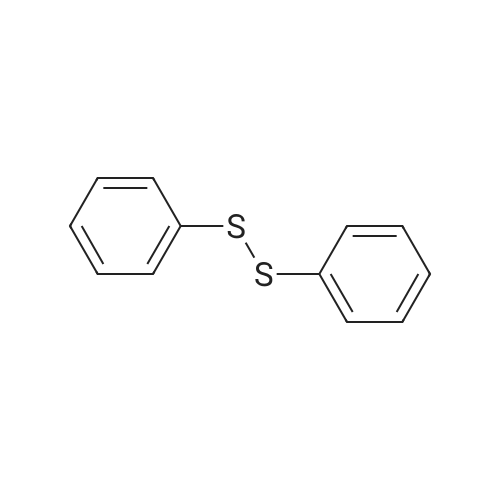
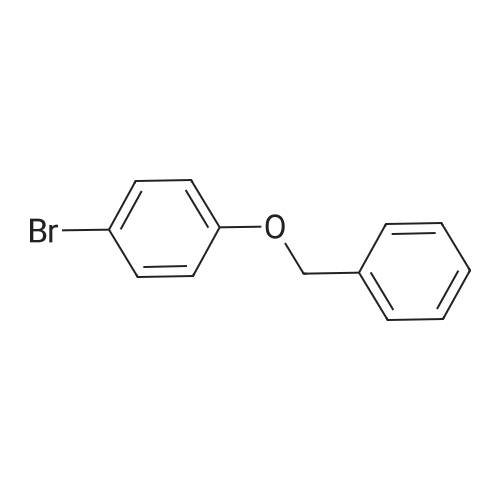
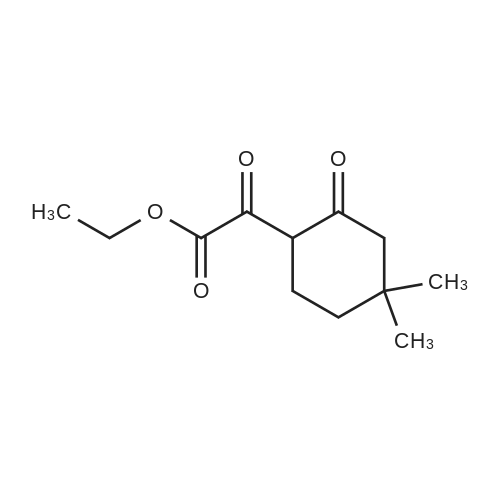
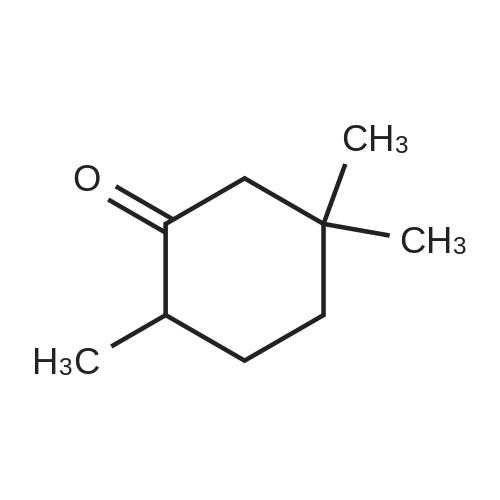
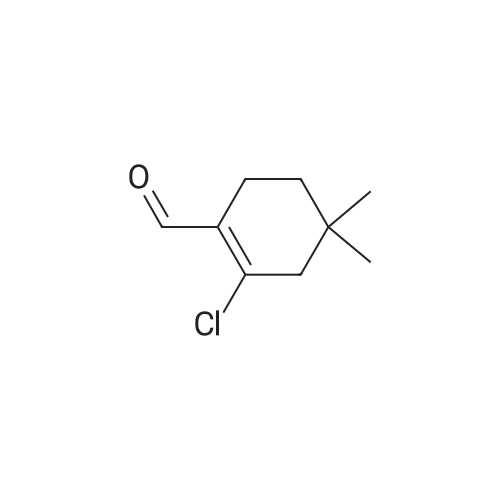
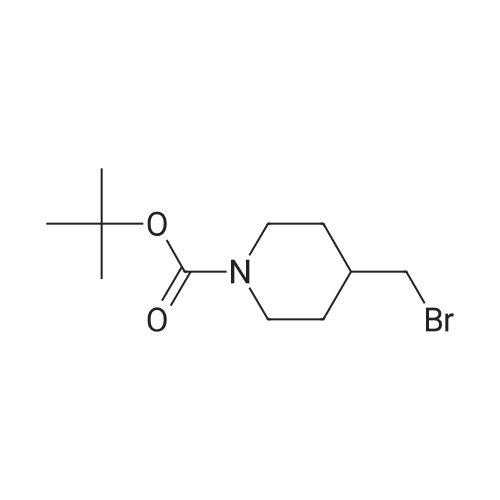
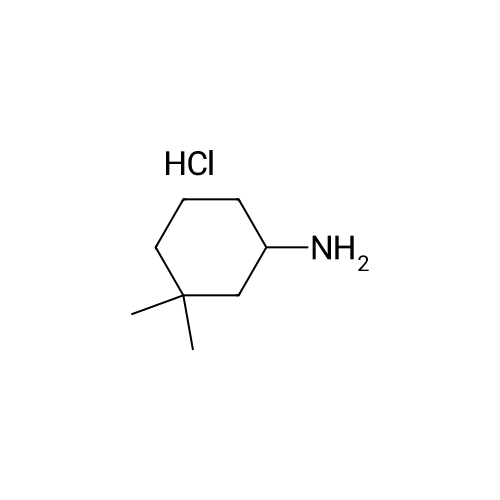
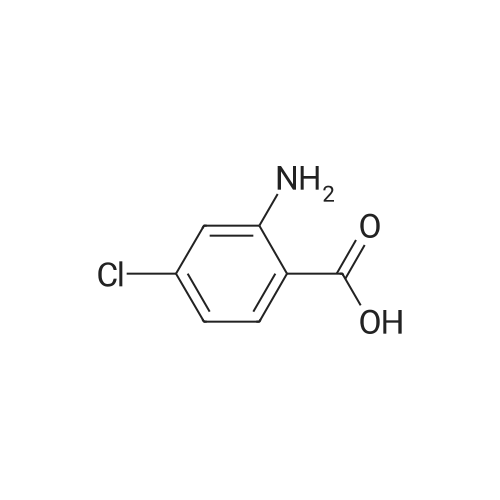
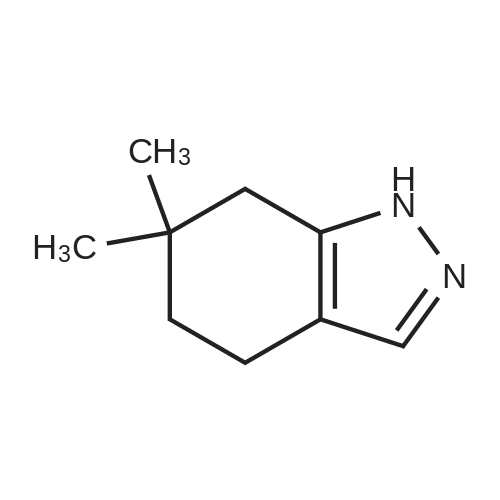
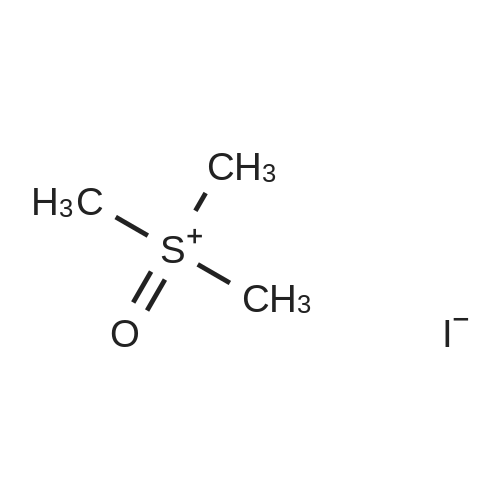
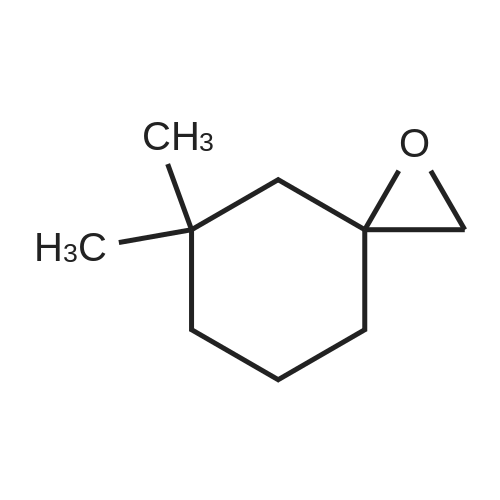
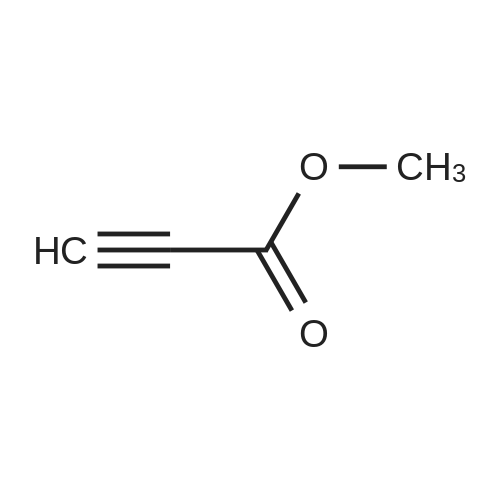
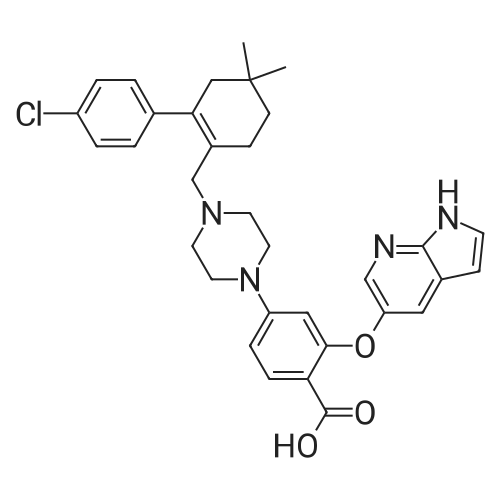
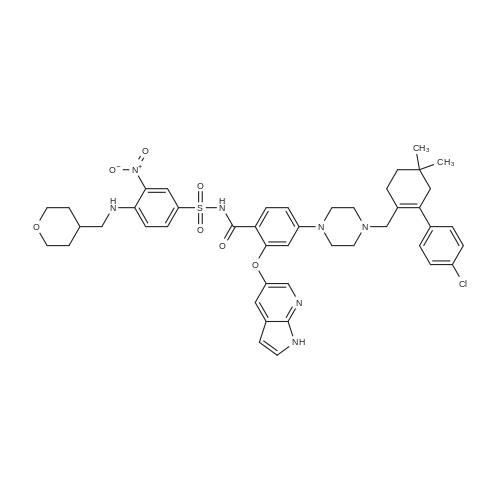
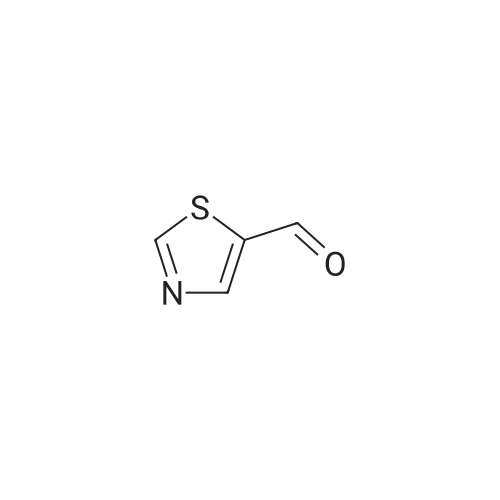
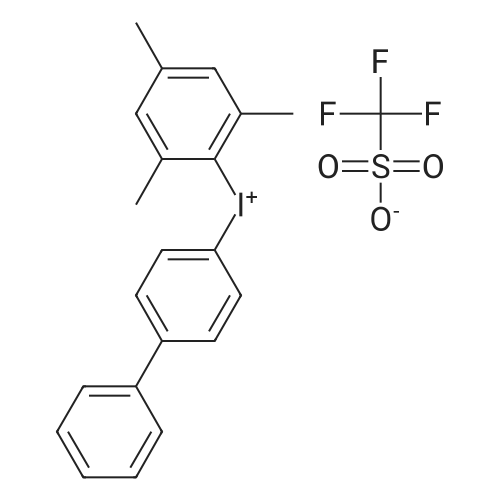
| 92% |
Stage #1: carbonic acid dimethyl ester With sodium hydride In tetrahydrofuran Reflux; Inert atmosphere;
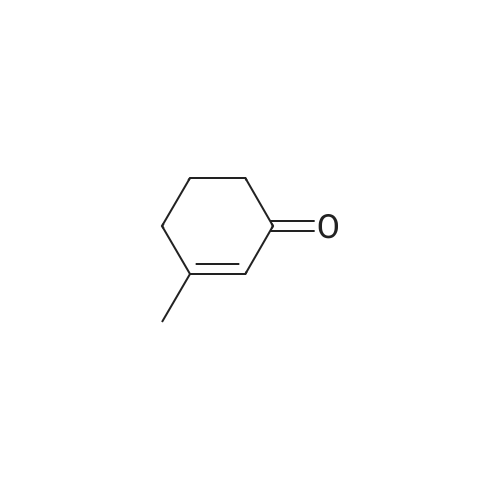
Stage #2: 3,3-dimethylcyclohexan-1-one In tetrahydrofuran for 4h; Reflux; |
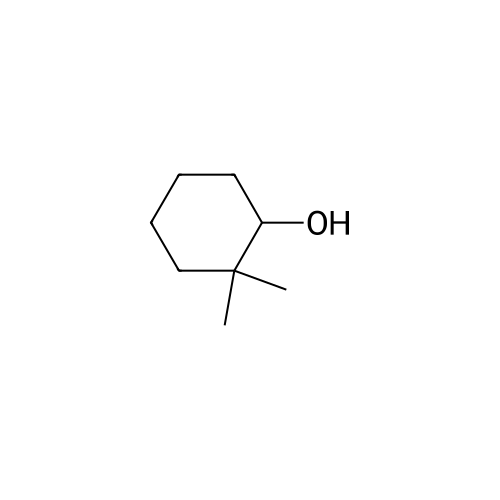
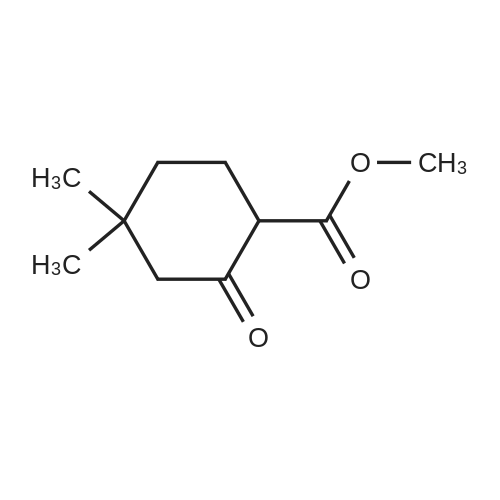
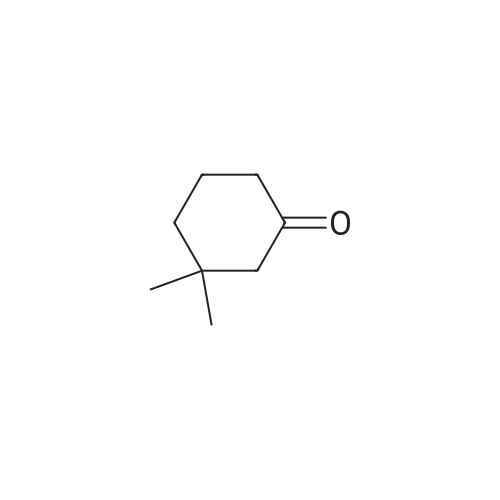
1.8 Step 8: 4,4-Dimethyl-2-oxocyclohexanecarboxylic acidMethyl ester
To 15 mL of anhydrous THF was added sodium hydride (1.25 g, 31.3 mmol, 2.0 eq) andDimethyl carbonate (6.5 g,72.0 mmol, 4.6 eq), refluxed under nitrogen, and 8 mL of 3,3-dimethylcyclohexanone (2.0 g,16.0 mmol, 1.0 eq) in THF. The reaction was refluxed for 4 h. To the reaction solution was added 5 mL of methanol, followed by addition of 40 mL of water, DCM (30 mL x 3). The organic phase was dried over anhydrous sodium sulfate, spin-dried, PE / EA (v / v) = 100 / Color liquid product 2.8g, yield 92.0%. |
| 92% |
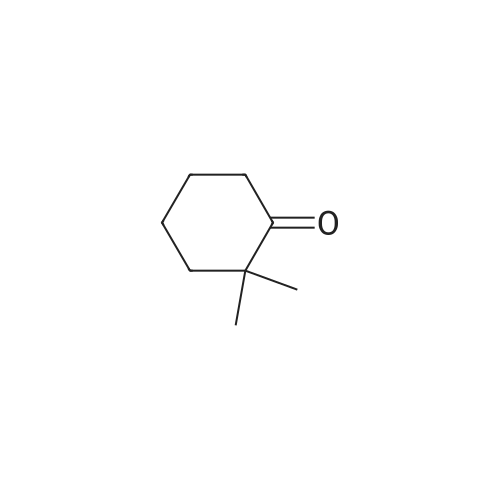
Stage #1: 3,3-dimethylcyclohexan-1-one With lithium dipropan-2-ylazanide In tetrahydrofuran at -20 - 0℃; for 1h;
Stage #2: carbonic acid dimethyl ester In tetrahydrofuran at -20 - 0℃; for 2h; |
1.1; 2.1 Step 1: Synthesis of methyl 4,4-dimethyl-2-cyclohexanonecarboxylate
General procedure: In the reaction flask, dissolve 100g (0.79mol) of 3,3-dimethylcyclohexanone in 500mL tetrahydrofuran, cool to -20°C to -30°C, and add 1mol/L LDA (lithium diisopropylamide) dropwise. ) 870 mL (0.87 mol) of tetrahydrofuran solution, heated to 0°C, stirred for 1 hour, cooled to -20°C, added dropwise 93 g (1.03 mol) of dimethyl carbonate, raised to 0°C, reacted for 2 hours, TLC monitored the reaction to complete , Add 1M dilute hydrochloric acid to adjust PH=6-7, separate the liquids, extract the aqueous phase twice with ethyl acetate, combine the organic phases, dry with sodium sulfate, and concentrate under reduced pressure.get134.3g of methyl 4,4-dimethyl-2-cyclohexanonecarboxylate,The yield was 92% |
| 92% |
With sodium hydride In tetrahydrofuran for 4h; Inert atmosphere; Reflux; |
2.2 The second step is to prepare 4,4-dimethyl-2-oxocyclohexanecarboxylic acid methyl ester
Add sodium hydride (1.25g, 31.3mmol, 2.0eq) and dimethyl carbonate (6.5g, 72.0mmol, 4.6eq) to 15mL of anhydrous THF, reflux under nitrogen protection, and add 8mL 3,3-bis of methylcyclohexanone (2.0 g, 16.0 mmol, 1.0 eq) in THF solution . The reflux reaction was completed for 4h. Add 5mL methanol to the reaction solution, then add 40mL water, extract with DCM (30mL*3), dry the organic phase with anhydrous sodium sulfate, spin dry through the column, PE/EA(v/v)=100/1, get nothing The color liquid product is 2.8g, and the yield is 92.0%. |
| 85% |
Stage #1: carbonic acid dimethyl ester With sodium hydride In tetrahydrofuran at 80℃; for 0.5h; Inert atmosphere;
Stage #2: 3,3-dimethylcyclohexan-1-one In tetrahydrofuran for 2.5h; Inert atmosphere; |
Methyl 4,4-dimethyl-2-oxocyclohexane-1-carboxylate (9)
A solution of dimethyl carbonate (3.3 mL, 0.039 mol) and NaH (1.24 g, 0.052 mol) in THF (24 mL) was heated to 80 °C for 30 min.Then 3,3-dimethylcycloheaxanone (2.0 g, 0.016 mol) was added and stirred for 2.5 h under nitrogen atmosphere. After reaction completion, the reaction mass was cooled to about 0 °C, quenched with methanol (10 mL), followed by water (25 mL), and the resultant mixture was acidified to about pH 1 with 3 M HCl. The compound was extracted with dichloromethane, dried over sodium sulfate and concentrated to afford compound 9 as:15 Pale yellow liquid; yield 85%; 1H NMR (400 MHz, DMSO-d6): δ 0.91 (s, 6H), 1.32-1.36 (t, 2H, J1 = 6.3 Hz, J2 = 6 Hz), 2.03 (s, 2H), 2.17-2.19 (t, 2H, J1 = 6.3 Hz, J2 = 6 Hz), 3.71 (s, 3H), 12.09 (s, 1H, enol-OH); IR (ATR) (υ cm-1): 821, 1065, 1231, 1441, 1617, 1657, 1712, 1746, 2922, 2952; LCMS (ESI) m/z for C10H16O3: 184.9 Da ([M + H]+). |
| 85% |
With sodium hydride In tetrahydrofuran for 2.5h; Inert atmosphere; |
Methyl 4,4-dimethyl-2-oxocyclohexane-1-carboxylate (4)
Dimethyl carbonate (3.3 mL, 0.039 mol) and NaH (1.24 g, 0.052 mol) in THF (24 mL) were heated to about 80 °C for 30 min. Then, 3,3-dimethylcyclohexanone (2.0 g, 0.016 mol) was added and stirred for 2.5 h under nitrogen atmosphere. After reaction completion by TLC (10% methanol in chloroform), the reaction mass was cooled to about 0 °C and methanol followed by water was added. Then, the resultant reaction mixture was acidified to pH 1 using 3 M HCl and the product was extracted with dichloromethane, dried over sodium sulfate and concentrated under reduced pressure to afford 4. Yield: 85%, pale yellow liquid. 1H NMR (400 MHz, DMSO-d6, δ ppm): 0.91 (s, 6H), 1.32-1.36 (t, 2H, J1 = 6.3 Hz, J2 = 6 Hz), 2.03 (s, 2H), 2.17-2.19 (t, 2H, J1 = 6.3 Hz, J2 = 6 Hz), 3.71 (s, 3H), 12.09 (s, 1H, enol-OH); IR (ATR, ν cm-1): 821, 1065, 1231, 1441, 1617, 1657, 1712, 1746, 2922, 2952; LCMS (ESI) m/z for C10H16O3 [M + H]+: 184.9 Da. |
| 81% |
With sodium hydride In tetrahydrofuran for 6h; Heating; |
|
| 81% |
With sodium hydride In tetrahydrofuran for 2h; Heating / reflux; |
2
Sodium hydride (60%, 1.95g, 81.2 mmol) is suspended in THF (120 ml), dimethyl carbonate (17 ml, 198.0 mmol) is added and the mixture is heated to reflux. 3,3- Dimethylcyclohexanone (5.Og, 39.6 mmol, see Preparation 1 ) in THF (60 ml) is dropwise added and this mixture is refluxed for 2h. Once at room temperature, the reaction mixture is poured on saturated solution of ammonium chloride (125 ml). After successive extractions with ethyl ether, the organic phase is washed with water and brine, dried over magnesium sulfate, filtered and the solvent evaporated under reduced pressure. 5.94g of the final compound are obtained as an oil, pure enough to perform the next synthetic step. Yield= 81%.1H NMR (300 MHz, CHLOROFORM-D) δ ppm 1.0 (s, 6 H) 1.4 (t, J=6.6 Hz, 2 H) 2.1 (s, 2 H) 2.2 (m, 3 H) 3.8 (s, 3 H) |
| 80% |
With sodium hydride In tetrahydrofuran for 2h; Reflux; |
|
| 70% |
With sodium hydride In tetrahydrofuran for 4h; Inert atmosphere; Reflux; |
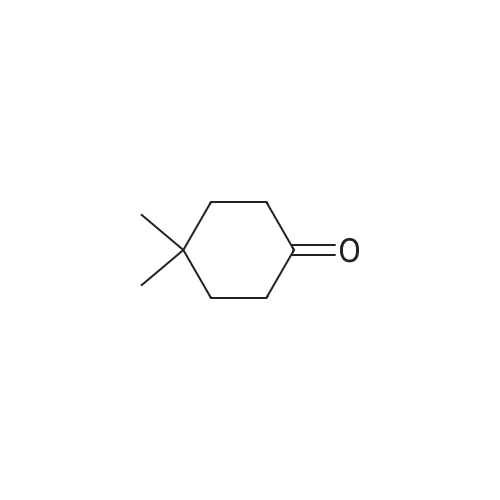
1.1 Step 1 Synthesis of Compound 5.
Sodium hydride (1.9 g, 79.3 mmol) and dimethyl carbonate (14.3 g, 158.6 mmol) were added to a solution of anhydrous THF (15 mL). 3,3-Dimethylcyclohexanone (5.0 g, 39.6 mmol) in THF was added dropwise at reflux, and after the addition was completed,Continue to reflux for 4 h. Cool to room temperature and add methanol to quench the reaction.Add water and dichloromethane to extract and collect the organic phase.Separated and purified by column chromatography to obtain 4.2 g of a colorless liquid product.The yield was 70%. |
| 65% |
With potasssium hydride; sodium hydride In tetrahydrofuran Heating; |
|
|
With sodium hydride |
|
|
With sodium hydride In benzene for 3h; Heating; Yield given; |
|
|
With sodium hydride In tetrahydrofuran for 6h; Reflux; |
A solution of dimethyl carbonate (7.01 mL, 83 mmol) and NaH (2.61 g of 95%, 103 mmol) in 35 THF was heated to reflux. A solution of 3,3-dimethylcyclohexanone (4.2 g, 33.3 mmol) (House, J. Org. Chem. 1968, 33, 949-956) in 15 mL THF was added via cannula (2 mL rinse), and the reaction was refluxed for a further 6 h. Cooled to 0° C., added MeOH dropwise until fizzing stopped, then added H2O very cautiously. Added CH2Cl2, then acidified with 3M HCl until pH of aqueous layer was 1 while stirring vigorously. Separated layers, washed aq. with CH2Cl2 (3×), dried combined organics over Na2SO4, filtered and concentrated to afford desired product primarily as the keto tautomer, which was used without further purification. 1H NMR (400 MHz, CDCl3) δ 3.74 (s, 3H), 3.73 (m, 3H), 2.24 (m, 2H), 2.04 (s, 2H), 1.37 (t, J=6.4 Hz, 2H), 0.94 (s, 6H). |
|
With sodium hydride In tetrahydrofuran Reflux; |
3
To a mixture of sodium hydride (7.7g, 0.19mol) in THF (200 ml), dimethylcarbonate (39.5 ml, 0.47mol) is added and heated to reflux. 3,3-Dimethylcyclohexanone (11.8g, 0.09mol, see Preparation 2) in THF (100 ml) is dropwise added and heated with stirring for 2h. After cooling until room temperature, the reaction mixture is poured on a saturated solution of ammonium chloride (300 ml) and extracted with ether. After usual work-up, 17.9g of the final compound are obtained as an oil.1H NMR (200 MHz, CHLOROFORM-d) δ ppm 0.87 - 1.00 (m, 6 H) 1.33 - 1.47 (m, 2 H) 1.57 - 1.75 (m, 1 H) 2.04 (s, 2 H) 2.15 - 2.30 (m, 2 H) 3.72 - 3.78 (m, 3 H) |
|
With sodium hydride In tetrahydrofuran Reflux; |
|
|
With sodium hydride In tetrahydrofuran Reflux; |
|
|
With sodium hydride In tetrahydrofuran for 3h; Reflux; Inert atmosphere; |
|






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping