
[ 6342-56-9 ]
[ 4637-24-5 ]
[ 74-88-4 ]
[ 180869-36-7 ]
Yield
Reaction Conditions
Operation in experiment
82%
With sodium methylate; thiourea In methanol
a 2-Methylthiopyrimidine-4-carboxaldehyde dimethyl acetal Pyruvic aldehyde dimethyl acetal (60 mL, 459 mmol) and N,N-dimethyl formamide dimethyl acetal (60 mnL, 459 mmol) were stirred together at 100° C. for 18 h. The mixture was cooled. Methanol (300 mL), thiourea (69.6 g) and sodium methoxide (231 mL, 25 wt percent in MeOH) were added to the above mixture and stirred at 70° C. for 2 h. After cooling, iodomethane (144 mL) was added dropwise and the mixture was stirred 3 h. at room temp. After diluting with EtOAc and H2 O, the organic phase was separated, dried (Na2 SO4),and concentrated to yield the title compound as a brown oil (75.5 g, 82percent yield). 1 H NMR (CDCl3): d 8.17 (d, 1H), 6.77 (d, 1H), 5.15 (s, 1H), 3.40 (s, 6H).
82%
With sodium methylate; thiourea In methanol
a 2-Methylthiopyrimidine-4-carboxaldehyde dimethyl acetal Pyruvic aldehyde dimethyl acetal (60 milliliter (hereinafter "mL"), 459 millimole (hereinafter "mmol")) and N,N-dimethyl formamide dimethyl acetal (60 mL, 459 mmol) were stirred together at 100° C. for 18 hours (hereinafter "h"). The mixture was cooled. Methanol (300 mL), thiourea (69.6 g) and sodium methoxide (231 mL, 25 wt percent in MeOH) were added to the above mixture and stirred at 70° C. for 2 h. After cooling, iodomethane (144 mL) was added dropwise and the mixture was stirred 3 h. at room temp. After diluting with EtOAc and H2 O, the organic phase was separated, dried (Na2 SO4), and concentration to yield the title compound as a brown oil (75.5 gram (hereinafter "g"), 82percent yield). 1 H NMR (CDCl3): d 8.17 (d, 1H), 6.77 (d, 1H), 5.15 (s, 1H), 3.40 (s, 6H).
82%
With sodium methylate; thiourea In methanol
a 2-Methylthiopyrimidine-4-carboxaldehyde dimethyl acetal Pyruvic aldehyde dimethyl acetal (60 mL, 459 mmol) and N,N-dimethyl formamide dimethyl acetal (60 mL, 459 mmol) were stirred together at 100° C. for 18 h. The mixture was cooled. Methanol (300 mL), thiourea (69.6 g) and sodium methoxide (231 mL, 25 wt percent in MeOH) were added to the above mixture and stirred at 70° C. for 2 h. After cooling, iodomethane (144 mL) was added dropwise and the mixture was stirred 3 h. at room temp. After diluting with EtOAc and H2 O, the organic phase was separated, dried (Na2 SO4),and concentrated to yield the title compound as a brown oil (75.5 g, 82percent yield). 1 H NMR (CDCl3): d 8.17 (d, 1H), 6.77 (d, 1H), 5.15 (s, 1H), 3.40 (s, 6H).
Reference:
[1] Patent: US5977103, 1999, A,
[2] Patent: US6046208, 2000, A,
[3] Patent: US5756499, 1998, A,
83
[ 6342-56-9 ]
[ 4637-24-5 ]
[ 180869-36-7 ]
Yield
Reaction Conditions
Operation in experiment
93%
With sodium methylate; thiourea; methyl iodide In methanol; water
a) 2-Methylthiopyrimidine-4-carboxaldehyde dimethyl acetal Pyruvaldehyde dimethyl acetal (19.2 mL, 159.1 mmol) and N,N-dimethylformamide dimethyl acetal (21.12 mL, 159.1 mmol) were combined in a 500 mL flask. After heating at 100° C. 4.5 h, thiourea (11.0 g, 144.5 mmol), NaOMe (25 wt. percent solution in MeOH, 39.7 mL, 173 mmol) and 30 mL of MeOH were added and heating was continued at 65° C. After 18 h, the solution was cooled to 25° C. and MeI (10.8 mL, 173 mmol) was added over 5 min (exothermic). After 3 h, the solution was diluted with 250 mL of H2O and extracted with EtOAc (3*100 mL). The organics were combined, dried (Na2SO4) and concentrated to give the title compound (26.8 g, 93percent) as a brown oil.
Reference:
[1] Patent: US6218537, 2001, B1,
84
[ 6342-56-9 ]
[ 4637-24-5 ]
[ 17356-08-0 ]
[ 74-88-4 ]
[ 180869-36-7 ]
Reference:
[1] Journal of Medicinal Chemistry, 2015, vol. 58, # 3, p. 1067 - 1088
[2] Patent: WO2016/7966, 2016, A2, . Location in patent: Paragraph 0024
[3] Patent: US2003/225082, 2003, A1, . Location in patent: Page 8-9
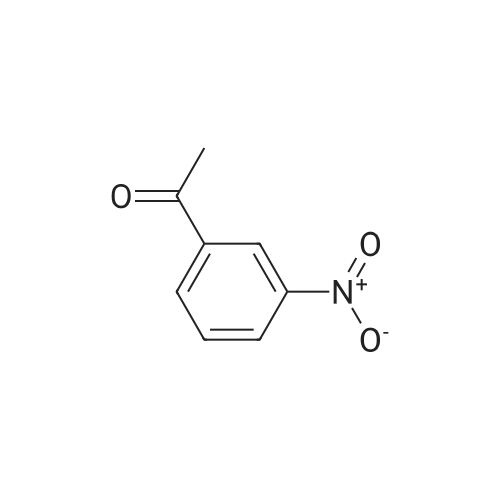
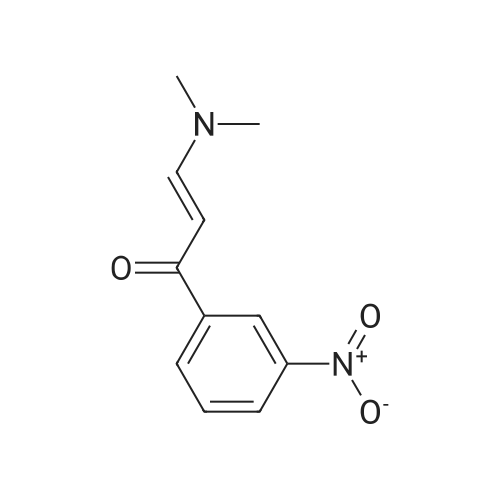
In a 250 mL round bottom flask was added 16.5 g (100 mmol) of 3-nitroacetophenone, 26.5 mL of N,N-dimethylformamide dimethyl acetal (200 mmol) and 100 mL of xylene. The mixture was heated to 140°C for 12 hours. Then the reaction mixture was cooled to room temperature and the formed precipitate was filtered off, washed with petroleum ether and diethyl ether followed by purification by chromatography using ethyl acetate/cyclohexene (25/75) to afford 19.6 g of a yellow solid. Yield: 89percent. 1H NMR (200 MHz, DMSO-d6) δ 8.59 (t, 1H), 8.42 – 8.23 (m, 1H), 7.81 (d, J = 12.1 Hz, 1H), 7.70 (t, J = 7.9 Hz, 1H), 5.89 (d, J = 12.1 Hz, 1H), 3.17 (s, 1H), 2.95 (s, 1H). 13C NMR (50 MHz, DMSO-d6) δ 182.8, 155.2, 147.9, 141.6, 133.4, 129.8, 125.1, 121.5, 90.3, 44.7, 37.3.
79%
Heating / reflux; Neat (no solvent)
3-Nitroacetophenone (5.0 g, 30.3 mmol) in dimethylformamide-dimethylacetal (10 mL) is heated at reflux overnight. The reaction mixture is cooled to room temperature and evaporated to remove the volatiles. The residue is slurried in ethyl ether and the suspension is filtered and washed with ether to give 10.5 g (79percent) of 3-(dimethylamino)-1-(3-nitrophenyl)-2-propen-1-one, 104-105° C.
79%
Heating / reflux
3-Nitroacetophenone (5.0g, 30.3 mmol) in dimethylformamide-dimethylacetal (10 mL) was heated at reflux overnight. The reaction mixture was cooled to room temperature and evaporated to remove the volatiles. The residue was slurried in ethyl ether and the suspension was filtered and washed with ether to give 10.5 g (79percent) of 3-(dimethylamino)-1-(3-nitrophenyl)-2-propen-1-one, 104-105° C.
79%
Heating / reflux
3-Nitroacetophenone (5.0g, 30.3 mmol) in dimethylformamide-dimethylacetal (10 mL) was heated at reflux overnight. The reaction mixture was cooled to room temperature and evaporated to remove the volatiles. The residue was slurried in ethyl ether and the suspension was filtered and washed with ether to give 10.5 g (79percent) of 3-(dimethylamino)-1-(3-nitrophenyl)-2-propen-1-one, 104-105° C.
79%
Heating / reflux
Step 1: 3-(Dimethylamino)-1-(3-nitrophenyl)-2-propen-1-one 3-Nitroacetophenone (5.0 g, 30.3 mmol) in dimethylformamide-dimethylacetal (10 mL) is heated at reflux overnight. The reaction mixture is cooled to room temperature and evaporated to remove the volatiles. The residue is slurried in ethyl ether and the suspension is filtered and washed with ether to give 10.5 g (79percent) of 3-(dimethylamino)-1-(3-nitrophenyl)-2-propen-1-one, 104-105° C.
11.34 g
for 21 h; Reflux
10 g of 3-nitroacetophenone (60.6 mmol, 1 equiv.), 35.63 g of N, N-dimethylformamide dimethylacetal (181 mmol, 3 equiv.), and 50 ml of xylene were refluxed overnight (21 hours), a little raw material was not reacted and the reaction was stopped. The reaction solution was cooled and concentrated to remove xylene. 150 ml of petroleum ether was added to the residue, and a solid precipitated under stirring, followed by filtration to obtain 11.34 g of 3-dimethylamino-1-(3-nitrophenyl)-prop-2-en-1-one as a yellow solid.
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2017, vol. 27, # 8, p. 1780 - 1783
[2] Patent: US2007/219183, 2007, A1, . Location in patent: Page/Page column 11
[3] Bioorganic and Medicinal Chemistry Letters, 2009, vol. 19, # 10, p. 2735 - 2738
[4] Bioorganic and Medicinal Chemistry Letters, 2009, vol. 19, # 24, p. 6890 - 6892
[5] Patent: US2006/63784, 2006, A1, . Location in patent: Page/Page column 43
[6] Patent: US2006/63785, 2006, A1, . Location in patent: Page/Page column 48-49
[7] Patent: US2007/219186, 2007, A1, . Location in patent: Page/Page column 22
[8] Journal of Medicinal Chemistry, 1998, vol. 41, # 15, p. 2858 - 2871
[9] Patent: WO2009/90548, 2009, A2, . Location in patent: Page/Page column 38-39
[10] Patent: EP3287456, 2018, A1, . Location in patent: Paragraph 0154
Reference:
[1] Chemical and Pharmaceutical Bulletin, 2000, vol. 48, # 1, p. 131 - 139
89
[ 367-21-5 ]
[ 4637-24-5 ]
[ 162012-67-1 ]
Yield
Reaction Conditions
Operation in experiment
62%
Stage #1: With acetic acid In toluene at 105℃; for 2 h; Stage #2: at 125℃; for 3 h;
Compound 2-amino-4-fluoro-5-nitrobenzonitrile (7.2 g, 40 mmol) was dissolved into toluene (70 mL), DMF-DMA (N,N-dimethylformamide dimethyl acetal, 4.7 g, 40 mmol) and acetic acid (1 mL) was added, stirring to react at 105°C for 2 h, after concentration by evaporation, acetic acid (140 mL) and 3-chloro-4-fluoroaniline (6.9 g, 48 mmol) were added, stirring toreact at 125°C for 3 h. The reaction mixture was cooled to room temperature, poured into ice-water, after the pH value was adjusted to 9 with ammonia, then ethyl acetate 40 mL was added, stirred for 1 h, filtrated, dried to deliver the product N-(3-chloro-4-fluorophenyl)-7-fluoro-6-nitro-4-quinazolinamine (8.3 g, 62percent yield). 1H-NMR (d-DMSO, 400 MHz, δ ppm): 10.61 (br, 1 H), 9.68 (d, J = 10.8 Hz, 1 H), 8.81 (s, 1 H), 8.10-8.13 (d, J= 7.6 Hz, 1 H), 8.05-8.07 (d, J = 8 Hz, 1 H), 7.81-7.85 (m, 1 H), 7.50-7.54 (d, J = 8 Hz, 1 H).
Stage #1: With pyrrolidine In N,N-dimethyl-formamide at 90℃; for 18 h; Stage #2: for 1 h; Reflux Stage #3: With sodium carbonate In water
To a solution of 1-bromo-4-methoxy-2-methyl-3-nitro-benzene (31 g, 0.126 mol) in DMF (150 mL) was added dimethylformamide dimethylacetal (27 mL) and pyrrolidine (10.5mL, 0.127 mol). The mixture was heated at 90°C for 18h and cooled to r.t. The mixture was diluted with water and extracted with CH2CI2. The organic phase was dried over Na2S04i filtered and concentrated. The residue was dissolved into HOAc (50 mL) and added dropwise to a solution of Fe (20.5 g, 0.37 mol) in boiling HOAc (150 mL). The mixture was refluxed for 1h and cooled to r.t., water added and the mixture was neutralised with Na2C03, and extracted with CH2CI2.The organic phase was dried over Na2S04, filtered and concentrated. The residue was purified by Prep-TLC (PE/EtOAc =5/1) to give the title compound (13 g, 44percent). H NMR CDCI3400 MHz δ: 8.41 (brs, 1 H), 7.18-7.08 (m, 2H), 6.49-6.43 (m, 2H), 3.86 (s, 3H).
44%
Stage #1: With pyrrolidine In N,N-dimethyl-formamide at 90℃; for 18 h; Stage #2: for 1 h; Reflux
To a solution of 1-bromo-4-methoxy-2-methyl-3-nitro-benzene (31 g, 0.126 mol) in DMF (150 mL) was added dimethylformamide dimethylacetal (27 mL) and pyrrolidine (10.5 mL, 0.127 mol). The mixture was heated at 90° C. for 18 h and cooled to r.t. The mixture was diluted with water and extracted with CH2Cl2. The organic phase was dried over Na2SO4, filtered and concentrated. The residue was dissolved into HOAc (50 mL) and added dropwise to a solution of Fe (20.5 g, 0.37 mol) in boiling HOAc (150 mL). The mixture was refluxed for 1 h and cooled to r.t., water added and the mixture was neutralised with Na2CO3, and extracted with CH2Cl2. The organic phase was dried over Na2SO4, filtered and concentrated. The residue was purified by Prep-TLC (PE/EtOAc=5/1) to give the title compound (13 g, 44percent). 1H NMR CDCl3400 MHz δ: 8.41 (brs, 1H), 7.18-7.08 (m, 2H), 6.49-6.43 (m, 2H), 3.86 (s, 3H).
Stage #1: at 150℃; for 1 h; Stage #2: at 150℃; for 5 h;
In a 1000 ml three-necked flask, 6.3 g (0.05 mmol) of 2-acetylthiophene was added,(N, N-dimethylbutyl) benzylammonium chloride (202 g, 0.5 mmol)Dimethylformamide dimethylacetal (22 g, 0.15 mmol) was added, and the mixture was stirred at 150 ° C for 1 hour.Then, 1.7 g (0.05 mmol) of hydroxylamine was added, and the reaction was stirred at 150 ° C for 5 hours.6.8 g (0.05 mmol) of aminoguanidine hydrochloride and the appropriate amount of water were added and the reaction was stirred at 150 ° C for 1 hour,12.3 g (0.05 mmol) of N- [3- [3- (dimethylamino) -1-oxo-2-propenyl] -phenyl] -N-methylacetamide was added,The reaction was stirred at 100 ° C for 5 hours, cooled to 10 ° C,Add 100 ml of dichloromethane to extract, dry over anhydrous magnesium sulfate, filter,The methylene chloride was evaporated to give an oil which was recrystallized from methanol to give 11 g of product as a yellow needle, yield 58percent.
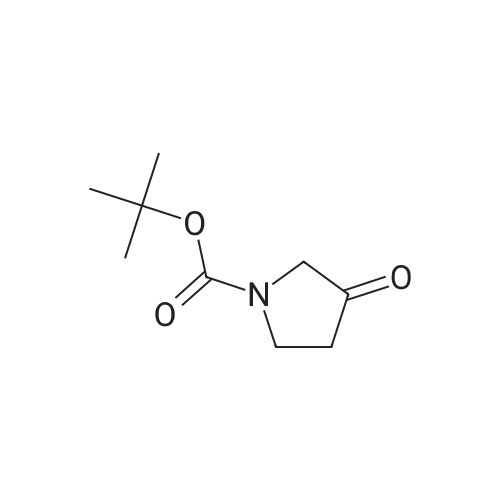
[00377] Step 1: Synthesis of tert-butyl 3-((dimethylamino)methylene)-4- oxopyrrolidine– 1-carboxylate. A suspension of tert-butyl 3-oxopyrrolidine-1-carboxylate (10 g, 54.05 mmol) in 1,1-dimethoxy-N,N-dimethylmethanamine (120 mL) was stirred at 105°C for 1 h., cooled down to room temperature, the solvent was removed in vacuo and the residue was purified by chromatographic column on silicagel (DCM/MeOH = 60/1 to 20/1) to give tert-butyl 3-((di-methyl- amino)methylene)-4-oxopyrrolidine-1-carboxylate (10.78 g, 83percent yield). ESI-LCMS (m/z): 241.2[M+1]+;1HNMR (400 MHz, CDCl3) δ ppm: 7.26 (s, 1H), 4.57 (s, 2H), 3.86 (s, 2H), 3.09 (s, 6H), 1.48 (s, 9H).
46%
at 100℃; for 17 h;
Preparation 31 3-Dimethylaminomethylene-4-oxo-pyrrolidine-1-carboxylic acid tert-butyl ester A solution of tert-butyl 3-oxopyrrolidine-1-carboxylate (25.0 g, 135 mmol), dimethylformamide dimethylacetal (27 mL, 202 mmol) and 1,4-dioxane (170 mL) was heated at 100° C. for 17 hours. The dioxane was removed in vacuo. The resulting red solid was triturated with hexanes (180 mL) for one hour, and filtered. The solid was rinsed with hexanes (2*80 mL) and air dried. Chromatography on silica gel eluting with dichloromethane:methanol 39:1 then 19:1 to give the title compound (15.04 g, 46percent).
46%
at 100℃; for 17 h;
Preparation 31 3-Dimethylaminomethylene-4-oxo-pyrrolidine-1-carboxylic acid tert-butyl ester A solution of tert-butyl 3-oxopyrrolidine-1-carboxylate (25.0 g, 135 mmol), dimethylformamide dimethylacetal (27 mL, 202 mmol) and 1,4-dioxane (170 mL) was heated at 100° C. for 17 hours. The dioxane was removed in vacuo. The resulting red solid was triturated with hexanes (180 mL) for one hour, and filtered. The solid was rinsed with hexanes (2*80 mL) and air dried. Chromatography on silica gel eluting with dichloromethane:methanol 39:1 then 19:1 to give the title compound (15.04 g, 46percent).
41 g
at 105℃; for 0.666667 h;
The 3-oxo-pyrrolidine-1-carboxylic acid tert-butyl ester (40g, 216mmol) was dissolved in 300ml DMF-DMA was heated at 105 deg. C 40 minutes. The solution was cooled and evaporated under reduced pressure, hexane was added, and filtered to give an orange solid 41g, and dried, it was used for the next reaction without purification.
41 g
at 105℃; for 0.666667 h;
(0086) 3-oxopyrrolidine-1-tert-butyl carboxylate (40g, 216mmol) was dissolved in 300mL DMF-DMA, heated for 40 minutes at 105°C. Cooled and concentrated to dry under vacuum, treated with hexane, filtered to obtain orange solid 41g, dried, the product can be used in the reaction of the next step without purification.
4.7 g
at 100℃; for 1.5 h;
A solution of N-Boc-3-pyrrolidinone (CAS Number 101385-93-7; 5.0 g, 27.0 mmol) in DMF- DMA (32.17 g, 270 mmol) was heated at 100°C for 1.5 h. The resulting mixture was cooled to rt and concentrated under reduced pressure. The obtained residue was triturated with n-pentane (100 ml). The resulting solid was dried under high vacuum yielding tert-butyl 3-((dimethylamino)methylene)-4- oxopyrrolidine-l-carboxylate (4.700 g, 19.58 mmol). LCMS: Method 1, 1.556 min, MS: ES+ 241.43.
Reference:
[1] Patent: WO2016/44641, 2016, A2, . Location in patent: Paragraph 00377
[2] Heterocycles, 2002, vol. 56, # 1-2, p. 257 - 264
[3] Patent: US2013/237538, 2013, A1, . Location in patent: Paragraph 0308
[4] Patent: US2013/237537, 2013, A1, . Location in patent: Paragraph 0292-0293
[5] Patent: WO2007/97931, 2007, A2, . Location in patent: Page/Page column 32
[6] Bioorganic and Medicinal Chemistry Letters, 2013, vol. 23, # 19, p. 5361 - 5366
[7] Bioorganic and Medicinal Chemistry Letters, 2015, vol. 25, # 24, p. 5767 - 5771
[8] Patent: CN105622612, 2016, A, . Location in patent: Paragraph 0126; 0127
[9] Journal of Medicinal Chemistry, 2016, vol. 59, # 23, p. 10498 - 10519
[10] Patent: EP3144310, 2017, A1, . Location in patent: Paragraph 0085-0086
[11] Patent: WO2017/158381, 2017, A1, . Location in patent: Page/Page column 73
[12] Patent: US9951069, 2018, B1, . Location in patent: Page/Page column 14-15
95
[ 79099-07-3 ]
[ 4637-24-5 ]
[ 157327-41-8 ]
Yield
Reaction Conditions
Operation in experiment
100%
at 80℃; for 24 h;
A mixture of N,N-dimethylformamide dimethyl acetal (18 g, 20 mL, 151 mmol) and l-(tert-butoxycarbonyl)piperidin-4-one (30 g, 151 mmol) in DMF (240 mL) was stirred at 80 °C for 24 h. The mixture was concentrated in vacuo to give the title compound as a yellow solid (38.4 g, 100percent). MS (ESI, pos. ion) m/z: 255 [M + H]+.
100%
at 80℃; for 24 h;
N, N-dimethylformamide dimethyl acetal (18 g, 20 mL, 151 mmol) and1- (tert-butoxycarbonyl) piperidin-4-one (30 g, 151 mmol)N, N-dimethylformamide (240 mL)The mixture was heated and stirred at 80 ° C for 24 hours.The reaction was complete and concentrated under reduced pressure to give the title compound as a yellow solid (38.4 g, 100percent).
94.4%
at 95℃; for 1.5 h;
S1. Add N- t-butoxycarbonyl-4-piperidone (1) (12g, 60mmol) in 250ml flask, 150ml DMF-DMA, reaction at 95 1.5h, TLC showed the reaction was complete, concentrated to give 3 - ((N, N- dimethylamino) - methylene) -N- tert-butoxycarbonyl-4-piperidone (2) (white solid, 14.4g), yield 94.4percent.
82%
for 20 h; Reflux
Preparation 21 t-Butyl-3-[(dimethylamino)methylidene]-4-oxopiperidine-1-carboxylate A solution of N-Boc-4-piperidone (10 g, 50.19 mmol) and DMF-dimethylacetal (20.16 ml, 150.57 mmol) in 1,4-dioxane (100 ml) was heated at reflux for 20 hours. The reaction mixture was concentrated and the residue was eluted through a flash column (silica gel 60, 230-400 mesh, 7percent MeOH in EtOAc) to give the title compound as an orange oil which crystallized on standing (10.51 g, 82percent).
82%
for 20 h; Reflux
Preparation 21 t-Butyl-3-[(dimethylamino)methylidene]-4-oxopiperidine-1-carboxylate A solution of N-Boc-4-piperidone (10 g, 50.19 mmol) and DMF-dimethylacetal (20.16 ml, 150.57 mmol) in 1,4-dioxane (100 ml) was heated at reflux for 20 hours. The reaction mixture was concentrated and the residue was eluted through a flash column (silica gel 60, 230-400 mesh, 7percent MeOH in EtOAc) to give the title compound as an orange oil which crystallized on standing (10.51 g, 82percent).
76%
at 80℃; for 18 h; Inert atmosphere
Step 1DMF dimethyl acetal (5.82 mL, 0.044 mol) was added to a stirred solution of tert-butyl 4-oxopiperidine-l-carboxylate (8.73 g, 0.044 mol) in DMF (80 mL) and the reaction mixture was heated to 80 °C under N2 for 18 h. After cooling, the DMF was removed under reduced pressure and the residue was partitioned between EtOAc and H20. The organic layer was washed with H20 and saturated brine, then dried over MgS04 and evaporated to afford tert- butyl 3-((dimethylamino)methylene)-4-oxopiperidine-l-carboxylate (8.44 g, 76percent).
63.8%
for 1.5 h; Reflux
N-tert-butoxycarbonyl-4-piperidone (58, 5.8 g, 31.4 mmol) wasdissolved in N,N-dimethylformamide dimethyl acetal (45.0 mL),and the solution was heated under reflux for 1.5 h and concentrated.The residue was triturated with hexane, filtered, andwashed with hexane to give 59 as a yellow powder (5.1 g, 63.8percent):mp 135e136 C; To a solution of 59 (5.0 g, 20.8 mmol) in EtOH(200.0 mL) were added guanidine carbonate (15.0 g, 84.0 mmol)and sodium acetate (13.7 g, 167.0 mmol), and the solution washeated under reflux for 48 h. The reaction mixturewas filtered, andthe insoluble material was extracted with CHCl3 and washed withwater. The organic layer was dried over anhydrous MgSO4 andevaporated. The resultant solid was triturated with 2-propanol,filtered, and washed with 2-propanol and Et2O to give a colorlesspowder. It was dissolved in TFA (50.0 mL) at 0 C, and the solutionwas stirred at room temperature for 1 h and concentrated. Theresidue was dissolved in 2-propanol and treated with concentrated HCl (4.0 mL). The precipitated solidwas filtered andwashed with 2-propanol and Et2O to give 60a (4.2 g, 81.6percent) as a colorless powder: Mp 258e260 C; Compound 57g was obtained from 60a in thesame way as 57f.
60%
for 15 h; Reflux
A solution of N-t-Butoxycarbonyl-4-piperidone (10.0 g, 50.19 mmol) and N,N-dimethylformamide dimethylacetal (20.16 mL, 150.57 mmol) in 1,4-dioxane (100 mL) was heated at reflux for 15 hours. The solvent was removed in vacuo and the residue was eluted through a flash column (silica gel 60, 230-400 mesh, 8percent MeOH in EtOAc) to obtain the title compound as an orange oil which crystallized on standing (7.64 g, 60percent).
40%
for 6 h; Reflux
Stage 1: tert-Butyl 3-((dimethylamino)methylene)-4-oxopiperidine-1-carboxylate A mixture of tert-butyl 4-oxopiperidine-1-carboxylate (20.0 g, 100.38 mmol, 1.0 eq.) and N,N-dimethylformamide dimethylacetal (80 ml) was refluxed for 6 hours. After monitoring by thin-layer chromatography, the reaction mixture was concentrated under reduced pressure and the residue was taken up in DCM (300 ml), washed with water (200 ml) and sat. NaCl solution (200 ml), dried over sodium sulfate, concentrated and purified by column chromatography (silica gel, 2.5percent MeOH in DCM). Yield: 40percent (10.2 g, 40.16 mmol)
Reference:
[1] Patent: WO2014/89324, 2014, A1, . Location in patent: Paragraph 0308
[2] Patent: CN104016979, 2017, B, . Location in patent: Paragraph 0905; 0906; 0907
[3] Patent: CN105669672, 2016, A, . Location in patent: Paragraph 0027; 0031; 0032
[4] Patent: US2013/237538, 2013, A1, . Location in patent: Paragraph 0252
[5] Patent: US2013/237537, 2013, A1, . Location in patent: Paragraph 0236-0237
[6] Patent: US6169093, 2001, A,
[7] Patent: WO2011/103196, 2011, A1, . Location in patent: Page/Page column 82-83
[8] Heterocycles, 2002, vol. 56, # 1-2, p. 257 - 264
[9] European Journal of Medicinal Chemistry, 2014, vol. 79, p. 399 - 412
[10] Patent: US9296747, 2016, B1, . Location in patent: Page/Page column 22-23
[11] Patent: US2012/71461, 2012, A1, . Location in patent: Page/Page column 136
[12] Patent: WO2005/105759, 2005, A1, . Location in patent: Page/Page column 81
[13] Patent: EP1806347, 2007, A1, . Location in patent: Page/Page column 17
[14] Patent: WO2007/146122, 2007, A2, . Location in patent: Page/Page column 44
[15] Patent: US2004/147561, 2004, A1, . Location in patent: Page 44
[16] Patent: EP1803710, 2007, A1, . Location in patent: Page/Page column 17
[17] Patent: WO2012/85167, 2012, A1, . Location in patent: Page/Page column 51
[18] Patent: WO2014/113191, 2014, A1, . Location in patent: Paragraph 0157; 0158
[19] Patent: WO2016/164200, 2016, A1, . Location in patent: Page/Page column 24
[20] Journal of Medicinal Chemistry, 2016, vol. 59, # 23, p. 10498 - 10519
[21] Patent: WO2016/177655, 2016, A1, . Location in patent: Page/Page column 175; 200
[22] Patent: WO2018/83106, 2018, A1, . Location in patent: Page/Page column 20; 21
Stage #1: at 100℃; for 48 h; Stage #2: With sodium methylate In methanol for 118 h; Heating / reflux
1) 4-Acetyl-2-methylthiopyrimidine A mixture of 3, 3-dimethylbutan-2-one (25.15 g) and N, N-dimethylformamide dimethylacetal (126 mL) was stirred at an external temperature of 100°C for 48 hours. After air cooling, the low boiling point components generated during the reaction were evaporated under reduced pressure, and methanol (400 mL), thiourea (28.92 g) and sodium methoxide (15.39 g) were added to the residue thus obtained. The mixture was heated to reflux for 118 hours. After air cooling, sodium methoxide (10.26 g) was added to the reaction solution, methyl iodide (17.8 mL) was added dropwise to the mixture over 5 minutes under ice cooling, and the mixture was stirred for 5 hours. Water and ethyl acetate were added to a residue obtained by evaporating the reaction solvent under reduced pressure, and the mixture was partitioned. The organic layer was washed with water and saturated saline, and then dried over anhydrous sodium sulfate. After separation by filtration, the solvent was evaporated under reduced pressure, and an aqueous 3 N hydrochloric acid solution (400 mL) was added to the residue thus obtained and stirred for 15 hours at room temperature. Ethyl acetate was added to the reaction solution and the mixture was partitioned, and the organic layer was dried over anhydrous sodium sulfate. After separation by filtration, a residue obtained by evaporating the solvent under reduced pressure was purified by silica gel column chromatography (ethyl acetate-hexane), to obtain 4-acetyl-2-methylthiopyrimidine (26.34 g, 82percent) as a solid. 1H-NMR(400MHz, CDCl3)δ: 2.63(3H, s), 2.70(3H, s), 7.51(1H, d, J=4.9Hz), 8.74(1H, d, J=4.9Hz). ESI-MSm/z: 169(M+H)+.
With hydrazine hydrate In tetrahydrofuran; methanol; N,N-dimethyl-formamide
6-Bromo-5-fluoroindole N,N-Dimethylformamide dimethylacetal (8.5 mL, 60 mmol) was added in one portion to a stirred solution of 3-bromo-4-fluoro-6-methylnitrobenzene (11.8 g, 50 mmol) in N,N-dimethylformamide (30 mL) at room temperature under Ar. The mixture was heated to 120° C., stirred for 16 h then concentrated in vacuo to leave a crude oil. The oil was crystallized [methanol-dichloromethane (4:1)] to give a purple solid (4.5 g). The solid was dissolved in methanol/tetrahydrofuran (1:1; 30 mL) and Raney Nickel.(R). (1 g) was added. The mixture was cooled to 0° C. and hydrazine hydrate (0.8 mL, 16 mmol) was added in one portion. The mixture was stirred at 0° C. for 90 min then a further aliquot of hydrazine hydrate (0.8 mL) was added. The mixture was stirred at 0° C. for 30 min then filtered through celite.(R). and the filter cake was washed with tetrahydrofuran. The filtrate was concentrated in vacuo and purified by column chromatography [SiO2; heptane-dichloromethane (4:1)] to give the product (1.7 g, 16percent) as an off-white solid: IR νmax (nujol)/cm-1 3395, 2925, 2855, 1570, 1469, 1451, 1408, 1314, 1145, 1105, 865, 763 and 502; NMR δH (400 MHz, CDCl3) 7.85 (1H, br. s), 7.55 (1H, d, J 5.5 Hz), 7.34 (1H, d, J 9 Hz), 7.23 (1H, t, J 2.8 Hz), 6.49-6.51 (1H, m).
Reference:
[1] Patent: US6380238, 2002, B1,
100
[ 4637-24-5 ]
[ 187834-88-4 ]
[ 280110-69-2 ]
Yield
Reaction Conditions
Operation in experiment
81%
Stage #1: at 50℃; for 4 h; Stage #2: With toluene-4-sulfonic acid In toluene at 100℃; for 18 h;
To a solution of 9.0 g of 4-Hydrazinocarbonyl-piperidine-1-carboxylic acid tert-butyl ester (see reference WO 9703986 A1 19970206) (37 mmoles, 1 eq.) in 40 ml of THF, were added 8.1 ml of dimethylformamide dimethyl acetal (55.4 mmoles, 1.5 eq.). The reaction mixture was then stirred at 50° C. for 4 hours under nitrogen. The solvent was removed under reduced pressure, the residue dissolved in 40 ml of toluene, and 400 mg of para toluene sulfonic acid were added. The mixture was then heated at 100° C. under nitrogen for 18 hours, the volatiles were removed under reduced pressure and the residue was partitioned between methylene chloride and an aqueous solution of sodium bicarbonate. The organic phase was dried over magnesium sulfate and filtered. The volatiles were removed under reduced pressure and the residue was purified by column chromatography on silica gel using methylene chloride/methanol as eluant (98:2 v/v to 95:5 v/v) to afford 8.07 g of the title compound as a white solid (81percent). 1H NMR (400 MHz, CD3OD): δ 1.42 (s, 9H), 1.70 (m, 2H), 2.05 (m, 2H), 2.50 (s, 3H), 3.00 (m, 2H), 3.15 (m, 1H), 4.05 (m, 2H); LCMS: m/z APCl+, 268 [MH]+; Found; C, 58.26percent; H, 7.96percent; N, 15.78percent; C13H21N3O3 requires C, 58.41percent, H, 7.92percent, N, 15.72percent
81%
Stage #1: at 50℃; for 4 h; Stage #2: at 100℃; for 18 h;
To a solution of ferf-butyl 4-(hydrazinocarbonyl)-1-piperidinecarboxylate (see WO 00/39125, preparation 27) (9.0 g, 37 mmol), in tetrahydrofuran (40 ml), was added dimethylformamide dimethyl acetal (8.1 ml, 55.4 mmol). The reaction mixture was stirred at 500C for 4 hours, under nitrogen. The solvent was then removed under reduced pressure, the residue was dissolved in toluene (40 ml), and para-toluenesulfonic acid (400 mg, 2.1 mmol) was added. The reaction mixture was heated at 1000C, under nitrogen, for 18 hours, after which time the cooled reaction EPO <DP n="48"/>mixture was concentrated under reduced pressure and the residue was partitioned between dichloromethane (200 ml) and an aqueous solution of sodium bicarbonate (150 ml). The organic phase was separated and dried over magnesium sulfate, filtered and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel using dichloromethane/methanol as eluant (98:2 v/v to 95:5 v/v) to yield the title compound (8.07 g, 81percent) as a white solid.1H NMR (400MHz1 CD3OD): δ 1.42 (s, 9H), 1.70 (m, 2H), 2.05 (m, 2H1), 2.50 (s, 3H), 3.00 (m, 2H), 3.15 (m, 1 H), 4.05 (m, 2H); LCMS: m/z APCI+ 268 [MH]+
To a solution of ethyl acetoacetate (2.6 g, 20 mmol) in ethanol (10 mL) was added N,N-Dimethylformamide dimethyl acetal (2.5 g, 21 mmol). The mixture was allowed to stir at 60 °C for 3 h, then cooled to room temperature and concentrated to give a crude product (3.6 g, 97percent), which was used without further purification.
Reference:
[1] Journal of Heterocyclic Chemistry, 2012, vol. 49, # 5, p. 1038 - 1043
[2] Patent: WO2017/151786, 2017, A1, . Location in patent: Paragraph 00230
In N,N-dimethyl-formamide; at 80 - 140℃; for 0.5h;
The procedure was adapted from that reported J. Org. Chem. 1978, 43, 2536. Dimethylfomamide-dimethyl acetal (4 mL, 30.05 mmol) was added to a warmed (80°) suspension of 6-methyl-5-nitropyrimidine-2,4(lH,3H)-dione (3g, 17.54 mmol) in dimethylformamide (10 ml). The resultant mixture was heated to 140° for 30 min then allowed to cool to room temperature. The solid was collected by filtration and washed with ethyl acetate then dried in vacuo to afford 6-[(e)-2-(dimethylamino)vinyl]-5- nitropyrimidine-2,4(lH,3H)-dione (2.62 g, 66percent).
In DMF (N,N-dimethyl-formamide); at 140℃; for 18h;
Example 1 Preparation of 1-Cyclohexyl-2- H-BENZOIMIDAZOLE-5- carboxylic-acid (Compound 201); Step 1: TRANS-3- (2-DIMETHYLAMINO-VINYL)-4-NITRO-BENZOIC acid methyl ester (Compound 3); [0215] A 100 mL flask fitted with a 15 cm Vigreux head was charged with 10 g (49.7 mmol) of <strong>[24078-21-5]3-methyl-4-nitro-benzoic acid methyl ester</strong>, 12.5 mL of DMF and 14.8 g (124.2 mmol) of N, N-dimethylformamide dimethylacetal. The reaction vessel was immersed in a 140 C oil bath for 18 h under argon while the forming methanol distilled away. Upon cooling to room temperature the dark red content of the flask solidified. The solid was transferred to a 250 mL flask using DMF which was removed by evaporation. The residue was triturated with petroleum ether to give 11. 81g of enamine as dark red solid. MS: 251.10 (M+H+) ; H¹-NMR (CDCL3): No.(PPM) 8.11 (d, 1H, AR-H2), 7.80 (d, 1H, Ar-H5), 7.53-7. 50 (dd, 1H, AR-H6), 7.06 (d, 1H, CH=), 5.76 (d, 1H, CH=), 3.93 (s, 3H, OCH3), 2.93 (s, 6H, (CH3) 2N).
100.3 g (91%)
In N-methyl-acetamide;
(a) Methyl trans-3-[beta-(dimethylamino)vinyl]-4-nitrobenzoate (6a) A solution of 86.4 g (0.443 mole) of <strong>[24078-21-5]methyl 3-methyl-4-nitrobenzoate</strong> and 158.2 g (1.329 moles) of N,N-dimethylformamide dimethyl acetal in 500 ml of dimethylformamide is heated at 130 C. for 6 hours. The dimethylformamide is then distilled off under a high vacuum and the residue is dried at 20 C. in vacuo. Crude yield: 100.3 g (91%); C12 H14 N2 O4 (250.3). NMR (DMSO): delta=2.96 (s, 6H), 3.96 (s, 3H), 5.8 (d, J=14 Hz, 1H), 7.1 (d, J=14 Hz, 1H), 7.57 (dd, 1H), 7.85 (d, J=8.5 Hz, 1H) and 8.15 (s, 1H) ppm.
A solution of <strong>[881-03-8]2-methyl-1-nitronaphthalene</strong> (10.0 g, 53.4 mmol) and N,N-dimethyl formamide dimethyl acetal (14.3 mL, 107 mmol) in anhydrous DMF (18 mL) was refluxed at 160 oC for 48 hours. The mixture was cooled to room temperature and 50 mL of hexanes was added. After vigorously stirring for 30 minutes, a solid was collected, washed with additional hexanes, and dried to provide the title compound.
for 3h;Reflux; Inert atmosphere; Schlenk technique;
A mixture of <strong>[59576-26-0]1-(4-methylpyridin-2-yl)ethanone</strong> (0.68 g, 5.00 mmol) and N,N-dimethylformamide-dimethylacetal (5.0 mL, excess) was heated to reflux for 3 h. The excess N,N-dimethylformamide-dimethylacetal was removed under vacuum to afford an orange solid which was recrystallized using CH2Cl2/n-hexane to give yellow solid of 3-(dimethylamino)-1-(4-methylpyridin-2-yl)prop-2-en-1-one (0.60 g). Then, a mixture of 3-(dimethylamino)-1-(4-methylpyridin-2-yl)prop-2-en-1-one (0.60 g, 3.15 mmol) and hydrazine monohydrate (1.8 mL) in ethanol (10 mL) was heated at 60 °C for 30 min, and then cooled to room temperature. Water (40 mL) was added to form the precipitate. A pure white precipitate of 4-methyl-2-(1H-pyrazol-3-yl)pyridine (Hmpypz) was collected after filtering and drying in 80percent yield (2.50 mmol, 0.40 g). 1H NMR (500 MHz, CDCl3, delta [ppm]): 11.80 (s, 1H), 8.54 (t, J = 5.5 Hz, 1H), 7.66 (d, J = 2.0 Hz, 1H), 7.58 (s, 1H), 7.06 (d, J = 5.0 Hz, 1H), 6.78 (s, 1H), 2.40 (s, 3H). IR (cm-1): v = 3443 (w), 3125 (m), 3050 (w), 2908 (w), 2838 (w),1713 (w),1611 (s),1555 (m), 1486 (w), 1395 (m), 1308 (w), 1130 (m), 1090 (s), 1043 (s), 999 (m), 925 (m), 846 (m), 771 (m), 557 (w), 526 (m), 439 (m). HRMS (m/z), found: [M + H]+ 160.0804; molecular formula C9H9N3, requires [M + H]+ 160.0796.
1,1-dimethoxy-N,N-dimethylmethanamine (100 g, 839 mmol, 1.02 equiv.) and 1,1-dimethoxypropan-2-one (97 g, 821 mmol) were added and stirred at 110 C. for 3 hours. The produced methanol was removed by a Dean-Stark apparatus. After the solution was cooled to room temperature, the remaining volatile materials were removed in vacuo to provide 130 g of the crude product, (E)-4-(dimethylamino)-1,1-dimethoxybut-3-en-2-one (1) (130 g, 143 g theoretical, 91%). LC-MS m/z 283 (M+1). Reference: WO 2006/0097341A1 (incorporated by reference), pg 67.
79%
In iso-butanol;
(a) (E)-4-(Dimethylamino)-l,l-dimethoxybut-3-en-2-one - prepared as described in Maury ei al, J. Heterocyclic Chem., 1978, 15, p. 1041
68%
In iso-butanol; for 20h;Reflux;
(3E)-4-(Dimethylamino)-1,1-dimethoxybut-3-en-2-one (C10) A solution of N,N-dimethylformamide dimethyl acetal (147 g, 1.23 mol) and 1,1-dimethoxyacetone (146 g, 1.24 mol) in 2-butanol (1 L) was heated at reflux for 20 hours. After removal of solvent in vacuo, the residue was distilled under vacuum to provide the product as an oil. Yield: 145 g, 0.837 mol, 68%. Boiling point: 132-140 C./0.15 torr. NMR and MS data were obtained using the product of a reaction run under similar conditions. LCMS m/z 174.0 (M+1). 1H NMR (400 MHz, CDCl3) delta 2.77 (br s, 3H), 3.02 (br s, 3H), 3.30 (s, 6H), 4.47 (s, 1H), 5.23 (br d, J=12.6 Hz, 1H), 7.63 (d, J=12.6 Hz, 1H).
68%
at 110℃; for 3h;Inert atmosphere;
A stuffed solution of 1,1-dimethoxy-N,N-dimethylmethanamine (5 g, 42 mmol) was mixed with 1,1-dimethoxypropan-2-one (4.95 g, 42 mmol) under nitrogen atmosphere. The reaction mixture was heated to 110 C for 3 h. The methanol produced was removed by Dean-Stark apparatus. After completion the solution was cooled to rt and volatiles were removed under reduced pressure to obtain oily residue. The crude material was purified by column chromatography (60-120 silica gel, 0-5% Methanol/DCM) to afford 2.3g of the title compound (68%). LCMS: mlz=174 [M+1]. ?H-NMR (400MHz, CDC13) oe: 7.6 1-7.58 (d, 1H), 5.19-5.16 (d, 1H), 4.43 (s, 1H), 3.26 (s, 6H), 3.10-3.08 (d, 3H), 2.79 (s, 3H), 1.15 (s, 6H).
at 100℃; for 16h;
Step 1: (E)-4-(dimethylamino)-1,1-dimethoxybut-3-en-2-one, 57 is prepared as follows: N,N-dimethylformamide dimethylacetal (67.21 g) and 1,1-dimethoxypropan-2-one (66.62 g), are heated together at 100 C. for 16 h. Residual methanol is removed in vacuo and the product used crude in the next step.
at 100℃; for 16h;
2-Aminopyrimidine-4-carboxaldehyde dimethylacetal.; A solution of 5.5 ml (41 mmol, 1 eq.) of dimethylformamide dimethyl acetal and 5.0 ml (41 mmol, 1 eq.) pyrvuric aldehyde dimethyl acetal was heated at 100 C. for 16 h. Methanol was removed in vacuo to afford an oil. A solution of NaOH (1.8 g, 45 mmol, 1.1 eq.) in 5 mL of H2O was added to a solution of 4.3 g (45 mmol, 1.1 eq.) of guanidine HCl in 10 mL of H2O. The resulting solution was added to the above described oil. The resulting mixture was stirred at r.t. for 48 h. Filtration afforded 2.5 g (50%) of 2-aminopyrimidine-4-carboxaldehyde dimethylacetal.
at 80℃;
Equimolar amounts of dimethyformamide dimethylacetal (15 mL, 0.11 mol) and 1,1- Dimethoxy-propan-2-one (14 mL, 0.11 mol) were combined and heated to 80 C overnight. After cooling to room temperature, volatile materials were evaporated. 4-Dimethylamino-l,l- dimethoxy-but-3-en-2-one resulted as a dark brown liquid (20 g) without further purifications. 1HNMR: (300 MHz, CDCl3), delta 7.70 (d, IH), 5.30 (d, IH), 4.54 (s, IH), 3.37 (s, 6H), 3.08 (s, 3H), 2.83 (s, 3H). [0115] To a solution of the above compound (15g, 86.6 mmol) in ethanol (50 mL) was added trifluoroacetamidine (10.7 g, 95.2 mmol) and heated to reflux overnight. It was then cooled down to room temperature and concentrated in vacuo. The residue was purified on silica gel column with 100% CH2Cl2 to give 4-dimethoxymethyl-2-trifluoromethyl-pyrimidine as a light yellow liquid (8.5 g) as (Rf=0.2 using 100% CH2Cl2). 1HNMR: (300 MHz, CDCl3), delta 8.94 (s, IH), 7.75 (s, IH), 5.33 (s, IH), 3.46 (s, 6H).
at 100℃; for 18h;
Protocol F: (Scheme 6) A mixture of pyruvaldehyde dimethylacetal (125 mmol) and dimethylformamide dimethylacetal (437.5 mmol) was stirred under nitrogen atmosphere at 100C for 18 hours. The reaction mixture was cooled to room temperature and evaporated to dryness under reduced pressure.
at 110℃; for 3h;
Example 1 (E)-4-(dimethylamino)-1,1-dimethoxybut-3-en-2-one (1): 1,1-dimethoxy-N,N-dimethylmethanamine (100 g, 839 mmol, 1.02 equiv.) and 1,1-dimethoxypropan-2-one (97 g, 821 mmol) were added and stirred at 110 C. for 3 hours. The produced methanol was removed by a Dean-Stark apparatus. After the solution was cooled to room temperature, the remaining volatile materials were removed in vacuo to provide 130 g of the crude product, (E)-4-(dimethylamino)-1,1-dimethoxybut-3-en-2-one (1) (130 g, 143 g theoretical, 91%). LC-MS m/z 283 (M+1). Reference: WO 2006/0097341A1, pg 67.
at 110℃; for 3h;
1, l -dimethoxy-N,N- dimethylmethanamine ( 100 g, 839 mmol, 1 .02 equiv.) and l , l -dimethoxypropan-2-one (97 g, 821 mmol) were added and stirred at 1 10C for 3 hours. The produced methanol was removed by a Dean-Stark apparatus. After the solution was cooled to room temperature, the remaining volatile materials were removed in vacuo to provide 1 30 g of the crude product, (E)-4-(dimethylamino)-l ,l -dimethoxybut-3-en-2-one (1 ) ( 1 30 g, 143 g theoretical, 91 %). LC-MS m/z 283 (M+ I ). Reference: WO20060097341 A I , pg 67.
at 100℃; for 5h;
Step 1 (E)-4-(dimethylamino)-1,1-dimethoxybut-3-en-2-one A solution of 1,1-dimethoxypropan-2-one (12 g, 101.58 mmol, 1.00 equiv) and (dimethoxymethyl)dimethylamine (12 g, 100.70 mmol, 0.99 equiv) was stirred at 100 C. for 5 h. The reaction mixture was cooled to room temperature and used in the next step directly. LCMS (method D, ESI): RT=0.94 min, m/z=174.0 [M+H]+.
at 105℃; for 3h;
502.1 g l,l-dimethoxypropan-2-one (4,25 mol) and 506.4 g l,1-dimethoxy-N,N-dimethyl-methanamine (4.25 mol) were mixed in a 2 L flask and stirred at 105C for 3 hours. The formed McOH was removed continuously via distillation. When MeOll formation stopped (at 65C head temperature) the reaction mixture was vacuum distilled (decreasing the pressure slowly to 30 mbar) to remove side products and unreacted starting materials. The crude product was distilled at 0.1 mbar. Fractions were collected between 107-118C headtemperature (bath temperature 160-165C) to give a yellow oil.?H NMR (500 MHz, DMSO-d6): 7.59 (d, 1H), 5.17 (d, lH), 4.42 (s, 1H), 3.25 (s, 6H), 3.09 (s, 3H), 2,78 (s, 3H).
at 105℃; for 3h;
502.1 g l,l-dimethoxypropan-2-one (4.25 mol) and 506.4 g l ,l-dimethoxy-N,N-dimethyl- methanamine (4.25 mol) were mixed in a 2 L flask and stirred at 105 C for 3 hours. The formed MeOH was removed continuously via distillation. When MeOH formation stopped (at 65 C head temperature) the reaction mixture was vacuum distilled (decreasing the pressure slowly to 30 mbar) to remove side products and unreacted starting materials. The crude product was distilled at 0.1 mbar. Fractions were collected between 107-118 C head temperature (bath temperature 160-165 C) to give a yellow oil. 1H NMR (500 MHz, DMSO-dg): 7.59 (d, 1H), 5.17 (d, 1H), 4.42 (s, 1H), 3.25 (s, 6H), 3.09 (s, 3H), 2.78 (s, 3H)
at 105℃; for 3h;
502.1 g 1,1 -dimethoxypropan-2-one (4.25 mo 1) and 506.4 g 1,1 -dimethoxy-N,N-dimethyl25 methanamine (4.25 mol) were mixed in a 2 L flask and stirred at 105 C for 3 hours. The formed MeOH was removed continuously via distillation. When MeOH formation stopped (at 65 C head temperature) the reaction mixture was vacuum distilled (decreasing the pressure slowly to 30 mbar) to remove side products and unreacted starting materials. The crude product was distilled at 0.1 mbar. Fractions were collected between 107-118 C headtemperature (bath temperature 160-165 C) to give a yellow oil. 1H NMR (500 MHz, DMSO-d6) oe: 7.59 (d, 1H), 5.17 (d, 1H), 4.42 (s, 1H), 3.25 (s, 6H), 3.09 (s, 3H), 2.78 (s, 3H)
at 105℃; for 3h;
502.1 g l,l-dimethoxypropan-2-one (4.25 mol) and 506.4 g l ,l-dimethoxy-N,N-dimethyl- methanamine (4.25 mol) were mixed in a 2 L flask and stirred at 105 C for 3 hours. The formed MeOH was removed continuously via distillation. When MeOH formation stopped (at 65 C head temperature) the reaction mixture was vacuum distilled (decreasing the pressure slowly to 30 mbar) to remove side products and unreacted starting materials. The crude product was distilled at 0.1 mbar. Fractions were collected between 107-118 C head temperature (bath temperature 160-165 C) to give a yellow oil. 1H NMR (500 MHz, DMSO-de) delta: 7.59 (d, 1H), 5.17 (d, 1H), 4.42 (s, 1H), 3.25 (s, 6H), 3.09 (s, 3H), 2.78 (s, 3H)
at 105℃; for 3h;
Step M: (E)-4-(Dimethylamino)- 1 , 1 -dimethoxy-but-3-en-2-one (0190) 502.1 g l,l-dimethoxypropan-2-one (4.25 mol) and 506.4 g 1 ,1-dimethoxy-NN-dimethyl- methanamine (4.25 mol) were mixed in a 2 L flask and stirred at 105 C for 3 hours. The formed methanol was removed continuously via distillation. When methanol formation stopped (at 65 C head temperature) the reaction mixture was vacuum distilled (decreasing the pressure slowly to 30 mbar) to remove side products and unreacted starting materials. (0191) The crude product was distilled at 0.1 mbar. Fractions were collected between 107-118 C head temperature (bath temperature 160-165 C) to give a yellow oil. 1H NMR (500 MHz, (0192) DMSO-de) delta: 7.59 (d, 1H), 5.17 (d, 1H), 4.42 (s, 1H), 3.25 (s, 6H), 3.09 (s, 3H), 2.78 (s, (0193) 3H)
at 100℃; for 2h;Large scale;
14.8 Kg Compound 1 was dissolved in 14.9 KgDimethylformamide dimethyl acetal.The mixture was magnetically stirred at 100 degrees Celsius for 2 hours.The reaction was concentrated in vacuo to give 20.4 Kg of crude compound 2 which was used directly in the next step.
at 110℃; for 3h;
Example 100A (E)-4-(dimethylamino)-1,1-dimethoxybut-3-en-2-one 1,1-Dimethoxy-N,N-dimethylmethanamine (15 g) and 1,1-dimethoxypropan-2-one (14.9 g) were mixed in a 250 mL flask and the mixture was stirred at 110 C. for 3 hours. Thin layer chromatography showed the starting material was consumed. The formed methanol was removed continuously via distillation. The reaction mixture was distilled under high vacuum (decreasing the pressure slowly to 30 mbar) to remove by-products and starting materials. The remaining crude product was distilled at 0.1 mbar. Fractions were collected between 107-118 C. head temperature (bath temperature 160-165 C.) to provide the title compound. 1H NMR (400 MHz, dimethyl sulfoxide-d6) delta ppm 2.78 (s, 3H), 3.09 (s, 3H), 3.26 (s, 6H), 4.42 (s, 1H), 5.18 (d, J=12.35 Hz, 1H), 7.59 (d, J=12.79 Hz, 1H).
at 90℃; for 16h;
A mixture of l,l-dimethoxypropan-2-one II-l (10 g, 85 mmol) and l,l-dimethoxy-A,iV- dimethylmethanamine III-l (10.1 g, 85 mmol) was heated at 90 C for 16 h. After completion of the reaction, the reaction mass was concentrated and the crude product (£)-4-(dimethylamino)-l,l- dimethoxybut-3-en-2-one (11 g, 63.5 mmol, 75 % yield) obtained after concentration was used in next step without any purification. -NMR (400 MHz, CHLOROFORM-D) d 7.72 (d, J = 12.8 Hz, 1H), 5.33 (d, J = 12.2 Hz, 1H), 4.57 (s, 1H), 3.36-3.43 (m, 6H), 3.05-3.10 (m, 3H), 2.86 (d, J = 6.1 Hz, 3H).
With acetic acid; at 90 - 95℃; for 2h;Product distribution / selectivity;
Example-2: Preparation of 3-dimethylamino-l-pyridin-3-ylpropenoneMethod A: 3-Acetylpyridine (25g, 0.20mol), NN-dimethylformamide dimethyl acetal (37g, 0.3 1 mol) and acetic acid (2.5ml) were heated at 90-95 C for 2 hours with continuous removal of methanol. After completion of reaction, excess of methanol formed in the reaction was distilled out under vacuum. The resulting residue was diluted with water and extracted with dichloromethane ( 125 ml). The organic layer was washed with water and distilled out completely under vacuum to get 34g (92%) of the title compound having purity 99.1 % by HPLC.
90%
In o-xylene; at 130℃; for 20h;
Preparation of 3-(dimethylamino)-1-(pyridine-3-yl) prop-2-en-1-one (compound 4): Acetylpyridine (11 mL, 0.1 mol) and N,N'-dimethylformamide dimethylacetal (27 mL, 0.2 mol) were dissolved in o-xylene (35 mL). The mixture was heated to 130 C. with stirring for 20 hours. During the reaction the methanol formed was removed by a trap connected to the reflux condenser. Upon cooling to room temperature, hexane (20 ml) was added to the mixture. The precipitated solid was collected by suction filtration and washed with hexane (200 mL). The desired product was obtained (15.4 g, yield: 90%) as a white solid; 1H NMR (500 MHz, CDCl3) delta b 2.95 (s, 3H), 3.18 (s, 3H), 5.68 (d, 1H), 7.36 (m, 1H), 7.84 (d, 1H), 8.18 (d, 1H), 8.67 (m, 1H), 9.10 (s, 1H). LC-MS (m/z) calculated, 176.1, found 177.1 [M+H]+.
85%
In neat (no solvent); for 23h;Reflux;
3-Acetylpyridine 3 (100g, 0.19mol) was added to dimethylformamide dimethyl acetal (156.5g, 1.27mol), and the mixture was reacted under reflux for 23h. After the reaction mixture was cooled to 0C, a mixture of diethyl ether and hexane (3:2, v/v) (500mL) was added, and the whole mixture was stirred for 4h. The resulting solid was filtered and washed with a mixture of diethyl ether and hexane (500mL, 3/2, v/v) to give 3-dimethylamino-1-(3-pyridyl)-2-propen-1-one 4 (120g, 85%).
83.3%
In 5,5-dimethyl-1,3-cyclohexadiene; for 21h;Reflux;
10 g of 3-acetylpyridine (82 mmol, 1.0 equiv.), 29.3 g of N, N-dimethylformamide dimethylacetal (246 mmol, 3.0 equiv), and 50 ml of xylene were refluxed overnight (21 hours). A little raw material was not reacted and the reaction was stopped. The reaction solution was cooled and concentrated to remove xylene. 150 ml of petroleum ether was added to the residue, and a solid precipitated under stirring, followed by filtration to obtain 12.0 g of 3-dimethylamino-1-pyridin-3-yl-prop-2-en-1-one as a yellow solid, 83.3%.
72%
In ethanol; at 20℃; for 12h;Reflux;
General procedure: N,N-DMF/DMA was added to a solution of 3-acetyl pyridine (6a) (1.2 g, 10 mmol) in ethanol (20 ml) at room temperature, the reaction mixture was refluxed in an oil bath for 12 h. The reaction was monitored by TLC and the solvent was evaporated under the vacuum. The mixture was cooled to 0 C and added ether. The crude product was removed by filtration and recrystallized from ethanol to afford compound 7a as light yellow solid (1.26 g, 72%).
50.8 - 52%
In ethanol; at 80℃; for 16 - 18h;Heating / reflux;
Step 1: 3-Dimethylamino-1-pyridin-3-yl-propenone: 3-Acetylpyridine (2.5 g, 20.64 mmol) and N,N-dimethyl-formamide dimethylacetal (3.20 ml, 24 mmol) were refluxed in ethanol (10 mL) overnight. The reaction mixture was cooled to room temperature and evaporated under reduced pressure. Diethyl ether (20 mL) was added to the residue and the mixture was cooled to 0 C. The mixture was filtered to give 3-dimethylamino-1-pyridin-3-yl-propenone (1.9 g, 10.78 mmol) as yellow crystals. (Yield: 52%.) This material was used in subsequent steps without further purification.; Step-2: A stirred solution of 3-acetyl pyridine (10.0 g, 82.56 mmol) and N,N-dimethylformamide dimethyl acetal (12.8 g, 96.00 mmol) in ethanol (40 mL) was refluxed for 16 h. It was then cooled to rt and concentrated under reduced pressure to get a crude mass. The residue was taken in ether (10 mL), cooled to 0 C. and filtered to get the corresponding enamide (7.4 g, 50.8%) as a yellow crystalline solid.
3-Acetylpyridine (100 g, 0.19 mol) was added to dimethylformamide dimethylacetal (156.5 g, 1.27 mol), and the mixture was reacted under reflux for 23 hours. After the reaction mixture was cooled to 0 C., a mixture of diethyl ether and hexane (3:2, v/v) (500 ml) was added and the whole mixture was stirred for 4 hours. The resulting solid was filtered and washed with a mixture of diethyl ether and hexane(500 ml, 3/2, v/v) to give 3-dimethylamino-1-(3-pyridyl)-2-propen-1-one (120 g). Rf=0.46 (Methylene chloride:Methanol=9:1). 1H-NMR(CDCl3)=3.04(s, 3H), 3.24(s, 3H), 5.83(s, 1H), 5.89(s, 1H), 7.48-7.55 (m, 1H), 7.89-7.95(m, 1H), 8.27-8.32(m, 1H), 9.00-9.02(s, 1H).
for 6h;Reflux;
A mixture of 30 g of 3-acetylpyridine and 60 g of N, -dimethylformamidedimethylacetal was stirred for 6 hours under reflux with heat. After cooling down to room temperature, the mixture was concentrated under reduced pressure. 3-dimethylamino-l- (pyridin-3-yl ) - propenone (43 g) was obtained. 1 H-N R (CDC13) delta: 9.09-9.07 (1H, m) , 8.68-8.65 (1H, m) , 8.19 (1H, dt), 7.85 (1H, d) , 7.38-7.34 (1H, m) , 5.68 (1H, d) , 3.19 (3H, s), 2.96 (3H, s).
60.2 g
at 25 - 80℃; for 2h;Inert atmosphere;
3-Acetylpyridine (44.1 g) and dimethylformamide dimethylacetal (112.8 g) was charged to a round bottom flask at 25-35C under nitrogen atmosphere. The reaction mass was heated to 78-80C and stirred for 2 his. Then the reaction mass was concentrated under vacuum at below 60C. Diisopropylether (200 mL) was added to the reaction mass and stirred for an hour. i1tered the reaction mass and washed with mixture ofdiisopropylether and hexane. The compound obtained was dried under vacuum to get the title compound (60.2 g).
In toluene; at 120℃; for 0.25h;Microwave irradiation;
The mixture of N, N-dimethylformamide dimethylacetal(0.02 mol), acetylpyridine (0.01 mol), and toluene (3.5 mL) in a10 mL glass vial well equipped. The reaction mixturewas irradiatedin the microwave synthesizer at 120 C for 15 min. After completionof the reaction (indicated by TLC), the mixture was cooled down to room temperature. Petroleum ether (2 mL) was added to the reaction mixture and kept stirring for 20 min. The resultant solid was collected by filtration and washed with hexane to get compound (3). Melting point 83-85 C, ES-MS (M1) found (m/z): 177.1
at 110℃;
4-Pyridin-3 - yl-pyrimidin-2- ylamine (7)[0042] A mixture of 3-acetylpyridine (20.72 g, 171.0 mmol) and N, N-dimethylformamide dimethyl acetal (60 mL, 450 mmol) is heated at 110 0C. After reaction overnight, the reaction mixture is cooled to room temperature and concentrated. Ethyl ether (20 mL) and hexanes (60 <n="17"/>P A T E N TGNF Docket No.: P1286PC10mL) are added to the residue. The resulting solid is collected by filtration, washed with hexanes, and dried to afford 3-dimethylamino-l-pyridin-3-yl-propenone which is used for next reaction without further purification. A mixture of 3-dimethylamino-l-pyridin-3-yl-propenone (17.6 g, 100 mmol) and guanidine carbonate (10.8 g, 60.0 mmol) in 2-butanol (100 mL) is heated at reflux. After 24 hours, the reaction mixture is cooled to room temperature and concentrated. The residue is triturated in water (50 mL). The solid is collected by filtration, washed with water and dried to afford the desired product 4-pyridin-3-yl-pyrimidin-2-ylamine as a white solid. C29H31N7O Exact MS: 172.07. Found MS m/z 173.1 (M+l). 1H NMR (DMSO-J6): delta 9.23 (s, IH), 8.69 (m, IH), 8.37 (m, 2H), 7.54 (d, IH), 7.20 (d, IH), 6.80 (s, 2H).
for 16h;Heating / reflux;
Reactant 3 can be obtained by the following procedures. A mixture of 3- acetylpyridine (2.47 mol) and N,N-dimethylformamide dimethylacetal (240 mL) is heated at reflux for 16 h. The solvent is removed in vacuo and hexanes (100 mL) is added to the residue to crystallize a solid. The solid is recrystallized from methylene chloride-hexanes to give 3- dimethylamino-l-(3-pyridyl)-2-propen-l-one 3. 1H NMR (400MHz, d-chloroform) delta 9.08 (d, 7 = 2.4 Hz, IH), 8.66 (m, IH), 8.20 (m, IH), 7.87 (m, IH), 7.37 (m, IH), 5.68 (d, 7 = 16.4 Hz, IH), 3.18 (s, 3H), 2.97 (s, 3H).
for 16h;Heating / reflux;
Reactant 3 can be obtained by the following procedures. A mixture of 3-acetylpyridine (2.47 mol) and N,N-dimethylformamide dimethylacetal (240 mL) is heated at reflux for 16 h. The solvent is removed in vacuo and hexanes (100 mL) is added to the residue to crystallize a solid. The solid is recrystallized from methylene chloride-hexanes to give 3-dimethylamino- l-(3-pyridyl)-2-propen-l-one 3.1H NMR (400MHz, ^-chloroform) delta 9.08 (d, / = 2.4 Hz, IH), 8.66 (m, IH), 8.20 (m, IH), 7.87 (m, IH), 7.37 (m, IH), 5.68 (d, J= 16.4 Hz, IH), 3.18 (s, 3H), 2.97 (s, 3H).
for 16h;Heating / reflux;
To a solution of 3-amino-4-methyl-benzoic acid methyl ester (0.6 mol) in nBuOH (50 mL) is added 70% nitric acid (2.7 mL) to form the nitrate salt followed by condensation with aqueous cyanamide solution (50%wt., 7 mL, 0.09 mol). The resulting mixture is heated at reflux for 16 h and cooled to ambient temperature followed by addition of diethyl ether (100 mL). After cooling at OC for 30 min, filtration and washing with 1 :1 = methanol : diethyl ether (120 mL) affords 3-guanidino-4-methyl-benzoic acid methyl ester nitrate 2.[00105] To 3-guanidino-4-methyl-benzoic acid methyl ester nitrate 2 (0.02 mol) in nBuOH (40 mL) is added 3 (0.02 mol) and sodium hydroxide flakes (0.02 mol). The resulting mixture is heated at reflux for 12 h to yield ester 4. 1 N aq. NaOH (20 mL) is added to the nBuOH solution of ester 4 and heated at reflux for 30 min. After cooling to it, 1 N (aq) HCl (20 mL) is slowly added to the mixture with vigorous stirring. The product is collected by filtration and washed with water to afford acid S. 1H NMR (400MHz, d6-DMSO) delta 9.28 (d, J =1.8 Hz, IH), 9.08 (s, IH), 8.7 (dd, 7= 4.7, 1.5 Hz, IH), 8.55 (d,J = 5.1 Hz, IH), 8.46 (dt,J= 8, 1.8 Hz, IH), 8.31 (s, IH), 7.65 (dd, J= 7.8, 1.5 Hz, IH), 7.54 (dd,J= 7.7, 4.7 Hz, IH), 7.49 (dd,J= 5.2 Hz, IH), 7.37 (d, J = 7.9 Hz , IH), 3.08 (s, 3H). MS (m/z) (M+l)+: 307.2. [00106] Reactant 3 can be obtained by the following procedures. A mixture of 3- acetylpyridine (2.47 mol) and N,N-dimethylformamide dimethylacetal (240 mL) is heated at reflux for 16 h. The solvent is removed in vacuo and hexanes (100 mL) is added to the residue to crystallize a solid. The solid is recrystallized from methylene chloride-hexanes to give 3- dimethylamino-l-(3-pyridyl)-2-propen-l-one. 1H NMR (400MHz, d-chloroform) delta 9.08 (d, J = 2.4 Hz, IH), 8.66 (m, IH), 8.20 (m, IH), 7.87 (m, IH), 7.37 (m, IH), 5.68 (d,J = 16.4 Hz, IH), 3.18 (s, 3H), 2.97 (s, 3H).
6.9 g
for 24h;Reflux;
5 g of 3-acetylpyridine and 25 ml of DMF-DA were placed in a 50 ml flask and stirred under reflux for 24 hours. After the reaction is completed, the excess pressure of the pump is removed, and the remaining yellowish solid is left in the bottle; then 50 ml of the n-hexane solution is added, and the mixture is stirred at room temperature to wash off the excess reactant, and the filter cake is collected by filtration and dried. This gave 6.9 g of a yellow product.
In N,N-dimethyl-formamide; at 125 - 135℃; for 24h;
Compound 2 (17.9 g) and DMF-DMA (59.4 g) was dissolved in DMF (190 mL) and degassed. The mixture was heated to 125-13 5 C for 24 hours. The mixture was concentrated to 4O mLtotal volume under vacuum and cooled to 10-15 C. Water (234 mL) was added to crystallize compound 3. Compound 3 was then isolated by filtration, washed with water (61 mL), and dried to give 22.2 g of 3 in 93.0% yield, 99.1 LCAP, and 96.9 wt%.
[0621] A mixture of XI-2 (13 g, 95.6 mmol, 1 eq) and 18.2 mL of N,N-dimethylformamide dimethyl acetal was heated at 50° C. for 2 hrs. During the second hour, all the volatiles was removed. The residue was cooled to rt., diluted with 100 mL of anhydrous N,N-dimethylformamide, and then treated carefully with batch wise portions of sodium hydride (5 g, 124.3 mmol, 1.3 eq, 60percent oil dispersion; caution: vigorous evolution of hydrogen). The mixture was heated at 80° C. for 2.5 hrs, and then ice-cooled, treated cautiously with 25 mL of 2-propanol, and then maintained at 0-5° C. overnight. The solid were collected, and then dissolved in 10 mL of hot water. The solution was filtered, the filtrate was ice-cooled and then treated dropwise with concentrated hydrochloric acid to pH=7.0. After storage at 0-5° C. for 3 hrs, the precipitated solids were collected, washed with ice-cold water, and dried in vacuum to give XI-3 (3 g, 32percent yield). 1H NMR (DMSO-d6, 300 MHz): delta 8.90 (s, 1H), 8.49 (d, J=7.6 Hz, 1H), 7.51-7.43 (m, 2H), 6.61 (d, J=7.6 Hz, 1H).
32%
A mixture of X-2 (13 g, 95.6 mmol, 1 eq) and 18.2 mL of N,Ndimethylformamide dimethyl acetal was heated at 50°C for 2 hrs. During the second hour, all the volatiles was removed. The residue was cooled to rt., diluted with 100 mL of anhydrous N,Ndimethylformamide, and then treated carefully with batch wise portions of sodium hydride (5 g, 124.3 mmol, 1.3 eq, 60percent oil dispersion; caution: vigorous evolution of hydrogen). The mixture was heated at 80°C for 2.5 hrs, and then ice-cooled, treated cautiously with 25 mL of 2-propanol, and then maintained at 0-5°C overnight. The solid were collected, and then dissolved in 10 mL of hot water. The solution was filtered, the filtrate was ice-cooled and then treated dropwise with concentrated hydrochloric acid to pH=-7.0. After storage at 0-5°C for 3 hrs, the precipitated solids were collected, washed with ice-cold water, and dried in vacuum to give X-3 (3 g, 32percent yield). ?H NMR (DMSO-d6, 300 MHz): oe 8.90 (s, 1H), 8.49 (d, J=7.6 Hz, 1H), 7.51-7.43 (m, 2H), 6.61 (d, J =7.6 Hz, 1H).
A solution of <strong>[24078-21-5]3-methyl-4-nitro-benzoic acid methyl ester</strong> (24.99 g, 128.1 mmol) and N,N-dimethylformamide dimethyl acetal (40.0 mL, 300 mmol) was heated at 1400C for 22.5 h. After cooling to rt, the reaction mixture was concentrated and the residue was crystallized from MeOH to give a purple solid.This solid was dissolved in THF (500 mL) and water (500 mL), and sodium periodate (62.62 g, 292.8 mmol) was added followed by additional sodium periodate (15.6 g, 72.9 mmol) two hours later. After stirring at rt for an additional 1 h, the reaction mixture was filtered through Celite washing with EtOAc (2 L). The filtrate was washed with saturated NaHCO3 (600 mL) and the organic layer was dried over Na2SO4. After filtration, the filtrate was concentrated and the residue was passed through a pad of silica gel, washing with CH2Cl2/hexanes (75%- 100%). The filtrate was concentrated and dried to give 3-formyl-4-nitro-benzoic acid methyl ester as yellowish solid. MS (EI): cal'd 210.0 (MH+), exp 210.2 (MH+).
Intermediate 35; 4-Bromo-6-fluoro-lH-indole; l-Bromo-5-fluoro-2-methyl-3-nitrobenzene (2.00 g, 8.55 mmol) and(dimethoxymethyl)dimethylamine (5.66 mL, 42.7 mmol) in dry DMF (20 mL) was refiuxed under N2 for 8 h, then rt. over night. The mixture was diluted with DCM and extracted 5 times with water. The organic layer was dried, filtered and concentrated under reduced pressure. The residue was dissolved in AcOH (10 mL) and added drop wise to a boiling mixture of Fe(s, fine powder) in AcOH (10 mL). The mixture was refiuxed for 40 min., partitioned between DCM and saturated aq. Na2CO3/brine (the mixture was filtered through celite before phase separation). The water layer was extracted once more with DCM. The organic layers were combined, dried and concentrated. Purification was performed by flash column chromatography (DCM/hexane 1:3) and afforded the title compound (660 mg, 39percent) as a yellow oil. MS (ESI+) for C8H5BrFN m/z 214 (M+H)+.
27%
Example 41 N4-(5-Cyclobutyl- 1 H-pyrazol-3-yl)-N2-((6-fluoro- 1 H-indol-4-yl)methyl)pyrimidine-2,4-diamine Formate (1-88) step 1 : To a solution of l-bromo-5-fluoro-2-methyl-3-nitrobenzene (4.69 g, 20 mmol) in 1,4-dioxane (25 mL) at RT was slowly added DMF dimethylacetal (13.3 mL) and pyrrolidine (1.7 mL). The solution was heated at 100 °C for 18 h, then concentrated in vacuo to give a dark residue. To the residue was added HOAc (30 mL) and iron powder (11 g, 200 mmol) then the mixture was heated to reflux for 1 h, cooled to RT, neutralized by addition of 50percent aq. NaOH and extracted with EtOAc (2 x 200 mL). The combined organic extracts were dried (MgSO i), filtered and concentrated in vacuo. The residue was purified by Si02 chromatography eluting with an EtO Ac/petroleum ether gradient (5 to 30percent EtOAc) to afford 1.16 g (27percent) of 4-bromo-6-fluoro-lH-indole (224) -as brown solid: MS (ESI) m/z = 213.9 [M+l] +.
Intermediate 73; 4-Bromo-5-methoxy-lH-indole; 2-Bromo-3-rnethyl-4-nitro-phenol, (100 g, 0.43 mol, Intermediate 72,) was dissolved in acetone (500 mL), grinded K2CO3, 119 g (0.86 mol) and methyl iodide, 83 g (0.59 mol) were added and the reaction mixture was heated at reflux for one hour. The suspension was <n="121"/>filtered and the solvent was removed at reduced pressure to give a brown spontaneously crystallizing oil that was used directly in the next synthetic step. Quantitative yield. The crude methoxy ether, 106 g (0.43 mol) was dissolved in dry DMF (350 mL), dimethylformamid dimethylacetal [DMFDMA], 103 g (0.87 mol) was added and the reaction was heated and stirred at 900C for two days. During the next three days were each day a portion of DMFDMA, 20 g (0.17 mol) added while the mixture was continued to be heated. The solvent was removed at reduced pressure and the black/red oily residue was dissolved in HOAc (300 mL). The viscous solution was carefully added to a well stirred suspension of iron powder, 72 g (1.3 mol) in warm HOAc (700 mL) at such rate the exothermic reaction allowed. The thick reaction mixture was heated at reflux for one hour, the solids were filtered of and the solvent was removed at reduced pressure. The black residue was dissolved in warm CHCI3 (700 mL), heptane (600 mL) and 50 g of silica gel was added, the mixture was filtered through a pad of silica, washed with 50/50 CHC13/heptane and the solvent was again removed at reduced pressure. The black residue was chromatographed on a column of silica with petroleum ether/EtOAc 90/10 as eluent to give 14.9 g (15percent) of the target compound as a olive green solid. MS (ESI+) for CgH8BrNO m/z 226/228 (M+H)+.
1-(2,6-Dichlorophenyl)-3-dimethylaminopropenone[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With pyridinium p-toluenesulfonate; for 1.5h;Heating / reflux;
To a solution of 2,6-dichloroacetophenone (2 g) in dry dimethylformamide dimethyl acetal (10 ml) was added pyridinium 4-toluene sulfonate (0.2 g). The mixture was stirred under nitrogen and heated to reflux for 90 minutes. An azeotrope of dimethylformamide dimethylacetal/methanol was distilled under nitrogen to complete loss of 2,6-dichloroacetophenone by thin layer chromatography. The cold mixture was evaporated to give a solid. The solid was triturated with 10percent diethyl ether in light petroleum (b.p. 40-60°C). filtered and washed with the same to give the title compound. m.p. 98-100°C.
A solution of 1-(4-trifluoromethoxy-phenyl)-ethanone (20.4 g, 100 mmol) in dimethylformamide dimethylacetal (20 ml, 150 mmol) was heated to 100° C. for 24 h. The solvent was removed under reduced pressure and the crude product crystallized from ether/n-pentane to give 23.1 g (89 mmol, 89percent) of the title compound as yellow solid. MS: 260.1 (M+H)+.
(E)-3-dimethylamino-1-(4-trifluoromethoxy-phenyl)-propenone[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
at 100℃; for 23.5h;
C] (E)-3-Dimethylamino-1-(4-trifluoromethoxy-phenyl)-propenone (Following the procedure described in Gammill, R. B. A new and efficient synthesis of 3-halogenated 4H-1-benzopyran-4-ones. Synthesis (1979), (11), 901-3) A solution of 20.4 g (100.0 mmol) 1-(4-trifluoromethoxy-phenyl)-ethanone in 20 ml (150.0 mmol) of dimethylformamide dimethylacetal were heated at 100° C. for 23.5 h. The reaction was evaporated and precipitated from ether/n-pentane to give 23.1 g of the title compound as a yellow solid.
N'-(6-Dimethylaminomethylene-7-oxo-4,5, 6, 7-tetrahydro-benzothiazol-2-yl)-N, N-dimethyl- formamidine A mixture of 2-amino-5, 6-dihydro-4H-benzothiazol-7-one (0.5 g, 3 mmol) and N, N- dimethylformamide dimethyl acetal (1.96 mL, 1. 76 g, 14.8 mmol) in ETOH (2mL) was heated under microwave irradiation (300 W, 150 °C) for 30 min. The mixture was concentrated under vacuum before purification by flash column chromatography (50 percent EtOAc: hexane) to afford the title product as a tan solid (0.49 g, 59 percent)
1-(2-chloro-pyridin-3-yl)-3-dimethylamino-propenone[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
at 85℃; for 1.5h;
Step 1. Preparation of 1-(2-Chloro-pyridin-3-yl)-3- dimethylamino-propenone; 1- (2-Chloro-pyridin-3-yl)-ethanone ([Kuo, D. L. Tetrahedron, 48,42, 9233-9236](21.7 g, 139 mmol) in 46 mL N,N- dimethylformamide, dimethyl acetal (42 g, 350 mmol) was heated under a drying tube at 85 °C for 1.5 h and concentrated. The residue was purified by suction filtration chromatography (using 150 g silica in a Buchner funnel, with rapid collection of fractions eluting with 10: 1 and then 5:1 CH2Cl2/IpOH) to provide the title compound as a yellow solid. MS m/z = 211 [M+H]+. Calc'd for C10H11ClN2O: 210.66.
Preparation 32 : 4-Fluoro-2-nitrobenzaldehyde; To a solution of 2-nitro-4-fluorotoluene (5.0g, 32.2mmol) in DMF (5mL) was addedDMF.DMA (12.84mL, 97mmol) and the reaction heated to 1350C (bath temp) for 2Oh. The mixture was cooled to rt before being added to a stirring solution of sodium periodate (6.9g, 97mmol) in water/DMF (23mL: 12mL) via canula. The reaction was stirred at rt for 3h then filtered and the precipitate washed with toluene (20OmL). The filtrate was separated and the organics washed with water (3xl00mL) and dried (MgSO4) before removing the solvent in vacuo. Purification by column chromatography (SiO2, 9:1 hexane/EtOAc) afforded the title compound. deltaH(CDCl3): 10.36 (IH, s), 8.03 (IH, dd), 7.81 (IH, dd), 7.49 (IH, m).
a) 4-(Dimethylaminomethylene-thiocarbamoyl)-piperazine-l-carboxylic acid tert-butyl ester; A mixture of 122 mmol N,N-dirnethylformamide dimethyl acetal and 6.11 mmol 4- thiocarbamoyl-piperazine-1-carboxylic acid tert-butyl ester (prepared from tert-butyl 1- piperazinecarboxylate, lj'-thiocarbonyldiimidazole and ammonia according to the procedure of J. Med. Chem. 1998, 41, 5037-5054) was heated at 110 0C for 3 h. The reaction mixture was then concentrated in vacuo and the residue was resuspended in ethyl acetate/ tetrahydrofuran ( 1:1) and washed with brine. The organic phase was dried over sodium sulphate and concentrated in vacuo to afford the title compound as a light yellow crystalline solid (yield 95%). MS (m/e): 301.4 (M+H+, 100%).
A solution of l-(benzo[b]thiophen-2-yl)ethanone (1 g) in dimethoxy-N,N-dimethyl- methanamine (10 mL) was refluxed for 6 h and then cooled to rt. The precipitate was filtered and washed with diethyl ether to afford the title compound (l.lg, 84percent).
3-amino-6-benzyloxy-7-methoxynaphthalene-2-carbonitrile[ No CAS ]
N'-(7-benzyloxy-3-cyano-6-methoxynaphthalen-2-yl)-N,N-dimethylformamidine[ No CAS ]
[ 98446-49-2 ]
[ 4637-24-5 ]
(8-Benzyloxy-7-methoxybenzo[g]quinazolin-4-yl)-(2,4-dichloro-5-methoxy phenyl)amine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In acetic acid; toluene;
EXAMPLE 30 (8-Benzyloxy-7-methoxybenzo[g]quinazolin-4-yl)-(2,4-dichloro-5-methoxy phenyl)amine A mixture of 0.12 g (1.7 mmol) of 3-amino-6-benzyloxy-7-methoxynaphthalene-2-carbonitrile and 0.5 mL of N,N-dimethylformamide dimethyl acetal in 2.0 mL of toluene is heated under reflux for 4 hours. The solvent is evaporated and the residue is dried on high vacuum to yield N'-(7-benzyloxy-3-cyano-6-methoxynaphthalen-2-yl)-N,N-dimethylformamidine as an off-white solid. A mixture of N'-(7-benzyloxy-3-cyano-6-methoxynaphthalen-2-yl)-N,N-dimethylformamidine and 0.092 g (0.48 mmol) of <strong>[98446-49-2]2,4-dichloro-5-methoxyaniline</strong> in 4 mL of glacial acetic acid is heated under reflux for 7 hours, then cooled to room temperature and stirred overnight. The resulting solid is collected by filtration, washed with ether and dried to yield 0.05 g of (8-benzyloxy-7-methoxybenzo[g]quinazolin-4-yl)-(2,4-dichloro-5-methoxyphenyl)amine as a yellow solid, mp 275-280 C. 1H-NMR (d6-DMSO+trifluoroacetic acid): delta 9.25 (s, 1H); 8.95 (s, 1H); 8.28 (s, 1H); 7.86 (s, 2H); 7.59-7.39 (m, 7H); 5.37 (s, 2H); 4.04 (s, 3H); 3.90 (s, 3H).
A solution of <strong>[24078-21-5]3-methyl-4-nitro-benzoic acid methyl ester</strong> (24.99 g, 128.1 mmol) and N,N-dimethylformamide dimethyl acetal (40.0 mL, 300 mmol) was heated at 14O0C for 22.5 h. After cooling to rt, the reaction mixture was concentrated and the residue was crystallized from MeOH to give a purple solid. This solid was dissolved in THF (500 mL) and water (500 mL), and sodium periodate (62.62 g, 292.8 mmol) was added followed by additional sodium periodate (15.6 g, 72.9 mmol) two hours later. After stirring at rt for an additional 1 h, the reaction mixture was filtered through Celite washing with EtOAc (2 L). The filtrate was washed with saturated NaHCO3 (600 mL) and the organic layer was dried over Na2SO4. After filtration, the filtrate was concentrated and the residue was passed through a pad of silica gel, washing with CH2Cl2/hexanes (75%- 100%). The filtrate was concentrated and dried to give 3-formyl-4-nitro-benzoic acid methyl ester as yellowish solid. MS (EI): cal'd 210.0 (MH+), exp 210.2 (MH+).
at 140℃; for 22.5h;
Compounds from 6-carboxybenzothiophenes 3-Form) 4-nitro-benzoic acid methyl ester. A solution of <strong>[24078-21-5]3-methyl-4-nitro-benzoic acid methyl ester</strong> (24.99 g, 128.1 mmol) and N, N-dimethylformamide dimethyl acetal (40.0 mL, 300 mmol) was heated at 140 C for 22.5 h. After cooling to rt, the reaction mixture was concentrated and the residue was crystallized from MeOH to give a purple solid. This solid was dissolved in THF (500 mL) and water (500 mL), and sodium periodate (62.62 g, 292.8 mmol) was added followed by additional sodium periodate (15.6 g, 72.9 mmol) two hours later. After stirring at rt for an additional 1 h, the reaction mixture was filtered through Celite washing with EtOAc (2 L). The filtrate was washed with saturated NaHCO3, (600 mL) and the organic layer was dried over Na2SO4. After filtration, the filtrate was concentrated and the residue was passed through a pad of silica gel, washing with CH2CI2/hexanes (75%-100%). The filtrate was concentrated and dried to give 3-fonnyl-4-nitro- benzoic acid methyl ester as yellowish solid. MS (El) : cal'd 210.0 (MH+), exp 210.2 (MH+).
4-(1H-lmidazol-2-yl)piperidine dihydrochloride[ No CAS ]
[ 551-16-6 ]
[ 4637-24-5 ]
[2S-(2alpha,5alpha,6beta)]-6-[[[4-(1H-imidazol-2-yl)-1-piperidinyl]methylene]amino]-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid hydrochloride trihydrate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
26%
With sodium methylate; N-ethyl-N,N-diisopropylamine; In methanol; chloroform; water;
EXAMPLE 22 [2S-(2alpha,5alpha,6beta)]-6-[[[4-(1H-Imidazol-2-yl)-1-piperidinyl]methylene]amino]-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid hydrochloride trihydrate A solution of the dihydrochloride salt of 4-(1H-imidazol-2-yl)piperidine (0.448 g, 0.002 M) in methanol (15 ml) which contained sodium methoxide (0.004 M) was stirred at room temperature for about 5 minutes and dimethylformamide dimethyl acetal (10 ml) was added and the mixture was heated at 80° C. for 8 hrs. The excess of dimethylformamide dimethylacetal was removed to give 4-(1H-imidazol-2-yl)-1-piperidine carboxaldehyde dimethylacetal. This acetal was then dissolved in 6 ml of chloroform. To a mixture of <strong>[551-16-6]6-aminopenicillanic acid</strong> (0.388 g, 0.0018 M) and diisopropylethylamine (0.2 ml) in CHCl3 (20 ml) was added the chloroform solution of the above piperidine carboxaldehyde dimethylacetal at 0° C. The reaction was stirred at 0° C. for 1 hr and room temperature for 3 hrs. After this period, the solvent was removed and the residue dissolved in water (15 ml). The aqueous solution, washed with ethylacetate, was acidified to pH 3 with dil. HCl and purified on Sephadex LH 20 to give [2S-(2alpha,5alpha,6beta)]-6-[[[4-(1H-imidazol-2-yl)-1-piperidinyl]methylene]amino]-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid hydrochloride trihydrate (0.245 g, 26percent yield).
1,4,5,6-tetrahydro-2-[(4-piperidinyl)methyl]-pyrimidine[ No CAS ]
[ 551-16-6 ]
[ 4637-24-5 ]
[ 90747-87-8 ]
Yield
Reaction Conditions
Operation in experiment
7%
With N-ethyl-N,N-diisopropylamine; In methanol; dichloromethane; water;
EXAMPLE 47 [2S-(2alpha,5alpha,6beta)]-6-[[[4-[(1,4,5,6-tetrahydro-2-pyrimidinyl)methyl]-1-piperidinyl]methylene]amino]-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid hydrochloride A solution of 1,4,5,6-tetrahydro-2-[(4-piperidinyl)methyl]-pyrimidine (0.9 g, 0.005 M) in methanol (5 ml) and dimethylformamidedimethylacetal (15 ml) was heated at 90° C. for 8 hrs. The reaction was then stripped to dryness to yield 4-[(1,4,5,6-tetrahydro-2-pyrimidyl)methyl]-1-piperidinecarboxaldehyde dimethylacetal. The solution of the piperidine carboxaldehyde dimethylacetal in methylene chloride (5 ml) was added to a mixture of <strong>[551-16-6]6-aminopenicillanic acid</strong> (0.97 g, 0.0045 M) and diisopropylethylamine (0.76 ml) in methylene chloride (10 ml) at 0° C. The reaction was stirred at 0° C. for 1 hr and room temperature for 3 hrs. The solvent was removed and water (20 ml) was added. After being washed with ethyl acetate, the aqueous solution, acidified to pH 3 to about 3.5, was purified on Sephadex LH 20 columns to give [2S-(2alpha,5alpha,6beta)]-6-[[[ 4-[(1,4,5,6-tetrahydro-2-pyrimidinyl)methyl]-1-piperidinyl]methylene]amino]-3,3-dimethyl-7oxo-4-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid hydrochloride (0.14 g, 7percent yield).
base 4[2-(imidazol-1-yl)ethyl]piperidine[ No CAS ]
[ 551-16-6 ]
[ 4637-24-5 ]
[2S-(2alpha,5alpha,6beta)]-6-[[[4-[2-(1H-imidazol-1-yl)ethyl]-1-piperidinyl]methylene]amino]-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid hydrate hydrochloride[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With N-ethyl-N,N-diisopropylamine; In dichloromethane; water;
EXAMPLE 3 [2S-(2alpha,5alpha,6beta)]-6-[[[4-[2-(1H-Imidazol-1-yl)ethyl]-1-piperidinyl]methylene]amino]-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid hydrate hydrochloride The free base 4[2-(imidazol-1-yl)ethyl]piperidine (5.1 g, 28.7 mM) and dimethylformamide dimethyl acetal (23 ml) was heated at 100° C. for 6 hours. Removal of the excess of dimethylformamide dimethyl acetal gave 4-[2-(imidazol-1-yl)ethyl]-1-piperidine carboxaldehyde dimethylacetal (6.32 g, 87percent) which was dissolved in CH2 Cl2 (27 ml) and added to the mixture of <strong>[551-16-6]6-aminopenicillanic acid</strong> (5.6 g, 258 mM) and diisopropylethylamine (3.41 ml) in CH2 Cl2 (100 ml) at 0° C. The reaction was stirred at 0° C. for 1 hr. and room temperature for 3 hrs. The solvent was then removed and water (100 ml) was added. After being washed with ethylacetate, the aqueous solution, acidified to pH 3, was purified on Sephadex LH 20 columns to give [2S-(2alpha,5alpha,6-beta)]-6-[[[4 -[2-(1H-imidazol-1-yl)ethyl]-1-piperidinyl]methylene]amino]-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid hydrate hydrochloride (1.8 g, 16percent yield).
dihydrochloride salt of 4-[(4,5-dihydro-1H-imidazol-2-yl)methyl]piperidine[ No CAS ]
[ 551-16-6 ]
[ 4637-24-5 ]
[ 90747-45-8 ]
Yield
Reaction Conditions
Operation in experiment
62%
With sodium methylate; N-ethyl-N,N-diisopropylamine; In methanol; chloroform; water;
EXAMPLE 19 [2S-(2alpha,5alpha,6beta)]-6-[[[4-[(4,5-Dihydro-1H-Imidazol-2-yl)methyl]-1-piperidinyl]methylene]amino]-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid hydrochloride A mixture containing the dihydrochloride salt of 4-[(4,5-dihydro-1H-imidazol-2-yl)methyl]piperidine (8.5 g, 0.035 M) and methanol (160 ml) which contained sodium methoxide (0.071 M) was stirred at room temperature for 10 minutes and dimethylformamide dimethyl acetal (170 ml) was added and heated at 90° C. for 6 hrs. The excess of dimethylformamide dimethyl acetal was removed to give 4-[(4,5-dihydro-1H-imidazol-2-yl)methyl]piperidine carboxaldehyde dimethyl acetal. To the mixture of <strong>[551-16-6]6-aminopenicillanic acid</strong> (6.3 g, 0.029 M) and diisopropylethylamine (5.1 ml) in dried chloroform was added the solution (20 ml CHCl3 and 5 ml MeOH) of the above piperidine carboxaldehyde dimethylacetal at 0° C. The reaction was stirred at 0° C. for 1 hr and at room temperature for 3 hrs. After this period, the solvent was removed in vacuo and dissolved in 150 ml of water. The aqueous solution, washed with ethyl acetate was acidified to pH 3 with dilute hydrochloric acid and purified on Sephadex LH20 columns to give [2S-(2alpha,5alpha,6beta)]-6-[[[4-[(4,5-dihydro-1H-imidazol-2-yl)methyl]-1-piperidinyl]methylene]amino]-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid hydrochloride (8.1 g, 62percent yield).
dihydrochloride salt of 4-[2-(4,5,6,7-tetrahydro-1H-1,3-diazepin-2-yl)ethyl]piperidine[ No CAS ]
[ 551-16-6 ]
[ 4637-24-5 ]
[ 90747-54-9 ]
Yield
Reaction Conditions
Operation in experiment
38%
With sodium methylate; N-ethyl-N,N-diisopropylamine; In methanol; chloroform; water;
EXAMPLE 28 [2S-(2alpha,5alpha,6beta)]-6-[[[4-[2-(4,5,6,7-tetrahydro-1H-1,3-diazepin-2-yl)ethyl]-1-piperidinyl]methylene]amino]-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid hydrochloride The dihydrochloride salt of 4-[2-(4,5,6,7-tetrahydro-1H-1,3-diazepin-2-yl)ethyl]piperidine (1.6 g, 5.8 mM) in methanol (12 ml) which contained MeONa (12 mM) and dimethylformamide dimethyl acetal (20 ml) was heated at 90° C. for 8 hrs. The excess of dimethylformamide dimethylacetal was removed in vacuo to give the 4-[2-(4,5,6,7-tetrahydro-1H-1,3-diazepin-2-yl)ethyl] 1-piperidinecarboxaldehyde dimethylacetal. To the mixture of <strong>[551-16-6]6-aminopenicillanic acid</strong> (1.1 g, 5 mM) and diisopropylethylamine (0.96 ml) in 20 ml dry CHCl3 was added the solution (CHCl3 8 ml: MeOH 1 ml) of the above piperidine-carboxaldehyde dimethylacetal at 0° C. The reaction was stirred at 0° C. for 1 hr and then at room temperature for 3 hrs. The solvent was then removed, and water added (30 ml). The aqueous solution, washed with ethylacetate, was acidified to pH 3 with dilute aqueous HCl and purified on Sephadex LH 20 column to yield [2S-(2alpha,5alpha,6beta)]-6-[[[4-[2-(4,5,6,7-tetrahydro-1H-1,3-diazepin-2-yl)ethyl]-1-piperidinyl]methylene]amino]-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid hydrochloride (0.85 g, 38percent yield)
4-[3-(2-Nitro-1H-imidazol-1-yl)propyl]-1piperidinecarboxaldehyde[ No CAS ]
[ 551-16-6 ]
[ 4637-24-5 ]
[ 90747-40-3 ]
Yield
Reaction Conditions
Operation in experiment
26%
With hydrogenchloride; N-ethyl-N,N-diisopropylamine; In 1,4-dioxane; chloroform; water;
EXAMPLE 16 [2S-(2alpha,5alpha,6beta)]-6-[[[4-[3-(2-nitro- 1H-imidazol-1-yl)propyl]piperidinyl]methylene]amino]-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid hydrochloride 0.66 molar hydrate A solution of 4-[3-(2-nitro-1H-imidazol-1-yl)propyl]-1-piperidinecarboxaldehyde (3.25 g, 12.2 mM) in dioxane (90 ml) and 1 N HCl (90 ml) was heated at 100° C. for 4 hrs. After the reaction, the solvent was removed in vacuo, and the residue, basified with 1 N NaOH to pH 10, was extracted with methylenechloride (3*40 ml). The extracts, dried over Na2 SO4 were stripped to give an oil (2.62 g, 11 mM). Without further purification, the resulting crude product (2.62 g) and dimethyl formamide dimethylacetal (30 ml) was heated at 100° to about 110° C. for 10 hrs. The excess of dimethyl formamide dimethylacetal was removed at reduced pressure to give 4-[3-(2-nitro-1H-imidazol-1-yl)propyl]-1-piperidine carboxaldehyde dimethylacetal. This acetal in dry chloroform (30 ml) was added at 0° C. to a mixture of <strong>[551-16-6]6-aminopenicillanic acid</strong> (2.07 g, 9.8 mL) and diisopropylethylamine (1.69 ml). This reaction mixture was stirred at 0° C. for 1 hr. and then at room temperature for 3 hrs. After this period, the solvent was removed in vacuo, the residue was dissolved in water (50 ml). The resulting aqueous solution, washed with ethylacetate (2*20 ml), was acidified to pH 3 and purified on Sephadex LH 20 columns to give [2S-(2alpha,5alpha,6beta)]-6-[[[4-[3-(2-nitro-1H-imidazol-1-yl)propyl]piperidinyl]methylene]amino]-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid hydrochloride 0.66 molar hydrate (2.3 g, 4.6 mM, 26percent yield).
Compound (A): iV,N-dimethylformamide dimethyl acetal (2.75 g, 23 mmol) and pyrrolidine (1.64 g, 23 mmol) was added to a solution of nitronaphtalin (3.75 g, 20 mmol) in DMF (1 ImI). The solution was refluxed (1100C) for 3 hr under nitrogen gas and allowed to cool in room temperature. The volatile components were evaporated and the residue was dissolved in dichloromethane (6 mL) and methanol (48 mL). The solution was concentrated to a volume of 46 mL on a steam bath and cooled to 50C. Filtration and washing of the product with cold methanol afforded 4.2 g of dark red crystals. The mother liquor was evaporated and the residue was recrystallized from methanol to give an additional 0.6 g of red solid. Yield: 4.8 g (89 %). Data in agreement with the literature
Compound 137 (2.03 g) was dissolved in ethyl acetate (10 mL) and N,N-dimethylformamide (2 mL) and added dropwise with N,N-dimethylformamide dimethylacetal (1.47 mL) and the mixture was stirred at room temperature overnight. The reaction solution was added with hexane (20 mL) and allowed to stand still overnight. The precipitate was separated by filtration and vacuum-dried to obtain 2.37 g (yield 92%) of the target compound (Compound 138). Rf value (silica gel plate, hexane:ethyl acetate = 1:2): 0.29. 1H-NMR (400MHz, CDCl3) delta: 3.04 (3H, s), 3.21 (3H, s), 7.68 (1H t, J=8.1Hz), 8.18 (1H, s), 8.26 (1H, d, J=8.1Hz), 8.37 (1H, d, J=8.1Hz), 8.71 (1H, s). MS(ESI)m/z:258.1([M+H]+).
Intermediate 3;(2.pound.)-1-(4-bromo-2-hydroxyphenyl)-3-(dlmethylamlno)prop-2-en-1-one.; To a solution of i-(2-bromo-6-hydroxyphenyI)ethanone (35.8 g, 167 mmol) in dry benzene (800 mL) was added N.N-dimethylformamide dimethylacetal (44 mL, 333 mmol) and <n="23"/>the solution was heated to reflux for 4 h. The reaction mixture was then evaporated to dryness, dissolved in CHCI3 (300 rrL) and filtered through SiO2 (63-100 mum, 200 mL) to give after evaporation the title compound (31,9 g, 71percent) as a bright yellow solid. LC/MS data; 270.0 (M+Hf (Calculated for C11H12BrNO2 270.13). (calc. monoisotopic mass is 269.01, calc. monoisotopic mass (M+Hf = 270.01). 1H NMR data (DMSO-d6): 14.96 (s, 1H, OH), 7.93 (d, 1H, J=12.0 Hz, =CH), 7.87 (d, 1H, J = 9.0 Hz, Ar-H), 7.03 (d, 1H, J = 2.2 Hz, Ar-H)1 6,99 (dd, 1H, J1=S-O Hz7 J2=2.2 Hz, Ar-H), 5.94 (d, 1H, J=12.0 Hz, =CH), 3.21 (s, 3H, CH3), 3.00 (S, 3H1 CH3).
In n-heptane; dichloromethane; water; N,N-dimethyl-formamide;
A. Pyrazolo[1,5-a]pyridine-4-carboxylic acid ethyl ester (777A) To a clear solution of 2-methyl-nicotinic acid ethyl ester (50.6 g, 306 mmol) in anhydrous methylene chloride (200 mL) was added O-mesitylenesulfonylhydroxylamine (73 g, 337 mmol; prepared according to a literature procedure described in Krause; J. G., Synthesis 1972, 140); in portions at 0° C. The solution obtained was stirred at 0° C. for 15 min, and then at rt for 1 h. Concentration under reduced pressure gave a yellow solid. After the solid was dissolved in anhydrous DMF (300 mL), N,N-dimethylformamide dimethyl acetal (122 mL, 918 mmol) was added at 0° C. The mixture was stirred at 0° C. for 15 min, and then at 90° C. for 3 h. The reaction mixture was concentrated under reduced pressure, mixed with H2O (200 mL), and extracted with Et2O (3*180 mL). The combined organic solutions were washed sequentially with brine (50 mL), 1N aqueous HCl (50 mL), and brine (50 mL), and dried over Na2SO4. Filtration through a SiO2 column, which was then eluted with 30percent EtOAc in heptane, gave compound 777A (43.3 g, 74percent) as a yellow solid. HPLC: 80percent at 3.20 min (retention time) plus 20percent corresponding methyl ester at 2.83 min (retention time) (YMC S5 ODS-A column 4.6*50 mm eluding with 10-90percent aqueous methanol over 4 mincontaining 0.2percent phosphoric acid, 4 mL/min, monitoring at 220 nm). MS (ES): m/z 191 [M+H]+.
Example 35; This example describes the synthesis of Aldehyde 10: To a stirred solution of 9 (4.94 g, 22.3 mmol) and DMF (22.3 mL) was added DMF'DMA (8.18 g, 9.59 mL, 68.6 mmol). After heating at 135°C for 16 h, the dark red solution was cooled to 0°C and added to a rapidly stirred solution OfNaIO4 (14.7 g, 68.6 mmol) in H2O (46 mL) and DMF (23 mL) at 0°C. The reaction flask was washed with DMF (23 mL) at 0°C and added to NaIO4 mixture. The reaction was stirred at O0C for 4 h then allowed to warm to rt. After an additional 18 h, the orange solution was filtered over a pad of celite-.(R). and rinsed with EtOAc (200 mL) to remove precipitate. The filtrate was then washed with H2O (3 x 150 mL) and sat. aq. NaCl (3 x 150 mL). The dried extract (MgSO4) was concentrated in vacuo and purified by chromatography over silica gel, eluting with 40percent EtOAc / Hexanes to give the known aldehyde 10 (3.44 g, 14.8 mmol, 67percent). 1H NMR (400 MHz, CDCl3) delta 10.3 (s, IH), 8.04 (dd, J= 1.0, 8.2 Hz, IH), 7.95 (dd, J= 1.0, 8.0 Hz, IH), 7.55 (t, J= 8.0 Hz, IH); 13C NMR (100 MHz, CDCl3) delta 188.6, 148.4, 138.6, 132.9, 132.4, 123.4, 121.9.
a) (E)-4-Dimethylamino-1,1-dimethoxy-but-3-en-2-one A mixture of N,N-dimethylformamide dimethylacetal (86.0 g, 584 mmol) and methylglyoxal 1,1-dimethylacetal (85.6 g, 724 mmol) in isobutanol (500 mL) was heated at 100 C. overnight. The mixture was then cooled and evaporated. Purification by distillation afforded the title product (49.9 g, 48%) as an orange liquid. Bp 123-124 C. at 0.9 mbar. MS: m/e=174.4 [M+H]+.
4-bromoacetophenone (20.0 g; 0.10 mole) and N,N-dimethylformamide dimethylacetal (28.5 mL; 0.20 mole) were mixed together in DMF (12 mL) and heated to 1100C for 4 hours. The methanol and water that were generated during the reaction were distilled (6.2 mL). The mixture was cooled to 25C. Methyl t-butyl ether (100 mL) and methylhydrazine (21.2 mL; 0.40 moles) <n="70"/>were added and the mixture was stirred over night. The reaction mixture was washed with 1 M aqueous ammonium chloride (3 x 40 ml_) and water (40 ml_). The organic phase was dried by azeotropic distillation using a Dean-Stark apparatus. As an alternative to distillation, the solution was dried through an anhydrous magnesium sulfate cartridge. The solution was filtered through a silica gel cartridge (60 g). The product was flushed from the cartridge with methyl t-butyl ether. The fraction(s) containing product were combined and concentrated to about 70 ml_ by distillation. Heptane (120 ml_) was added and distillation was continued until the pot temperature reached 98.4 0C. About 100 ml_ of distillate was collected. The mixture was cooled to 40 0C. The mixture was seeded and the temperature was maintained at 40 0C for 30 minutes while crystallization was initiated. The mixture was slowly chilled to 0 0C over 90 minutes. The mixture was held at 0 0C for 30 minutes. The mixture was filtered and the solid was washed (3 x) with chilled (00C) heptane. The solid was dried on the filter. A cream-colored, crystalline solid (16.3 g; 68% yield) was obtained. The NMR data of the title compound are as per alternative 1
A N,N'-dimethylformamide (15 mL) solution of 4-bromoacetophenone (10.60 g, 53.25 mmols) and N,N'-dimethylformamide dimethyl acetal (2.5 equivalents) was heated at 125 degrees Celcius for 3 hours. The dark red solution was cooled to room temperature. The volatiles were removed by rotary evaporation providing a red viscous oil. To this substance was added anhydrous N,N'-dimethylformamide (15 mL) and methyl hydrazine (7.6 g, 160 mmols, 3 equivalents). The mixture was stirred at room temperature for 1 hour and then heated at 75 degrees Celcius for 4 hours. The volatiles were removed by rotary evaporation and the crude residue was taken up in a small volume of methylene chloride. This red solution was applied to a cartridge of silica gel. The cartridge was eluted with a 20:80 mixture of ethyl acetate and hexanes, respectively. The appropriate fractions were combined and concentrated to produce 12.5 g of a white solid. 1 H NMR (400 MHz, CHLOROFORM-c/) delta ppm 3.87 - 3.95 (m, J=2.22 Hz, 3 H) 6.29 - 6.36 (m, 1 H) 7.31 (dd, J=8.36 Hz, 2 H) 7.52 - 7.56 (m, 1 H) 7.62 (dd, J=2.05 Hz, 2 H).
General procedure for the synthesis of NiTo a stirred solution of 1-(4-bromophenyl)ethan-1-one (30.0 g, 150 mmol) in DMF (18 mL) was added DMFDMA (43.5 mL, 300 mmol), the resulting mixture was stirred at 110 C for 4 hours. After cooling to room temperature, MTBE (150 mL) was added to the mixture, then MeNHNH2 (78.9 g, 600 mmol) was added into above mixture, the resulting mixture was stirred at 25 C for 17 hours. The mixture was diluted with EtOAc (100 mL), filtered and the filter cake was washed with EtOAc (150 mL). The combined organic layer was washed with water (200 mLx3) and brine (200 mL), dried over anhydrous Na2SO4 and concentrated under reduced pressure to afford a residue. The residue was purified by flash column chromatography (eluted: PE/EtOAc=8/1 to 4/1) to afford Ni.
A mixture of l-fluoro-2-methyl-3-nitrobenzene and Lambda/,Lambda/-dimethylformamide dimethyl acetal (2.75 eq.) was heated to 135C for 12 h. The reaction mixture was cooled to RT and added dropwise to a solution Of NaIO4 (2.75 eq.) in water/DMF (1 :1, 1.1 M). After the addition is finished, the reaction mixture was stirred at RT for 3 h. Then, it was filtered, and the solid was washed with toluene. The filtrate was transferred to a separatory funnel, and the layers were separated. The organic layer was washed with water and dried (Na2SO4). Evaporation of the solvent under reduced pressure gave a crude that was purified by flash chromatography on silica using a gradient of EtO Ac/Petroleum ether (1 :30) to afford (23%) the title compound. MS (ES+) C7H4FNO3 required: 169, found: 170 (M+H)+.
1.3. Preparation ofl-(2-Bromo-5-fluorophenyl)-3-(dimethylamino)prop-2-en-l-one[0229] A solution of l-(2-bromo-5-fluorophenyl)ethanone (15.7 g, 72.2 mmol) in dimethylformamide dimethyl acetal (24.0 mL, 181 mmol) was heated to 8O0C for 3 h. The reaction mixture was allowed to cool to room temperature and was then cooled to about O0C. Water (100 mL) was slowly added to the reaction mixture while the internal temperature was maintained below about 2O0C. The resulting biphasic mixture was further diluted with MTBE and water. The aqueous phase was extracted with MTBE, and the combined organic phases were washed with water and brine, dried over Na2SO4, and concentrated in vacuo to give l-(2-bromo-5-fluorophenyl)-3-(dimethylamino)prop-2-en- 1-one as a dark orange solid (19.6 g, 100%). This material was used without further purification in the next reaction step. 1H-NMR (500 MHz, CDCl3) delta 7.51 (d, J = 8.8, 4.9 Hz, IH), 7.02-7.12 (m, 2H), 6.93 (dt, J = 8.2, 3.0 Hz, IH), 5.28 (d, J = 12.5 Hz, IH), 3.12 (br s, 3H), 2.89 (s, 3H). Alternatively, a standard aqueous workup was employed to isolate the title compound as a brown oil, which crystallized upon standing (250 g, 96% yield, 97% purity by HPLC).
Step A: A solution of <strong>[870089-49-9]tert-butyl 3-acetylazetidine-1-carboxylate</strong> (Preparation 85, 20.6 g, 0.103 mol) in N-N-dimethylformamide dimethyl acetal was refluxed for 45 hours. The reaction mixture was evaporated and then co-evaporated with toluene (40OmL) to leave a residue that was used in step B without further purification.
Stage A.2: N'-{6-[1-Dimethylamino-meth-(E)-ylidene]-7-oxo-4,5,6,7-tetrahydro-benzothiazol-2-yl}-N,N-dimethyl-formamidineA suspension of <strong>[17583-10-7]2-amino-5,6-dihydro-4H-benzothiazol-7-one</strong> (3.5 g, 20.81 mmol) in dimethoxymethyldimethylamine (12 mL, 90 mmol) was heated at 100° C. with stirring for 65 h. The RM was then evaporated to dryness in vacuo and the residue was suspended in EtOAc. After 1 h at 4° C., the solid was filtered off, washed with EtOAc and then dried under high vacuum at 60° C. to give the pure title product as brown crystals. LC: tR 3.25 min (method D). MS: M+H=279. 1H-NMR in DMSO-d6: 8.40 (s, 1H); 7.23 (s, 1H); 3.15 (s, 3H); 3.05 (s, 6H); 2.97 (s, 3H); 2.91 (t, 2H); 2.67 (t, 2H).
In N,N-dimethyl-formamide; at 80 - 140℃; for 0.916667h;
In a 1000 mL two-necked flask, 119 g of compound 1-1 and 400 mL were addedN, N-dimethylformamide,Oil bath 80 heating and stirring 25min after adding 160mLN, N-dimethylformamide dimethyl acetal,After heating to 140 C for 30 min,Ice bath cooling,Precipitation of solid, filter, filter cake with ethyl acetate,Ether washed and dried to give the target compound 1-2.
2-(pyridin-2-yl)-6,7-dihydro-2H-pyrazolo[4,3-c]pyridin-4(5H)-one[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
22%
To a solution of <strong>[50607-30-2]piperidine-2,4-dione</strong> (2.00 g, 17.7 mmol) and 2-hydrazinylpyridine (1.93 g, 17.7 mmol) in water (19.7 mL) and ethanol (157 mL). The reaction was stirred at room temperature overnight. The reaction mixture was concentrated under reduced pressure. The residue was dried azeotropically with ethanol/toluene to give a yellow powder. Dimethylformamide (7.07 mL) was added followed by 1,1-dimethoxy-N,N-dimethylmethanamine (7.05 mL, 53.0 mmol), and the reaction was heated at 90° C. for 60 minutes. The reaction was cooled to room temperature and concentrated under reduced pressure. The residue was dissolved in methanol, concentrated onto Celite®, and purified by flash column chromatography using 1-15percent methanol/dichloromethane as eluent to give Compound CE1 as a yellow solid (0.870 g, 22percent).
To a solution of <strong>[50607-30-2]piperidine-2,4-dione</strong> (2.00 g, 17.7 mmol) in dimethylformamide (70.7 mL) was added 1,1-dimethoxy-N,N-dimethylmethanamine (2.47 mL, 18.6 mmol). The reaction was stirred at 90 C. for 4 hours, then evaporated to dryness under reduced pressure to give Compound C1 as a brown oil (3.17 g, 85%): 1H NMR (300 MHz, DMSO-d6) delta 7.94 (s, 1H), 3.24 (s, 2H), 3.13 (s, 2H), 2.87 (s, 3H), 2.71 (s, 3H).
(E)-N'-(4-chloro-5-nitropyridin-2-yl)-N,N-dimethylformamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
68%
In methanol; at 75℃; for 20h;
General procedure: The compounds were prepared as described in the literature with minor modifications. To a solution of the corresponding 4-chloro-nitropyridin-2-amine (10 or 11, 1.0 mmol) in methanol (10 mL per 1.0 mmol starting material) and dimethylformamide(1.0 mL) was added dimethylformamide dimethyl acetal (DMFDMA, 1.0 mL) and heated to 75 C. After a reaction time of 16-20 h,the solvent was evaporated and the residue was stirred under water-petroleum ether (50percent, 10 mL) until full crystallization. The product was filtered under reduced pressure, washed with petroleum ether (310 mL), and dried at 70 C to afford amidine 16 or 17.
(Z)-3-((dimethylamino)methylene)piperidine-2,4-dione[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
77.4%
In toluene; at 20℃; for 12h;
To a solution of <strong>[50607-30-2]piperidine-2,4-dione</strong> (1.13 g, 10.0 mmol) in toluene (20 mL) was added N, N-dimethylformamide dimethyl acetal (2.24 g, 18.8 mmol) at room temperature Stir for 12 hours.The solvent was evaporated under reduced pressure and the crude product was purified by column chromatography (methanol / ethyl acetate (v / v) = 1/10) to give a yellow solid (1.30 g, 77.4percent).
(E)-1-(2-bromo-5-fluorophenyl)-3-dimethylaminopropenone[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
95%
at 110℃; for 5h;Inert atmosphere;
General procedure: Dimethylformamide dimethylacetal (7 vol) was added under nitrogen atmosphere to corresponding substituted acetophenones (1a-c) (1g) at ambient temperature. The reaction medium was heated to 110 °C and stirred for 6?8 h. Completion of the reaction was monitored by TLC. After completion of the reaction, volatiles were removed under reduced pressure. The residue obtained was dissolved in ethyl acetate(2 vol) and hexane (8 vol) was added drop wise to it at room temperature. The resultant reaction mixture was stirred for 1h at the same temperature. The solid formed was filtered,washed with hexane and dried under vacuum. (E)-1-(2-Bromo-5-fluoro-phenyl)-3-dimethylaminopropenone(2a) This compound was prepared from 2-bromo-5-fluoro acetophenone(1a) (5g, 23.04 mmol) with dimethylformamidedimethyl acetal (35 mL). It was obtained as pale yellow solid (5.9 g, 95percent). MP: 107.8?108.6 °C. IR (ATR, cm?1) upsilon:585.78 (C-Br), 1022.41 (C-F), 1068.13 (C-O), 1164.81 (CN N), 1424.39 (C-C), 1456.96 (C-C), 1485.03 (C-C), 1547.82(C-C), 1639.92 (C=O), 3055.26 (C-H). 1H NMR (DMSOd6,300 MHz) delta (ppm): 2.84 (3H, s, ?N?CH3), 3.31 (3H, s,?N?CH3), 5.16 (1H, d, J = 12.6 Hz, =C?H?CO), 7.15?7.17(2H, m, Ar?H), 7.21 (1H, d, J = 13.0 Hz, =CH?N?(CH3)2), 7.64 (1H, dt, J1 = 13.8 Hz, J2 = 5.1 Hz, Ar?H). 13C NMR(DMSO-d6, 100MHz) delta (ppm): 37.48 (?N?CH3), 45.01(?N?CH3), 95.1 (?ene?CH), 113.79 & 113.86 (d, 3J C?F =7.8 Hz , ArC?Br), 115.84 & 116.07 (d, 2J C-F = 23.2 Hz,Ar-C), 117.43 & 117.66 (d, 2J C?F = 22.4 Hz, Ar?C),134.99 & 135.07 (d, 3J C-F = 8 Hz, Ar?C), 146.6 (Ar?C),154.8 (?ene?CH), 160.35 & 162.80 (d, 1J C-F = 244.8 Hz,ArC?F), 187.8 (?C=O). LC-MS (ESI, m/z): 274.42[M+2 H]
4-((dimethylamino)methylene)dihydrofuran-3(2H)-one[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In 1,4-dioxane; at 110℃; for 2h;
Step 1 - 4-((Dimethylamino)methylene)<strong>[22929-52-8]dihydrofuran-3(2H)-one</strong> (0554) [00355] To a solution of 1,1-dimethoxy-N,N-dimethylmethanamine (48.4 g, 407 mmol) in dioxane (150 mL) was added <strong>[22929-52-8]tetrahydrofuran-3-one</strong> (7.00 g, 81.3 mmol). The reaction mixture was stirred at 110 °C for 2 hrs. On completion, the mixture was concentrated in vacuo. The residue was triturated with petroleum ether to give the title compound. 1H NMR (400 MHz, DMSO-d6) delta = 7.15 (s, 1H), 5.00 (s, 2H), 3.86 (s, 2H), 3.01 (s, 6H).
N-((dimethylamino)methylene)-3,5-bis(trifluoromethyl)benzamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In N,N-dimethyl-formamide; at 20 - 30℃;
Charged 3,5-bis(trifluoromethyl)benzonitrile (100 gm, 1 meq.), 2 (N) Sodium hydroxide (21.7 gm, 1.3 meq.) into 400 ml methanol and stirred the reaction mass at 20 - 30°C for 2-3 hrs. Added Dimethyl sulphoxide (100 ml) followed by dilute 30percent H202 solution (100 gm diluted in 300m1 water, 2.1 eq.) at 20 -35°C. Stirred the mass for 2-3 hrs. at 20-30°C and confirmed reaction completion by HPLC. Reaction mass quenched by addition of water (500 ml, S Vol). Filtered the slurry and washed the wet cake by water (500 ml, SVol). Dried under vacuum at 50-60°C to get 100 gm 3, 5-bis (trifluoromethyl) benzamide (Purity 99 percent). Charged 3, 5-bis (trifluoromethyl) benzamide, prepared according to procedures a or b, as described above, (100 gm, 1 meq) into N, N Dimethyl formamide (100 ml, 1 vol). Added N, N Dimethyl formamide dimethyl acetal (70 gm, 1.5 meq) at 20-30°C and stirred the reaction mass for 2-3 hrs. Reaction completion for the formation N-((dimethylamino)methylene)-3,5- bis(trifluoromethyl)benzamide was confirmed by HPLC. Cooled reaction mass to 10-15°C and added acetic acid (S00 ml, S vol.) followed by hydrazine hydrate (31 gm, 1.5 meq). Reaction mass was heated to S0-SS°C and stirred for 2-3 hrs. at same temparature. Reaction was monitored by HPLC. Cooled reaction mass to 20-30°C and quenched by addition of water (2.S L, 2S vol.). Filtered the slurry and washed the wet cake by water (S00 ml, S vol.). Dried under vacuum at 50-60°C to get 3 -(3,5 -bis(trifluoromethyl)phenyl)- 1 H-i ,2,4-triazole (94 gr, Purity 97percent).
5-amino-2-(1-cyclopropyl-1H-pyrazol-4-yl)-N-[(dimethylamino)methylidene]benzenesulfonamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
2-Chloro-5-nitrobenzenesulfonamide (674 mg, 2.85 mmol) and 1 -cyclopropyl-4-(4, 4,5,5- tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 /-/-pyrazole (1 .00 g, 4.27 mmol) were dissolved in n- propanol (34 ml) and bis(triphenylphosphine)palladium(ll) dichloride (CAS 13965-03-2) (100 mg, 142 muetaetaomicronIota) and triphenylphosphine (37.3 mg, 142 muetaetaomicronIota) were added. The reaction was purged with argon for 5 minutes and aq. potassium carbonate (5.7 ml, 1.0 M, 5.7 mmol) was added. The reaction was heated at 100°C for 3h. Afterwards the mixture was filtered over Celite and the solvent was removed under reduced pressure. Ethyl acetate and water were added. The phases were separated and the aqueous phase was extracted with ethyl acetate. The combined organic phases were dried over Whatmanfilter and the solvent was removed under reduced pressure. The crude was used in the next step without further purification. (0458) 2-(1 -Cyclopropyl-1 /-/-pyrazol-4-yl)-5-nitrobenzenesulfonamide (1.17 g, 3.79 mmol) and 1 ,1 -dimethoxy-/V,/V-dimethylmethanamine (1 .0 ml, 7.6 mmol) were dissolved in DMF (25 ml) and the reaction was stirred at room temperature until completion of the reaction. The solvent was removed under reduced pressure and the crude was used without further purification in the next step. (0459) 2-(1 -Cyclopropyl-1 /-/-pyrazol-4-yl)-/V-[(dimethylamino)methylidene]-5- nitrobenzenesulfonamide (1.84 g, 5.06 mmol) was dissolved in THF (30 ml) and the flask was flushed with nitrogen. Palladium on charcoal (10percent loading, 53.9 g, 506 muetaetaomicronIota) was added and the flask was evacuated and subsequently flushed with hydrogen (1 bar). Stirring was continued at room temperature until completion of the reaction. The reaction mixture was filtered over Celite and the solvent was removed under reduced pressure. The crude was used without further purification in the next step (1.3 g, 53percent purity, 75percent yield over 3 steps). (0460) LC-MS (Method A): Rt = 0.70 min; MS (ESIpos): m/z = 334 [M+H]+






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +
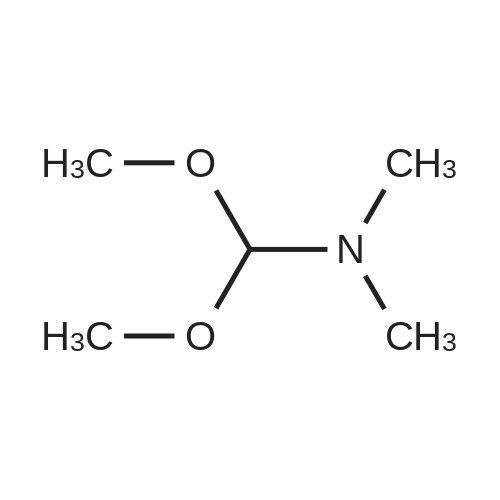

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping